

ABSTRACT
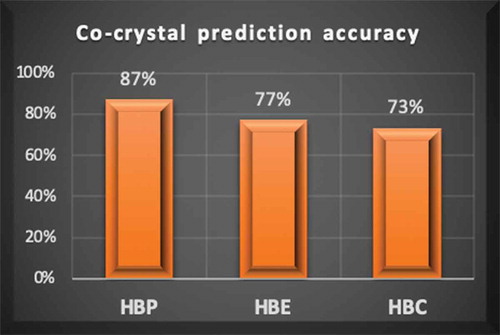
Two different structure-informatics based methods and one approach based on hydrogen-bond interaction energies were evaluated for their abilities to predict the experimental outcomes of attempted co-crystallisations between two known drug molecules, Nevirapine and Diclofenac, and a series of potential co-formers. The hydrogen-bond propensity (HBP) tool gave the correct result in 26 out of 30 cases, whereas a hydrogen-bond coordination (HBC) method predicted the correct outcome in 22 out of 30 cases. Finally, calculated hydrogen-bond energies (HBE) using a simple electrostatic model, gave the correct result in 23 out of 30 experiments. In those cases, where the crystal structure of a co-crystal of either Nevirapine or Diclofenac was known, we also examined how well the three methods predicted which primary hydrogen-bond interactions were present in the crystal structure. HBP correctly predicted 6 out of 6 cases, HBC could not predict any of the synthon formations correctly, and HBE successfully predicted 1 out of 6 cases.
GraphicalAbstract
Acknowlegements
We would like to acknowledge Dr. Mariusz Krawiec, Dr. Dongyue Xin and Dr. Nina C. Gonnella from Boehringer Ingelheim for their support and input.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data of this article can be accessed here.