
Abstract
Mechanically interlocked molecules (MIMs) of all shapes and sizes are available to Chemists, driving forward progress across a broad range of research areas. Yet, some classes of MIMs remain relatively elusive. This Review summarises the properties of multiply threaded rotaxanes and the methods that have been developed to prepare them, outlining some of the pitfalls and solutions that have been uncovered along the way.
Introduction
Increasingly elegant synthetic methods have become available to assemble mechanically interlocked molecules (MIMs) over the past few decades, allowing researchers to investigate and exploit the unique properties of mechanical bonds (Citation 1 ). As might be expected, the class of MIMs that has been the most easily accessed, rotaxanes, has also been the most frequently studied – articles concerning their synthesis and applications have accounted (Citation 1 ) for over two thirds of the publications in the area.
Rotaxanes are made up (Figure ) of ring and dumbbell components. Although the components are not covalently linked to one another, they are kinetically trapped together and cannot be separated without breaking a covalent bond. The components remain entangled in this way because of ‘stoppers’ (Citation 2 ) – sterically bulky groups that are too large to pass through the aperture of the ring component – that are present at the ends of each dumbbell. In their most simple form, therefore, rotaxanes are formed from just one ring component and one dumbbell component, i.e. a [2]rotaxane. The numerical prefix [n] indicates the total number of independent components present in the MIM. Higher order [n]rotaxanes are created either by (i) encircling a long dumbbell with multiple rings, (ii) threading multiple dumbbells through the aperture of a sufficiently large ring, or (iii) a combination of the two. [n]Rotaxanes made up of more than one ring component are classified (Figure ) as type 1.m, referred to here as being multiply encircled, whereas their multiply threaded counterparts, also known as ‘molecular sheafs’, are classified as type m.1, where n = m + 1 (Citation 3 ).
Figure 1. (colour online) Schematic representations of [2]- and [3]rotaxanes illustrating the two types of three-component rotaxanes that result from either doubly encircled dumbbells (type 1.2) or doubly threaded rings (type 2.1).
![Figure 1. (colour online) Schematic representations of [2]- and [3]rotaxanes illustrating the two types of three-component rotaxanes that result from either doubly encircled dumbbells (type 1.2) or doubly threaded rings (type 2.1).](/cms/asset/b4f963f5-acb7-4bf5-b96a-a169018c3f2a/gsch_a_1433832_f0001_oc.jpg)
In general, the template-directed synthesis strategies developed for the preparation of [2]rotaxanes can be extrapolated to the preparation of multiply encircled [n]rotaxanes with relative synthetic ease. The length of the dumbbell and the number of recognition motifs present in its structure can be increased and the stoichiometry of rings or ring-forming precursors can be scaled accordingly, without necessarily having to adjust the structure of the ring or the stoppers. As a result, reliable methods have been developed to prepare multiply encircled threads, which have allowed for the preparation of long but precise [n]rotaxane oligomers or mechanically interlocked polymers (Citation 4 ) The potential for cooperative (Citation 5 ) noncovalent interactions between the rings has been exploited to aid (Citation 4 ) in their syntheses, as well as to alter the physical properties of the long dumbbell. For example, aromatic interactions between neighbouring rings can rigidify oligomeric structures (Citation 4 ), increasing the persistence length of the rotaxane relative to the non-interlocked dumbbell.
On the other hand, the preparation of multiply threaded rings presents challenges: (i) the ring component may have to participate in mutually stabilising reactions with two or more components simultaneously (Citation 6 ); (ii) any noncovalent bonding interactions between components must compete with destabilisation brought about by steric overcrowding in the multicomponent system; (iii) the ring must be sufficiently large to accommodate multiple dumbbells while being small enough to remain mechanically interlocked; and, similarly, (iv) large stoppers may be required to prevent slippage (Citation 7 ) of the large ring. It is not entirely straightforward, therefore, to extend the design elements of a [2]rotaxane to a multiply threaded [n]rotaxane.
This Review aims to provide a survey of the successful syntheses of multiply threaded rotaxanes and a discussion of their properties. An overview is given of some template-directed synthesis strategies before moving on to individual examples. Only a small number of multiply threaded [n]rotaxanes have been synthesised, so this review encompasses examples of all recognition motifs used to date. Rotacatenanes (Citation 8 ) and main chain oligocatenanes (Citation 9, 10 ), although also multiply threaded, are considered beyond the scope of the present article, as are macrobicyclic [3]rotaxanes (Citation 11 ) and similar handcuff-type (Citation 12 ) systems.
Template-directed synthesis strategies
The full extent of strategies available to assemble [2]rotaxanes have been comprehensively reviewed elsewhere (Citation 1 ). Three general template-directed approaches have been employed (Figure ) successfully in the synthesis of multiply threaded rotaxanes, placing different demands on the structural features of the precursors and giving rise to differently functionalised MIMs. A ‘clipping’ reaction can be performed by making (Figure ) a ring from an acyclic precursor, while simultaneously gathering and encircling dumbbell components within its cavity. Alternatively, a m:1 inclusion complex (Citation 6 ) made up of linear building blocks and a preformed ring can be covalently captured (Figure ) in a ‘stoppering’ reaction. In order to bring all of the clipping or stoppering precursors together prior to the covalent bond formation, the ring (or acyclic ring precursor) must be able to associate with two other components simultaneously through noncovalent bonding interactions – i.e. the ring must be multivalent. This multivalency can be achieved either through (i) multiple directional interactions, (ii) non-directional noncovalent interactions, e.g. solvophobic effects, or, (iii) by complexation of a multivalent metal ion that can bring together more than two ligands.
Figure 2. (colour online) Schematic representations of the synthetic strategies used to prepare multiply threaded rotaxanes. Hashed black lines illustrate attractive noncovalent bonding interactions that may be required between components. Purple spheres represent metal ions.
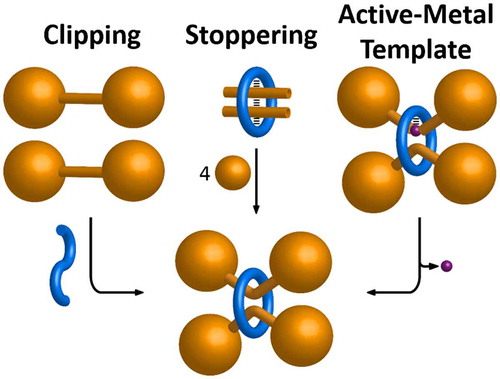
Both the clipping and stoppering strategies are regarded as passive template approaches – that is, the supramolecular association of components is not directly coupled to the covalent bond forming step. Active template (Citation 11, 13 ) approaches, on the other hand, employ molecular recognition elements that play a direct role in mediating covalent bond formation. A transition metal ion held endotopically within the macrocyclic cavity gathers (Figure ) a reactive half-dumbbell on either face of the ring, catalysing bond formation through the annulus. There is no need for a traditional recognition motif to be part of the half-dumbbell structure. Instead, the key structural feature of the half-dumbbell is a reactive end group, which coordinates to the metal template, and is transformed during the reaction. The structure can be assembled, therefore, without any significant attractive interactions remaining between the ring and the dumbbell in the resulting MIM. Moreover, the transition metal centre returns to its original state after mediating the covalent bond formation, so it can turn over, acting as a catalyst to form multiple mechanically interlocked dumbbells. This active metal template approach constitutes (Figure ) the third template strategy to make multiply threaded rotaxanes. Mechanically interlocked dumbbells can be formed through the centre of vacant or an already-threaded rings by the same mechanism, provided that there is sufficient space to access the metal ion.
Multivalent rings
Ammonium templates
Secondary dialkylammoniums have long been exploited (Citation 14 ) as template motifs in the dumbbells of [2]rotaxanes. Their positive charge, polarised N–H bonds, and relatively small size make them ideal hydrogen bond donors to interact with endotopic hydrogen bond acceptors present in a complementary ring component.
Leigh and Winpenny reported on hybrid organic-inorganic rotaxanes (Citation 15 ) that are synthesised by clipping large heterometallic rings (Citation 16 ) around secondary ammonium dumbbells. The heterometallic rings are formed reversibly from several trivalent CrIII ions and one or two divalent metal ions (such as CuII), which are connected to one another by μ2-bridging fluoride and bridging pivalate (κO:κO′ coordination) anions. A doubly threaded [4]rotaxane 1, comprising (Figure ) two dumbbells encircled by two rings, is formed when a long dumbbell, containing two dialkylammonium sites separated by 12 methylene units, is used as a template for the heterometallic assembly of CrIII and CuII centres. The template effect of the secondary ammonium arises from its hydrogen bonding interactions with the endotopic fluoride ligands that bridge the metal centres of the ring component. [4]Rotaxane 1 has been isolated in a yield of 37%, which is impressively high given that it is formed by the assembly of 98 components – 24 metal ions, 44 pivalate groups, 28 fluoride ions, and two dumbbells. The assembly is even more remarkable given that it occurs in preference to the formation of a structurally simpler [3]rotaxane, which would form if the ring-forming equilibrium were weighted toward smaller heterometallic rings suited to encircling a single dumbbell. Indeed, when the reaction is performed using CoII as the divalent cation in place of CuII, smaller rings are favoured and a [3]rotaxane predominates.
Figure 3. (colour online) Hybrid organic-inorganic [4]rotaxane 1 comprises two heterometallic rings and two bis(dialkylammonium) dumbbells.
![Figure 3. (colour online) Hybrid organic-inorganic [4]rotaxane 1 comprises two heterometallic rings and two bis(dialkylammonium) dumbbells.](/cms/asset/87725523-3db4-4a06-ab0f-7bedbdbd263b/gsch_a_1433832_f0003_oc.jpg)
Figure 4. (colour online) A DNA pseudo[3]rotaxane is assembled by (a) hybridisation of ssDNA regions present in the central ring and dumbbell components. (b) The association of components is visible by AFM. (Reprinted with permission from (Citation 20 ). Copyright © 2010 Macmillan Publishers Limited, part of Springer Nature.).
![Figure 4. (colour online) A DNA pseudo[3]rotaxane is assembled by (a) hybridisation of ssDNA regions present in the central ring and dumbbell components. (b) The association of components is visible by AFM. (Reprinted with permission from (Citation 20 ). Copyright © 2010 Macmillan Publishers Limited, part of Springer Nature.).](/cms/asset/8b1dc105-04f1-4e86-8513-818d933fe2a8/gsch_a_1433832_f0004_oc.jpg)
Figure 5. (colour online) (a) A schematic representation of the sequential formation of [2]- and [3]rotaxanes through an active template mechanism. (b) Stick representation of the X-ray crystal structure of doubly threaded [3]rotaxane 16, where x = 1 and y = 1.
![Figure 5. (colour online) (a) A schematic representation of the sequential formation of [2]- and [3]rotaxanes through an active template mechanism. (b) Stick representation of the X-ray crystal structure of doubly threaded [3]rotaxane 16, where x = 1 and y = 1.](/cms/asset/d1a7b7de-a8fe-458d-8b9c-9aa3ff3ae1c4/gsch_a_1433832_f0005_oc.jpg)
Figure 6. (colour online) Doubly threaded internal alkyne rotaxanes have been prepared (a) by Saito et al. using Glaser homocoupling conditions and (b) Anderson et al. under both Glaser and Cadiot coupling conditions. (c) Partial stick and shape filling representation of the X-ray crystal structure of doubly threaded polyyne [3]rotaxane 19, where z = 1.
![Figure 6. (colour online) Doubly threaded internal alkyne rotaxanes have been prepared (a) by Saito et al. using Glaser homocoupling conditions and (b) Anderson et al. under both Glaser and Cadiot coupling conditions. (c) Partial stick and shape filling representation of the X-ray crystal structure of doubly threaded polyyne [3]rotaxane 19, where z = 1.](/cms/asset/1b1c81d7-9842-48c0-92b4-69393916a512/gsch_a_1433832_f0006_oc.jpg)
The structural formula of 1 was confirmed (Figure ) by single crystal X-ray diffraction, which also revealed a driving force behind the surprising formation of a doubly threaded MIM. In contrast to six-coordinate CoII, the five-coordinate environment of the CuII disrupts the continuous series of μ2-bridging fluoride ligands, resulting in a fluoride ions that is only bound to one metal centre. This singly coordinated fluoride can participate in a bifurcated N–H···F···H–N hydrogen bonding interaction with two ammonium groups simultaneously, i.e., it introduces an element of multivalency to the ring–dumbbell interaction. The large ring, therefore, has a propensity to accommodate (Figure ) two of the ammonium sites in its cavity, one above the other.
Liu et al. prepared an all-organic MIM in a stoppering reaction (Scheme ) that also made use of secondary ammonium template sites (Citation 17 ). The hetero[7]rotaxane 2, which is composed of two dumbbells and five rings, incorporates a doubly threaded ring. In this case, the multivalency present in the doubly threaded ring arises from its large bis(p-phenylene-34-crown-10) (BPP34C10) polyether structure, which possesses two distinct binding sites to participate in hydrogen bonding with an ammonium through N–H···O contacts. Azide-containing 1:1 inclusion complex 3 is reacted with preformed alkyne-containing 2:1 inclusion complex 4 to covalently capture the MIM through a copper-catalysed alkyne–azide cycloaddition (Citation 18 ) (CuAAC) reaction, before methylation of the newly formed triazole groups affords 2.
Scheme 1. (colour online) The CuAAC reaction of 1:1 inclusion complex 3 and 2:1 inclusions complex 4 produces hetero[7]rotaxane 2. The central BPP34C10 ring, which is doubly threaded, is prevented from dissociating by cascade stoppering.
![Scheme 1. (colour online) The CuAAC reaction of 1:1 inclusion complex 3 and 2:1 inclusions complex 4 produces hetero[7]rotaxane 2. The central BPP34C10 ring, which is doubly threaded, is prevented from dissociating by cascade stoppering.](/cms/asset/8dc4abbd-035e-42b5-82eb-59115997843d/gsch_a_1433832_f0007_oc.jpg)
By using ‘cascade stoppering’ (Citation 2 ), the multicomponent assembly elegantly overcomes one of the problems associated with making multiply threaded rotaxanes – namely that a large, multivalent ring typically requires the synthesis of even larger stoppers. Inclusion complex 3 is easily prepared from benzo-21-crown-7 (B21C7) and a simple dialkylammonium guest. Although its terminal phenyl group itself is far too small to prevent dethreading of a BPP34C10 ring, it is sufficiently large to entrap the smaller B21C7 ring, which in turn prevents slippage of the large ring in a cascade of steric bulk.
DNA base pairing
The high fidelity of DNA base pairing allows multivalent interactions between DNA strands to be designed precisely (Citation 19 ). Famulok et al. have taken advantage of this programmable assembly to prepare (Figure (a)) a doubly threaded DNA sturcture (Citation 20 ). First, the authors synthesised a 126-base pair ring (purple, Figure (a)), which is made up predominantly of double-stranded DNA (dsDNA) with two short 13-nucleotide ‘gap regions’ of single-standed DNA (ssDNA). They also prepared two DNA dumbbells, each bearing a ssDNA motif that is complementary to one of the gap regions. When the three components are mixed in aqueous buffer, the complementary ssDNA regions present on the ring and dumbbells hybridise, giving rise (Figure (a)) to a doubly threaded structure.
Gel electrophoresis and atomic force microscopy (AFM) were used to characterise the three-component assembly – the AFM images confirmed (Figure (b)) the predicted doubly threaded structure was indeed present. The majority of viable structures in the AFM images clearly display the four loops of the dumbbell components surrounding the central ring component. Further experiments revealed, however, that the three components were not strictly part of a MIM, but rather a pseudo[3]rotaxane. The 168-base pair loops included at the ends of each dumbbell were not sufficiently large to prevent dethreading. After the addition of release oligomers, which hybridise with the gap regions of the ring and displace the dumbbells, the components of the pseudo[3]rotaxane dissociated over a period of 48 h at 0 °C. However, a solution to this dethreading problem was reported by the authors in the context of a [2]rotaxane (Citation 20 ). ‘Spherical’ stoppers were introduced by designing a dumbbell that incorporates two intersecting dsDNA loops at its termini. These stoppers have reduced flexibility and greater steric bulk, which much more effectively entraps the 126-base pair ring around the dumbbell as part of a fully-fledged MIM.
Solvophobic effects
It is well-established that cyclodextrins (CDs) accommodate lipophilic guests by virtue of hydrophobic interactions (Citation 21 ). Whereas the rims and exterior surfaces of the CDs are decorated primarily with hydrophilic alcohol functions, lipophilic CH groups are oriented towards their inner cavities, creating a hydrophobic environment. γ-CD comprises eight glucopyranosyl units linked head-to-tail in a cycle, creating an inner cavity of approximately 1 mm in diameter and 0.8 nm in depth that is large enough to accommodate two guests simultaneously(Citation 6 ). Anderson et al. took advantage of the large, well-defined cavity of γ-CD to prepare (Scheme ) doubly threaded [3]rotaxane 5 (Citation 22 ). First, [2]rotaxane 6 was prepared in a stoppering reaction. A 1:1 inclusion complex of γ-CD and a difunctional stilbene derivative were subjected to Pd-catalysed cross coupling in the presence of an iodoarene stopper precursor 7, giving rise to 6 in 17% isolated yield. Notably, the partially filled γ-CD ring of 6 has a higher binding affinity for a second aromatic guest when compared to the vacant cavity of the parent γ-CD molecule – 6 was found to bind a cyanaine dye guest with an association constant, K a = 1.0 ± 0.2 × 105 M−1 in H2O at 298 K, while γ-CD binds the same guest with K a = 87 ± 15 M−1 under the same conditions. This difference in binding strength is good illustration of γ-CD’s preference for encapsulation of two guests, reaching a more optimal ratio of guest volume to host size (Citation 23 ). Capitalising on this phenomenon, inclusion complex 8⊂6 was reacted (Scheme ) with 7 under Suzuki coupling conditions to afford doubly threaded [3]rotaxane 5 in 18% yield.
Scheme 2. (colour online) A hetero[3]rotaxane 5 is assembled in a Suzuki coupling reaction between [2]rotaxane 6 and dumbbell precursors 7 and 8.
![Scheme 2. (colour online) A hetero[3]rotaxane 5 is assembled in a Suzuki coupling reaction between [2]rotaxane 6 and dumbbell precursors 7 and 8.](/cms/asset/3f0917d3-64a7-4eb2-91e2-bddd4f02be07/gsch_a_1433832_f0008_oc.jpg)
The stoppering reaction covalently captures an otherwise labile pairing of stilbene and cyanine chromophores present in the preceding [2]rotaxane complex 8⊂6, ensuring that they are held together irreversibly and in close proximity to one another within the γ-CD ring. Anderson et al. observed that this enforced interaction impacted upon the optical properties of the chromophores. Excitation of the stilbene component of 5 is followed by quantitative energy transfer to the cyanine dye component, resulting in fluorescence with a photoluminescence quantum yield, ΦF, of 0.56. In contrast, the [2]rotaxane complex 8⊂6 fluoresces with a lower ΦF of 0.12.
Inouye et al. demonstrated a similar phenomenon in the context of a doubly alkynylpyrene-threaded [4]rotaxane 9 (Citation 24 ). The MIM was prepared (Scheme ) by stoppering a 2:2 inclusion complex (10)2⊂(γ-CD)2 that forms between pyrene derivative 10 and γ-CD in H2O. Dumbbell precursor 10 is a water soluble pyrene derivative funtionalised with alkyne-terminated polyether chains at the 1- and 6-positions. Inouye reported that a Sonogashira reaction couples the terminal alkyne groups and iodoarene stopper 7 together, affording 9 in an overall yield of 5%.
Scheme 3. (colour online) A 2:2 inclusion complex formed between γ-CD and pyrene derivative 10 gives rise to [4]rotaxane 9 after stoppering. (Reprinted with permission from (Citation 24 ). Copyright © 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim).
![Scheme 3. (colour online) A 2:2 inclusion complex formed between γ-CD and pyrene derivative 10 gives rise to [4]rotaxane 9 after stoppering. (Reprinted with permission from (Citation 24 ). Copyright © 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim).](/cms/asset/b2b8b9cc-5dca-4858-adb6-8d3061fe38fa/gsch_a_1433832_f0009_oc.jpg)
Both the inclusion complex (10)2⊂(γ-CD)2 and [4]rotaxane 9 exhibited photoluminescence features associated with pyrene excimers as a consequence of being sandwiched together by the γ-CD rings. However, the effect was essentially independent of concentration for the [4]rotaxane 9 – it displayed pure excimer emission at concentrations as low as 7.5 × 10−10 M−1! The inclusion complex (10)2⊂(γ-CD)2, on the other hand, displayed prominent characteristics of monomeric pyrene species in fluorescence spectra acquired at complex concentrations below 2.3 × 10−5 M−1. This observation highlights the ability of multiply threaded rotaxanes to enforce close interactions between their components, even under conditions that would cause their non-interlocked analogues to dissociate.
Monovalent rings
The above examples have illustrated three types of noncovalent bonding interactions and emphasised the benefits of using a multivalent ring to bring together components. Assembly of a multiply threaded MIMs can also be achieved by making use of transition metal templates (Citation 25 ), even with rings that possess a solitary molecular recognition site.
Passive transition metal templates
By judicious choice of the transition metal and ligand environments, a ring and two acyclic strands can be gathered and entwined as part of a single complex. Sauvage et al. exploited the octahedral coordination geometry of CoIII in this way to prepare (Scheme ) doubly threaded [3]rotaxane 11 from metal complex 12 (Citation 26 ). Bidentate ligands based on a 3,3ʹ-biisoquinoline (biiq) backbone were used in preference to more common bidentate motifs, such as 2,2ʹ-bipyridine or 1,10-phenanthroline derivatives. The biiq motif is necessary to allow macrocyclic linkages to be introduced into the ligand structure (i.e. aryl groups at the 8- and 8ʹ-positions) without overcrowding the coordination environment and preventing double threading. The heteroleptic complex 12 is prepared by mixing together a CoII salt, the ring, and the acyclic ligand a 1:1:2 stoichiometry, then oxidising the metal centre to CoIII to lessen the kinetic lability of the complex. It is then converted (Scheme ) into a [3]rotaxane by stoppering the azide end groups in a CuAAC reaction with a bulky alkyne. Sodium ascorbate present in the reaction mixture also reduces the kinetically inert CoIII back to labile CoII, and the CoII-containing [3]rotaxane, obtained in >70% yield, can be demetallated quantitatively by treating with a large excess of ethylenediaminetetraacetic acid, affording 11.
Scheme 4. (colour online) The octahedral coordination geometry of CoIII is exploited to bring together a bidentate ring and two bidentate ligand strands, which can be stoppered to produce a doubly threaded [3]rotaxane.
![Scheme 4. (colour online) The octahedral coordination geometry of CoIII is exploited to bring together a bidentate ring and two bidentate ligand strands, which can be stoppered to produce a doubly threaded [3]rotaxane.](/cms/asset/abb5fba9-7329-47d6-8bc5-ea3fe4faaa28/gsch_a_1433832_f0010_oc.jpg)
[3]Rotaxane 11 is only metastable, however. The 42-membered ring is sufficiently large for slippage to occur gradually, leading to complete dissociation of the MIM with a half-life of 206 h at 298 K. Sauvage et al. monitored this dissociation process by 1H NMR spectroscopy in order to measure kinetic parameters, observing that slippage is best described as a first-order process. Dissociation of the first dumbbell is the rate determining step, which precedes much more rapid slippage of the second dumbbell. A rather small enthalpy of activation, ΔH ‡, of 33 ± 3 kJ·mol−1 and a large negative entropy of activation, ΔS ‡, of −182 ± 55 J·K−1·mol−1 were measured for the process, suggesting the presence of a highly ordered transition state. These observations are consistent with slippage occurring through a strained co-conformation, as would be expected for a situation where the tris(tBu-phenyl)methine stopper of one dumbbell must squeeze through the aperture of the ring while minimising steric clash with the second dumbbell.
Saito et al. also exploited (Citation 27 ) a passive metal template approach to prepare doubly threaded [3]rotaxanes. They first prepared a [2]rotaxane bearing a phenanthroline unit oriented endotopoically as part of the ring. The [2]rotaxane was reacted with CuI and an acyclic ligand bearing another phenanthroline site in order to form a threaded tetrahedral complex, which was then covalently captured in a stoppering reaction. In performing a step-wise assembly sequence, they were able to install two constitutionally different dumbbells, giving rise to hetero[3]rotaxanes.
Active metal templates
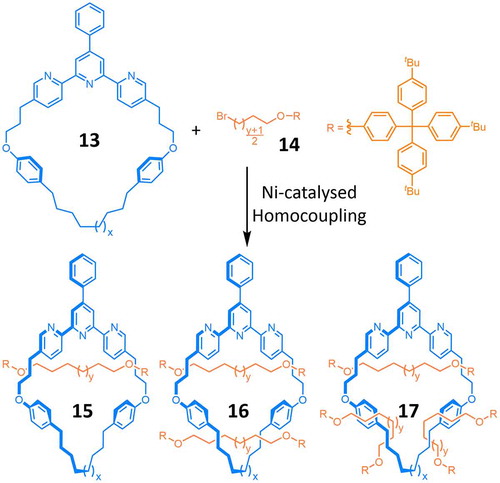
Leigh et al. reported on the active metal template synthesis of multiply threaded rotaxanes (Citation 28 ) using a Ni-catalysed homocoupling (Citation 13 ) of alkyl bromides as the key bond forming step. First, the 2,2ʹ:6ʹ,2″-terpyridine (terpy) binding site of the ring 13 coordinates a NiII centre, which is reduced to Ni0 in situ by activated Zn dust. In the presence of an excess of Zn, this Ni0–13 complex catalyses (Scheme ) the reductive dimerisation of two alkyl bromide half-dumbbells 14, forming an oligomethylene chain. If the two half-dumbbells add to the metal centre from opposite faces of the ring – a scenario which is favoured by steric repulsions between the large stoppering groups – the formation of this new C–C linkage also creates (Figure (a)) a new mechanical bond, producing [2]rotaxane 15 during the first cycle. The Ni ion remains complexed to the ring, so in the presence of an excess of 14, the catalytic cycle can be repeated (Figure (a)) and a second, and even a third(!), dumbbell is mechanically interlocked, producing [3]rotaxane 16 and [4]rotaxane 17, respectively. The doubly threaded character of a [3]rotaxane 16, where x = 1 and y = 1, could be confirmed unambiguously by X-ray crystallographic analysis (Figure (b)), as well as by characteristic changes in the proton resonances observed by 1H NMR spectroscopy. The X-ray analysis reveals that the two hexamethylene chains take up fully stretched zig-zag conformations in the solid state, allowing the dumbbells to be offset from one another, which minimises steric interactions between their tris(tBu-phenyl)methine stoppers.
Scheme 5. (colour online) A Ni ion coordinated within the cavity of ring 13 catalyses the formation of dumbbell components in an active template homocoupling of alkyl bromides.
The outcome of the Ni-catalysed active template reaction is highly depended on the size of the ring (Citation 28 ). A fine balance of steric factors govern (i) the accessibility of the metal centre, (ii) the propensity of the coupling reaction to form threaded dumbbells (rather than noninterlocked dumbbells), (iii) the maximum number of dumbbells that can be accommodated within the cavity, and (iv) the stability of the rotaxane intermediates and products towards unwanted slippage. The 35-membered ring (x = 1) produces [3]rotaxane in up to 51% alongside 20% of [2]rotaxane, employing 20 equivalents of a short half-dumbbell 14 (y = 1). By using 37-membered (x = 3) and 38-membered (x = 4) rings, the active metal template reaction is able to continue further to produce triply threaded [4]rotaxanes 17, in addition to [3]rotaxanes 16. Perhaps surprisingly, the yields of [3]- and [4]rotaxane (approximately 20 and 10%, respectively) are relatively constant when employing the 37-membered or 38-membered ring in reactions with half-dumbbells of various lengths (y = 1, 7, 19). Moreover, no traces of singly threaded rotaxane [2]rotaxane are isolated from the reaction mixtures when x = 3 or 4, despite the observation of the higher order species. This observation suggests that, while these intermediate Ni-complexed [2]rotaxanes are sufficiently long-lived to take part in subsequent active metal template cycles, the Ni-free species formed during the work up dissociate rapidly. Finally, when a 39-membered ring (x = 5) is employed, no mechanically interlocked products are isolated – the tris(tBu-phenyl)methine stoppers of any transiently formed pseudo[2]rotaxane are able to slip through the aperture of the ring during the reaction, even while the cavity is occupied by a Ni ion.
The active metal template approach to synthesise multiply threaded rotaxanes is not limited to the Ni-catalysed dimerization of alkyl bromides. Saito et al. exploited the active template Glaser coupling (Citation 13 ) of terminal alkynes by macrocyclic CuI-phenanthroline complexes. In the presence of I2 and base, the CuI catalyses an oxidative dimerisation of alkyne substrates that gives rise to butadiyne products. When a 37-membered ring component is employed in conjunction with an a alkyne half-dumbbell, a [3]rotaxane 18 is produced (Citation 29 ) (Figure (a)) in yields of up to 63% as a result of sequential active template reaction occurring (Figure (a)) through the macrocyclic cavity.
Properties and applications
Rotaxane formation has been exploited as a means of tuning properties of the mechanically interlocked components without changing their covalent backbones. As discussed above, a rather unique property of doubly threaded rotaxanes is their ability to enforce close contacts between two dumbbell components by ensnaring them inside the same molecular cavity – modulating the dumbbells’ physical and optical properties as a result. Doubly threaded [3]rotaxanes 5 and 9 both exemplify this characteristic. The hetero[3]rotaxane 5 exhibits an enhanced ΦF relative to its noninterlocked analogue and, unusually, solutions of homo[3]rotaxane 9 display excimer-type emission irrespective of changes in concentration. While [2]rotaxanes or other MIMs, such as catenanes, may also be exploited to restrict the relative orientations of π-systems, multiply threaded rotaxanes offer the possibility of shielding multiple chromophores (or other functional units) within a single macrocyclic cavity while still allowing for some degree of translational motion relative to one another.
Recently, Anderson et al. exploited the active metal template approach to sheathe extended polyyne dumbbells as part (Figure (b)) of doubly threaded rotaxanes 19 (Citation30). Polyynes are of interest for their unusual electronic properties (Citation 31 ), yet their stability decreases with increasing length. Anderson et al. have found that thermal stability can be enhanced by the presence of a ring encircling the polynne chain. Doubly threaded rotaxanes could be prepared from macrocyclic CuI-phenanthroline complexes with appropriate alkyne-terminated half-dumbbells according to Saito’s protocols. Higher yields are achieved, however, by employing an active template Cadiot (Citation 13 ) cross coupling of stoppered alkyne and alkynyl halide precursors instead. Two different sizes of ring (z = 1 and 3) were employed, confining two hexayne dumbbells together in [3]rotaxanes 19. X-ray crystallographic evidence reveals (Figure (c)) that the [3]rotaxane architecture imposes an C···C distance of just 3.29 Å between the polyyne chains at their closest point, which is shorter than the sum of the van der Waals radii (3.4 Å). As a consequence of this interaction, the normally observed bond-length alternation of the polyyne chains is perturbed. Moreover, in the solid state, the polyyne chains are oriented in a crossed geometry (74° angle), which apparently limits the interaction of the two π-systems.
Beyond modulating the electronic properties of π-systems (Citation 32 ), multiply threaded rotaxanes may find applications in catalysis (Citation 33 ). Metal binding sites used to template the assembly of MIMs can be exploited in synthesis by their coordination to a catalytically active metal centre, whereby the catalytic properties are tuned by the unique environment of the mechanically interlocked ligand (Citation 34 ). The potential for the components of the multiply threaded [n]rotaxane to undergo motion relative to one another, while preserving a sterically encumbered but flexible environment around the metal centre, would presumably impart unusual catalytic properties. There may also be potential for multiply threaded rotaxanes to contribute to advances in molecular machines and the design of functional materials. In the context of molecular machines, there is scope to direct the pirouetting motion of the ring through its interactions with the dumbbell components. Alternatively, the irreversible encapsulation of two guests within a single ring might open up opportunities in drug delivery (Citation 35 ) or it might be exploited in ‘slide-ring’ (Citation 36 ) materials. Of course, the likelihood of any of these applications coming to be is highly dependent on the development of synthetic methods that are robust and efficient, as well as on the properties of the multiply threaded rotaxanes being much more desirable than those of structurally simpler alternatives. It remains to be seen what the absolute limitations are in preparing multiply threaded rotaxanes, both in terms of synthetic accessibility as well as the extent to which more and more components can be trapped together. Will it ever be possible to mechanically interlock a bundle of several chains within a single ring? At present, it appears that even the most versatile of synthetic methods are rather restricted in the number of components that can be brought together in a rotaxane.
Conclusions
During the past decade, a modest toolbox of protocols has emerged for the construction doubly threaded rotaxanes. Multivalent rings associate with more than one dumbbell simultaneously, enabling effective template-directed synthesis strategies. Solvophobic forces, metal coordination and hydrogen bonding interactions, including DNA base pairing, have all been exploited as a primary driving force to gather a multivalent ring and two dumbbell components together. Active metal template protocols have also been developed, whereby a catalytically active metal centre held within a ring sequentially installs dumbbell components. This catalytic approach is the least demanding in terms of molecular recognition motifs, requiring only one metal binding site to be present in the ring and, perhaps as a consequence, it is the only approach so far to have given rise to triply threaded [4]rotaxanes. Preliminary investigations have revealed some of the consequences of forcing chromophores together within a single macrocyclic cavity by virtue of mechanical bonds, but otherwise there has been little use of the dynamic and flexible, yet crowded and restricted environments created by these MIMs. Now that multiply threaded rotaxanes are synthetically accessible, more attention can be directed towards investigating and exploiting their unique properties.
Disclosure statement
No potential conflict of interest was reported by the author.
Funding
This work was supported by the University of Durham and the EPSRC [grant number EP/N029992/1].
Related Research Data
References
- Xue, M. ; Yang, Y. ; Chi, X. ; Yan, X. ; Huang, F. Chem. Rev. 2015, 115 , 7398: Bruns, C.J. ; Stoddart, J.F. The Nature of the Mechanical Bond: From Molecules to Machines ; Wiley, Hoboken, NJ, 2017: De Bo, G. Chem. Sci. 2018, 9 , 15.
- Jiang, W. ; Winkler, H.D.F. ; Schalley, C.A. J. Am. Chem. Soc. 2008, 130 , 13852.
- Yerin, A. ; Wilks, E.S. ; Moss, G.P. ; Harada, A. Pure Appl. Chem. 2008, 80 , 2041.
- Avestro, A.-J. ; Belowich, M.E. ; Stoddart, J.F. Chem. Soc. Rev. 2012, 41 , 5881: Ke, C. ; Smaldone, R.A. ; Kikuchi, T. ; Li, H. ; Davis, A.P. ; Stoddart, J.F. Angew. Chem. Int. Ed. 2013, 52 , 381: Ke, C. ; Strutt, N.L. ; Li, H. ; Hou, X. ; Hartlieb, K.J. ; McGonigal, P.R. ; Ma, Z. ; Iehl, J. ; Stern, C.L. ; Cheng, C. ; Zhu, Z. ; Vermeulen, N.A. ; Meade, T.J. ; Botros, Y.Y. ; Stoddart, J.F. J. Am. Chem. Soc. 2013, 135 , 17019: Sun, S. ; Hu, X.-Y. ; Chen, D. ; Shi, J. ; Dong, Y. ; Lin, C. ; Pan, Y. ; Wang, L. Polym. Chem. 2013, 4 , 2224: Harada, A. ; Takashima, Y. ; Nakahata, M. Acc. Chem. Res. 2014, 47 , 2128: Nakazono, K. ; Ishino, T. ; Takashima, T. ; Saeki, D. ; Natsui, D. ; Kihara, N. ; Takata, T. Chem. Commun. 2014, 50 , 15341: Wilson, E.A. ; Vermeulen, N.A. ; McGonigal, P.R. ; Avestro, A.-J. ; Sarjeant, A.A. ; Stern, C.L. ; Stoddart, J.F. Chem. Commun. 2014, 50 , 9665: Cheng, C. ; McGonigal, P.R. ; Schneebeli, S.T. ; Li, H. ; Vermeulen, N.A. ; Ke, C. ; Stoddart, J.F. Nat. Nanotech. 2015, 10 , 547: Hou, X. ; Ke, C. ; Bruns, C.J. ; McGonigal, P.R. ; Pettman, R.B. ; Stoddart, J.F. Nat. Commun. 2015, 6 , 6884: Wei, P. ; Yan, X. ; Huang, F. Chem. Soc. Rev. 2015, 44 , 815: Hou, X. ; Ke, C. ; Stoddart, J.F. Chem. Soc. Rev. 2016, 45 , 3766: Lewis, J.E.M. ; Winn, J. ; Cera, L. ; Goldup, S.M. J. Am. Chem. Soc. 2016, 138 , 16329: Altmann, P.J. ; Pöthig, A. Angew. Chem. Int. Edit. 2017, 56 , 15733: Pezzato, C. ; Nguyen, M.T. ; Cheng, C. ; Kim, D.J. ; Otley, M.T. ; Stoddart, J.F. Tetrahedron 2017, 73 , 4849: Ngo, T.H. ; Labuta, J. ; Lim, G.N. ; Webre, W.A. ; D’Souza, F. ; Karr, P.A. ; Lewis, J.E.M. ; Hill, J.P. ; Arigab, K. ; Goldup, S.M. Chem. Sci. 2017, 8 , 6679: Evans, N.H. ; Akien, G.R. Supramol. Chem. 2018, 30 , 158.
- Hunter, C.A. ; Anderson, H.L. Angew. Chem. Int. Ed. 2009, 48 , 7488.
- Ueno, A. ; Takahashi, K. ; Osa, T. J. Chem. Soc. Chem. Commun. 1980, 921: Rao, K.S.S.P. ; Hubig, S.M. ; Moorthy, J.N. ; Kochi, J.K. J. Org. Chem. 1999, 64 , 8098: Kim, H.-J. ; Heo, J. ; Jeon, W.S. ; Lee, E. ; Kim, J. ; Sakamoto, S. ; Yamaguchi, K. ; Kim, K. Angew. Chem. Int. Ed. 2001, 40 , 1526: Badjić, J.D. ; Nelson, A. ; Cantrill, S.J. ; Turnbull, W.B. ; Stoddart, J.F. Acc. Chem. Res. 2005, 38 , 723: Liu, S. ; Shukla, A.D. ; Gadde, S. ; Wagner, B.D. ; Kaifer, A.E. ; Isaacs, L. Angew. Chem. Int. Ed. 2008, 47 , 2657: Barrow, S.J. ; Kasera, S. ; Rowland, M.J. ; del Barrio, J. ; Scherman, O.A. Chem. Rev. 2015, 115 , 12320: Lipke, M.C. ; Cheng, T. ; Wu, Y. ; Arslan, H. ; Xiao, H. ; Wasielewski, M.R. ; Goddard, W.A., III; Stoddart, J.F. J. Am. Chem. Soc. 2017, 139 , 3986: Wu, G. ; Olesinska, M. ; Wu, Y. ; Matak-Vinkovic, D. ; Scherman, O.A. J. Am. Chem. Soc. 2017, 139 , 3202.
- McGonigal, P.R. ; Li, H. ; Cheng, C. ; Schneebeli, S.T. ; Frasconi, M. ; Witus, L.S. ; Stoddart, J.F. Tetrahedron Lett. 2015, 56 , 3591.
- Amabilino, D.B. ; Ashton, P.R. ; Bravo, J.A. ; Raymo, F.M. ; Stoddart, J.F. ; White, A.J.P. ; Williams, D.J. Eur. J. Org. Chem. 1999, 1295: Barin, G .; Coskun, A .; Friedman, D. C .; Olson, M. A .; Colvin, M. T .; Carmielli, R .; Dey, S. K .; Bozdemir, O.A .; Wasielewski, M. R .; Stoddart, J. F . Chem. Eur. J . 2011, 17 , 213: Hayashi, R. ; Wakatsuki, K. ; Yamasaki, R. ; Mutoh, Y. ; Kasama, T. ; Saito, S. Chem. Commun. 2014, 50 , 204.
- Amabilino, D.B. ; Ashton, P.R. ; Reder, A.S. ; Spencer, N. ; Stoddart, J.F. Angew. Chem. Int. Ed. Engl. 1994, 33 , 1286: Wu, Q. ; Rauscher, P.M. ; Lang, X. ; Wojtecki, R.J. ; de Pablo, J.J. ; Hore, M.J.A. ; Rowan, S.J. Science 2017, doi:10.1126/science.aap7675.
- Lincheneau, C. ; Jean-Denis, B. ; Gunnlaugsson, T. Chem. Commun. 2014, 50 , 2857.
- Goldup, S.M. ; Leigh, D.A. ; McGonigal, P.R. ; Ronaldson, V.E. ; Slawin, A.M.Z. J. Am. Chem. Soc. 2010, 132 , 315: Wittenberg, J.B. ; Costales, M.G. ; Zavalij, P.Y. ; Isaacs, L. Chem. Commun. 2011, 47 , 9420.
- Kohn, D.R. ; Movsisyan, L.D. ; Thompson, A.L. ; Anderson, H.L. Org. Lett. 2017, 19 , 348.
- Aucagne, V. ; Hänni, K.D. ; Leigh, D.A. ; Lusby, P.J. ; Walker, D.B. J. Am. Chem. Soc. 2006, 128 , 2186: Saito, S. ; Takahashi, E. ; Nakazono, K. Org. Lett. 2006, 8 , 5133: Berná, J. ; Goldup, S.M. ; Lee, A.L. ; Leigh, D.A. ; Symes, M.D. ; Teobaldi, G. ; Zerbetto, F. Angew. Chem. Int. Ed. 2008, 47 , 4392: Crowley, J.D. ; Goldup, S.M. ; Lee, A.-L. ; Leigh, D.A. ; McBurney, R.T. Chem. Soc. Rev. 2009, 38 , 1530: Goldup, S.M. ; Leigh, D.A. ; McBurney, R.T. ; McGonigal, P.R. ; Plant, A. Chem. Sci. 2010, 1 , 383: Barran, P.E. ; Cole, H.L. ; Goldup, S.M. ; Leigh, D.A. ; McGonigal, P.R. ; Symes, M.D. ; Wu, J.Y. ; Zengerle, M. Angew. Chem. Int. Ed. 2011, 50 , 12280: Hoekman, S. ; Kitching, M.O. ; Leigh, D.A. ; Papmeyer, M. ; Roke, D. J. Am. Chem. Soc. 2015, 137 , 7656: Neal, E.A. ; Goldup, S.M. Angew. Chem. Int. Ed. 2016, 55 , 12488: Brown, A. ; Lang, T. ; Mullen, K.M. ; Beer, P.D. Org. Biomol. Chem. 2017, 15 , 4587: Denis, M. ; Goldup, S.M. Nat. Rev. Chem. 2017, 1 , 0061: Yamazaki, Y. ; Mutoh, Y. ; Saito, S. Chem. Lett. 2017, 47 , 904.
- Ashton, P. R .; Glink, P. T .; Stoddart, J. F .; Tasker, P. A .; White, A. J. P .; Williams, D. J . Chem. Eur. J . 1996, 2 , 729: Kim, K. Chem. Soc. Rev. 2002, 31 , 96: Aucagne, V. ; Leigh, D.A. ; Lock, J.S. ; Thomson, A.R. J. Am. Chem. Soc. 2006, 128 , 1784.
- Lee, C.-F. ; Leigh, D.A. ; Pritchard, R.G. ; Schultz, D. ; Teat, S.J. ; Timco, G.A. ; Winpenny, R.E.P. Nature 2009, 458 , 314.
- McInnes, E.J.L. ; Timco, G.A. ; Whitehead, G.F.S. ; Winpenny, R.E.P. Angew. Chem. Int. Ed. 2015, 54 , 14244.
- Zhang, Z.-J. ; Zhang, H.-Y. ; Wang, H. ; Liu, Y. Angew. Chem. Int. Ed. 2011, 50 , 10834.
- Hänni, K.D. ; Leigh, D.A. Chem. Soc. Rev. 2010, 39 , 1240.
- Zadegan, R.M. ; Norton, M.L. Int. J. Mol. Sci. 2012, 13 , 7149.
- Ackermann, D. ; Schmidt, T.L. ; Hannam, J.S. ; Purohit, C.S. ; Heckel, A. ; Famulok, M. Nat. Nanotech. 2010, 5 , 436.
- Crini, G. Chem. Rev. 2014, 114 , 10940.
- Klotz, E.J.F. ; Claridge, T.D.W. ; Anderson, H.L. J. Am. Chem. Soc. 2006, 128 , 15374.
- Mecozzi, S .; Rebek, J., Jr . Chem. Eur. J . 1998, 4 , 1016.
- Inouye, M. ; Hayashi, K. ; Yonenaga, Y. ; Itou, T. ; Fujimoto, K. ; Uchida, T. ; Iwamura, M. ; Nozaki, K. Angew. Chem. Int. Ed. 2014, 53 , 14392.
- Lewis, J.E.M. ; Beer, P.D. ; Loeb, S.J. ; Goldup, S.M. Chem. Soc. Rev. 2017, 46 , 2577.
- Prikhod’ko, A.I .; Durola, F .; Sauvage, J.P . J. Am. Chem. Soc . 2008, 130 , 448: Prikhod’ko, A. I .; Sauvage, J. P . J. Am. Chem. Soc . 2009, 131 , 6794.
- Hayashi, R. ; Mutoh, Y. ; Kasama, T. ; Saito, S. J. Org. Chem. 2015, 80 , 7536.
- Cheng, H.M. ; Leigh, D.A. ; Maffei, F. ; McGonigal, P.R. ; Slawin, A.M.Z. ; Wu, J. J. Am. Chem. Soc. 2011, 133 , 12298: Danon, J.J. ; Leigh, D.A. ; McGonigal, P.R. ; Ward, J.W. ; Wu, J. J. Am. Chem. Soc. 2016, 138 , 12643.
- Yamashita, Y .; Mutoh, Y .; Yamasaki, R .; Kasama, T .; Saito, S . Chem. Eur. J . 2015, 21 , 2139.
- Movsisyan, L.D. ; Franz, M. ; Hampel, F. ; Thompson, A.L. ; Tykwinski, R.R. ; Anderson, H.L. J. Am. Chem. Soc. 2016, 138 , 1366.
- Gulcur, M .; Moreno-Garcia, P .; Zhao, X. ; Baghernejad, M .; Batsanov, A.S. ; Hong, W. ; Bryce, M. R .; Wandlowski, T . Chem. Eur. J . 2014, 20 , 4653.
- Kwan, P.H. ; MacLachlan, M.J. ; Swager, T.M.J. Am. Chem. Soc. 2004, 126, 8638. Masai, H.; Terao, J.; Makuta, S.; Tachibana, Y.; Fujihara, T.; Tsuji, Y. J. Am. Chem. Soc. 2014, 136 , 14714.
- Blanco, V. ; Leigh, D.A. ; Marcos, V. Chem. Soc. Rev. 2015, 44 , 5341.
- Lewis, J.E.M. ; Galli, M. ; Goldup, S.M. Chem. Commun. 2017, 53 , 298.
- Loh, X.J. Mater. Horiz. 2014, 1 , 185: Tamura, A. ; Yui, N. J. Biol. Chem. 2015, 290 , 9442.
- Kato, K. ; Okabe, Y. ; Okazumi, Y. ; Ito, K. Chem. Commun. 2015, 51 , 16180: Choi, S. ; Kwon, T.-W. ; Coskun, A. ; Choi, J.W. Science 2017, 357 , 279.