
Abstract
A novel porphyrin derivative 1∙Zn was synthesised in order to mimic the self-assembly properties of natural light-harvesting antennas and its self-assembly behaviour in solution and in solid state were studied by NMR and X-Ray spectroscopies. The self-assembly of this molecule was triggered in apolar solvents and studied in solution by UV-Vis spectroscopy, suggesting it is able to form slipped face-to-face aggregates, or J-aggregates. The nanoscopic and microscopic morphology of the aggregates was elucidated by atomic force microscopy, revealing the formation of extended two-dimensional structures.
Introduction
Skilful organisation of organic molecules is an area of research of special interest as a bottom-up approach for self-assembled functional dyes that hold great promise in materials science applications (Citation1–6). In this context, it is not surprising that, in the area of supramolecular chemistry (Citation7–9), extensive efforts have been placed into mimicking the light-harvesting antennae (Citation10–16), chromophoric arrays employed by plants and bacteria for photosynthesis (Citation17, 18). Especially the chlorosome antennae, in the green photoprotic bacteria (e.g. Chloroflexus aurantiacus), is able to aggregate and exert its function without the presence of any protein. For this reason, bacteriochlorophylls (BChl) have become the main source of inspiration for semi-synthetic and artificial chlorins and porphyrins in mimicking bacterial light-harvesting systems (Citation11, 14, 19–25). For example, semi-synthetic derivatives from chlorophyll a have been self-assembled in chlorosomal-type aggregates (Citation26–31). Additionally, synthetic porphyrin analogues revealed to be good models for mimicking chlorosomes by employing the same self-assembly recognition groups that are found in BChl’s (Citation32–34). The self-assembly algorithm for this family of (semi)synthetic derivatives is made of three key points: a central metal ion, a hydroxy group and a keto function. Meticulous tailoring of these three groups enabled a given self-organisation with coordination to the central metal atom with the hydroxy of another molecule as main contact that, through a lateral hydrogen-bond with a keto group of a third molecule, organises the stacks together with π–π interactions (Citation26–34).
Herein we report a fully synthetic porphyrin 1∙Zn, a tetrapyrrolic macrocycle carrying a central zinc core (capable of ligation) and two hydroxymethyl groups that are trans to each other (Figure ). The design of this macrocycle should enable self-assembly of the porphyrins in stacks and, owing to the second hydroxymethyl group, should give rise to novel nanostructures, other than the usual nanorods (Citation29, 35–37). The self-assembly properties of 1∙Zn was evaluated by a variety of variable-temperature and concentration-dependent UV-Vis spectroscopic measurements. Moreover, the formation of different nanostructures was evaluated under different solvent conditions, given the explicit role that the solvent plays in the formation of self-assembled nanostructures (Citation38–42).
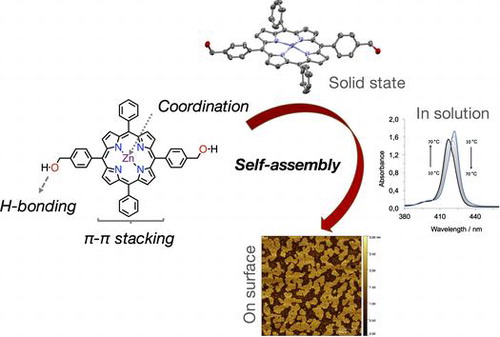
Figure 1. (Colour online) Chemical structure of porphyrin 1∙Zn along with its cartoon representation highlighting the relevant functional units.
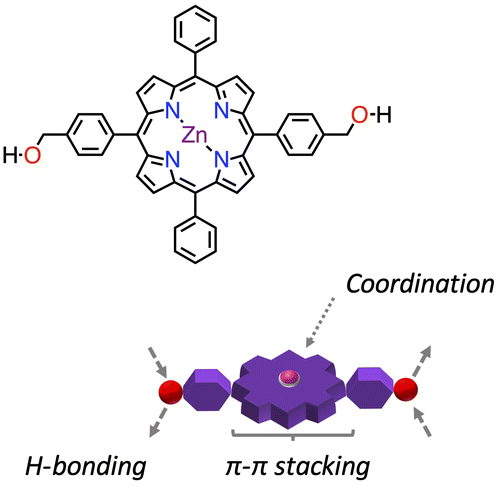
Figure 2. (Colour online) NMR spectroscopy experiments for porphyrin 1∙Zn (500 MHz, THF-d8, 298 K, c = 5.0 mM) – (a) 1H-NMR spectra; (b) 1H-NMR; 1H–1H correlation spectroscopy; (c) 1H-13C single quantum correlation and (d) 1H-13C multiple bond correlations.
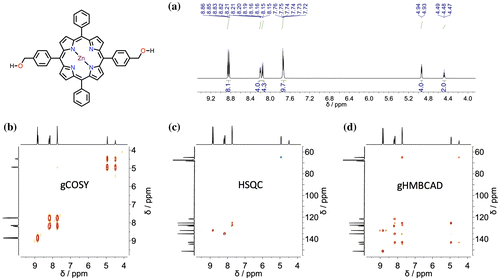
Figure 3. (Colour online) Crystallographic structure of porphyrin 1 (a) – thermal ellipsoids are at 50% level; H atoms were omitted for clarity. Molecular packing (b–d) of 1 and 1∙Zn is shown: (b,c) top and side view of 1 with blue dashes showing solvent (THF) filled tubes and red dashes showing the H-bonding pores; (d) helicoidal arrangement of the O atoms (50% thermal ellipoids). See the ‘Crystallographic data’ section in the Supporting Information for more details.
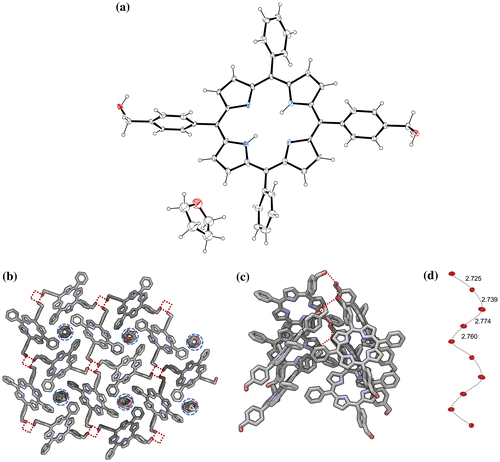
Figure 4. (Colour online) Crystallographic structures of porphyrin 1∙Zn (a) – thermal ellipsoids are at 50% level; H atoms were omitted for clarity. Molecular packing (b) of 1∙Zn is shown: two crystallographically independent porphyrin molecules with differently coordinated ligands: one bound to a THF molecule, while the other is bound to a H2O molecule. See the ‘Crystallographic data’ section in the Supporting Information for more details.
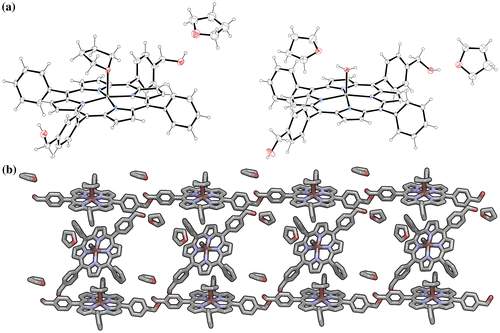
Figure 5. (Colour online) Absorbance profiles of 1∙Zn in different solvents that include THF (purple trace), CHCl3 (pink), MCH (with THF, blue trace) and MCH (with CHCl3, light blue); inset shows enlarged region where the Q bands absorb; on the right are shown the absorption profiles after addition of a polar protic solvent such as MeOH. Fluorescence profiles are presented in supporting information Figure S11.
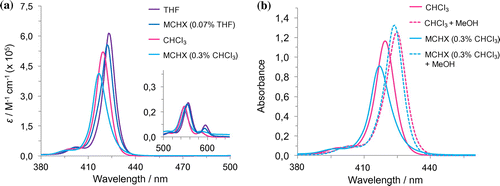
Figure 6. (Colour online) UV-Vis titration experiments for 1∙Zn with MeOH (a,c) for CHCl3 and MCH respectively and pyridine (b,d) for CHCl3 and MCH respectively.
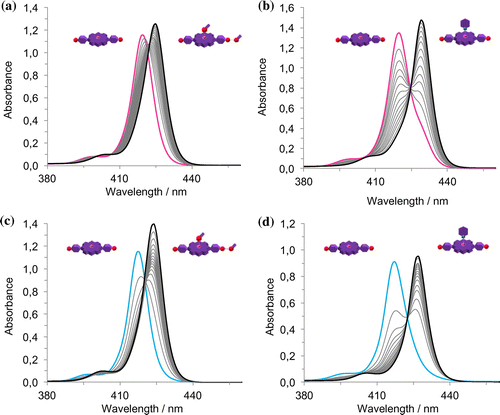
Figure 7. (Colour online) Absorption and fluorescence spectra of 1∙Zn. Variable-temperature UV-Vis experiment (left); variable-temperature fluorescence experiment (middle; λex = 419 nm) and concentration-depended experiment (right).
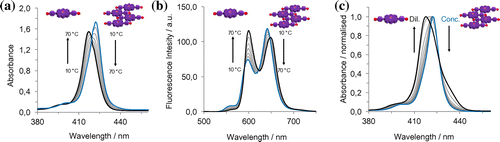
Figure 8. (Colour online) AFM height images in tapping mode with the profile of the white dashed lines. Samples were prepared by spin-coating a solution of 1∙Zn in CHCl3 (a) scale bars 5 and 2 μm, in MCH (0.3% CHCl3, scale bars 5 and 0.5 μm (b) and MCH (0.07% THF, scale bars 5 and 0.5 μm) (c).
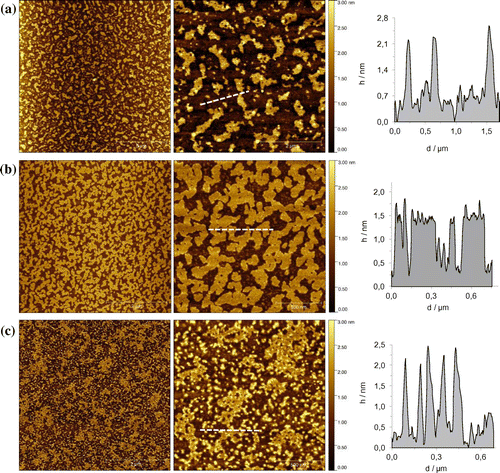
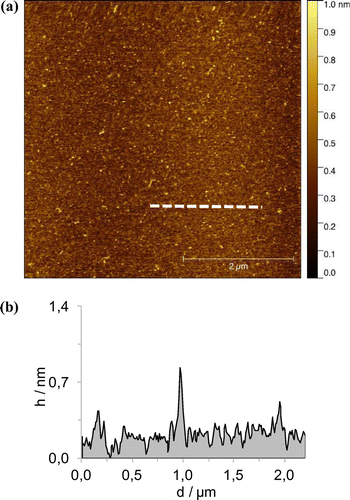
Figure 9. (Colour online) AFM height images in tapping mode (a) scale bar 2 μm with the profile of the white dashed line (b). Sample were prepared by spin-coating a solution of 1∙Zn in CHCl3 + MeOH.
Results and discussion
Synthesis
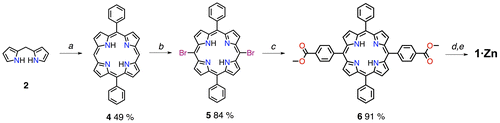
Initially, we wanted to develop a high-yielding route to synthesise the porphyrin derivative 1∙Zn (Scheme ). We reasoned that the best way to obtain the final benzyl alcohol would be the reduction of the porphyrin di-ester. In order to obtain this key intermediate, it was desirable to avoid tedious chromatographic separations of porphyrin mixtures obtained by statistical Alder-Longo or Lindsey reactions (Citation16, 43, 44). The best route to the 5,15 substituted porphyrin is certainly through the MacDonald [2 + 2] condensation approach (Citation45, 46). At first, the meso-unsubstituted dipyrromethane 2 was obtained by reaction of H2CO with a large excess of pyrrole (as solvent) containing TFA (trifluoroacetic acid) as catalyst. The dipyrromethane was reacted with benzaldehyde (under TFA catalysis) in order to obtain 5,15-diphenylporphyrin derivative (Citation47). The 10,20 meso-free positions were easily brominated with NBS (N-bromosuccinimide) in the presence of pyridine as acid scavenger (Citation48). The 5,15-dibromo-10,20-diphenylporphyrin 5 was then subjected to a Pd-catalysed Suzuki-Miyaura cross coupling (Citation49) reaction with the methyl pinacolboranebenzoate to obtain exclusively the trans-porphyrin di-ester 6. Reduction from ester to benzyl alcohol in the presence of LiAlH4 and subsequent metallation with Zn(OAc)2∙2H2O yielded the final porphyrin 1∙Zn in a good overall yield. Notably, this sequence of reactions gave products fairly different in polarity, making it easy to separate them by single column chromatography.
Scheme 1. (Colour online) Synthetic scheme for porphyrin 1∙Zn. Reagents and conditions: (a) (i) TFA, CH2Cl2, r.t. (ii) DDQ, reflux (iii) Et3 N; (b) NBS, CHCl3/Py, 0 °C; (c) methyl 4-pinacolboranebenzoate 5, Pd(PPh3)4 cat., K3PO4, THF, reflux; (d) LiAlH4, THF, 0 °C ➝ r.t.; (e) Zn(OAc)2, CHCl3, CH3OH, r.t. Abbreviations: TFA – trifluoroacetic acid; DDQ – 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone; NBS – N-bromosuccinimide; Py – pyridine; THF – tetrahydrofuran.
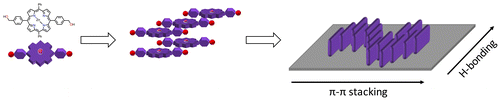
Scheme 2. (Colour online) Schematic representation of the aggregation of 1∙Zn in solution and when it is transferred onto the surface.
Characterisation
The porphyrin 1∙Zn was characterised by mono- (1H and 13C) and bi-dimensional (1H-1H and 1H-13C) NMR spectroscopies (Figure ). Notably, in a polar aprotic solvent (such as THF-d8) the O–H resonance appears as a triplet, due to the coupling to the benzylic –CH2– that, in turn, is a doublet (coupling constant 6 Hz). Additionally, the β-pyrrolic resonances manifest as two doublets, probably due to the diminished symmetry in THF-d8 induced by the Zn⋯O complexation. Protons that are part of the four phenyl rings (both unsubstituted and para-substituted ones) and are further away from the porphyrin macrocycle lie together in the 7.8–7.7 ppm region. Resonances from the phenyl rings close to the porphyrin macrocycle can be found at 8.2 ppm as a doublet of doublets (with coupling constants being 7 and 2 Hz for ortho- and meta- couplings, respectively) for the unsubstituted phenyl and at 8.15 ppm as a doublet (8 Hz for the only possible ortho- coupling). These findings were confirmed both by homonuclear (COSY) and heterenuclear correlations (on single quantum and multiple bond correlation, HSQC and HMBC, respectively).
Crystals suitable for X-ray spectroscopy were obtained for 1 (from THF/Cyclohexane vapour exchange) and 1∙Zn (slow evaporation of a THF solution). Solid structure and molecular packing of porphyrins 1 and 1∙Zn are shown in Figures and . In the asymmetric unit of crystals for 1, one full and two half porphyrin molecules were found. Remarkably, a chiral helicoidal arrangement of hydrogen bonds connects 32 neighbour porphyrins (dO−O = 2.749(8) Å), forming channels along the a axis occupied by the THF solvent molecules (Citation50–52). Finally, a slipped face-to-face stacking with an offset of 4.2 Å was observed, suggesting that this synthetic variant of chlorin forms J-aggregates. Although a similar ladder stacks are formed in an already reported bis[4-(hydroxymethyl)phenyl]porphyrin (Citation16), porphyrin 1 forms a different hydrogen-bonded arrays (no hydrogen bonding is present between hydroxyl and pyrrolic NH) and includes also solvents in the solid state structure, contrarily to already reported porphyrin. Molecule 1·Zn model (Figure ) shows two crystallographically independent porphyrin molecules with differently coordinated ligands: one bound to a THF molecule, while the other is bound to a H2O molecule (Supporting Information Figure S15). As also observed in the case of molecule 1, neighbour porphyrins show intermolecular hydrogen bonds connections (average dO−O = 3.126(7) Å). Interstitial voids are filled by three additional THF molecules tightly connected to available hydroxyl groups (from porphyrins and water).
Self-assembly in solution
The optical properties of 1∙Zn were investigated in THF, CHCl3 and the combination of the two with methylcyclohexane (MCH). The latter solvent was used to trigger the self-assembly of the organic nanostructures (Figure ).
At room temperature, 1∙Zn is dissolved in THF. The UV-Vis spectrum displays a sharp Soret band at λmax = 424 nm (S2 ← S0 transition) accompanied by Q-bands at λmax = 556 and 596 nm, typical of metal porphyrins where the increased symmetry causes the S1 ← S0 transition to reduce from four to two bands. In CHCl3, the absorption spectrum presents changes with respect to that observed from the THF solution, with the Soret band showing hypso- and hypo-chromic shift. Only slight changes were discerned with variable temperature (VT) and concentration-dependent (Supporting Information Figure S12), such as narrowing of the Soret band. This suggests the formation of weakly bonded supramolecular assemblies (Citation53).
To further investigate the formation of 1∙Zn assemblies in CHCl3, MeOH or pyridine were added to disrupt both hydrogen-bonding or axial zinc coordination interactions, respectively (Figure ). By adding MeOH, the Soret band is steadily bathochromically shifted from λmax = 420 to 425 nm, without the presence of a clear isosbestic point, most likely indicating the presence of sequential equilibriums, from disaggregation to the MeO⋯Zn complexation. The pyridine UV-Vis titration in contrast showed marked bathochromic shift from λmax = 420 to 429 nm with a clear isosbestic point, recalling an equilibrium between the formation aggregates and the porphyrin–pyridine adduct (Citation42). In order to further favour the formation of aggregates, a relatively concentrated solution of 1∙Zn in CHCl3 was diluted in the apolar and non-coordinating MCH solvent (0.3% CHCl3 content). The resulting solution presents a broader Soret band with λmax = 417 nm, clearly indicating the formation of aggregates. Successive addition of different aliquots of MeOH or pyridine showed analogous behaviour to that observed in CHCl3, namely the restoring of the monomeric porphyrin species. However, temperature-dependent UV-Vis studies displayed a strong narrowing of the Soret band accompanied by a clear disappearing of the blue-shifted band centred at around 435 nm (Supporting Information Figure S13). This result indicated the formation of weak aggregates, although stronger than in the case of CHCl3. Finally, in an attempt to further increase the content of apolar MCH, a more apolar solution of 1∙Zn could be prepared in a MCH-diluted THF solution (0.07% THF content). Notably, the absorption spectrum compared to that obtained in a THF solution, where 1∙Zn is molecularly dissolved, appears to be broadened exhibiting a loss in oscillator strength. Apparently, the energy of the absorption maximum does not shift and temperature-dependent and concentration-dependent studies were thus performed. VT-UV-Vis revealed that, by increasing temperature, the S-band of the aggregates at 422 nm decreases and the apparent monomer band at 417 nm was revealed. Concentration-depended studies showed the disappearance of the 422 nm band and the appearance of the absorption maximum at 417 nm, confirming the temperature-depended behaviour and the reversible nature of the aggregation phenomena (Figure ).
Taken all together, these data suggest the formation of a bathochromically shifted band that, when compared to that of the monomer, indicates the presence of slipped face-to-face stacking arrangements, known also as J-aggregates. This is in line with previous studies performed with zinc chlorins, as well as with their semisynthetic and synthetic variants (Citation12).
Self-assembly on surface
Prompted by these results, we carried out atomic force microscopy (AFM) studies to elucidate the morphology of the aggregates of 1∙Zn as deposited on a surface. In Figure are reported AFM height images of the 1∙Zn nanostructures as obtained from different solvents spin-coated onto a freshly cleaved mica surface. As it can be seen in the images, porphyrin 1∙Zn is able to form two-dimensional islands on the surface, with a typical height of 2 nm, in accordance with edge-on stacked height of the macrocycle onto the surface (Citation39, 42, 54). Notably, the size of the islands is depended on the solvent used for the solution. For example, samples obtained from CHCl3 (Figure (a)) show the formation of scattered islands, in agreement with the formation of weak non-covalent architectures that were observed by absorption experiments. From a MCH (0.3% CHCl3) solution (Figure 8(b)) extended aggregates were also observed. Finally, structures obtained from MCH (0.07% THF content) shown in Figure (c) present themselves as heterogeneous structures, including islands displaying different sizes. As observed from spectroscopic studies, the presence of THF in the sample is responsible both for H-bonding disruption and the Zn complexation, the latter disrupting the formation of extended stacked nanostructures. However, since the amount of THF is very low (0.07%) the formation of two-dimensional structures is still observed.
In order to appreciate the effect of H-bonding and zinc coordination, MeOH was added to the previously prepared CHCl3 or MCH solutions. A typical AFM image is shown in Figure , where the formation of two-dimensional structures in not present. Most likely the presence of MeOH disrupted the formation of the aggregates favouring the presence of individual molecules on the surface, confirming the results observed from the spectroscopic measurements.
Conclusions
In summary, we described the self-assembly behaviour of a zinc porphyrin carrying two hydroxymethyl groups. Absorption experiment in different solvent conditions hinted at the formation of slipped face-to-face stacks (J-aggregates). Owing to the second hydroxy group, the stacks are able to extend in another dimension as observed by the two-dimensional aggregates with AFM microscopy.
Supplemental material
CCDC-1442721 and 1442722 contain the supplementary crystallographic data for compounds 1 and 1∙Zn. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1080/10610278.2016.1158407
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
DB gratefully acknowledges the EU through the ERC Starting Grant ‘COLORLANDS’ project, the MIUR through the FIRB ‘Futuro in Ricerca’ (‘SUPRACARBON’, contract n° RBFR10DAK6). LĐ thanks the University of Trieste for his doctoral fellowship.
Supplemental Material
Download MS Word (3.6 MB)Acknowledgements
LÐ warmly thanks Ms. Francesca Arcudi (University of Trieste) for help with AFM measurements and useful discussion.
Related Research Data
References
- Aida, T.; Meijer, E.W.; Stupp, S.I. Science 2012, 335, 813–817.10.1126/science.1205962
- Maggini, L.; Bonifazi, D. Chem. Soc. Rev. 2012, 41, 211-241.10.1039/C1CS15031F
- Stupp, S.; Palmer, L. Chem. Mater. 2013, 26, 507–518.
- Busseron, E.; Ruff, Y.; Moulin, E.; Giuseppone, N. Nanoscale 2013, 5, 7098.10.1039/c3nr02176a
- Wei, P.; Yan, X.; Huang, F. Chem. Soc. Rev. 2015, 44, 815–832.10.1039/C4CS00327F
- Yang, L.; Tan, X.; Wang, Z.; Zhang, X. Chem. Rev. 2015, 115, 7196–7239.10.1021/cr500633b
- Lehn, J.-M. Science 2002, 295, 2400–2403.10.1126/science.1071063
- Lehn, J.-M. Proc. Nat. Acad. Sci. USA 2002, 99, 4763–4768.10.1073/pnas.072065599
- Whitesides, G.M. Science 2002, 295, 2418–2421.10.1126/science.1070821
- Balaban, T.S. Acc. Chem. Res. 2005, 38, 612–623.10.1021/ar040211z
- Miyatake, T.; Tamiaki, H. Coord. Chem. Rev. 2010, 254, 2593–2602.10.1016/j.ccr.2009.12.027
- Würthner, F.; Kaiser, T.E.; Saha-Möller, C.R. Angew. Chem. Int. Ed. 2011, 50, 3376–3410.10.1002/anie.201002307
- McHale, J.L. J. Phys. Chem. Lett. 2012, 3, 587–597.10.1021/jz3000678
- Hasobe, T. J. Phys. Chem. Lett. 2013, 4, 1771–1780.10.1021/jz4005152
- Sengupta, S.; Würthner, F. Acc. Chem. Res. 2013, 46, 2498–2512.10.1021/ar400017u
- Balaban, T.S.; Eichhöfer, A.; Lehn, J. J. Eur. J. Org. Chem. 2000, 2000, 4047–4057.10.1002/(ISSN)1099-0690
- Tamiaki, H.; Holzwarth, A.R.; Schaffner, K. J. Photochem. Photobiol. B Biol. 1992, 15, 355–360.
- Holzwarth, A.; Griebenow, K. J. Photochem. Photobiol. A Chem. 1992, 65, 61–71.10.1016/1010-6030(92)85032-P
- Holten, D.; Bocian, D.F.; Lindsey, J.S. Acc. Chem. Res. 2002, 35, 57–69.10.1021/ar970264z
- Bonifazi, D.; Kiebele, A.; Stöhr, M.; Cheng, F.; Jung, T.; Diederich, F.; Spillmann, H. Adv. Funct. Mater. 2007, 17, 1051–1062.10.1002/(ISSN)1616-3028
- Mohnani, S.; Bonifazi, D. Coord. Chem. Rev. 2010, 254, 2342–2362.10.1016/j.ccr.2010.05.006
- O’Sullivan, M.C.; Sprafke, J.K.; Kondratuk, D.V.; Rinfray, C.; Claridge, T.D.W.; Saywell, A.; Blunt, M.O.; O’Shea, J.N.; Beton, P.H.; Malfois, M.; Anderson, H.L. Nature 2011, 469, 72–75.10.1038/nature09683
- Babu, S.S.; Bonifazi, D. ChemPlusChem 2014, 79, 895–906.10.1002/cplu.201402047
- Neuhaus, P.; Cnossen, A.; Gong, J.Q.; Herz, L.M.; Anderson, H.L. Angew. Chem. Int. Ed. 2015, 54, 7344–7348.10.1002/anie.v54.25
- Kondratuk, D.V.; Perdigão, L.M.A.; Esmail, A.M.S.; O’Shea, J.N.; Beton, P.H.; Anderson, H.L. Nat. Chem. 2015, 7, 317–322.10.1038/nchem.2182
- Tamiaki, H.; Miyatake, T.; Tanikaga, R.; Holzwarth, A.R.; Schaffner, K. Angew. Chem. Int. Ed. 1996, 35, 772–774.10.1002/(ISSN)1521-3773
- Tamiaki, H.; Kimura, S.; Kimura, T. Tetrahedron 2003, 59, 7423–7435.10.1016/S0040-4020(03)01128-1
- Kunieda, M.; Tamiaki, H. J. Org. Chem. 2008, 73, 7686–7694.10.1021/jo8014402
- Huber, V.; Katterle, M.; Lysetska, M.; Würthner, F. Angew. Chem. Int. Ed. 2005, 44, 3147–3151.10.1002/(ISSN)1521-3773
- Huber, V.; Lysetska, M.; Würthner, F. Small 2007, 3, 1007–1014.10.1002/(ISSN)1613-6829
- Ganapathy, S.; Sengupta, S.; Wawrzyniak, P.K.; Huber, V.; Buda, F.; Baumeister, U.; Würthner, F.; de Groot, H.J.M. Proc. Nat. Acad. Sci. USA 2009, 106, 11472–11477.10.1073/pnas.0811872106
- Balaban, T.S.; Bhise, A.D.; Fischer, M.; Linke-Schaetzel, M.; Roussel, C.; Vanthuyne, N. Angew. Chem. Int. Ed. 2003, 42, 2140–2144.10.1002/anie.200250465
- Balaban, T.S.; Bhise, A.D.; Bringmann, G.; Bürck, J.; Chappaz-Gillot, C.; Eichhöfer, A.; Fenske, D.; Götz, D.C.G.; Knauer, M.; Mizoguchi, T.; Mössinger, D.; Rösner, H.; Roussel, C.; Schraut, M.; Tamiaki, H.; Vanthuyne, N. J. Am. Chem. Soc. 2009, 131, 14480–14492.10.1021/ja905628h
- Jochum, T.; Reddy, C.M.; Eichhofer, A.; Buth, G.; Szmytkowski, J.; Kalt, H.; Moss, D.; Balaban, T.S. Proc. Nat. Acad. Sci. USA 2008, 105, 12736–12741.10.1073/pnas.0802719105
- Balaban, T.S.; Linke-Schaetzel, M.; Bhise, A.D.; Vanthuyne, N.; Roussel, C.; Anson, C.E.; Buth, G.; Eichhöfer, A.; Foster, K.; Garab, G.; Gliemann, H.; Goddard, R.; Javorfi, T.; Powell, A.K.; Rösner, H.; Schimmel, T. Chem. Eur. J. 2005, 11, 2267–2275.10.1002/chem.200400664
- Huber, V.; Sengupta, S.; Würthner, F. Chem. Eur. J. 2008, 14, 7791–7807.10.1002/chem.v14:26
- Chappaz-Gillot, C.; Marek, P.L.; Blaive, B.J.; Canard, G.; Bürck, J.; Garab, G.; Hahn, H.; Jávorfi, T.; Kelemen, L.; Krupke, R.; Mössinger, D.; Ormos, P.; Reddy, C.M.; Roussel, C.; Steinbach, G.; Szabó, M.; Ulrich, A.S.; Vanthuyne, N.; Vijayaraghavan, A.; Zupcanova, A.; Balaban, T.S. J. Am. Chem. Soc. 2012, 134, 944–954.10.1021/ja203838p
- Jonkheijm, P.; van der Schoot, P.; Schenning, A.P.H.J.; Meijer, E.W. Science 2006, 313, 80–83.10.1126/science.1127884
- van Hameren, R.; Schon, P.; van Buul, A.M.; Hoogboom, J.; Lazarenko, S.V.; Gerritsen, J.W.; Engelkamp, H.; Christianen, P.C.M.; Heus, H.A.; Maan, J.C.; Rasing, T.; Speller, S.; Rowan, A.E.; Elemans, J.A.A.W.; Nolte, R.J.M. Science 2006, 314, 1433–1436.10.1126/science.1133004
- Cook, J.L.; Hunter, C.A.; Low, C.M.R.; Perez-Velasco, A.; Vinter, J.G. Angew. Chem. Int. Ed. 2007, 46, 3706–3709.10.1002/(ISSN)1521-3773
- Đorđević, L.; Marangoni, T.; Miletić, T.; Rubio-Magnieto, J.; Mohanraj, J.; Amenitsch, H.; Pasini, D.; Liaros, N.; Couris, S.; Armaroli, N.; Surin, M.; Bonifazi, D. J. Am. Chem. Soc. 2015, 137, 8150–8160.10.1021/jacs.5b02448
- Helmich, F.; Lee, C.C.; Nieuwenhuizen, M.M.L.; Gielen, J.C.; Christianen, P.C.M.; Larsen, A.; Fytas, G.; Leclère, P.E.L.G.; Schenning, A.P.H.J.; Meijer, E.W. Angew. Chem. Int. Ed. 2010, 49, 3939–3942.10.1002/anie.201000162
- Lindsey, J.S.; Schreiman, I.C.; Hsu, H.C.; Kearney, P.C.; Marguerettaz, A.M. J. Org. Chem. 1987, 52, 827–836.10.1021/jo00381a022
- Đorđević, L.; Marangoni, T.; De Leo, F.; Papagiannouli, I.; Aloukos, P.; Couris, S.; Pavoni, E.; Monti, F.; Armaroli, N.; Prato, M.; Bonifazi, D. Phys. Chem. Chem. Phys., 2016.
- Arsenault, G.P.; Bullock, E.; MacDonald, S.F. J. Am. Chem. Soc. 1960, 82, 4384–4389.10.1021/ja01501a066
- Senge, M.O. Chem. Commun. 1943, 2011, 47.
- Brückner, C.; Posakony, J.J.; Johnson, C.K.; Boyle, R.W.; James, B.R.; Dolphin, D. J. Porphyrins Phthalocyanines 1998, 2, 455–465.10.1002/(SICI)1099-1409(199811/12)2:6<455::AID-JPP67>3.0.CO;2-C
- DiMagno, S.G.; Lin, V.S.Y.; Therien, M.J. J. Org. Chem. 1993, 58, 5983–5993.10.1021/jo00074a027
- Bakar, M.B.; Oelgemöller, M.; Senge, M.O. Tetrahedron 2009, 65, 7064–7078.10.1016/j.tet.2009.06.037
- Hatada, M.H.; Tulinsky, A.; Chang, C.K. J. Am. Chem. Soc. 1980, 102, 7115–7116.10.1021/ja00543a044
- Hirasawa, K.; Yuge, H.; Miyamoto, T.K. Acta Crystallogr. C 2008, 64,, m97–m100.10.1107/S0108270107068059
- Weissbuch, I.; Lahav, M. Chem. Rev. 2011, 111, 3236–3267.10.1021/cr1002479
- Hoeben, F.J.M.; Wolffs, M.; Zhang, J.; De Feyter, S.; Leclère, P.; Schenning, A.P.H.J.; Meijer, E.W. J. Am. Chem. Soc. 2007, 129, 9819–9828.10.1021/ja072548c
- Ogi, S.; Sugiyasu, K.; Manna, S.; Samitsu, S.; Takeuchi, M. Nat. Chem. 2014, 6, 188–195.10.1038/nchem.1849