Abstract
The up‐regulation of transcobalamins [hitherto posited as indicating a central need for cobalamin (Cbl) in inflammation], whose expression, like inducible nitric oxide synthase (iNOS), is Sp1‐ and interferon‐dependent, together with increased intracellular formation of glutathionylcobalamin (GSCbl), adenosylcobalamin (AdoCbl), methylcobalamin (MeCbl), may be essential for the timely promotion and later selective inhibition of iNOS and concordant regulation of endothelial and neuronal NOS (eNOS/nNOS.) Cbl may ensure controlled high output of nitric oxide (NO) and its safe deployment, because: (1) Cbl is ultimately responsible for the synthesis or availability of the NOS substrates and cofactors heme, arginine, BH4 flavin adenine dinucleotide/flavin mononucleotide (FAD/FMN) and NADPH, via the far‐reaching effects of the two Cbl coenzymes, methionine synthase (MS) and methylmalonyl CoA mutase (MCoAM) in, or on, the folate, glutathione, tricarboxylic acid (TCA) and urea cycles, oxidative phosphorylation, glycolysis and the pentose phosphate pathway. Deficiency of any of the NOS substrates and cofactors results in ‘uncoupled’ NOS reactions, decreased NO production and increased or excessive O2−, H2O2, ONOO− and other reactive oxygen species (ROS), reactive nitric oxide species (RNIS) leading to pathology. (2) Cbl is also the overlooked ultimate determinant of positive glutathione status, which favours the formation of more benign NO species, s‐nitrosothiols, the predominant form in which NO is safely deployed. Cbl status may consequently act as a ‘back‐up disc’ that ensures the active status of antioxidant systems, as well as reversing and modulating the effects of nitrosylation in cell signal transduction. New evidence shows that GSCbl can significantly promote iNOS/eNOS NO synthesis in the early stages of inflammation, thus lowering high levels of tumour necrosis factor‐α that normally result in pathology, while existing evidence shows that in extreme nitrosative and oxidative stress, GSCbl can regenerate the activity of enzymes important for eventual resolution, such as glucose 6 phosphate dehydrogenase, which ensures NADPH supply, lactate dehydrogenase, and more; with human clinical case studies of OHCbl for cyanide poisoning, suggesting Cbl may regenerate aconitase and cytochrome c oxidase in the TCA cycle and oxidative phosphorylation. Thus, Cbl may simultaneously promote a strong inflammatory response and the means to resolve it.
- Cobalamin
- aquacobalamin
- methylcobalamin
- adenosylcobalamin
- glutathionylcobalamin
- nitrosylcobalamin
- transcobalamins
- inflammation
- selective promotion/inhibition nitric oxide synthases
- nitric oxide
- GSNO
- tumour necrosis factor alpha
- interferon
- IRF‐1
- tetrahydrobiopterin
- GTP
- arginine
- glutathione
- heme enzymes
- G6PDH
- NADPH
- succinyl CoA
Introduction
A Scarlet Pimpernel for the Resolution of Inflammation? Citation[1] proposed that vitamin B12, cobalamin (Cbl), in all its various forms, is central to the effectiveness of the immune inflammatory response, and that its deficiency, chronic, functional or ‘compartmental’, may largely contribute to the aetiology of systemic inflammatory response system (SIRS)/sepsis/septic shock, as well as autoimmune disease, central nervous system (CNS) disease, cancer, in particular haematological malignancy Citation[2], and the progression of AIDS. The hitherto unexplained elevation of Cbl carrier proteins, the transcobalamins (TC I, II and III), their receptors, and TC unsaturated B12 binding capacity (UBBC) in trauma, infections, chronic inflammatory conditions Citation[3–9] and some cancers Citation[2], Citation[9–13] was seen to signal a central need for Cbl as a principal regulator of inflammation. The initial hypothesis proposed that Cbl might exert a pivotal effect on inflammation via regulation of the redox sensitive transcription factor, NFκB Citation[14], which determines the expression of a diversity of genes encoding mediators of the pro‐ and anti‐inflammatory phases of the immune response: cytokines, chemokines and inducible enzymes, principally, cyclooxygenase (Cox II), inducible nitric oxide synthase (iNOS) Citation[15] and heme‐oxygenase (HO‐1) Citation[16]. Regulated expression of such genes by NFκB, a family of rel protein homo‐ and heterodimers (RelA/p65, RelB, cRel, p50, p52), ultimately determines cell survival or proliferation, tissue repair and apoptosis. Evidence for five interrelated mechanisms by which Cbl might regulate NFκB was put forward: (1) hormone‐like regulation of tumour necrosis factor‐α (TNFα), through scavenging of excess nitric oxide (NO) by Cbl, as well as through the selective inhibition by Cbl, in tandem with gluthathione, of iNOS; (2) Cbl‐quenching of NO radicals (RNIS) and reactive oxygen species (ROS), enhanced by Cbl's glutathione (GSH) sparing/promotional effect; (3) Cbl promotion of acetylcholine synthesis, central to the neuro‐immune cholinergic anti‐inflammatory pathway; (4) Cbl's promotion of cellular energy and respiration via the tricarboxylic acid (TCA) cycle and oxidative phosphorylation; (5) a bacteriostatic role of the TCS released by neutrophil secondary granules during phagocytosis, which also appears to modulate the inflammatory response Citation[1].
Recent in vitro explorations of some aspects of the original hypothesis have shown that, at least in the pro‐inflammatory phase, Cbl does not inhibit NFκB Citation[17], and that indeed certain Cbls have a slightly promotional, although not statistically significant, effect on NFκB Citation[17]. What direct/indirect effect Cbl may have on NFκB in the anti‐inflammatory resolution phase of the immune response remains to be explored in a temporal in vivo model Citation[18]. However, a totally novel in vivo finding of a strong promotional effect of Cbl, particularly glutathionylcobalamin (GSCbl), on iNOS, with simultaneous supportive promotion of endothelial NOS (eNOS), in the early stages of inflammation Citation[17] (further corroborated by an inversely related suppression of the glucocorticoid, annexin‐1, and lower, well‐regulated levels of TNFα) Citation[17], may be consistent with one of the original hypotheses, that the ubiquity of Cbl and GSH is due to their mutual regulation of NO, in a continuous scavenger–donor redox dance Citation[1]. Because NO produced by iNOS can ultimately inhibit iNOS Citation[19], Citation[20] in the resolution of inflammation, as well as NFκB at its conclusion Citation[21], Citation[22], a direct promotional effect of Cbl, particularly GSCbl, on iNOS induction Citation[17] would mean that Cbl does ultimately regulate NFκB, indirectly, via NO regulation. If this is so, Cbl status could be the fulcrum on which the entire immune system turns. The Return of the Scarlet Pimpernel will attempt to explore how Cbl might act as both a timely selective promoter and a selective inhibitor of iNOS, as well as a key regulator of all three NOS in general.
‘They seek him here. They seek him there…’ Cbl's multiple forms and multiple roles
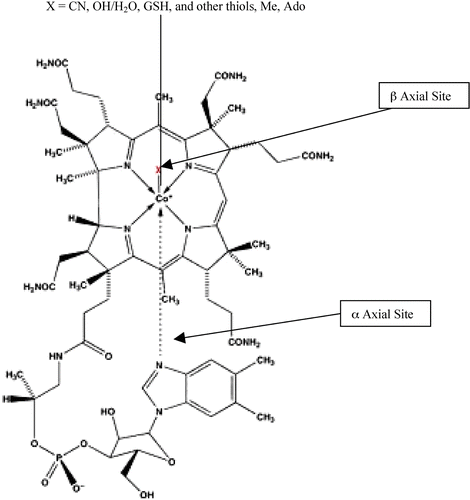
Cbl, C63–65H88O14N14PCo, vitamin B12 Citation[23], Citation[24], a red crystalline, water‐soluble substance (molecular weight 1357 kDa), comprises various polycyclic compounds, with a central cobalt atom set within a planar, tetrapyrrole (corrin) ring, that resembles that of the porphyrin of heme, except that it is less symmetrical. The upper β axial cobalt ligand is variable and can combine with H2O, OH, CN, GSH and other thiols, and with Me and Ado to form the coenzymes, methylcobalamin (MeCbl) and 51′‐deoxy‐5‐adenosylcobalamin (AdoCbl) Citation[10]. The latter two have a unique, covalent carbon–cobalt bond that gives Cbl its remarkable chemical and biological reactivity, and makes it one of the most potent physiological compounds, with a daily requirement of only 1 μg. The lower α axial ligand for the principal forms of the vitamin is a 5–6dimethylbenzimidazole, ‘false’, nucleotide base (DMBI) (). Cbl is nature's most complex non‐polymer molecule and the most complex of the vitamins and enzymatic cofactors known to date. It is synthesized by bacteria both in the soil and in the lumen of ruminants. Humans must derive Cbl from their diet, chiefly liver, kidneys, red meat, oysters, egg yolk and yeast extract. Absorption from food is also complex, as it involves the binding of Cbl in food by the Cbl transport protein TCI in saliva, gastric acid to separate Cbl from protein, and intrinsic factor in the ileum, as well as the transport protein, TCII Citation[25]. In the circulation there are, in fact, three transport proteins, TC I, II and III, with separate functions Citation[26]. Cbl also assumes different forms, the two principally known being MeCbl (75–90% of the body pool of circulating Cbl, transported chiefly on TCI) and the coenzyme AdoCbl (10–25% of endogenous Cbl, transported chiefly on TCII). TCIII appears to remove Cbl analogues or corrinoids, and cyanocobalamin (CNCbl) from the tissues and circulation and take them to the liver for excretion in bile, as corrinoids seem to interfere with the function of Cbl, whereas CNCbl, found mostly in the lungs of smokers Citation[11], is probably an excretory, detoxification product and is functionally inert. Cbl, which enters the circulation as OHCbl/H2OCbl, is transported by TCII, via the TCII endocytosis ion channel receptor (TCIIr), into all tissues and cells, where, after TCII degradation in the lysosomes, it is converted to MeCbl and AdoCbl, and largely retained for use intracellularly Citation[27], although some is exported on TCII and TCIII Citation[28]. MeCbl acts in the cytosol, AdoCbl in the mitochondria. Synthesized primarily by granulocytes, high concentrations of TCI are found in the reticuloendothelial system, in neutrophils, and in the liver. TCI is largely confined to the circulation, perhaps as a mobile store of Cbl (to complement the larger, long‐term storage of Cbl as MeCbl and AdoCbl in the liver and the pool of free Cbl in the kidneys). TCI does not have a specialist receptor, unlike TCII. Instead it is taken into the cell via a multipurpose receptor, the asialoglycoprotein receptor, a liver‐specific protein Citation[29]. TCII, a low molecular weight glycoprotein (43 kDa), with β globulin mobility, delivers MeCbl, AdoCbl and OHCbl/H2OCbl and other Cbls to the tissues. However, there is clearly some form of communication and flexibility between the TCS: TCII carries the larger fraction of Cbl present in portal vein blood than in hepatic and axillary vein blood. In disease, TCII sometimes holds the bulk of Cbl present in peripheral blood Citation[30], suggesting Cbl transfer from TCI as needed. High concentrations of TCI are also found in extracellular fluids: milk, saliva, tears, semen, amniotic and spinal fluid. Moreover, although every DNA synthesizing cell in the body contains receptors for TCII, the principal TC in tissues, there are nonetheless fine gradations of all three TCS present in cells, varying continuously in amount and intracellular location, according to the cell type and stage of maturation Citation[31].
Figure 1 Structure of vitamin B12 and its derivatives.
The elegance and ubiquity of such a fine‐tuned and flexible Cbl delivery system is further adapted during inflammation with a rapid response in the liver and granulocytes to produce marked elevations in the TCS, either I and/or II Citation[3], their receptors Citation[4] and UBBC Citation[4], Citation[5]. This is true for both chronic inflammation [for example, rheumatoid arthritis (RA) Citation[6], systemic lupus erythematosus Citation[7], diabetes, Crohn's disease Citation[8] and acute inflammation, including cancer‐associated inflammation, trauma and infections Citation[4], Citation[5]. The TCs and their UBBC are also increased in Cbl deficiency Citation[10], notably in the immune‐compromised (AIDS Citation[23], Citation[32] and cancer patients Citation[10–13], Citation[23], Citation[32]) and the elderly Citation[33] (who have a median of 40.5% Cbl deficiency) Citation[34], the same two groups who are also most susceptible and likely to succumb to SIRS/sepsis/severe sepsis and septic shock Citation[35]. Although TCS have been labelled ‘acute phase response proteins’, the possible significance of this has been either overlooked and no crisis function ascribed to them, or, as in the case of TC elevations in cancer, they have been negatively interpreted.
The mysterious go‐between: GSCbl and B12 coenzymes
The conversion of aquacobalamin (H2OCbl) on cell entry to the coenzymes MeCbl and AdoCbl is not straightforward. It apparently proceeds via the formation of the unusually stable intermediate Citation[36], Citation[37] GSCbl, the product of H2OCbl+ and excess reduced GSH only Citation[38–40].GSCbl is believed to be a major form of intracellular Cbl Citation[41], although this has not yet been proven unequivocally, and GSCbl's exact biological role, other than as a principal intermediate on the pathway to MeCbl and AdoCbl coenzyme formation Citation[42], remains to be fully explored. As befits its role as intermediate for MeCbl and AdoCbl formation, GSCbl may be a true go‐between and also act independently, both in the cytosol and mitochondria. (The possible significance of such GSCbl flexibility will be discussed in the Hypothesis section on the GSH, Cbl, NO triad relationship.) Recent chemical discoveries about GSCbl may point to its potential biochemical significance. The observed rate constant for the formation of GSCbl increases with decreasing pH, reaching a limit value at pH<6. Conversely, the equilibrium constant for the formation of GSCbl from H2OCbl+ and GSH in the pH range 4.50–6 increases with increasing pH Citation[38]. Intracellular pH is tightly regulated. However, unlike extracellular pH, which is a constant 7.3–7.4, intracellular pH varies according to location and vocation, from 7–7.3 in the cytosol to 5–6 in the endosome, with the pH also notably lowering in the lysosome, phagosome, and secretory granules to 5–6 or 5.5 Citation[43]. Thus, I would propose that the formation of GSCbl is so set up that, as in degrees of inflammation intracellular pH becomes even more acidic, GSCbl is formed increasingly rapidly, but in controlled amounts, presumably to conserve its steady availability over the crisis period. Then, as inflammation is resolved and cellular pH returns to normal, GSCbl formation equilibrium increases and the rate constant decreases. Nevertheless, even at the normal cytosolic pH of 7.4 and the normal body temperature of 37°C, conversion of H2OCbl+ to GSCbl will occur almost instantaneously, with a half‐life of 2.8 sec for the reaction with 5 mM GSH Citation[38]. (Levels of GSH in cells can range up to 10 mM.)
It may be an index of the previously posited special relationship between Cbl and GSH that the much larger than expected formation constant for GSCbl shows that thiolate forms of GSH are the first identifiable Cbl ligands to approach the remarkably high binding affinity of CN to H2OCbl Citation[38]. (Based on the pH dependence of KobsGSCbl in the pH region 4.5–6, an estimate of K GSCbl in the order of 5×109 M−1 is the closest formation constant to that of CN for H2OCbl≈1014 M−1Citation[38].) It is pertinent also that AdoCbl formation is four times greater from GSCbl than from H2OCbl or CNCbl Citation[42]. In coenzyme formation, GSCbl is postulated as interacting directly with the active sites of methionine synthase (MS) or methylmalonyl CoA mutase (MCoAM), and after reduction to Cob(I)alamin it is believed it may react respectively with s‐adenosylmethionine (SAM) or adenosine triphosphate (ATP) to form enzyme‐bound MeCbl and AdoCbl Citation[38]. It is not known whether some GSCbl is protein bound intracellularly, or whether any of it is exported on TCII and TCIII, as small amounts of MeCbl and AdoCbl are. However, both MeCbl and AdoCbl are largely protein bound, and in an unexpected manner. A huge surprise for B12 chemists when the crystal structures of the two enzyme‐bound cofactors were elucidated, was that the DMBI, in both enzyme‐bound MeCbl and AdoCbl, is ‘base‐off’, that is, the DMBI is no longer co‐ordinated to the cobalt, which is instead liganded or base‐on to the imidazole Nϵ2 in one of the two proteins' histidine residues (histidine 759 and A610 for MS and MCoAM, respectively). The DMBI had been expected to play a crucial allosteric role in the enzymes' catalysis, but instead is confined to functioning as an anchor sunk into a deep hydrophobic pocket in both enzymes Citation[1], Citation[44–46]. Some 16 Cbl‐dependent enzyme reactions are known to date, of which only two, possibly three, the latter is controversial Citation[1], Citation[47], Citation[48], occur in man. Yet, these two Cbl‐dependent enzymes control key metabolic pathways, whose far‐reaching relationships and often ‘hidden hand’ (of the Scarlet Pimpernel) consequences ensure the protection of every organ and system of the body.
As a methyl donor, MeCbl, via MS, reduces homocysteine to methionine, which then combines with ATP to form SAM, and ensures good methylation of DNA, RNA, protein, and successful DNA replication Citation[2], Citation[5], Citation[49]. (Therefore, Cbl, as well as folate, status should be critical for cancer chemoprevention Citation[2], Citation[5], Citation[49], Citation[50].) MS also reduces N5‐methyltetrahydrofolate (NMTHF) to H4folate (THF), thus ensuring its bioavailability for purine and δTMP synthesis Citation[2], Citation[50]. AdoCbl, via MCoAM, mediates the isomerization of methylmalonyl‐CoA to the energy‐rich thiol ester, succinyl CoA, the formation of which AdoCbl shares with α‐ketoglutarate (αKG). This means that Cbl acts at a critical stage in the Krebs or TCA cycle, as succinyl CoA represents a metabolic branch point wherein intermediates may enter or exit the cycle, leading ultimately to the release of guanosine triphosphate (GTP), a source of energy in gluconeogenesis and protein synthesis, and, in collaboration with the electron transport oxidative phosphorylation chain, to release of ATP. Succinyl CoA may also be converted to succinate or condensed with glycine to form δ‐aminolevulate, the initial step in porphyrin biosynthesis. The conversion of methylmalonyl‐CoA to succinyl CoA is also important for the catabolism of valine, isoleucine, methionine; the pyrimidine DNA‐specific nucleobase, thymine; odd‐chain fatty acids; and degradation of the side‐chain of cholesterol Citation[51]. Cbl is thus essential for cellular respiration and energy, and both protein synthesis and catabolism.
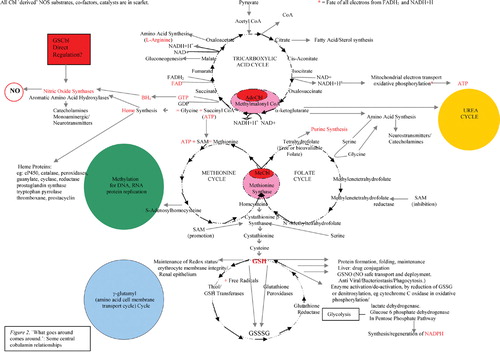
The web of complex inter‐relationships sustained by the two mammalian Cbl coenzymes is illustrated in . It may be seen from this that degrees of Cbl deficiency may result in malfunction on many levels. Perhaps then, it is not surprising that, although some remain sceptical, the Cbl chemical, biochemical, medical literature Citation[52], including clinical case histories, now supports claims, and increasing evidence, for the efficacy of Cbl in everything from cancer Citation[2], Citation[52], heart disease Citation[52], autism Citation[52], Alzheimer's disease Citation[52], multiple sclerosis Citation[52] and other neurological conditions Citation[52], AIDS Citation[52], SIRS/sepsis/septic and traumatic shock Citation[1], infertility Citation[52], depression Citation[52], circadian rhythm disorders Citation[52], autoimmune disease Citation[52], chronic fatigue syndrome Citation[52], eczema and other skin conditions Citation[52], allergies Citation[52], and, not least, growth and megaloblastic anaemia, as Cbl is critical to haemopoiesis Citation[52]. Patents granted or applied for include evidence that Cbl promotes anti‐inflammatory HO–1 while lowering inflammatory arachidonic acid metabolites, such as 12(R)‐HETE and 12(R)‐DiHETE (USP 5,674,505): also that Cbl, alone or combined with interferon or chemotherapeutic agents, is effective in viral, proliferative and inflammatory disease, including hepatitis C and B, herpes, vesticular stomatitis, autoimmune encephalomyelitis, multiple sclerosis and astrocyte gliomas (USP application 2005/0163751 A1). This pleiotropic character of Cbl's effects is curiously reminiscent of that of NO. Is this just a coincidence?
Figure 2 ‘What goes around comes around’: some central cobalamin relationships.
The NOCbl controversy
The fact that Cbl has some kind of rapid physiological impact on NO, which might prove beneficial in pathology involving unresolved inflammation, is suggested by various reports dating back to 1991. Large intravenous doses of OHCbl significantly increased systemic vascular resistance in normal conscious dogs Citation[53]. The relaxation of isolated vascular and visceral smooth muscle induced by NO and NO donors was reversed by OHCbl Citation[54]. Mice exposed to lipopolysaccharide (LPS) and given either CNCbl or OHCbl had a 30 and 40% increased survival, respectively (with zero survival for the control group) Citation[55]. OHCbl also blocked NO‐mediated inhibition of leukaemia cell proliferation Citation[56]. Such data has led to the popular view of Cbl's role in inflammation as a scavenger for excess NO, supposedly combining with it to form nitrosylcobalamin (NOCbl) Citation[55–58]. Although studies show this can happen, in a reversible manner, at the point of an electrode Citation[59], the existence of NOCbl has been much disputed by B12 chemists Citation[56], Citation[60–62], and an alternative mechanism for Cbl's inactivation of NO has been proposed, as NOCbl is not thought to be a stable enough complex to account for the inhibition/blockade of NO's biological effects by Cbl. Based on the observation that superoxide can, under certain conditions, lead to the rapid inactivation of NO, it has been proposed that Cbl (III)O2−, a superoxide species of Cbl that rapidly and spontaneously regenerates in aerobic solutions, interacts with NO by forming ONOO− (peroxynitrite) in a cyclic mechanism for its rapid inactivation Citation[60]. Although not implausible, this mechanism remains hypothetical, as Cbl (III)O2− is too unstable and difficult to purify for direct dose–response studies. A more recent study that apparently established rapid interaction between NO and the Cbl of MS, in the Co+1 state, Cbl (I), based on spectroscopic analysis, proposed that NO, at physiologically normal concentrations, may regulate carbon flow through the folate pathway by inhibiting MS and decreasing rates of methionine, serine, and de novo purine nucleotide synthesis. In this cell culture in vitro model, homocysteine (Hcy)Citation[63] was seen to act as an inhibitor of NO, a perhaps surprising finding given that NO is normally seen as anti‐atherogenic, yet by inactivating MS, NO raised the levels of supposedly pro‐atherogenic homocysteine. This hints at a complexity in the NO/Cbl relationship outside the study model's parameters.
Moreover, much of the data cited above for direct NO/Cbl interaction is handicapped by being largely based on purely chemical studies, or in vitro or isolated tissue studies that necessarily omit full physiological complexity. The view that Cbl just scavenges NO has always seemed too simplistic to the writer of this hypothesis. Combining with NO in a physiologically consequential way is more the province of heme and non‐heme iron, O2, superoxide, GSH and other thiols Citation[64]. If NOCbl has a physiological role, then maybe it is a very transient, possibly negligible, one, possibly less negligible in pathology, given the proposed impact of NO on MS Citation[63]. Nevertheless, it is still a question whether NOCbl exists at all endogenously. So far NOCbl has not yet been detected in vivo (although it has been given exogenously to mice as an effective anti‐cancer agent Citation[65]). Indeed, if Cbl were capable of continuous competition with heme or non‐heme iron or GSH as a successful rival for NO, it might prove dangerous to life. Whereas, the rodent, large mammal and human clinical literature demonstrates that Cbl in high, even extremely high, supra‐physiological doses is a life restorer in perilous situations, even capable of resurrecting the dead Citation[66]. (‘Des souris en état de mort apparènte parairent être réanimées par iv. 250 mg/kg OHCbl, sans autre mésure’ after cyanide poisoning Citation[66].) Apart from its traditional use as the treatment for subacute neuronal degeneration of the spinal cord and pernicious anaemia, a discovery that in its day was equivalent to finding a cure for cancer, Cbl has been shown to work safely in a variety of animal models: mice, rats Citation[55], guinea‐pigs Citation[67], dogs Citation[68], baboons Citation[69] and in a variety of extreme situations, from the trauma of radiation Citation[70] and electrocution Citation[71] to fatal injury Citation[68], anaphylactic Citation[67] and septic shock Citation[55]. In addition, Cbl has been used successfully for over 40 years in the intensive care unit in France (but also latterly in Spain, Italy, Germany, Hong Kong and China). The French case literature Citation[72–76] documents near‐miraculous recoveries from cyanide poisoning with the use of OHCbl as an antidote, given in extraordinarily high doses of 4–5 g, sometimes repeated on consecutive days. At these doses, the only side‐effects reported were a transient urticaria or red rash. Cyanide victims thus rescued are documented as recovering consciousness and cardiovascular function within 30 min of Cbl infusion, and walking out of the intensive care unit within 2 days, with liver and other vital organs intact. This is not the normal intensive care unit experience with such or similar extreme conditions, and may not therefore, this hypothesis maintains, be solely ascribed to the binding of cyanide by Cbl. The same supra‐physiological 5 g dose of OHCbl given to normal heavy smoking volunteers produced only modest transient rises in blood pressure and slight transient bradycardia Citation[77], conditions that might be welcomed as side‐effects in sepsis or shock treatment. Indeed, if Cbl were just an NO mop, it might be expected to cause dangerously high blood pressure and persistent vasoconstriction. Instead, few drugs can hope to emulate Cbl's proven pharmacological safety profile Citation[53], Citation[72–77].
So the Cbl/NO relationship has to be more complex and interesting than the crude idea of Cbl as just an NO mop. It is much more plausible, given the safety and efficacy literature, that Cbl should exert a central control over NO, in part through the regulation of all three NOS and through selective promotion and inhibition Citation[1], Citation[78] of iNOS, as and where it is needed. Because recent in vivo studies at the William Harvey Institute Citation[17], with other corroborative markers, show quite clearly that high‐dose OHCbl, and particularly GSCbl, promote iNOS mRNA in the early stages of LPS‐induced inflammation, and because mice given high‐dose Cbl to treat LPS‐induced sepsis show remarkable survival Citation[55], it is possible that Cbl first promotes iNOS NO production and later, in the resolution phase of inflammation, inhibits it, perhaps over and above iNOS inhibition by NO itself. Moreover, it may be that the high levels of iNOS NO production apparently promoted by Cbl, particularly by GSCbl Citation[17], in the pro‐inflammatory phase are essential for regulating inflammation and signalling entry into the resolution phase. The question is how?
Hypothesis
A well‐regulated, successful immune inflammatory response is biphasic Citation[79], Citation[80]. A pro‐inflammatory phase, entailing the tyrosine kinase phosphorylation cascade (MAPK) and the activation of key transcription factors, such as STAT‐1, NFκB, AP‐1, Sp1, IRF‐1, which up‐regulate the production of inflammatory cytokines, principally, TNFα, interleukin 1β, interleukin‐6, interferons α, β and γ, chemokines, adhesion molecules, growth factors, proteases, inducible enzymes such as HO‐1, Cox II, iNOS, phospholipase A2 as well as prostaglandins, particularly PGE‐2, and other lipid mediators, such as platelet activating factor, leukotrienes, thromboxanes, and tissue factor, which activate the extrinsic coagulation cascade. Such pro‐inflammatory factors radically change the redox environment at the site of inflammation, particularly in the macrophages Citation[81] and neutrophils Citation[82], so that it becomes more oxidant. Increased oxidative products, such as superoxide (O2−), singlet oxygen, hydroxyl radicals, NO and its species, interact to form other potentially lethal species, such as H2O2 and ONOO−. These free radicals collectively play vital roles in the immune response, acting both as signalling/acute response activation agents and as cytotoxic agents Citation[82–86]. For maximum lethality to the invader and minimum damage to the host they must be deployed in a focussed and balanced manner Citation[79], which has a natural time limit and often a distinct, spatially confined, or discrete intracellular location Citation[86]. Key antioxidant enzyme systems up‐regulated by decreasing pH, ensure that balance, focus, time and spatial limitation, signal specificity and efficacy are maintained Citation[84–88]. Superoxide dismutase (SOD), for example, removes excess superoxide using different catalytic transition metals, Cu, Zn, Mn, in differing cell environments — mitochondria, cytosol, phospholipid membrane Citation[89]. Catalase scavenges H2O2Citation[90]. GSH Citation[91], a major reductant and detoxifier, continuously recycled by selenium‐dependent GSH peroxidase (GPX), which also scavenges H2O2, is subsequently reduced again by GSH reductase Citation[91], while glutathione‐S‐transferase is also up‐regulated by the transcription factor AP‐1, which is under direct control by NO Citation[92]. HO‐1 breaks down heme to biliverdin Citation[16], which in turn yields the antioxidant, bilirubin Citation[93]. Heme peroxidases, such as myeloperoxidase, consume H2O2 in phagocytes and Cox II Citation[90]. Glucose 6 phosphate dehydrogenase (G6PDH) produces a steady stream of the reducing equivalent NADPH, needed for key enzyme catalysis Citation[94]. Thus, by a complex interplay of phosphorylating/redox‐sensitive signalling/response systems, the inflammatory phase of the immune response is normally self‐limiting. But rather than giving way in a see‐saw manner to the resolution phase, the evidence so far suggests that the anti‐inflammatory phase is engaged early on, but, initially, at a much lower level, its activity increasing as the peak of inflammation is reached and then declines. This may depend on early parallel activation of the cholinergic immune pathway Citation[95], in tandem with the sympathetic/adrenal neuro‐endocrine pathway, involving the release of glucocorticoids, such as Annexin 1 Citation[96], and the catecholamines dopamine, epinephrine/norepinephrine; also the deployment of ‘good’ eicosanoids, such as the ω‐3 fatty acid, eicosapentanoic acid, known to suppress TNFα and interleukin‐1 Citation[97], and various oxidized derivatives of eicosapentanoic acid, neuro‐protectins Citation[98], resolvins Citation[99], lipoxins Citation[100]. Eventually anti‐inflammatory cytokines interleukin‐4, interleukin‐10 Citation[101], interleukin‐13, and growth factors, epidermal growth factor (EGF) and transforming growth factor β1 (TGFβ1) Citation[102] are fully expressed, preceded by the inhibition of iNOS and a change in sense of pro‐inflammatory factors such as nuclear translocated NFκB, Cox II and interleukin‐6, which can paradoxically also signal repair and resolution in due course Citation[103].
In pathologies of unresolvable inflammation, chronic or acute, such as multiple sclerosis, RA, or sepsis, the redox balance is lost, either locally or systemically, with devastating results in the latter case. In sepsis, endogenous antioxidant enzyme systems can be depleted within hours, with a 46–83% loss of activity in SOD and GPX, just 12 hours into sepsis, and a 52% reduction in the somewhat more resistant liver catalase Citation[104]. With little to keep it in check, a fireball of reactive oxygen intermediates (ROS) and nitric oxide species (RNIS) fuels the pro‐inflammatory phase as it works in a feed‐forward, continuously amplifying loop of widespread endothelial and epithelial damage; a build‐up of fibrin impeding circulation in the microvasculature: unresponsive hypotension; and increasing O2− and ONOO− dramatically impairing cellular respiration and energy production, through increasing inhibition of aconitase in the Krebs cycle and complexes I to IV in the mitochondrial oxidative phosphorylation, electron transport chain Citation[105], all lead, if prolonged, to cell death and eventual multi‐organ failure.
It is commonly thought that the chief cause of such scenarios is NO overproduction by iNOS Citation[106–108], a view based largely on numerous in vitro studies, often with exogenous NO donors, (studies, which by definition, omit the effect of potential systemic iNOS/nNOS modulation systems, such as the increase in circulating and tissue entry TCS,) or on murine studies that do not discriminate between iNOS mRNA and the potential for variability in its redox products Citation[109], or that use even less specific evidence, such as serum/urine nitrite/nitrate. Although there is not much large mammal or human data for it Citation[106], such evidence as there is for this view, seems largely based on plasma and/or urine measurements of nitrite, and nitrate, in RA or septic shock patients, for example Citation[106–108], as direct NO and NOS assays in vivo are fraught with difficulty Citation[110]. But this evidence is equivocal, as nitrite and nitrate do not necessarily indicate simply formation of NO, but can equally well be derivatives of RNIS, such as ONOO−, or its protonated species, HNO Citation[111]. Indeed, a recent study of nitrite/nitrate excretion in the serum, urine, saliva and tears of RA and healthy age‐matched controls, both on a low nitrate/nitrite diet, found no significant differences, or relationships Citation[112]. Moreover, nitrite and nitrate are also products of protein catabolism Citation[113], which is dramatically increased in pathologies of acute unresolved inflammation, such as burn injury and sepsis Citation[114]. The fact that NOS/iNOS inhibitors can attenuate the hypotension of sepsis is also non‐specific evidence for putative NO damage. It can equally well be argued that iNOS inhibitors also inhibit the production of RNIS, which may be much more likely candidates as a cause of hypotension than NO. iNOS inhibitors will also considerably reduce the production of TNFα and interleukin‐1, and this too may be material, as high dose administration of TNFα is known to produce lethal hypotension Citation[115], which may have been automatically attributed to assumed high NO. Moreover, there is evidence that levels of NO have a direct regulatory correlation to levels of TNFα. In a murine model using Staphylococcus B, NO inhibitors increased sustained release of TNFα (and interferon‐γ), which increased enterotoxin toxicity of Staphylococcus BCitation[116]. Anti‐interferon‐γ monoclonal antibodies were more effective at NO reduction than anti‐TNFα monoclonal antibodies, but together they produced total NO inhibition Citation[116]. This suggests the existence of a regulatory loop by which NO inhibits the production of TNFα/interferon‐γ, which induces its own synthesis Citation[81], Citation[117], Citation[118]. This TNFα/NO relationship has been observed elsewhere. TGFβ‐1, usually expressed in the resolution of inflammation, is a potent suppressor of NO in vitro and in vivo, and TGFβ‐1 transgenic mice exposed to LPS show blunted production of NO, but an eight‐fold higher production of TNFα, as opposed to wild controls, with consequent increased mortality Citation[119]. The recent studies at the William Harvey Institute also demonstrate this high NO–lower TNFα relationship, as a result of high‐dose Cbl administration in mice exposed to LPS Citation[17]. Because Cbl has also been shown to exert direct hormonal‐like regulation of TNFα Citation[120], it is reasonable to conclude that such Cbl/TNFα regulation is the result of Cbl/NO regulation. The pleiotropic transcription factor Sp1, which directly promotes transcription of the TCII gene Citation[27] (up‐regulated in inflammation) and of iNOS expression Citation[81], is also involved in regulating TNFα transcription via the Sp1 binding site of its promoter, in response to iNOS NO production Citation[121]. Sp1 is moreover involved in the regulation of anti‐inflammatory TGFβ Citation[122], and epidermal growth factor Citation[123], the latter known to be directly Cbl status dependent Citation[120]. Thus, Sp1, the TC‐Cbl carrier promoter, is responsible for parallel activation of pro‐ and anti‐inflammatory factors, just as this hypothesis proposes Cbl may be. The emphasis, however, is on Sp1/Cbl‐NO regulation of TNFα, not suppression. There must be a necessary right level of TNFα for a successful immune response, as anti‐TNFα antibodies in the clinic increase mortality Citation[124]. Similarly too, although it may seem an old, discarded paradigm, there may be a right level of NO, higher in relation to TNFα, for a successful immune response outcome. Hence, even setting aside the detrimental impact of non‐selective NOS inhibitors on eNOS, this is an additional explanation for the negative outcome of iNOS suppression in sepsis with increased mortality in the clinic Citation[106], prefigured in animal models. iNOS knockout mice treated with LPS showed no significant survival over the wild‐type Citation[125]. Other iNOS –/– mice showed no defence against Gram‐positive bacteria, and equal mortality and vital organ damage as the wild‐type Citation[126]. Furthermore, macrophages derived from these iNOS –\– mice failed to restrain the replication of Listeria monocytogenes in vivo and lymphoma cells in vitro. Since iNOS is primed to be inhibited by NO feedback Citation[19–21], it should in effect be inhibited by putative NO overproduction in sepsis. Clearly, it is not. So, NO overproduction seems less plausible as the source of trouble.
This hypothesis, then, proposes a contrary scenario: it is not NO per se that is the problem in unresolved inflammation. Rather, the problem may be a malfunction of iNOS, resulting from degrees of mild, subclinical or ‘functional’ Cbl deficiency, which might also involve ‘compartmental’ Cbl deficiency, with the local inactivation of one of the two Cbl coenzymes, MS in the CNS, for example Citation[127], allied perhaps to other factors such as age, immune compromise, poor general nutritional status, and/or iNOS, TNFα, interferon, platelet activating factor, TC, gastric atrophy, or other, genetic polymorphisms. A polymorphism, in the iNOS gene, for example, which promotes greater NO production, has been shown to confer greater resistance to malaria Citation[128]. Polymorphisms in the NRAMP1 gene, an intracellular NO chaperone, may also impact significantly on immune resistance Citation[129]. [As an epidemiological aside, US statistics show a significant increase of 139% in sepsis diagnoses since the 1980s, with the increase especially notable in patients over 65 years of age (162%) Citation[106], the very group notable for a 40.5% median Cbl deficiency Citation[32]. What is also notable since the 1980s is the introduction and widespread, often indiscriminate, use of proton‐pump inhibitors and H2‐blocker drugs, both of which interfere seriously with acid‐dependent Cbl absorption Citation[130], Citation[131]. The effects of the latter may be amplified by a decline in consumption of liver and kidneys, the post‐war generation's staple, and, latterly, red meat and eggs, all key sources of Cbl.]
The numerous studies that show a detrimental effect of iNOS activation on pro‐inflammatory factors do not appear to have taken into consideration that when iNOS, the least tightly ‘coupled’ NOS isoform, malfunctions it can catalyse reactions that are partially or largely ‘uncoupled’ from the production of NO Citation[109], Citation[132–136], but result instead in an excess of superoxide, H2O2 OONO− and other RNIS Citation[137]. Consequently, levels of NO may not be high enough for correct signalling, cytocidal and resolution purposes. iNOS may then get stuck, like a needle in a groove, chronically producing increasingly even less NO, and increasingly more dysregulating O2−, other ROS and RNIS. Such RNIS, produced in relatively modest amounts under normal conditions, play a very specific role in cell signal transduction and regulation of enzyme synthesis and degradation, by reversible covalent modification of proteins and enzyme systems Citation[84–86]. Yet, if produced in increasingly excessive amounts, RNIS such as OONO− can also affect the other principal cell signalling system, responsible for cell cycle control, tyrosine phosphorylation Citation[138], Citation[139]. Nitration of tyrosine residues located near phosphorylation sites is irreversible, and impairs both the rate of phosphorylation and its reversibility Citation[140], contributing further to unresolved inflammation.
This hypothesis also proposes that, in the absence of deficiency, Cbl may be the ultimate supplier of the substrates and cofactors necessary for efficient, more coupled, iNOS/nNOS NO production, as opposed to excess O2−, OONO− and RNIS, and that it may consequently be the ultimate determinant of the NO/O2− balance thought to be critical in achieving NO regulation of ONOO−‐mediated signal specificity and response Citation[86], Citation[136]. Moreover, Cbl, as GSCbl, may itself act as an additional direct promoter of iNOS, while simultaneously also acting as a ‘back‐up disc’ to preserve or reactivate key antioxidant systems and enzymes that protect the host from damage during the immune inflammatory response. The resolution phase is ushered in by Cbl positively shifting the antioxidant balance, and having ensured effective, high levels of NO for a contained time span, so that NO eventually inhibits iNOS. A tantalizing clue to this proposed Cbl/NO relationship is to be found in the extraordinary capacity of liver for regeneration. In ancient mythology, Prometheus, the Titan who stole fire from Zeus for mankind, was punished by having a vulture devour his liver daily. Nightly, however, Prometheus's liver regenerated. This is scarcely myth: after resection of up to two‐thirds of human liver, complete regeneration can occur within 2 weeks. During this period, iNOS is continuously active, as in foetal gestation. Might this powerful, safe and miraculous deployment of high NO over a long period of time have anything to do with the fact that the liver, with up to 5 years' supply, is the largest depository of Cbl in the body? Let us now consider the possible mechanisms.
NOS: ‘in sickness and in health, for better or for worse’?
NO is produced by a family of NOS Citation[136], Citation[141], Citation[142], heme‐based enzymes that have a catalytic resemblance to cytochrome P450 and other heme‐based oxygenases. NOS have been divided into three main classes: two constitutive forms, involved in respiration, cell signal transduction and neuro‐transmission, respectively: membrane bound, particulate eNOS (NOS III), with a lipid anchor, targeted to the caveolae, the least active of the three isoforms, found primarily in smooth muscle and vascular endothelium; nNOS (NOS I), in the CNS/neurons and neuromuscular junctions; and iNOS (NOS II), produced in inflammatory immune responses by polymorphonuclear/granulocytes (PMN), primarily macrophages Citation[81], the latter two enzymes both active in the cytosol. This rather simple picture has been complicated of late by the discovery of multiple isoforms and splice variants of NOS, and their discrete localization within subcellular compartments Citation[86], Citation[136] – somewhat reminiscent of the varying forms of Cbl/TC distribution, localization and action. An example given is that of cardiac and skeletal muscle that coexpresses three different NOS isoforms in four different locations: a plasmalemal NOS, regulating force production and blood flow, a mitochondrial NOS, controlling respiration at the level of cytochrome c oxidase, a sarcoplasmic reticular NOS, involved in calcium homeostasis and other constitutive/inducible cytosolic NOS whose exact function/location had yet to be defined Citation[143]. μ, α, β, γ tissue‐specific isoforms of nNOS are also known Citation[144]. Moreover, iNOS, hitherto thought to be active only briefly during immune responses, or chronically and aberrantly in pathologies of unresolved inflammation, has been shown to be continuously active, or constitutive, in B‐lymphocytes Citation[145], and in certain locations, such as the myocardium Citation[146], retina Citation[147], liver Citation[148], bronchial epithelium Citation[149], and also in murine ileum Citation[150]. Since continuously expressed iNOS in the latter locations clearly has a positive protective function, removed from persistent pathology, (the lungs are continuously exposed to pathogens and iNOS NO in bronchial epithelium Citation[151], yet most people do not have asthma, for example,) iNOS must be very tightly regulated, with strong endogenous safeguards to prevent its malfunction.
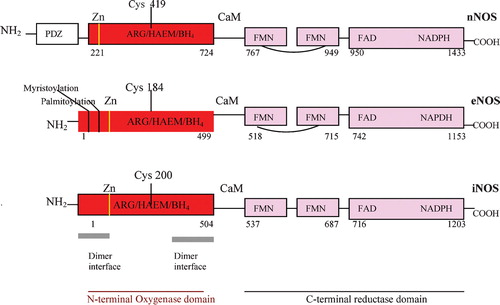
The three human NOS have a 51–57% homology, and share a fundamental bi‐domain structure: an N‐terminal oxygenase domain, with binding sites for zinc, iron protophorphyrin IX (heme), l‐arginine, (6R)‐5,6,7,8‐tetrahydrobiopterin (BH4), and a C‐terminal reductase domain, with binding sites for the two flavins, FAD, FMN, and for NADPH. These two domains are linked by a calmodulin (CaM) recognition site (). The crystal structures of the various NOS isoforms are gradually emerging Citation[152–157], and the truncated iNOS oxygenase domain, elongated in form, with an unusual αβ fold, has been memorably described as like a ‘baseball catcher's mitt’, with the heme in the palm of the mitt Citation[152]. Structurally, however, the NOS oxygenase domain differs from other heme oxygenases (cP450, peroxidase, catalase,) which have an α‐helical distal pocket, whereas the NOS distal pocket has a β‐sheet structure Citation[136].
Figure 3 Human neuronal nitric oxide synthase(nNOS), endothelial NOS (eNOS) and inducible NOS (iNOS) domain structure. The PDZ domain is named after homologous domains in three proteins: PSD‐95, DH/g, ZO‐1.
All three NOS monomers form dimers in their catalytically active states. Once the dimers are assembled, electrons donated by NADPH in the reductase domain are carried one by one by FAD to FMN and on to the oxygenase domain, where they reduce the heme iron and with BH4 supposedly catalyse oxidaton of l‐arginine's terminal, N‐guanidino via the intermediate, Ng‐hydroxyl‐l‐arginine (NHA) to NO and citrulline Citation[136], Citation[141], Citation[142]. This is a two‐step five‐electron oxidation reaction. Ca2+/CaM is required for electron flow from the reductase to the oxygenase domains. However, whereas iNOS, which contains tightly bound CaM, under normal basal physiological conditions, can produce NO at low ambient Ca2+ concentrations, and is thus largely Ca2+ independent, nNOS and eNOS are totally dependent on cellular Ca2+ influx Citation[136], Citation[141], Citation[142]. The amounts of NO produced by the constitutive and inducible isoforms also vary dramatically. Ca2+‐dependent eNOS and nNOS produce NO continuously in short puffs and low amounts, with some modest up‐regulation in inflammatory immune response, and up‐regulation or depression in pathologies of unresolved inflammation, such as tumour growth or sepsis: Ca2+‐independent iNOS, which is dependent for its expression on the transcription factors, NFκB, STAT‐1, AP‐1 and IRF‐1 Citation[141], Citation[158], can yield a 700–1000‐fold greater increase in NO in a very short space of time.
We now come to another great point of controversy, which may be material to this hypothesis: whether, in fact, NO is a direct product of the NOS Citation[109], or whether it is formed indirectly, after the formation of nitroxyl ion, NO−, or other RNI species, or s‐nitrosothiols Citation[141]. As with supposed iNOS over‐activation and NO overproduction in sepsis, NO production by NOS in general has been deduced from the generic breakdown products, nitrite, nitrate, or effects on heme proteins such as soluble guanylate cyclase induction, or oxidation of oxyhaemoglobin to methaemoglobin Citation[141]. Studies using specific NO electrodes or NO chemiluminescence assays have demonstrated that neither nNOS nor iNOS, at least, can produce NO in the absence of SOD, which reduces NO− to NO Citation[109], Citation[159], Citation[160]. SOD also, of course, scavenges O2− produced by both FAD/FMN in the reductase domain Citation[161], Citation[162], and in the oxygenase domains of eNOS/nNOS in the absence of BH4Citation[163], Citation[164]. Some evidence for NO− formation in BH4 depleted iNOS also exists Citation[165]. The general conclusion is that NOS may produce both NO and NO−in vivo, in varying ratios, under different conditions Citation[141]. The same is true of the NOS and O2− production, which may be contained or excessive. Depletion of l‐arginine, for example, increases NOS production of O2−, and subsequent H2O2Citation[166–168]. In the latter case, combinations of NO+O2−, or NO− and O2, result in excessive ONOO− and other RNIS Citation[136], Citation[169], redundant to the normal physiological needs for ONOO− and other RNIS for signalling/cytocidal purposes and thus likely to result in pathology. Such varying NO/NO−/O2− ratios will be determined by the redox environment, and, perhaps more critically, the availability of substrates and cofactors.
BH4: the Scarlet Pimpernel's butterfly
The requirement for NOS enzyme activity of the cofactor BH4 (6R‐5,6,7,8‐tetrahydrobiopterin) is absolute Citation[170]. The absence of BH4 results in no NO production, even with the l‐arginine substrate Citation[135]. Instead, BH4‐free NOS catalyses a partially uncoupled NADPH l‐arginine oxidation reaction, resulting in excess O2−Citation[164], and subsequent excess H2O2, and ONOO−. BH4‐free murine NOS, expressed in E. coli, yields citrulline and Nδ‐cyanoornithine, nitrite and nitrate, but no NO Citation[135]. (This is a good example of how these much‐relied on ‘NO’ markers, nitrite and nitrate, may mislead.) Moreover, BH4 depletion is associated with vascular pathology Citation[171]. Endothelial dysfunction due to eNOS inhibition, for example, has been reversed by BH4 administration to hypercholesterolaemic patients Citation[172]. The addition of BH4 to eNOS increases NO production and decreases O2−Citation[173]. Furthermore, degrees of BH4 availability have been shown to limit the onset of iNOS NO synthesis, in a dose‐dependent manner Citation[135]. Conversely, the addition of exogenous BH4 to cells leads to earlier LPS‐induced iNOS activity Citation[170]. The absence of BH4 also appears to inhibit cell growth and differentiation via the inhibition of NOS activity Citation[174].
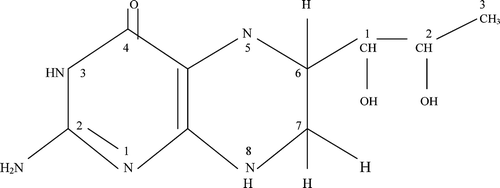
BH4 () is a pteridine, member of a class of pyrazino [2,3,d] pyrimidine compounds, which include molybdopterin and folate. Pteridines were initially discovered in the yellow pigments of the wings of butterflies by Sir Frederick Gowland Hopkins in 1889 Citation[175]. The structures of three of these pigments were elucidated in the 1940s, thus giving rise to the name pteridine, from the Greek ‘ptera’ or wing. The isolation in 1957 of the structure of fluorescent blue pigments in the eye of Drosophila melanogasterCitation[176], coincided with the discovery that the protozoan, Crithidia fasiculata required high doses of folic acid for growth and that biopterin could substitute wholly for it. Thus, it began to be appreciated that pteridines were not just pretty pigments, but had more complex biological functions, from light‐gathering molecules to enzyme cofactors in redox reactions and I‐carbon transfers Citation[175]. Xanthine oxidase, for example, uses molybdopterin to catalyse the last step in purine synthesis Citation[177]. BH4 is an essential reducing cofactor of aromatic amino acid hydroxylases (AAHs) in the metabolism of phenylalanine, tyrosine, tryptophan; epinephrine; the monoaminergic neurotransmitters: dopamine, norepinephrine, serotonin, and, en route to the latter, the sleep hormone, melatonin Citation[177]. The observation of high BH4 levels in tissues low in AAHs, blood, spleen, lungs, has led to other proposed roles: a function of BH4 in haematopoietic cell proliferation and differentiation, based on the specific appearance of BH4 during the differentiation of reticulocytes to erythrocytes, and its cell cycle‐dependent expression in various species, from mammalian thymocytes Citation[178] to the acellular slime mould Physarum, in which BH4 levels peak during S‐phase, and Drosophila embryos, which do not survive with BH4 mutation or inhibition Citation[179]. The exact role of BH4 in the NOS has proven to be somewhat elusive and mysterious. It had been thought that it may have at least two functions: first, allosteric, stabilizing the NOS dimeric structure and the high‐spin heme iron conformation; as well as promoting l‐arginine substrate binding (which now appears not to be affected by BH4Citation[156]), second, a redox role as a direct electron donor to the heme, promoting conversion of l‐arginine to the intermediate, NHA Citation[153], Citation[156].
Figure 4 Structure of 6R‐5,6,7,8‐tetrahydro‐L‐biopterin (BH4), the naturally occurring pterin cofactor of nitric oxide synthases and amino acid hydroxylases. The standard nomenclature for numbering the positions of the pteridine ring and the biopterin side chain is indicated.
BH4, which is constitutive in liver, neuronal tissue and macrophages, can be additionally induced by LPS, or cytokines, principally, interferon‐γ, TNFα and interleukin‐1, in macrophages, fibroblasts, endothelial and vascular smooth muscle cells Citation[175]. De novo BH4 synthesis requires the purine nucleotide, GTP, which is converted to BH4 in four enzymatic steps, by three separate enzymes Citation[175]. GTP cyclohydrolase (GTPCH), which is rate limiting, catalyses the first step, yielding neopterin as a byproduct Citation[175], a known marker of immune activation Citation[180]. GTP availability increases the GTPCH reaction velocity in a concentration‐dependent manner, and cytokine activation of cells significantly increases levels of GTP. The mechanism of this inflammatory‐induced up‐regulation is unknown Citation[175].
‘Cherchez le Scarlet Pimpernel’: interferons and TCS to the rescue
Now, remember what happens to Cbl in inflammation? When NFκB is activated, it cross‐talks to the transcription factor Sp1 Citation[181], constitutively expressed in the eNOS promoter and known to be essential for NO production Citation[141], and also expressed in the iNOS promoter region, along with the transcription factor IRF‐1, essential for iNOS/eNOS mRNA expression and NO production Citation[151]. Sp1 up‐regulates the production of TCII Citation[27], primed perhaps by interferons, particularly interferon‐β, which simultaneously increases TCIIr expression Citation[65] through further cross‐talk between IRF‐1 and Sp1. So more Cbl arrives intracellularly, where synthesis of GSCbl and its conversion to MeCbl and AdoCbl is up‐regulated by decreasing cellular pH Citation[38], Citation[42]. MeCbl binds to MS, which is then well armed to keep the folate pathway open and meet the increased needs in inflammation for purine nucleotide synthesis, essential for RNA synthesis and DNA replication. AdoCbl binds to MCoAM, and, as the cell's energy needs increase, AdoCbl, via MCoAM's impact on the TCA cycle and oxidative phosphorylation, ensures a steady stream of ATP, and the reducing equivalents, NADH+H+ and FADH2, for maximal efficiency of key defence enzymes. Each turn of the TCA cycle produces 1 mol GTP, and coupled to oxidative phosphorylation, 10 mol ATP Citation[51]. ATP may be converted to GTP, and vice versa, but the ATP/GTP ratio in cells is relatively constant, with total cell concentrations of adenine nucleotides, ATP+ADP+AMP, being four to six times greater than those of guanine nucleotides, GTP+GDP+GMP. ATP and GTP are derived from AMP and GMP via the common ribonucleotide precursor in the de novo purine nucleotide synthesis pathway, inosine 51‐monophosphate (IMP), an energy expensive pathway in terms of ATP consumed per mole of IMP produced. The conversion of IMP to AMP and GMP is tightly and mutually regulated: with IMP to GMP requiring ATP, and IMP to AMP requiring GTP, as energy sources. So when there is sufficient ATP in the cell, GMP is synthesized from IMP, and when sufficient GTP exists, AMP is synthesized from IMP Citation[177]. Thus, degrees of Cbl deficiency may ultimately impact on both levels of ATP and GTP, and increasingly depressed production of GTP will ultimately impact on BH4 status – IMP dehydrogenase inhibitors reduce BH4 synthesis Citation[175]. Finally, in turn, this will inhibit iNOS NO production. A deficiency of BH4 will also ultimately affect its catalytic recycling by AAHs, resulting in decreased catecholamine and monoaminergic neurotransmitter synthesis Citation[113]. Perhaps then the characteristic unresponsive hypotension of sepsis and septic or traumatic shock may be just as attributable to an increasing decline in catecholamine synthesis, as to the supposed overproduction of NO. Such LPS‐induced hypotension has been shown to precede iNOS activation in rodents Citation[182]. Furthermore, there may be an increase in oxidized and therefore ineffectual catecholamines such as adrenochrome Citation[183] and adrenolutin, which also disturb mental equilibrium Citation[184]. As Cbl is also critical to the synthesis of acetylcholine and the importance of the CNS and neuro‐immune cholinergic pathway in inflammation has been well demonstrated Citation[95], Citation[185], the characteristic mental obfuscation and loss of consciousness in sepsis Citation[186] may also be as much consequences of depressed GTP synthesis as of supposed NO overproduction. Such a Cbl/GTP deficiency would result in a decrease in acetylcholine and other neurotransmitters, an increase in toxic catecholamine metabolites, and unregulated cytokine production. Indeed, given the low–high and vice versa NO/TNFα regulatory correlation discussed earlier, together with established high levels of TNFα in sepsis Citation[187], and observed 83% increase in spinal fluid TNFα levels in Cbl deficiency Citation[188], the argument for NO overproduction in sepsis looks tenuous, whereas it has been shown that higher levels of ATP in septic patients have a positive significant correlation with survival Citation[189]. Declining ATP in sepsis would therefore correlate with a concomitant fall in levels of GTP, and consequently BH4 and consequently a decline in production of NO from iNOS, which would argue further against the popular view of increasing NO production in sepsis and other pathologies of unresolved inflammation.
Cbl, αKG and arginine: close collaborators and distant allies
The production of succinyl CoA in the TCA cycle is not, of course, solely dependent on Cbl. α‐Ketoglutarate, αKG, an important intermediate in the cycle, derived from citrate, is equally critical to succinyl CoA formation and the onward role of the cycle Citation[51]. αKG is a form of ornithine, and its production is important not just for the activity of the TCA cycle, but for that of the urea cycle, the major mechanism for the removal of ammonia (NH4) produced by protein catabolism Citation[113]. When αKG leaves the TCA cycle it is transaminated with glutamine to form glutamate, which can exit mitochondria and be further converted to other non‐essential amino acids. In the CNS, αKG is converted to the neurotransmitters, glutamate and GABA, γ‐aminobutyric acid. Glutamate can also be produced from αKG in mitochondria by the mitochondrial enzyme glutamate dehydrogenase in the presence of NADH or NADPH and ammonia Citation[113]. The amino group thus incorporated into glutamine is used by aminotranferases to form other amino acids. Glutamate is a precursor of ornithine, in turn a precursor of arginine. In the urea cycle, which depends on a supply of glutamate, via αKG from the TCA cycle, ornithine and arginine are continuously interconverted, and the arginine formed there is not available for protein synthesis and NOS NO synthesis Citation[113]. The synthesis of arginine for the latter begins in the intestinal mucosa, which is rich in glutamine and enzymes that convert glutamate via ornithine to citrulline. The final conversion of citrulline to arginine occurs in the renal proximal tubular cells, which lack the arginase of the urea cycle, and are responsible for 60% of net arginine synthesis Citation[190]. Since citrulline is also a by‐product of NO formation, most cells have the capacity to synthesize arginine to some degree, so that there is also in effect a citrulline–NO–arginine cycle, analogous to the recycling of arginine in the urea cycle Citation[190]. Yet this recycling provides only about 50% of the l‐arginine cofactor required by NOS to produce NO. The other 50% is supplied by arginine production from protein catabolism and synthesis Citation[190]. Although no studies evaluate the role of the latter in regulating arginine availability for iNOS NO synthesis in different cell types, it is probably an important factor, as it is in arginine homeostasis, a balance kept by dietary arginine, endogenous arginine production and degradation Citation[190]. In sepsis, where there is supposed iNOS NO overproduction, plasma arginine falls by as much as 50–60%, and the reason for this is not understood. Although high levels of iNOS/NO can inhibit protein synthesis, and inflammation induces both arginases and plasma arginine transporter up‐regulation Citation[190], thus potentially increasing plasma arginine clearance, there may be a different explanation, consistent with the hypothesis that in pathologies of unresolved inflammation, not enough NO is produced from NOS, but instead excess RNIS and ROS, so that the redox balance is lost, leading to increasing bioenergetic failure. High metabolic rates and increased catabolism without corresponding anabolism, characteristic of sepsis, mean that the liver and kidneys have to deal with the clearance of increasing levels of ammonia, the final breakdown product of protein catabolism. Glutamine, which constitutes 50% of circulating amino acids, and serves as an ammonia transporter, is diverted, as a result of lactic acidosis, from the intestinal mucosa to the kidneys – breakdown of the intestinal mucosa integrity is an early symptom of sepsis Citation[191]. The uptake of glutamine by the liver, needed for the urea cycle, is suppressed and more is directed to the kidneys to conserve bicarbonate required for the formation of urea Citation[113]. If the redox balance is not regained, persistent lactic acidosis and protein catabolism mean that the already dysregulated liver and kidneys start to fail. High ammonia concentrations lead to increasing liver sequestration of αKG as glutamate from the TCA cycle. This in turn reduces further the already reduced synthesis of ATP, leading to coma and death.
Thus, it is possible to see why there are two sources of succinyl CoA in the TCA cycle: Cbl functions as the back‐up disc for αKG. If Cbl status is replete, fluctuations in αKG will be minimized, homeostasis of the TCA cycle ensured, in turn ensuring homeostasis of the urea cycle, and the availability of arginine outside the urea cycle for protein synthesis and NOS NO production. This is why the 5 g supra‐physiological doses of Cbl used for 40 years by the French as an antidote to cyanide poisoning work so dramatically, restoring metabolism in hours and reversing damage to vital organs, so that liver function is normal at discharge, just 2 days after fatal doses of cyanide Citation[66], Citation[72–76]. Cbl does this not just by binding to CN, but by performing all its normal metabolic tasks, which include the supply of all the NOS substrates and cofactors: the purine nucleotides, FAD and FMN, via MS; BH4 via GTP; arginine by supporting αKG and keeping the onward momentum of the TCA cycle rolling (consequently decreasing glycolysis and reversing lactic acidosis); NADPH, as we will see, via Cbl's impact on G6PDH and the pentose phosphate pathway; and last but not least, even heme synthesis, the organic portion of which requires eight residues each of glycine and succinyl CoA Citation[192]. Cbl's critical role in heme synthesis particularly demonstrates the pivotal role Cbl may play in inflammation control, not only because many heme proteins such as cP450, catalase, HO‐1, have anti‐inflammatory functions, but because inducible HO‐1 promoted by Cbl (USP 5,674,505), continually breaks down heme, via biliverdin, to the potent antioxidant, bilirubin. However, the simultaneous up‐regulation of the TCs in inflammation ensures both the rapid regeneration of heme, and thus of key heme proteins, and a consequent increased potential production of bilirubin, as needed. (That AdoCbl via MCoAM is rate regulating in particular for the synthesis of the heme of haemoglobin has been specifically observed with heme staining of transparent 2 day old zebrafish larvae using o‐dianisidine. T. Penberthy, pers. commun.)
eNOS is depressed in sepsis Citation[107], Citation[193] and other pathologies of unresolved inflammation, and the evidence for alleged iNOS overexpression under these conditions is largely indirect or equivocal. iNOS may in fact be depressed as well as malfunctioning. Moreover, murine iNOS knockout models show that suppression of iNOS offers no protection against organ damage and mortality from LPS. iNOS expression and NO production are also dependent on the expression of interferons and the transcription factor, IRF‐1, in the iNOS promoter. Might this be because interferons up‐regulate TCS and their receptors, increasing and sustaining the arrival of more Cbl intracellularly, so that subsequently these increased local levels of Cbl, as AdoCbl and MeCbl, can ensure the assembly of all substrates and cofactors to enhance NO production, not only via iNOS, but eNOS, as seen in recent studies? Citation[17].
NO: friend or foe?
If it were not for the fact that it is a colourless compound, NO itself might well deserve the soubriquet ‘Scarlet Pimpernel’. Unknown until the last 25 or so years, the smallest but potent working mammalian molecule, NO, a paramagnetic gas, is produced enzymatically in diverse locations throughout the body and being both lipophilic and hydrophilic is easily diffusible towards its intracellular targets where exact specificity is achieved by the redox environment Citation[64]. For example, in the ryanodine receptor/Ca2+ release channel (RyR), NO nitrosylates only one out of 50 free cysteines per RyR, to alter the Ca2+ CaM/RyR interaction, sensitizing the channel to positive or negative Ca2+ regulation Citation[194]. This extraordinary precision can only happen at a restricted O2 concentration. NO, a second messenger for post‐translational modification, is responsible for a wide diversity of critical functions: immune regulation and anti‐microbial defence; neurotransmission and cerebral blood flow; smooth muscle relaxation; platelet aggregation or inhibition; exchange of gases in tissues; bronchodilation; glomerular filtration; gut peristalsis; penile erection; cardiac contractility; modulation of ligand‐gated receptors (N‐methyl‐d‐aspartate receptor) the Ca2+‐dependent potassium channel, the cardiac Ca2+ release channel, cyclic nucleotide cation gated channels, Janus kinases, tyrosine phosphatases; peptide hormone release, and more Citation[86], Citation[195–197].
To effect all of these physiological processes, NO assumes three redox forms, the free radical NO• itself, nitrosonium (NO+) and the nitroxyl anion (NO−). So the term NO is a collective description. These varying redox forms of NO have particular affinities for particular biological targets: NO• reacts with oxygen, superoxide O2− and redox metals; NO− with SH groups and metals; NO+ undergoes addition and substitution reactions with nucleophiles in aromatic compounds and electron‐rich bases Citation[64]. Fluctuations between these redox forms and their target products are of central importance to physiological homeostasis. Principally NO's strong affinity for transition metals results in its combination with the target heme iron, to form very stable catalytic Fe2+–NO iron–nitrosyl complexes, and/or combine with critical thiol residues, to form s‐nitrosothiols, or s‐nitrosylate proteins Citation[64], Citation[86], Citation[138]. Such combinations can simultaneously activate some enzymes and deactivate others, modifying protein function or initiating gene expression. Both Cox I and II Citation[138], Citation[198] and soluble guanylate cyclase and thence the second messenger cGMP Citation[199], Citation[200] are activated by NO binding to their Fe3+, whereas cytochrome c oxidase, complex IV of the oxidative phosphorylation, electron transport chain, is deactivated by NO/Fe3+ binding Citation[201] as are cP450 enzymes Citation[202], indoleamine 2, 3‐dioxygenase (IDO) Citation[203], important in bacteriostasis, and, indeed, NOS itself Citation[19–21]. Enzymes with catalytic thiols deactivated by s‐nitrosylation include glyceraldehyde phosphate dehydrogenase, inhibited by NO‐promoted ADP‐ribosylation Citation[204], Citation[205], protein kinase C Citation[206], γ‐glutamylcysteinyl synthetase Citation[207], alcohol and aldehyde dehydrogenase Citation[208], aldolase Citation[208], cathepsin B Citation[208], O6‐methylguanine‐DNA‐methyltransferase Citation[208], neutrophil NADPH oxidase Citation[86], Citation[138] and at least seven members of the caspase family, involved in apoptotic signal transduction and cytokine maturation Citation[86], Citation[209] and one of the most important antioxidant enzymes, GPX Citation[210]. Conversely, s‐nitrosylation of tissue plasminogen activator activates its vasodilatory and anti‐platelet effects Citation[211].
When the redox balance is maintained and oxidative stress is not extreme or persistent, such NO‐derived enzymatic activation and deactivation will occur reversibly, in a complementary manner, both simultaneously and/or in a relay system, preventing NO toxicity. But persistent or extreme oxidative stress may render NO toxic by shifting the balance between NO redox species and their target reactants and end products, from high output of s‐nitrosothiols, resistant to O2 and O2− interaction, the principal form in which NO is neutralized for safe transport to tissues Citation[64] via haemoglobin, myoglobin, albumin Citation[212] and GSH Citation[213–215], to increased production of RNIS such as the reactive intermediate, ONOO−, via increased reactions with H2O2 and O2−, and other ROS, which can prolong oxidative stress indefinitely, leading to increasing enzymatic malfunction, which will, of course, include malfunction of all three NOS.
If we assume that the NOS malfunction in extreme or persistent oxidative stress produces less NO and more O2−, then because the relative fluxes of NO and O2− modulate the oxidation of critical enzymatic and other protein thiols by ONOO− and other RNIS, and because ONOO− and O2−, rather than NO, inactivate aconitase Citation[216] in the TCA cycle, and also inactivate NADH dehydrogenase and succinate dehydrogenase, complexes I, II and III of the oxidative phosphorylation, electron transport chain Citation[217], as well as deregulating poly‐ADP‐ribosyl transferase Citation[204], Citation[205], the stage is thus set for terminal cell function dysregulation and energetic failure. Of course, normally this does not happen due to powerful antioxidant defence systems, which are well primed and in place before the conveniently delayed induction of NO by iNOS peaking at 6 hours. But sometimes these systems fail, and when they do, this hypothesis posits, it is because they have lost their ‘back‐up disc’, as a result of insufficient, or non‐functional, Cbl status. Moreover, it seems that there is a fine hierarchical balance relationship between key anti‐oxidant enzymes, which can be equally detrimental if disturbed, even though some of the enzymes may remain functional. With certain anti‐oxidant enzymes more is not necessarily better. Indeed, it may be worse: five‐ and 10‐fold increases in Mn‐SOD and Fe‐SOD, respectively, sensitize E. coli to paraquat toxicity Citation[218], Citation[219]; Cu/Zn‐SOD transfectants of mouse epidermal cells JB6 possess increased sensitivity to DNA strand breakage and growth inhibition in the presence of O2− and H2O2 from xanthine/xanthine oxidase Citation[220], whereas a concomitant increase in catalase in a double transfectant can correct this Citation[220]. Similarly, in protection from ischaemia‐reperfusion injury, a combination of Cu/Zn‐SOD and catalase worked better than either alone Citation[221]. Again, it has been observed in several systems that an increase in Cu/Zn‐SOD is accompanied by an increase in GPX Citation[222], with a high ratio of activity of GPX over Cu/Zn‐SOD related to notably increased growth potential and resistance to killing by paraquat in NIH‐3T3 transfectants Citation[223]. Catalase scavenges H2O2 produced by Cu/Zn‐SOD catalysis, but GPX, the primary H2O2 scavenger, additionally can destroy hydroperoxides, which catalase cannot, so the activity status of GPX is crucial and superior in the hierarchy, and ultimately that is dependent on the status of GSH, which apparently depends on the activity of GSH reductase, as well as the availability of cysteine, derived from serine, which provides the carbon skeleton, with supply of the sulphur dependent in turn on the reduction of homocysteine by MeCbl.
GSH, Cbl and NO: an eternal triangle?
GSH, C10H17N3O6S, is actually a tripeptide, γ‐glutamylcysteinylglycine, and so it has a less obvious debt to AdoCbl, via AdoCbl support of αKG in the TCA cycle, which ensures a supply of glutamate. An additional debt to MeCbl is in the supply of glycine, formed from the breakdown of serine in a reaction requiring pyridoxal phosphate and THF Citation[113], the latter dependent on Cbl status for its availability Citation[224]. As ubiquitous as Cbl and NO, GSH is a major reductant and detoxifier, involved in drug conjugation, and is a cofactor modulating O2− production in many key enzyme systems, including glycolytic enzymes Citation[91], Citation[214], Citation[225–227]. This function of GSH is of particular importance in the NOS. GSH is essential for leukotriene synthesis Citation[113], amino acid transport Citation[113], maintenance of erythrocyte membrane integrity Citation[113], and reduction of peroxides formed during oxygen transport Citation[91]. GSH is the principal thiol involved in the correct formation and degradation of protein disulphide bridges and through thiol–disulphide exchanges ensures proteins are folded into their native conformation Citation[228]. Lysozyme, ribonuclease, albumin and insulin are notable examples of important proteins requiring GSH for formation Citation[228]. GSH also ensures the reversibility of mixed disulphides GSSG, involved in the reactivity of proteins Citation[228]. The functionally correct ratio of S‐H groups to S‐S is ensured by very high levels of GSH in cells, of up to 10 mM. In erythrocytes, for example, this results in a 100:1 ratio of GSH to GSSG Citation[91]. Thus, the maintenance of very high levels of GSH, important in every respect, is critical for good immune function and the resolution of inflammation. GSH and GPX rapid depletion in sepsis has already been noted, a depletion that affects both arms of the immune system, with decreased lymphocyte response to mitogens in the relative absence of GSH Citation[229]. Given the shift in redox balance entailed by the inflammatory immune response, the recycling of GSH by GSH reductase is liable to be ultimately inadequate without de novo GSH synthesis, and, as seen in pathologies of unresolvable inflammation, even this can fail. The GSH‐sparing/promotional role of Cbl in preventing this appears to have been completely overlooked Citation[1].
Yet, over 50 years ago it was observed that in pernicious anaemia/Cbl deficiency, in both rats and humans, levels of GSH in blood are very considerably depressed, and that these low levels return to normal promptly, and without exogenous GSH or GSH precursor administration, simply by treating the Cbl deficiency. Moreover, an initial overshoot of GSH was observed on correction of the Cbl deficiency Citation[230], Citation[231]. That this is peculiar to Cbl was proved by studies showing that the normal reduction of S‐S groups remains unaffected in iron or folic acid deficiency Citation[231]. Contemporaneously, it was also shown that a combination of GSH and Cbl was synergistically more powerful than either alone in the reactivation of a range of enzymes with active S‐H groups in the E. coli mutant 113‐3 Citation[232]. It was noted, however, that certain other enzymes were conversely deactivated by GSH alone Citation[232]. These studies were, of course, performed in the era before the discovery of NO's biochemical role. With hindsight, given the NO‐analogous effect of Cbl and GSH on enzyme activation/deactivation, one can see that these GSH/Cbl effects, mediated perhaps by possible Cbl oxidation and reduction of thiols Citation[232–236], are part of bacterial defence against NO generated by macrophages in the phagocytic burst. In fact, as the studies used a combination of 50 mM GSH to 50 μg Cbl, it is likely perhaps that GSCbl was formed in the cultures. A noteworthy observation of these studies was that the GSH/Cbl combination was most effective at regenerating activity in aged enzymes Citation[232], thus simulating the effects of possible GSCbl on enzymes during oxidative stress, characteristic of inflammation, and again, with hindsight, perhaps indicating the potential importance of the controlled but increased rate of GSCbl formation in inflammation. Pertinently, enzymatic activity of G6PDH with added substrate was assayed in a range of pH, at 37°C, with Cbl and GSH, and found to be least active at pH 8.5, and most active at pH 6.5 – this is interestingly coincident with the pH variable for GSCbl's rate constant in inflammation. Most crucially from this viewpoint, the combination of GSH/Cbl was able to restore the activity of both lactate dehydrogenase and G6PDH Citation[232], key enzymes in the pentose phosphate pathway, the latter responsible, with glycolysis and the TCA cycle, for the synthesis of the reducing equivalent, NADPH Citation[94]. The activity of G6PDH is essential for the maintenance of erythocyte GSH in its reduced state via the formation of NADPH Citation[94] and, of course, NADPH is essential to the function of key enzyme systems from cP450 to GSH reductase, GPX and the NOS. An irreversible decline in NADPH production during inflammation will have serious consequences, and will certainly result in increasingly less coupled NOS reactions and less NO. Other enzymes activated by Cbl/GSH include: pyruvic transaminase, stearic acid oxidase, glycine and alamine deaminase, cysteine desulphurase, serine dehydrase, maltase, lactase, lysine and ornithine and glutamic decarboxylase. Citation[232] This last is of obvious significance for supply of αKG, arginine, glutamate, glutamine, etc. Moreover, the range of enzymes that responded suggested that this Cbl+GSH enzymatic activation was a general effect, not restricted to a particular class.
These early studies all provide forgotten evidence for an intimate co‐dependent relationship between GSH and Cbl. The nexus of this co‐dependence is in what has been described as a SAM ‘switch’ for the two alternating fates of homocysteine Citation[127] (). Homocysteine in the MeCbl MS‐dependent methionine cycle is used to regenerate methionine, which reacts with ATP – from the AdoCbl MCoAM supported TCA cycle – to yield SAM, the universal methyl donor. If there is an excess of methionine, some of it will be converted to ammonia and α‐ketobutyrate, which is decarboxylated to yield propionyl CoA Citation[113] then converted to succinyl CoA and thence re‐enters the TCA energy generation cycle. Some excess methionine will also be utilized in gluconeogenesis Citation[113]. This shuttling of excess methionine back to the TCA cycle to repay the ATP debt for SAM shows the complementary, co‐operative functions of the two Cbl coenzymes, everywhere apparent given the universal roles of MeCbl/AdoCbl, respectively, promoted by SAM and ATP. Levels of SAM are hence ultimately determined by Cbl status in general, as much as by dietary intake of methionine. If Cbl status is good, ATP and SAM will be abundant. High levels of SAM activate the SAM ‘switch’ that links Cbl to the synthesis of GSH, as SAM inhibits the provision of methyl folate for MS reduction of homocysteine to methionine, and increases cystathionine β‐synthase, which transulphurates homocysteine to cystathionine, a precursor of cysteine and thence GSH Citation[127].
This ultimate dependence of GSH status on Cbl status will have a direct impact on NO regulation because cellular thiol status has a major impact on NO species formation. The induction of iNOS is thus linked to the induction of GSH synthesis Citation[237]. In the presence of thiol, s‐nitrosation is preferential to O‐ or N‐nitrosation Citation[238], and, in health and a healthy immune response, s‐nitrosothiols predominate over other nitrosated forms. Moreover, GSH and other thiols limit NO/O2− interactions that produce excessive RNIS and ROS resulting in lipid peroxidation, excess S‐H oxidation and other detrimental consequences Citation[64], Citation[84], Citation[85], Citation[240]. For example, ONOO− formed by NO/O2− interaction can react with GSH to give s‐nitroglutathione (GSNO2), which decomposes spontaneously to NO Citation[241]. So the normal Cbl‐GSH‐dependent preponderance of RS‐NOs over RNIS favours the positive aspects of NO regulation over the pathologies of NO dysregulation. The brilliantly host advantageous deployment in the Cbl–GSH–NO relationship of the potentially dangerous dual aspects of NO can be well illustrated by what happens in bacteriostasis/phagocytosis. Many bacteria contain similar defence systems to eukaryotes Citation[86], such as SOD, GSH Citation[242]/GPX and Cbl, in addition to an NO reductase, and G6PDH. During bacteriostasis and phagocytosis, however, neutrophils secrete large quantities of TCI, along with myeloperoxidase in the secondary granules. These TCIs act as magnets that leach Cbl from bacteria [243] (analogously to lactoferrin and iron), which means that bacteria are effectively disarmed, bereft of the back‐up system that sustains their G6PDH and other enzymatic activity, in particular, their GSH/GPX, which is rapidly consumed in the increased acidity of the phagocytic burst and exposure to high levels of NO/ONOO−/O2−/H2O2 from the host. This lethal assault also affects the host's defences, with a 25% utilization of GSH in the first 10 min of phagocytosis Citation[82]. However, the host is well armed to regenerate and synthesize GSH de novo, with significant amounts of AdoCbl on TCIs, in situ, not to mention incoming extra supplies from the phagocytosed pathogen. It has also been noted that s‐nitrosothiols, such as GSNO, or s‐nitrosocysteine, have a significantly more potent virustatic, parasiticidal, and bactericidal activity than NO itself, with trans‐s‐nitrosation implicated Citation[81]. The regeneration of GSH, ultimately dependent on Cbl, is critical in this respect. One way or another it seems, the effects of NO cannot be subtracted from the effects of GSH and Cbl and when they are in balance, when Cbl status is sufficient, NO, GSH and Cbl may continuously regulate the NOS.
Acknowledgements
The author wishes to thank Hilary Boyd for help with the preparation of the figures and the manuscript and Professor Mauro Perretti for helpful discussion.
Related Research Data
References
- Wheatley C. A Scarlet Pimpernel for the resolution of inflammation? The role of supra‐therapeutic doses of cobalamin, in the treatment of systemic inflammatory response syndrome (SIRS), sepsis, severe sepsis, and septic or traumatic shock. Med Hypotheses 2006; 67: 124–42
- Wheatley C. A unified theory of the causes of monoclonal gammopathy of unknown significance (MGUS) and multiple myeloma, with a consequent treatment proposal for long‐term control and possible cure. J Orthomol Med 2002; 17: 7–16
- Olesen H., Andersen M. P., Amris A. Serum vitamin B12 binding capacity in patients with anaemia. Scand J Haematol 1968; 5: 235–40
- Seetharam B., Li N. Transcobalamin II and its cell surface receptors. Vitam Horm 2000; 59: 337–66
- Ghosh K., Mohanty D., Rana K. S., et al. Plasma transcobalamins in haematological disorders. Folia Haematol Int Mag Klin Morphol Blutforsch 1986; 113: 766–75
- Christensen P. A., Brynskov J., Gimsing P., Petersen J. Vitamin B12 binding proteins (transcobalamin and haptocorrin) in serum and synovial fluid of patients with rheumatoid arthritis and traumatic synovitis. Scand J Rheumatol 1983; 12: 268–72
- Lasser U., Kierat L., Grob P., et al. Transcobalamin II, a serum protein reflecting autoimmune disease activity, its plasma dynamics, and the relationship to established serum parameters in systemic lupus erythematosus. Clin Immunol Immunopathol 1985; 36: 345–57
- Rachmilewitz D., Ligumsky M., Rachmilewitz B., et al. Transcobalamin II level in peripheral blood monocytes – a biochemical marker in inflammatory diseases of the bowel. Gastroenterology 1980; 78: 43–6
- Nexø E., Olesen H. Intrinsic factor, transcobalamin, and haptocorrin. B12, biochemistry and medicine., D Dolphin. Wiley‐Interscience, New York 1982; 57–85
- Macdonald C., Bessent R., Adams J. Transcobalamin‐binding capacities in vitamin B12‐related diseases. Am J Clin Pathol 1981; 75: 677–83
- Carmel R., Eisenberg L. Serum B12 and transcobalamin abnormalities in people with cancer. Cancer 1977; 40: 1348–53
- Carmel R., Hollander H. Extreme elevations of transcobalamin II levels in multiple myeloma and other disorders. Blood 1978; 51: 1057–63
- Rachmilewitz B., Sulkes A., Rachmilewitz M., Fuks Z. Serum transcobalamin II levels in breast carcinoma patients. Isr J Med Sci 1981; 17: 874–8
- Baeuerle P., Baltimore D. Meeting review. NFκB ten years after. Cell 1996; 87: 13–20
- Kleinwort H., Schwarz P. M., Förstermann U. Regulation of the expression of inducible nitric oxide synthase. J Biol Chem 2003; 384: 1343–64
- Wagener F. A. D. T. G., Volk H‐D., Willis D. Different faces of the heme–heme oxygenase system in inflammation. Pharmacol Rev 2003; 55: 551–71
- Wheatley C., Perretti M., D'Acquisto F., Sampaio A. L. F., et al. (in preparation). 2006
- Perretti M., Wheatley C., D'Acquisto F., Sampaio A. L. F., et al. (in preparation). 2007
- Griscavage J. M., Fukuto J. M., Komori Y., Ignarro L. J. Nitric oxide inhibits neuronal nitric oxide synthase by interacting with the heme prosthetic group. J Biol Chem 1994; 269((34))21644–9
- Albakri Q. A., Stuehr D. J. Intracellular assembly of inducible NO synthase is limited by nitric oxide – mediated changes in heme insertion and availability. J Biol Chem 1996; 271((10))5414–21
- Peng H‐B., Spiecker M., Liao J. K. Inducible nitric oxide: an autoregulatory feedback inhibitor of vascular inflammation. J Immunol 1998; 161: 1970–6
- Mathews J. R., Botting C. H., Panico M., Morriss H. R., Hay R. T. Inhibition of NFκB binding by nitric oxide. Nucleic Acids Res 1996; 24: 2236–42
- Markle H. V. Cobalamin. Crit Rev Clin Lab Sci 1996; 33: 247–56
- Chemistry and biochemistry of B12., R Banerjee. Wiley Interscience, New York 1999; 1–921
- Nexø E. Cobalamin binding proteins. Vitamin B12 and B12 proteins, B Kräutler, D Arigoni, B. T Golding. Wiley, La Jolla 1998; 461–75
- Beck W. S. Biological and medical aspects of vitamin B12. B12, biochemistry and medicine., D Dolphin. Wiley‐Interscience, New York 1982; 1–30
- Seetharam B. Receptor‐mediated endocytosis of cobalamin (vitamin B12). Annu Rev Nutr 1999; 19: 173–95
- Ostray F., Gams R. A. Cellular fluxes of vitamin B12. Blood 1997; 50: 877–88
- Moestrup S. K. Cellular surface receptors important for vitamin B12 nutrition. Vitamin B12 and B12 proteins., B Kräutler, D Arigoni, B. T Golding. Wiley, La Jolla 1998; 477–89
- Carmel R. The distribution of endogenous cobalamin among cobalamin‐binding proteins in the blood in normal and abnormal states. Am J Clin Nutr 1985; 41: 713–9
- Zittoun J., Marquet J., Zittoun R. The intracellular content of the three transcobalamins at various stages of normal and leukaemic myeloid cell development. Br J Haematol 1975; 31: 299–310
- Herbert V. B12 deficiency in AIDS. J Am Med Assoc 1988; 260: 2837
- Carmel R., Eisenberg L. Serum B12 and transcobalamin abnormalities in people with cancer. Cancer 1977; 40: 1348–53
- Lindenbaum J., Rosenberg I. H., Wilson P. W., et al. Prevalence of cobalamin deficiency in the Framingham elderly population. Am J Clin Nutr 1994; 60: 2–11
- Alberti C., Brun‐Buisson C., Burchardi H., et al. Epidemiology of sepsis and infection in ICU patients from an international multicentre cohort study. Intensive Care Med 2002; 28: 108–21
- Brasch N. E., Hsu T., Doll K. M., Finke R. G. Synthesis and characterization of isolable thiolatocobalamin complexes relevant to coenzyme B12‐dependent ribonucleotide triphosphate reductase. J Inorg Biochem 1999; 76: 197–209
- Suto R. K., Brasch N. E., Anderson O. P., Finke R. G. Synthesis, characterization, solution stability and X‐ray crystal structure of the thiolatocobalamin γ‐glutamylcysteinylcobalamin, a dipeptide analog of glutathionylcobalamin: insights into the enhanced Co‐S bond stability of the natural product glutathionylcobalamin. Inorg Chem 2001; 40: 2686–92
- Xia L., Cregan A. G., Berben L. A., Brasch N. E. Studies on the formation of glutathionylcobalamin: any free intracellular aquacobalamin is likely to be rapidly and irreversibly converted to glutathionylcobalamin. Inorg Chem 2004; 43: 6848–57
- Dubnoff J. W. A cobalamin glutathione complex. Biochem Biophys Res Commun 1964; 16((5))484–8
- Adler N., Medwick T., Poznanski T. J. Reaction of hydroxocobalamin with thiols. J Am Chem Soc 1966; 88((21))5018–20
- Jacobsen D. W., Lee‐Denison C., Luce K., Green R. Glutathionylcobalamin (GSCbl) is found in cultured and ascites leukemia‐L1210 cells. Fed Proc 1987; 46: 1005
- Pezacka E., Green R., Jacobsen D. W. Glutathionylcobalamin as an intermediate in the formation of cobalamin coenzymes. Biochem Biophys Res Comm 1990; 169((2))443–50
- Kim J. H., Demaurex N., Grinstein S. Intracellular pH: measurement, manipulation and physiological regulation. Handbook of biological physics, W. N Koning, H. R Kaback, J. S Lolkema. Elsevier Science, Amsterdam 1996; 2: 447–72
- Luschinsky Drennan C., Huang S., Drummond J. T., Matthews R. G., Ludwig M. L. How a protein binds B12: a 3.0Å X‐ray structure of B12‐binding domains of methionine synthase. Science 1994; 266: 1669–74
- Mancia F., Keep N. H., Nakagawa A., et al. How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl‐coenzyme A mutase at 2Å resolution. Structure 1996; 4((3))339–49
- Sirovatka J. M., Finke R. G. Coenzyme B12 chemical precedent studies: probing the role of the imidazole base‐on motif found in B12‐dependent methylmalonyl‐CoA mutase. J Am Chem Soc 1997; 119: 3057–67
- Poston J. M. β‐leucine and the β‐keto pathway of leucine metabolism. Adv Enzymol 1986; 58: 173–89
- Stabler S. P., Lindenbaum J., Allen R. H. Failure to detect β‐leucine in human blood or leucine 2, 3‐aminomutase in rat liver using capillary gas chromatography‐mass spectrometry. J Biol Chem 1988; 263((12))5581–8
- Metz J., Kelly A., Chapin Swett V., Waxman S., Herbert V. Deranged DNA synthesis by bone marrow from vitamin B12 deficient humans. Br J Haematol 1968; 14: 575–91
- Mason J. B., Levesque T. Folate: effects on carcinogenesis and the potential for cancer chemoprevention. Oncology 1996; 10: 1727–43
- Beattie D. S. Bioenergetics and oxidative metabolism. Textbook of biochemistry, with clinical correlations, T. M Devlin. Wiley‐Liss, New York 2006; 531–80
- Parker J. N., Parker P. M. Vitamin B12, a medical dictionary, bibliography, and annotated research guide to Internet references. 2004; 1–344
- Riou B., Gérard J. L., Drieu la Rochelle C., et al. Hemodynamic effects of hydroxocobalamin in conscious dogs. Anaesthesia 1991; 74: 552–8
- Rajanayagam M. A. S., Li C. G., Rand M. J. Differential effects of hydroxocobalamin on NO‐mediated relaxations in rat aorta and anococcygeus muscle. Br J Pharmacol 1993; 108: 3–5
- Greenberg S. S., Zie J., Zatarain J. M., et al. Hydroxocobalamin (vitamin B12) prevents and reverses endotoxin‐induced hypertension and mortality in rodents: role of nitric oxide. J Pharmacol Exp Ther 1995; 273: 257–65
- Kruszyna R., Kruszyna H., Smith R. P., Thron C. D., Wilcox D. E. Nitrite conversion to nitric oxide in red cells and its stabilisation as a nitrosylated valency hybrid of hemoglobin. J Pharmacol Exp Ther 1987; 241: 307–13
- Rochelle L. G., Morana S. J., Kruszyna H., et al. Interactions between hydroxocobalamin and nitric oxide (NO): evidence for a redox reaction between NO and reduced cobalamin and reversible NO binding to oxidized cobalamin. J Pharmacol Exp Ther 1995; 275: 48–52
- Brouwer M., Chamulitrat W., Ferruzzi G., Sauls D. L., Weinberg J. B. Nitric oxide interactions with cobalamins: biochemical and functional consequences. Blood 1996; 88((5))1857–64
- Zheng D., Yan L., Birke R. L. Electrochemical and spectral studies of the reactions of aquacobalamin with nitric oxide and nitrite ion. Inorg Chem 2003; 41: 2548–55
- Kruszyna H., Magyar J., Rochelle L. G. Spectroscopic studies of nitric oxide (NO) interactions with cobalamins: reaction of NO with superoxocobalamin(II) likely accounts for cobalamin reversal of the biological effects of NO. J Pharmacol Exp Ther 1998; 285: 665–71
- Wolak M., Stochel G., Hamza M., Van Eldik R. Aquacobalamin (vitamin B12a) does not bind NO in aqueous solution. Nitrite impurities account for observed reaction. Inorg Chem 2000; 39: 2018–9
- Zheng D., Birke R. L. The reaction of nitric oxide with glutathionylcobalamin. J Am Chem Soc 2002; 124: 9066–7
- Danishpajooh I. O., Gudi T., Yongchang C., et al. Nitric oxide inhibits methionine synthase activity in vivo and disrupts carbon flow through the folate pathway. J Biol Chem 2001; 276((29))27296–303
- Stamler J. S., Singel D. J., Loscalzo J. Biochemistry of nitric oxide and its redox activated forms. Science 1992; 258: 1898–902
- Bauer J., Morrison B. H., Grane R., et al. Effects of interferon β on transcobalamin II‐receptor expression and anti‐tumor activity of nitrosylcobalamin. J Natl Cancer Inst 2002; 94((13))1010–9
- Mushett C. W., Kelly K. L., Boxer G. E., et al. Antidotal efficacy of vitamin B12 (hydroxocobalamin) in experimental cyanide poisoning. Proc Soc Exp Biol Med 1952; 81: 234–7
- Traina V. Vitamin B12 as an anti‐anaphylactic. Nature 1950; 166: 78–9
- Badyuzin I. S. The anti‐shock activity of large doses of cyanocobalamin. Byull Eksp Biol Med 1965; 60: 62–4
- Posner M. A., Rodkey F. L., Tobey R. E. Nitroprusside‐induced cyanide poisoning: antidotal effect of hydroxocobalamin. Anaesthesia 1976; 44: 330–5
- Mitrofanov V. G. The pathogenesis of traumatic shock and its complication with radiation sickness. 1959, [in Russian]
- Lazarev N. V., Vishniakov S. M. Problem of increasing resistance of the organism to operative trauma by medical means. Vestn Khir 1957; 79((11))19–23, [in Russian]
- Yacoub M., Faure H., Morena M., et al. L'intoxication cyanhydrique aiguë. Données actuelles sur le métabolisme du cyanure et le traitement par hydroxocobalamine. J Europ de Toxicol 1974; 7: 22–9
- Motin J., Bouletrean P., Rouzious J. M. Intoxication cyanhydrique grave traitée avec succès par hydroxocobalamine. J Med de Strasbourg 1970; I: 717–22
- Tassan H., Joyon D., Richard T., et al. Intoxication au cyanure de potassium traitée par l'hydroxocobalamine. Ann Fr Anesth Reanim 1990; 9: 383–5
- Brouard A., Blaisot B., Bismuth C. Hydroxocobalamine in cyanide poisoning. J Toxicol Clin Exp 1987; 7: 155–68
- Hall A. H., Rumack B. H. Hydroxocobalamin/sodium thiosulfate as a cyanide antidote. J Emerg Med 1987; 5: 115–21
- Forsyth J. C., Mueller P. D., Becker C. E., et al. Hydroxocobalamin as a cyanide antidote: safety, efficacy and pharmacokinetics in heavily smoking normal volunteers. Clin Toxicol 1993; 31: 277–94
- Wheatley C. A novel approach to the treatment of septic shock?. J Nutr Environ Med 2004; 14((1))56–7
- Nathan C. Points of control in inflammation. Nature, Inflammation 2002; 420: 846–52
- Henson P. M. Editorial: Get the balance right. Dampening inflammation. Nature Immunol 2005; 6: 1177–81
- MacMicking J., Xie Q., Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol 1997; 15: 323–50
- Hamers M. N., Roos D. Oxidative stress in human neutrophilic granulocytes: host defence and self‐defence. Oxidative stress., H Sies. Academic Press, London 1985; 351–81
- Flohé L., Beckmann R., Giertz H., . Oxygen‐centered free radicals as mediators of inflammation. Oxidative stress., H Sies, et al. Academic Press, London 1985; 403–36
- Deora A. A., Lauder H. M. Role of nitric oxide and other radicals in signal transduction. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 251–63
- Darley‐Usmar V. M., Patel R. P., O'Donnell V. B., Freeman B. A. Antioxidant actions of nitric oxide. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 265–76
- Stamler J. S., Lamas S., Fang F. C. Meeting review. Nitrosylation: the prototypic redox‐based signaling mechanism. Cell 2001; 106: 675–83
- Sies H. Oxidative stress: introductory remarks. Oxidative stress., H Sies. Academic Press, London 1985; 1–8
- Oxidative stress and the molecular biology of antioxidant defenses., J. G Scandalios. Cold Spring Harbour, New York 1997
- Wolin M. S., Mohazzab‐H K. M. Mediation of signal transduction by oxidants. Oxidative stress and the molecular biology of antioxidant defenses., J. G Scandalios. Cold Spring Harbour, New York 1997; 21–48
- Chance B., Sies H., Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev 1979; 59: 527–605
- Meister A. On the biochemistry of glutathione. Glutathione centennial, molecular perspectives and clinical implications., N Tamiguchi, T Higashi, Y Sakanoto, A Meister. Academic Press, New York 1989; 1–21
- Peunora N., Enikolopov G. Amplification of calcium‐induced gene transcription by nitric oxide in neuronal cells. Nature 1993; 364: 450–3
- Greenberg D. A. Bilirubin and the brain. Sci Med 2003; 9: 96
- Schwarz N. B. Carbohydrate metabolism II: special pathways and glycoconjugates. Textbook of biochemistry, with clinical correlations., T. M Devlin. Wiley‐Liss, New York 2006; 637–60
- Borovikova L. V., Lvanona S., Zhang M., et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000; 405: 458–62
- Damazo A. S., Yona S., D'Acquisto F., Flower R. J., Oliani S. M., Perretti M. Critical protective role for Annexin I gene expression in the endotoxemic murine microcirculation. Am J Pathol 2005; 166: 1607–17
- Endres S., Ghorbani R., Kelley V. E., et al. The effect of dietary supplementation with n‐3 polyunsaturated fatty acids on the synthesis of interleukin‐1 and tumor necrosis factor by mononuclear cells. New Engl J Med 1989; 320: 265–71
- Serhan C. N., Gotlinger K., Hong S., et al. Anti‐inflammatory actions of neuroprotectin DI/protectin DI and its natural stereoisomers: assignments of dihydroxy‐containing docosatrienes. J Immunol 2006; 176: 1848–59
- Serhan C. N., Clish C. B., Brannon J., et al. Novel functional sets of lipid‐derived mediators with anti‐inflammatory actions generated from omega‐3 fatty acids via cyclooxygenase 2–nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med 2000; 192: 1197–204
- Ramstedt U., Janet N. G., Wigzell H., Serhan C. N., Samuelson B. Action of novel eicosanoids lipoxin A and B on human natural killer cell toxicity: effects on intracellular cAMP and target cell binding. J Immunol 1985; 135: 3434–8
- Ajuebor M. N., Das A. M., Virag L., Flower R. J., Szabó C., Perretti M. Role of resident peritoneal macrophages and mast cells in chemokine production and neutrophil migration in acute inflammation: evidence for an inhibitory loop involving endogenous IL‐10. J Immunol 1999; 162: 1685–91
- Wahl S. M. Regulation of tissue inflammation, repair, and fibrosis by transforming growth factor beta. Epidermal growth factors and cytokines., T. A Luger, T Schwarz. Marcel Dekker, New York 1994; 241–52
- Lawrence T., Gilroy D. W., Colville‐Nash P. R., Willoughby D. A. Possible new role for NFκB in the resolution of inflammation. Nature Med 2001; 7((12))1291–7
- Llesuy S., Evelson P., Gonzalez‐Flecha B., et al. Oxidative stress in muscle and liver of rats with septic syndrome. Free Radic Biol Med 1994; 16: 445–51
- Poderoso J. J., Carreras M. C., Lisdero C., et al. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 1996; 328((1))85–92
- Ruetten H., Thiemermann C. Nitric oxide and septic shock. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 747–57
- Vincent J. L., Zhang H., Szabo C., Preiser J. C. Effects of nitric oxide in septic shock. Am J Respir Crit Care Med 2000; 161: 1781–5
- Evans T. J., Cohen J. Mediators: nitric oxide and other toxic oxygen species. Pathology of septic shock., E. T Rietschel, H Wagner. Springer, Berlin 1996; 189–208
- Schmidt H. W., Hofmann H., Schindler U., et al. No •NO from NO synthase. Proc Natl Acad Sci 1996; 93: 14492–7
- Vallance P. Assessment of nitric oxide in humans. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 923–4
- Fukuto J. M., Cho J. Y., Switzer C. H. The chemical properties of nitric oxide and related nitrogen oxides. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 23–40
- Weinberg J. B., Lang T., Wilkinson W. E., Pisetsky D. S., St Clair W. E. Serum, urinary and salivary nitric oxide in rheumatoid arthritis: complexities of interpreting nitric oxide measures. Arthritis Res Ther 2006; 8: R140
- Coomes M. W. Amino acid metabolism. Textbook of biochemistry, with clinical correlations., T. M Devlin. Wiley‐Liss, New York 2006; 743–87
- Kaeseler Andersen S., Gjedsted J., Christiansen C., Tonnesen E. The roles of insulin and hyperglycemia in sepsis pathogenesis. J Leukocyte Biol 2004; 75: 413–21
- Kilbourn R. G., Jubran A., Gross S. S., et al. Reversal of endotoxin mediated shock by NG‐methyl‐l‐arginine, an inhibitor of nitric oxide synthesis. Biochem Biophys Res Commun 1990; 172: 1132–8
- Florquin S., Amraoui Z., Dubois C., Decuyper J., Goldman M. The protective role of endogenously synthesized nitric oxide in staphylococcal enterotoxin B‐induced shock in mice. J Exp Med 1994; 180: 1153–8
- Kamijo R., Harada H., Matsuyama M., et al. Requirement for transcription factor IRF‐1 in NO synthase induction in macrophages. Science 1994; 263: 1612–5
- Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J 1992; 6: 3051–64
- Vodovotz Y., Kopp J. B., Takeguchi H., et al. Increased mortality, blunted production of nitric oxide, and increased production of TNF‐alpha in endotoxemic TGF‐beta1 transgenic mice. J Leukocyte Biol 1998; 63: 31–9
- Peracchi M., Bamonti Catena F., Pomati M., et al. Human cobalamin deficiency: alterations in serum tumour necrosis factor‐α and epidermal growth factor. Eur J Haematol 2001; 67: 123–7
- Wang S., Wang W., Wesley R. A., Danner R. L. A Sp1 binding site of the TNFα promoter functions as a nitric oxide response element. J Biol Chem 1999; 274: 33190–3
- Pardali K., Kurisaki A., Moren A. Role of Smad proteins and transcription factor Sp1 in p21 (Waf1/Cip1) regulation of TGFβ. J Biol Chem 2000; 275: 29244–56
- Merchant J. L., Shiotani A., Mortensen E. R., et al. Epidermal growth factor stimulation of the human gastrin promoter requires Sp1. J Biol Chem 1995; 270: 6314–9
- Reinhart K., Karzai W. Anti‐tumor necrosis factor therapy in sepsis: update on clinical trials and lessons learned. Crit Care Med 2001; 29: S121–5
- Laubach V. E., Shesely E. G., Smithies O., Sherman P. A. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide‐induced death. Proc Natl Acad Sci 1995; 92: 10688–92
- MacMicking J. D., Nathan C., Hom G., et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 1995; 81: 641–50
- McCaddon A., Regland B., Hudson P., Davies G. Functional vitamin B12 deficiency and Alzheimer disease. Neurology 2002; 58: 1395–9
- Hobbs M. R., Udhayakumar V., Levesque M., et al. A new NOS2 promoter polymorphism associated with increased nitric oxide production and protection from severe malaria in Tanzanian and Kenyan children. Lancet 2002; 360: 1468–75
- Nathan C. Natural resistance and nitric oxide. Cell 1995; 82: 873–6
- Force R. W., Nahata M. C. Effect of histamine H2‐receptor antagonists on vitamin B12 absorption. Ann Pharmacother 1992; 10: 1283–6
- Ruscin M. J., Page R. L., Valuck R. J. Vitamin B12 deficiency associated with histamine 2‐receptor antagonists and a proton‐pump inhibitor. Ann Pharmacother 2002; 36: 812–6
- Katusic Z. S. Superoxide anion and endothelial regulation of arterial tone. Free Radic Biol Med 1996; 20: 443–8
- Kinoshita H., Tsutsui M., Milstien S., Katusic Z. S. Tetrahydrobiopterin, nitric oxide and regulation of cerebral vascular tone. Prog Neurobiol 1997; 52: 295–302
- Mayer B., Hemmens B. Biosynthesis and action of nitric oxide in mammalian cells. Trends Biochem Sci 1997; 22: 477–81
- Rusche K. M., Spiering M. M., Marletta M. A. Reactions catalyzed by tetrahydrobiopterin‐free nitric oxide synthase. Biochemistry 1998; 37: 15503–12
- Alderton W. K., Cooper C. E., Knowles R. G. Nitric oxide synthases: structure, function, and inhibition. Biochem J 2001; 357: 593–615
- Ma X. L., Gao F., Liu G‐L., et al. Opposite effects of nitric oxide and nitroxyl on postischemic myocardial injury. Proc Natl Acad Sci 1999; 96: 14617–22
- Stamler J. S. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell 1994; 78: 931–6
- Wolin M. S. Mechanisms through which reactive nitrogen and oxygen species interact with physiological signaling systems. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 277–92
- Kong S‐K., Bin Yim M., Stadtman E. R., Boon Chock F. Peroxynitrite disables the tyrosine phosphorylation regulatory mechanism: lymphocyte‐specific tyrosine kinase fails to phosphorylate nitrated cdc2(6‐20) NH2 peptide. Proc Natl Acad Sci 1996; 93: 3377–82
- Kleinert H., Boissel J. P., Schwarz P. M., Förstermann U. Regulation of the expression of nitric oxide synthase isoforms. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 105–28
- Groves J. T., Wang C. C‐Y. Nitric oxide synthase: models and mechanisms. Curr Opin Chem Biol 2000; 4: 687–95
- Moncada S. Cited in Stamler JS, Lamas S, Fang FC. Meeting review. Nitrosylation: the prototypic redox‐based signaling mechanism. Cell 2001; 106: 675–83
- Wang Y., Newton D. C., Robb G. B., et al. RNA diversity has profound effects on the translation of neuronal nitric oxide synthase. Proc Natl Acad Sci 1999; 96: 12150–5
- Mannick J. B., Asano K., Izumi K., Kieff E., Stamler J. S. Nitric oxide produced by human B lymphocytes inhibits apoptosis and Epstein–Barr virus reactivation. Cell 1994; 79: 1137–46
- Casadei B., Sears C. E. Nitric oxide‐mediated regulation of cardiac contractility and stretch responses. Prog Biophys Mol Biol 2003; 82: 67–80
- Palamalai V., Darrow R. M., Organisciak D. T., Miyagi M. Light‐induced changes in protein nitration in photoreceptor rod outer segments. Mol Vis 2006; 12: 1543–51
- Koniaris L. G., McKillop L. A., Schwartz S. I., Zimmers T. A. Liver regeneration. J Am Coll Surg 2003; 197: 634–9
- Guo F. H., De Raeve H. R., Stuehr D. J., et al. Continuous nitric oxide synthesis by inducible nitric oxide synthase in normal human airway epithelium in vivo. Proc Natl Acad Sci 1995; 92: 7809–13
- Hoffman R. A., Zhang G., Nussler N. C., et al. Constitutive expression of inducible nitric oxide synthase in the mouse ileal mucosa. Am J Physiol 1997; 272: G383–92
- Gaston B., Reilly J., Drazen J. M., et al. Endogenous nitrogen oxides and bronchodilator s‐nitrosothiols in human airways. Proc Natl Acad Sci 1993; 90: 10957–61
- Crane B. R., Arvai A. S., Gachui R., et al. The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science 1997; 278: 425–31
- Crane B. R., Arvai A. S., Ghosh D. K., et al. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science 1998; 279: 2121–6
- Fischmann T. O., Hruza A., Niu X. D., et al. Structural characterization of nitric oxide synthase isoforms reveals striking active‐site conservation. Nat Struct Biol 1999; 6: 233–42
- Li H., Raman C. S., Glaser C. B., et al. Crystal structures of zinc‐free and ‐bound heme domain of human inducible nitric oxide synthase. Implications for dimer stability and comparison with endothelial nitric oxide synthase. J Biol Chem 1999; 274: 21276–84
- Raman C. S., Li H., Martasek P., Kral V., Masters B. S., Poulos T. L. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell 1998; 95: 939–50
- Tierney D. L., Huang H., Martasek P., et al. ENDOR studies of l‐arginine and N‐G‐hydroxyl‐l‐arginine bound to all three holo‐nitric oxide synthase isoenzymes. J Am Chem Soc 2000; 122: 5405–6
- Ganster R. W., Geller D. A. Molecular regulation of inducible nitric oxide synthase. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 129–56
- Hobbs A. J., Fukuto J. M., Ignarro L. J. Formation of free nitric oxide from l‐arginine by nitric oxide synthase; direct enhancement of generation by superoxide dismutase. Proc Natl Acad Sci 1994; 91: 10992–6
- Murphy M. E., Sies H. Reversible conversion of nitroxyl anion to nitric oxide by superoxide dismutase. Proc Natl Acad Sci 1991; 88: 10860–4
- Miller R. T., Martasek P., Roman L. J. Involvement of the reductase domain of neuronal nitric oxide synthase in superoxide anion production. Biochemistry 1997; 36: 15277–84
- Xia Y., Zweier J. L. Direct measurement of nitric oxide generation from nitric oxide synthase. Proc Natl Acad Sci 1997; 94: 12705–10
- Vasquez‐Vivar J., Hogg N., Martasek P., et al. Tetrahydrobiopterin‐dependent inhibition of superoxide generation from neuronal nitric oxide synthase. J Biol Chem 1999; 274: 26736–42
- Vasquez‐Vivar J., Kalyanaraman B., Martasek P., et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci 1998; 95: 9220–5
- Rusche K. M., Spiering M. M., Marletta A. A. Reactions catalysed by tetrahydrobiopterin‐free nitric oxide synthase. Biochemistry 1998; 37: 15503–12
- List B. M., Klosch B., Volker C., et al. Characterization of bovine endothelial nitric oxide synthase as a homodimer with down‐regulated uncoupled NADPH oxidase activity; tetrahydrobiopterin binding kinetics and role of haem in dimerization. Biochem J 1997; 323: 159–65
- Stroes E., Hijmering M., Van Zandvoort M., et al. Origin of superoxide production by endothelial nitric oxide synthase. FEBS Lett 1998; 438: 161–4
- Xia Y., Roman L. J., Masters B. S., Zweier J. L. Inducible nitric oxide synthase generates superoxide from the reductase domain. J Biol Chem 1998; 273: 22635–9
- Beckman J. S., Beckman T. W., Chen J., Marshall P. A., Freeman B. A. Apparent hydroxyl radical production by peroxynitrite; implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci 1990; 87: 1620–4
- Gross S., Levi R. Tetrahydrobiopterin synthesis. J Biol Chem 1992; 267: 25722–9
- Cosentino F., Patton S., d'Uscio L. V., et al. Tetrahydrobiopterin alters superoxide and nitric oxide release in prehypertensive rats. J Clin Invest 1998; 101: 1530–7
- Stroes E., Kastelein J., Cosentino F., et al. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest 1997; 99: 41–6
- Wever R. M. F., van Dam T., van Rijn H. J. M., de Groot P. G., Rabeliak T. J. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthesis. Biochem Biophys Res Comm 1997; 237: 340–4
- Chen X., Reynolds E. R., Ranganayakulu G., O'Donnell J. M. A maternal product of the Punch locus of Drosophila melanogaster is required for precellular blastoderm nuclear division. J Cell Sci 1994; 107: 3501–13
- Gross S. S., Jones L. J., Hattori Y., Raman C. S. Tetrahydrobiopterin: an essential cofactor of nitric oxide synthase with an elusive role. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 167–86
- Visconti M., Karmer P. Fluoresziende Stoffe aus Drosophila melanogaster. Helv Chim Acta 1957; 40: 986
- Cory G. J. Purine and pyrimidine nucleotide metabolism. Textbook of biochemistry, with clinical correlations., T. M Devlin. Wiley‐Liss, New York 2006; 790–822
- Zhuo S., Fan S., Kaufman S. Effects of depletion of intracellular tetrahydrobiopterin in murine erythro‐leukemia cells. Exp Cell Res 1996; 222: 163–70
- Werner‐Felmayer G., Goldferer G., Werner E. R., Gröbner P., Wachter H. Pteridine biosynthesis and nitric oxide synthase in Physarum polycephalum. Biochem J 1994; 304: 105–11
- Wachter H., Fuchs D., Reibnegger G., Werner E. R. Neopterin as a marker for activation of cellular immunity: immunologic basis and clinical application. Adv Clin Chem 1989; 27: 81–141
- Krehan A., Ansuini H., Böcher O., et al. Transcription factors, Ets‐1, NFκB, and Sp1 are major determinants of the promoter activity of the human protein kinase CK2α gene. J Biol Chem 2000; 275: 18327–36
- Szabo C., Mitchell J. A., Thiemermann C., Vane J. R. Nitric oxide‐mediated hyporeactivity to noradrenaline precedes the induction of nitric oxide synthase in endotoxin shock. Br J Pharmacol 1993; 108((3))786–92
- Hoffer A. The effect of adrenochrome and adrenolutin on the behaviour of animals and the psychology of man. Int Rev Neurobiol 1962; 4: 307–71
- Hoffer A. Adrenochrome and blood plasma. Am J Psychiatr 1958; 114: 752–3
- Tracey K. J. The inflammatory reflex. Nature, Inflammation 2002; 420: 853–9
- Bolton C. F., Young G. B., Zochodne D. W. Neurological changes during severe sepsis. Current topics in intensive care., H Burchardii, G Dobb, J Biou, R. F Dellinger. WB Saunders, London 1994; 180–218
- Cohen J. The immunopathogenesis of sepsis. Nature, Inflammation 2002; 420: 885–9
- Scalabrino G., Carpo M., Bamonti F., et al. High tumor necrosis factor‐α levels in cerebrospinal fluid of cobalamin‐deficient patients. Ann Neurol 2004; 56: 886–90
- Brealey D., Brand M., Hargreaves I., et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360: 219–23
- Morris S. M. Regulation of arginine availability and its impact on NO synthesis. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 187–97
- Deitch E. A., Specian R. D., Berg R. D. Endotoxin‐induced bacterial translocation and mucosal permeability: role of xanthine oxidase, complement activation and macrophageal products. Crit Care Med 1991; 19: 785–91
- Awad W. M. Iron and heme metabolism. Textbook of biochemistry, with clinical correlations., T. M Devlin. Wiley‐Liss, New York 2006; 824–47
- Wang P., Zheng F., Chaudry I. H. Endothelium‐dependent relaxation is depressed at the macro‐ and micro‐circulatory levels during sepsis. Am J Physiol 1995; 269: R988–94
- Sun J., Xin C., Eu J. P., Stamler J. S., Meissner G. Cysteine 3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc Natl Acad Sci 2001; 98: 11158–62
- Schmidt H. H. H. W., Walter U. NO at work. Cell 1994; 78: 919–25
- Ignarro L. J. Biological actions and properties of endothelium‐derived nitric oxide formed and released from artery and veins. Circ Res 1989; 65: 1–21
- Ignarro L. J. Introduction and overview. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 3–19
- Salvemini D., Misko T. P., Masferrer J. L., et al. Nitric oxide activated cycloxygenase enzymes. Proc Natl Acad Sci 1993; 90: 7240–4
- Fukoto J. M., Wallace G. C., Hszieh R., Chandhuri G. Chemical oxidation of N‐hydroxyguanidine compounds. Release of nitric oxide, nitroxyl and possible relationship to the mechanism of biological nitric oxide generation. Biochem Pharmacol 1992b; 43: 607–13
- Murad F. The nitric oxide‐cyclic GMP signal transduction system for intracellular and intercellular communication. Rec Prog Horm Res 1994; 49: 239–48
- Boveris A., Poderoso J. J. Regulation of oxygen metabolism by nitric oxide. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 355–68
- Khatsenko O. G., Gross S. S., Rifkind A. B., Vane J. R. Nitric oxide is a mediator of the decrease in cytochrome P450‐dependent metabolism caused by immunostimulants. Proc Natl Acad Sci 1993; 90: 11147–51
- Thomas S. R., Mohr D., Stocker R. Nitric oxide inhibits indolamine 2, 3‐dioxygenase activity in interferon‐primed mononuclear phagocytes. J Biol Chem 1994; 269: 14457–64
- Molina J., Vedia L., MacDonald B. R., Reep B., et al. Nitric oxide‐induced s‐nitrosylation of glyceraldehyde‐3‐phosphate dehydrogenase inhibits enzymatic activity and increases endogenous ADP‐ribosylation. J Biol Chem 1992; 267: 24929–32
- Zhang J., Snyder S. H. Nitric oxide stimulates auto‐ADP‐ribosylation of glyceraldehyde‐3‐phosphate dehydrogenase. Proc Natl Acad Sci 1992; 89: 9382–5
- Gopalakrishna R., Hai Chen Z., Gundimeda U. Nitric oxide and nitric‐oxide generating agents induce a reversible inactivation of protein kinase C activity and phorbol ester binding. J Biol Chem 1993; 268: 27180–5
- Stamler J. S. S‐nitrosothiols and bioregulatory actions of nitrogen oxides through reactions with thiol groups. Curr Top Microbiol Immunol 1995; 196: 19–36
- Laval F., Wink D. A. Inhibition by nitric oxide of the repair protein, O6‐DNA‐methyltransferase. Carcinogenesis 1994; 15: 443–7
- Melino G., Bernassola F., Knight R. A., et al. S‐nitrosylation regulates apoptosis. Nature 1997; 388: 432–3
- Asahi M., Fujii J., Suzuki K., et al. Inactivation of glutathione peroxidase by nitric oxide. Implication for cytotoxicity. J Biol Chem 1995; 270: 21035–9
- Stamler J. S., Simon D. I., Jaraki O., et al. S‐nitrosylation of tissue‐type plasminogen activator confers vasodilatory and antiplatelet properties on the enzyme. Proc Natl Acad Sci 1992; 89: 8087–91
- Stamler J. S., Jaraki O., Osborne J., et al. Nitric oxide circulates in mammalian plasma primarily as an s‐nitroso adduct of serum albumin. Proc Natl Acad Sci 1992; 89: 7674–7
- Stamler J. S., Simon D. I., Osborne J. A., et al. S‐nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci 1992; 89: 444–8
- Singh S. P., Wishnok J. S., Keshive M., Deen W. M., Tannenbaum S. R. The chemistry of the s‐nitroso‐glutathione/glutathione system. Proc Natl Acad Sci 1996; 93: 14428–33
- Ji Y., Akerboom T. P. M., Sies H., Thomas J. A. S‐Nitrosylation and S‐glutathionylation of protein sulfhydryls by S‐nitroso glutathione. Arch Biochem Biophys 1999; 362: 67–78
- Hausladen A., Fridovichi I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem 1994; 269: 29405–8
- Radi R., Rodriguez M., Castro L., Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys 1994; 308: 89–95
- Scott M., Meshnik S., Eaton J. Superoxide dismutase‐rich bacteria. Paradoxical increase in oxidant toxicity. J Biol Chem 1987; 262: 3640–5
- Bloch C., Ausubel F. Paraquat‐mediated selection for mutations in the manganese‐superoxide dismutase gene sodA. J Bacteriol 1986; 168: 795–8
- Amstad P., Peskin A., Shah G., et al. Paraquat‐mediated selection for mutations in the manganese‐superoxide dismutase gene sodA. Biochemistry 1991; 30: 9305–13
- Mao G., Thomas P., Lopaschuk G., Poznansky M. Superoxide dismutase (SOD)‐catalase conjugates. Role of hydrogen peroxide and the Fenton reaction in SOD toxicity. J Biol Chem 1993; 268: 416–20
- Amstad P., Moret R., Cerutti P. Glutathione peroxidase compensates for the hypersensitivity of Cu, Zn superoxide dismutase overproducers to oxidant stress. J Biol Chem 1994; 269: 1606–9
- Kelner M., Bagnell R. Alteration of endogenous glutathione peroxidase, manganese superoxide dismutase, and glutathione transferase activity in cells transfected with a copper‐zinc superoxide dismutase expression vector. Explanation for variations in paraquat resistance. J Biol Chem 1990; 265: 10872–95
- Staver P. J. Physiology of folate and vitamin B12 in health and disease. Nutr Rev 2004; 62: S3
- Mannervik B., Widersten M., Board P. G. Glutathione‐linked enzymes in detoxication reactions. Glutathione centennial, molecular perspectives and clinical implications., N Tamiguchi, T Higashi, Y Sakanoto, A Meister. Academic Press, New York 1989; 23–34
- Mannervik B., Carlberg I., Larson K. Glutathione: General Review of Mechanism of Action. Glutathione chemical, biochemical and medical aspects, coenzymes and cofactors, D Dolphin, O Avramovic, R Poulson. Wiley, New York 1989; 3A: 476–517
- Wink D. A., Cook J. A., Kim S. Y., et al. Superoxide modulates the oxidation and nitrosation of thiols by nitric oxide‐derived reactive intermediates. Chemical aspects involved in the balance between oxidative and nitrosative stress. J Biol Chem 1997; 272: 11147–51
- Brigelius R. Mixed disulfides: biological functions and increase in oxidative stress. Oxidative stress., H Sies. Academic Press, London 1985; 243–72
- Hamilos D. L., Wedner H. J. The role of glutathione in lymphocyte activation. I. Comparison of inhibitory effects of buthionine sulfoximine and 2‐cyclohexene‐1‐one by nuclear size transformation. J Immunol 1985; 135: 2740–7
- Ling C. T., Chow B. F. Effect of vitamin B12 on the levels of soluble sulfhydryl compounds in blood. J Biol Chem 1953; 202: 445–56
- Register U. D. The effect of vitamin B12 on liver and blood non‐protein sulfhydryl compounds. J Biol Chem 1954; 206: 705–9
- Dubnoff J. W., Bartron E. The effect of B12 on enzyme activity in E. coli mutant 113‐3. Arch Biochem Biophys 1956; 61: 99–110
- Peel J. L. Vitamin B12 derivatives and the CO2‐pyruvate exchange reaction: a reappraisal. J Biol Chem 1962; 237: PC263–5
- Aronovitch J., Grossowicz N. Cobalamin catalyzed oxidation of sulfhydryl groups. Biochem Biophys Res Comm 1962; 8: 416–20
- Schrauzer G. N., Windgassen R. J. On cobaloximes with cobalt‐sulfur bonds and some model studies related to cobamide‐dependent methyl‐group‐transfer reactions. J Am Chem Soc 1967; 89: 3607
- Jacobsen D. W., Pezacka E. H., Brown K. L. The inhibition of corrinoid‐catalyzed oxidation of mercaptoethanol by methyl iodide: mechanistic implications. J Inorg Biochem 1993; 50: 47–63
- Kuo P., Abe K. Y., Schroeder R. A. Interleukin‐I‐induced nitric oxide production modulates glutathione synthesis in cultured rat hepatocytes. Am J Physiol 1996; 271: C851–62
- Kharitonov V. G., Sundquist A. R., Sharma V. S. Kinetics of nitrosation of thiols by nitric oxide in the presence of oxygen. J Biol Chem 1995; 270: 28158–64
- Clementi E., Brown G. C., Feelisch M., Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S‐nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci 1998; 95: 7631–6
- Arteel G. E., Briviba K., Sies H. Mechanisms of antioxidant defense against nitric oxide/peroxynitrite. Nitric oxide: biology and pathobiology., L. J Ignarro. Academic Press, San Diego 2000; 343–54
- Quijano C., Alvarez B., Gatti R. M., Angusto O., Radi R. Pathways of peroxynitrite oxidation of thiol groups. Biochem J 1997; 322: 167–73
- Morris S. L., Walsh R. C., Hauser J. N. Identification and characterization of some bacterial membrane sulfhydryl groups which are targets of bacteriostatic and antibiotic action. J Biol Chem 1984; 259: 13590–4
- Gilbert H. S. Proposal of a possible function for granulocyte vitamin B12 binding proteins in host defense against bacteria. Blood 1974; 44: 926