
ABSTRACT
The synthesis and characterisation of tripodal ligand 1 containing 1,3,5-benzenetricarboxamide core and diethyl 4-(phenylethynyl)pyridine-2,6-dicarboxylate is described. The crystal structure of the intermediate tripodal molecule 7 exhibits an unusual staggered hydrogen-bonded chain motif instead of the anticipated helical assembly. We studied the basic photophysical properties of 1 in solvents of various polarity including CH3OH, CH3CN, DMSO, and CHCl3. The self-assembly experiments between 1 and Eu(CF3SO3)3⋅6H2O confirmed the formation of metal-ligand self-assembly species in the solution of CH3CN. It was also shown that the excitation of the Eu(III)-centred emission in such species occurred through the energy transfer from the ligand acting as an antenna to the metal centre. The binding constant values were evaluated using the nonlinear regression analysis software SPECFIT®, and their values correspond to those previously observed for the assemblies between 2,6-dipicolinic acid derivatives and lanthanide ions. This work describes the synthesis of tripodal 1,3,5-benzenetricarboxamide ligand with dipicolinic acid units and its binding with Eu(III) ions in solution. The photophysical properties of the target tripodal ligand were studied in solvents of various polarity. The self-assembly experiments between the ligand and Eu(III) ions in solution confirmed the formation of luminescent metal-ligand self-assembly species.
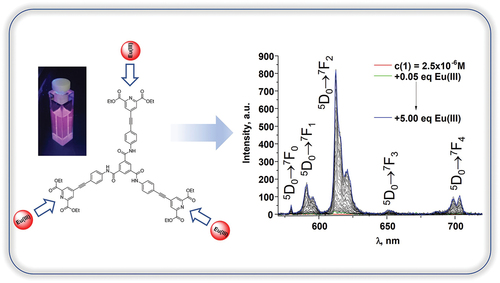
Graphical abstract
KEYWORDS:
Introduction
Over the last several decades significant progress has been made towards the understanding of fundamental principles as well as finding applications of supramolecular systems [Citation1]. Within this field of chemistry, the ligands based on 1,3,5-benzenetricarboxamide motif were of particular interest due to their ability to form helical supramolecular polymers through hydrogen bonding interaction of the three amide groups and π-stacking of the aromatic cores [Citation2]. The incorporation of metal ions into such systems enables for extending their supramolecular network and brings new functional properties to the resulting metal containing supramolecular polymer or metallogel [Citation3]. Amongst various metal ions lanthanides (Ln) occupy a special place thanks to their unique magnetic and optical properties that allow them to work as shift reagents in NMR, in catalysis, lasers and in colour television screens, as magnetic resonance imaging agents, etc. [Citation4]. In this work we are interested in the optical properties of Ln ions and their incorporation into the supramolecular polymer network. The direct excitation of Ln luminescence is known to be inefficient due to the low extinction coefficient of the Laporte forbidden f-f transitions. One way to overcome this is to use the antenna effect where the ligand coordinated to the Ln ion absorbs the energy of the excitation source, this energy is then transferred to the metal ion from where the emission occurs [Citation5]. Over the decades various types of ligands shown their efficiency towards the excitation of Ln-centred emission [Citation6] and one of the approaches was to use 2,6-dipicolinic acid (H2dpa) as it forms stable and highly emissive complexes with Eu(III) and Tb(III) [Citation7]. This approach was taken further by various research groups and involved reactions with the carboxylic groups [Citation8] including incorporation of chiral centres [Citation9] and derivatisation of the 4-position of the pyridine ring [Citation10]. The latter approach featured incorporation of various electron-donating groups allowing for the shift of the absorption band and extending the use of such ligands to the near-infrared emitters (Nd(III), Yb(III) [Citation11]) as well as enabling two-photon excitation of such molecules [Citation12].
Here we coupled 1,3,5-benzenetricarboxamide core with 2,6-dipicolinic acid through the incorporation of 4-ethynylphenyl group (, 1) using Sonogashira coupling reaction [Citation13] and utilised the protected version of the molecule (diethyl pyridine 2,6-carboxylate). The spectroscopic properties of 1 were investigated in various solvents and its self-assembly with Eu(III) ions in the solution evaluated using non-linear regression analysis [Citation14] that allowed us to propose the presence of 1:1, 2:1 and 3:1 (M:L) species in the solution and calculate the values of their binding constants. It is worth noting that similar titrations could have been done with Tb(III) ion as the design of the ligand anticipates the sensitisation of this lanthanide ion too [Citation7]. However, here we only looked at the self-assembly properties with Eu(III) due to its unique photophysical properties dictated by the electronic structure of this ion [Citation5].
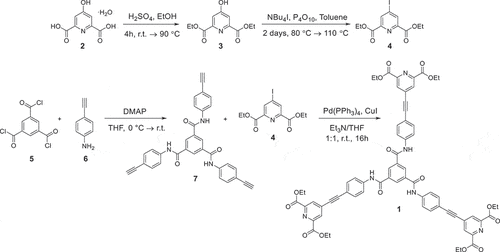
Scheme 1. Synthesis of ligand 1.
Results and discussion
Synthesis of 1
The synthesis of the ligand 1 was achieved in four steps () starting from the Fischer esterification of the carboxylic groups of the chelidamic acid (2) during 4 hours in presence of sulphuric acid in ethanol resulting in the compound 3 with 73% yield. It was then characterised using 1H, 13C NMR (Figures S1,S2) along with IR and ESI mass-spectrometry analyses (Figures S3,S4). The substitution of the hydroxyl group in the 4 position of pyridine was then achieved by the reaction of 3 with phosphorus pentoxide as oxidising agent and tert-butylammonium iodide as an iodide source following the procedure published in [Citation15]. The reaction mixture was refluxed for two days and resulted in the quantitative yield of the compound 4 (Figures S5-S8).
Figure 1. 1H NMR of the ligand 1 (DMSO-d6, 400 MHz).
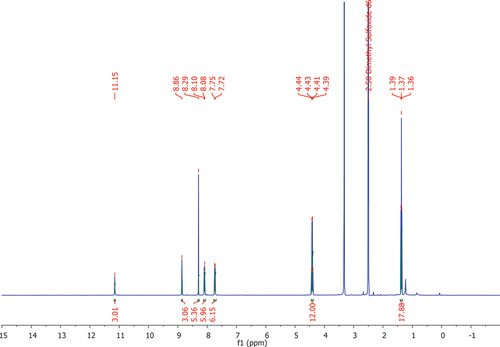
Next the tripodal ligand 7 was obtained (Figures S9-S12) by reacting 5 with three equivalents of 6 in the presence of 4-dimethylaminopyridine (DMAP) in tetrahydrofuran (THF) in 49% yield. This reaction was also performed in dichloromethane or THF in the presence of pyridine or triethylamine. However, these conditions did not result in the significant amount of product possibly due to lower basicity of these bases compared to DMAP that also acts as nucleophilic catalyst. Finally, the target ligand 1 was obtained following the general procedure for Sonogashira cross-coupling reaction [Citation15]. Further optimisation of this reaction is required as the overall yield of the product 1 was only 12%. The formation of the product was confirmed following 1H NMR () along with further analysis using 13C NMR, IR and ESI mass-spectrometry analysis (Figures S13-S15). The presence of the proton signals corresponding to ethoxy groups around 1.37 and 4.42 ppm as well as pyridine and benzene rings protons in the aromatic region confirms the formation of 1 (). Simultaneously with this the presence of carbonyl group vibrations corresponding to the 1,3,5-benzenetricarboxamide core as well as 4-(phenylethynyl)pyridine-2,6-dicarboxylate binding unit at around 1673 and 1739 cm–1 further confirms successful synthesis of the target ligand (Figure S14) along with the presence of 1193.3555 [M+ Na]+ signal in the ESI-MS spectrum (Figure S15).
Description of crystal structure of 7
Single crystals of compound 7 were obtained by slow evaporation of their solution in acetonitrile and were analysed by single crystal X-ray diffraction. The diffraction data were solved and refined in the triclinic space group P-1. The asymmetric unit confirms the expected molecular structure, containing one complete molecule of 7 and two lattice acetonitrile molecules, as shown in .
Figure 2. (A) Structure of 7 with heteroatom labelling scheme. ADPs are rendered at the 50% probability level. (B) The combined hydrogen bonding and π···π overlap between two molecules of 7. (C) The extended structure of 7 showing linking of closely held dimers by additional amide···amide hydrogen bonding. Selected hydrogen atoms are omitted for clarity.
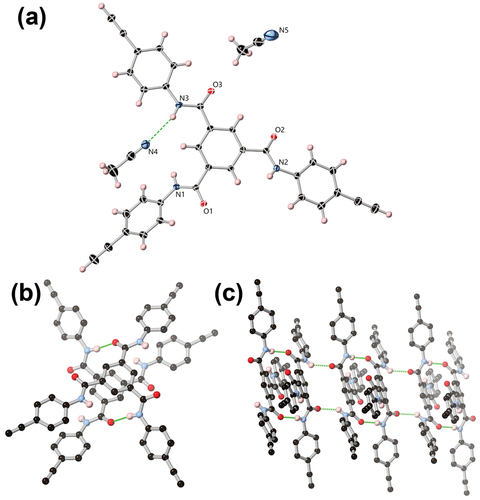
A low symmetry conformation is adopted by 7 instead of the idealised pseudo-threefold symmetric conformer, such that two amide groups are oriented in a cis conformation relative to each other, with the phenyl rings attached to N2 and N3 near-planar to the central ring (with interplanar angles 13.7° and 3.2°, respectively). The amide group N1 is more twisted, with the attached phenyl ring bent at a 49.3° angle relative to the central tricarboxamide ring.
As anticipated, the prevailing intermolecular interactions in 7 originate from the hydrogen bonding amide groups. Interestingly, while acetonitrile is a relatively weak hydrogen bond acceptor at the soft nitrile nitrogen atoms compared to the harder amide oxygen atoms, one of the three amides N3 donates a hydrogen bond to a lattice acetonitrile molecule N4 at a relatively long (N···N) distance of 3.348(2) Å and D-H···A angle of 163.14(13)°. Nitrogen atoms N1 and N2 donate hydrogen bonds to amide oxygen atoms O1 and O2 respectively, with shorter N···O distances of 2.863(2) and 2.875(2) Å and N-H···O angles of 144.31(9)° and 167.20(9)°, respectively. Both types of amide···amide contacts are reciprocated by symmetry equivalent molecules of 7 such that the extended structure consists of a linear chain of alternating dimers as shown in . The N1···O1 contacts link molecules of 7 which also feature π-π contacts at the phenyl cores, with parallel rings separated by 3.34 Å and laterally offset by 0.87 Å. In contrast, the two molecules linked by N2···O2 hydrogen bonds are laterally offset by 6.2 Å at the cores and show only minor edge-to-face type contacts between the outer rings. The resulting assembly is a staggered one-dimensional hydrogen bonding chain oriented parallel to the a axis where, unlike the typical helical 1,3,5-benzenetricarboxamide stacking motif, the normal vector of the central ring is offset by 46.44(3)° to the direction of propagation.
Compound 7 would not be forming stable chelates with lanthanides and the geometry of its donor oxygen atoms would not be able to satisfy the coordination environment of these ions. Also, there is no need for 1 to minimise destabilising electron-electron repulsion between donor atoms by the formation of the chelate [Citation16]. Hence, we did not investigate its interaction and focused on the self-assembly studies between 1 and Eu(III).
Self-assembly studies between 1 and Eu(CF3SO3)3 in solution
As the ligand 1 was synthesised and characterised we next recorded and evaluated its absorption, luminescence and excitation spectra in various solvents such as DMSO, CH3CN, CH3OH and CHCl3. Due to the low yield and low solubility of 1 its stock solutions were prepared in DMSO (c1 = 1.025 × 10–4 M). This solution was then gradually diluted into the fluorescence cell containing DMSO and each time its absorbance, fluorescence and excitation spectra were recorded in the concentration range of 5.00 × 10–5 M to 1.40 × 10–6 M (Figure S16). The main absorption maximum for the ligand 1 in DMSO was detected at 340 nm (ε340 = 65,771 ± 1487 M–1 × cm–1, Figures S16A,D). Upon excitation at 340 nm the emission of 1 occurred with maximum centred at 460 nm. It was observed that with increased concentration of the solution emission intensity of 1 enhanced linearly until c1 = 5.00 × 10–6 M after which its rate slowed with the following gradual decrease observed from the concentration of 1.35 × 10–5 M indicating the presence of inner filter effect. This is also reflected in the shape of the excitation spectrum that starts to correspond to the absorption spectrum only below the concentration 1.10 × 10–5 M (Figure S16C). Similar studies were then performed from the stock solution of 1 in DMSO upon its dilution into CH3CN (ε335 = 80,733 ± 928 M–1 × cm–1; Figure S17), CH3OH (ε335 = 78,449 ± 916 M–1 × cm–1; Figure S18) or CHCl3 (ε340 = 78,539 ± 717 M–1 × cm–1; Figure S19) in the fluorescence cell which allowed to choose c1 = 2.50 × 10–6 M for the self-assembly titration studies with Eu(III) ensuring the presence of single molecules of ligand 1 in the solution. Slight red shift (~5 nm) of the absorption maximum was observed when going from more polar CH3OH, CH3CN or DMSO solvents to less polar CHCl3. Upon excitation of ligand 1 the emission spectra were observed with maxima centred at 470 nm in CH3OH, 460 nm in DMSO, 454 nm in CH3CN and 421 nm in CHCl3 indicating the stabilisation of the excited state by the polar solvent molecules [Citation17].
The self-assembly studies between 1 and Eu(III) ion were then performed in CH3CN as commonly used polar aprotic solvent. Here we anticipated the coordination of both hard oxygen and soft nitrogen atoms of pyridine-2,6-dicarboxylate to the Eu(III) ions forming stable chelates [Citation10bb]. We monitored the changes in the absorbance, fluorescence and Eu(III)-centred emission of 1 (c1 = 2.50 × 10–6 M) upon gradual addition of Eu(CF3SO3)3⋅ 6H2O from 0 to 5 equivalents ().
Figure 3. The changes in (A) the absorption and (B) fluorescence spectra of 1 (c1 = 2.50 × 10–6 M) upon addition of Eu(CF3SO3)3⋅6H2O (0→5.00 equiv.) in CH3CN (λex = 335 nm); Inset (B): the photograph of the ligand solution at the start of the titration emitting the blue light and at the end of the titration indicating the presence of red Eu(III)-centred emission (λex = 365 nm).
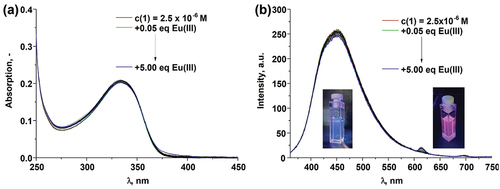
Figure 4. (A) The changes in the Eu(III)-centred emission spectra of 1 (c1 = 2.50 × 10–6 M) upon addition of Eu(CF3SO3)3⋅6H2O(0→5.00 equiv.) in CH3CN (λex = 335 nm); (B) speciation-distribution diagram; (C) experimental binding isotherms (∙∙∙) and their corresponding fit (–) obtained using non-linear regression analysis program SPECFIT®.
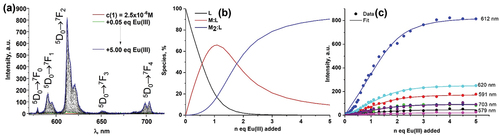
The absorption spectrum of 1 with the main absorbance band centred at 335 nm did not show any significant changes upon addition of Eu(CF3SO3)3⋅6H2O (). Similarly to this only minor decrease of the ligand-centred luminescence at 450 nm was observed with simultaneous appearance of Eu(III)-centred emission band at 616 nm (∆J = 2; 5D0 → 7F2) with low intensity (). These even though minor changes occurred up until the addition of 2.00 equivalents of Eu(III) ion where the formation of plateau was observed.
The binding interaction between 1 and Eu(III) was also monitored using delayed luminescence spectroscopy where gradual increase of Eu(III)-centred emission was observed until the addition of 2.00 equivalents of Eu(III) where again the plateau started to form (). The presence of 5D0 → 7F0 transition band in the emission spectrum () is an example of the breakdown of the selection rules as according to the Judd-Ofelt theory it is strictly forbidden. It also indicates that within metal-ligand assemblies formed in the solution Eu(III) occupies Cnv, Cn or Cs symmetry [Citation5b].
The energy transfer from the ligand to the Eu(III) ion was confirmed by monitoring the excitation spectra during the course of titration at λem = 616 nm (Figure S20A). The presence of the band centred at 335 nm and repeating the shape of the absorption spectrum confirms that the excitation of the Eu(III)-centred emission is the result of the energy transfer from the ligand to the metal centre. The life-times of the Eu(III)-centred emission were monitored during the course of the titration (Figure S20B) and remained monoexponential with the values around 1.33 ± 0.03 ms (Table S1) indicating the presence of only one type of coordination environment around Eu(III) ion within the metal-ligand species formed in the solution. The observed value of Eu(III)-centred life-time decay is long, and its comparison to the life-times of Eu(III) complexes with similar ligand structure does not allow us to assign it to a specific metal/ligand system as it was found to vary in a wide range [Citation11].
The changes observed during the course of the titration in absorbance, fluorescence and Eu(III)-centred emission spectra were also analysed using non-linear regression analysis program SPECFIT [Citation18].
The binding models corresponding to the formation of 1:1, 2:1 and 3:1 metal-ligand species were proposed and binding constants for such species were calculated for the changes observed in absorbance and fluorescence spectra (). However, only the presence of 1:1 and 2:1 metal-ligand species could be modelled for the changes observed in the Eu(III)-centred emission spectra ().
Table 1. Binding constants obtained by fitting the changes in absorption, fluorescence and Eu(III)-centred emission spectra for the titrations of 1 (c1 = 2.50 × 10–6 M) with Eu(CF3SO3)3⋅6H2O (0→5.00 equiv.) in CH3CN at 298 K.
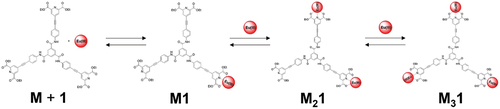
The recalculated spectra, speciation diagrams and binding isotherms with their fits are presented in ; S21–23 and confirming the goodness of the fit for the proposed binding model that is shown on each of the three diethyl 4-(phenylethynyl)pyridine-2,6-dicarboxylate binding units of 1 to Eu(III) ion is shown (1:1, 2:1 and 3:1; M:L species). The proposed coordination implies the presence of three identical binding sites for the Eu(III) ion corroborating with the experimental data where the presence of mono-exponential Eu(III)-centred emission decay was observed (Table S1).. Here the coordination of
Scheme 2. Schematic representation of the proposed binding between Eu(III) and ligand 1 in solution (M:L species).
Conclusion
The synthesis of tripodal ligand 1 based on 1,3,5-benzenetricarboxamide coupled with 4-(phenylethynyl)pyridine-2,6-dicarboxylate was achieved. The composition of the ligand was confirmed using conventional techniques including absorption and fluorescence spectroscopy in the solvents such as CH3OH, CH3CN, DMSO and CHCl3. Intermediate tripodal molecule 7 was found to crystallise in the triclinic space group P-1 and packing differently to often observed helical motif characteristic for many 1,3,5-benzenetricaroboxamide systems [Citation2]. Here a staggered one-dimensional hydrogen bonding chain oriented parallel to a axis was observed. We have used 1 to study its interaction with Eu(CF3SO3)3⋅6H2O in CH3CN. The analysis of the changes in the absorption, fluorescence and Eu(III)-centred emission spectra allowed us to calculate the binding constants for the proposed binding model including the presence of 1:1, 2:1 and 3:1 (M:L) species with the logβ values ~7, 13 and 18 which is corresponding to the binding constants values for the similar systems [Citation7, Citation10b]. This ligand 1 has a potential for further investigation of its interaction with other lanthanide ions including these emitting in the NIR region (for example Nd(III), Yb(III)) as well as due to its potential use as two-photon excited luminescence probe [Citation12].
Experimental
General
Methods and materials
All chemicals were purchased from commercial sources and used without further purification. Chelidamic acid, tert-butyl ammonium iodide, benzene-1,3,5-tricarbonyl trichloride, 4-ethynyl aniline, tetrakis(triphenylphosphine)palladium(0), sulphuric acid, hydrochloric acid, phosphorus pentoxide, sodium hydrogen carbonate, sodium chloride, magnesium sulphate, potassium thiosulphate, ethanol, dichloromethane, toluene, tetrahydrofuran were purchased. CDCl3 and DMSO-d6 for NMR analysis were also purchased. The 1H-NMR spectra were recorded at 400 MHz and the 13C-NMR spectra were recorded at 100 MHz using an Agilent 400 MR equipped with a 5 mm OneNMR probe for proton and multinuclear detection. Chemical shifts were reported in ppm using the deuterated solvent as the internal reference, J in Hz; data were processed with Bruker Topspin 2.1®. All the NMR spectra were carried out at 293 K. Mass spectrometry was carried out using HPLC grade solvents. Electrospray mass spectra were determined on a Micromass LCT spectrometer and high resolution mass spectra were determined relative to a standard of leucine enkephalin. Infrared spectra were recorded on a Perkin Elmer Spectrum One FT-IR spectrometer equipped with universal ATR sampling accessory. Melting points were determined using an Electrothermal IA9000 digital melting point apparatus.
Single-crystal X-ray crystallography
Structure and refinement parameters are given in .
Table 2. Crystallographic data and structural refinement for 7.
All diffraction data were collected using a Bruker APEX-II Duo dual-source diffractometer with Mo Kα radiation (λ = 0.71073 Å). Datasets were collected using ω and φ scans with the sample coated in NVH immersion oil and maintained at a constant temperature of 100 K using a Cobra cryostream. The data was reduced and processed using the Bruker APEX-3 suite of programs [Citation19]. Multi-scan absorption corrections were applied using SADABS [Citation20]. The diffraction data were solved using SHELXT and refined by full-matrix least squares procedures using SHELXL-2015 within the OLEX-2 GUI [Citation21–23]. All non-hydrogen atoms were refined with anisotropic displacement parameters. All carbon-bound hydrogen atoms were placed in calculated positions and refined with a riding model, with isotropic displacement parameters equal to either 1.2 or 1.5 times the isotropic equivalent of their carrier atoms.
Photophysical measurements
All measurements in solution were performed at 298 K either in DMSO, CHCl3, CH3OH and CH3CN (spectroscopical grade, Aldrich) as indicated on the caption to the relevant Figures. UV-Visible absorption spectra were measured in 1 cm quartz cuvettes on a Varian Cary 50 spectrophotometer. Baseline correction was applied to all spectra. Emission (fluorescence, phosphorescence and excitation) spectra and lifetimes were recorded on a Varian Cary Eclipse Fluorimeter.
Phosphorescence lifetimes of the Eu(III) (5D0) excited state were recorded in time-resolved mode. The life-time values are averages of three independent measurements, which were made by monitoring the emission decay at 616 nm, which corresponds to the maximum of the Eu(III) 5D0→7F2 transition, enforcing a 0.1 ms delay, and were analysed using Origin Pro 8.5®.
Spectrophotometric titrations and binding constants
The formation of the luminescent 1:1, 2:1 and 3:1 (M:L, where M = Eu(III) and L = 1) species was ascertained by UV-Visible and luminescence titrations of a solution of 1 prepared in DMSO (c1(stock) = 1.03 × 10–4 M) and diluted into 2.7 mL of CH3CN in the fluorescence cell (c1 = 2.50 × 10–6 M) with Eu(CF3SO3)3⋅6 H2O (c = 2.54 × 10–4 M; 0→5 equiv.) prepared in CH3CN. The data were fitted using the non-linear regression analysis program, SPECFIT® [Citation18].
Synthesis
4-Hydroxypyridine-2,6-dicarboxilate (3)
Chelidamic acid monohydrate (2, 2.50 g, 12.4 mmol, 1.00 equiv.) was suspended in ethanol (40 mL) and sulphuric acid (3.14 mL, 57.2 mmol, 4.60 equiv.) was added slowly to the solution at room temperature. The solution was heated up to 90 °C and was refluxed for 4 hours. The solvents were evaporated, and water (10 mL) was added. The resulting solution was neutralised with saturated sodium hydrogen carbonate solution and then extracted with dichloromethane (3 x 30 mL). The combined organic layers were washed with brine (50 mL) and dried over magnesium sulphate. Evaporation of the solvents gave chelidamic acid diethylester (3, 2.17 g, 9.07 mmol, 73%) as white solid. Mp: 91 °C-93 °C. Rf = 0.36 (dichlormethane/methanol 20:1). 1H NMR (400 MHz, DMSO-d6, δH): 1.33 (t, 6H, J = 7.1 Hz, -CH2CH3), 4.35 (q, 4 H, J = 7.1 Hz, -CH2CH3), 7.57 (s, 2 H, arom.) ppm. 13C NMR (100 MHz, DMSO-d6, δC): 14.13 (-CH3), 61.46 (-CH2CH3), 115.17 (2C, C1), 149.26 (2C, C2), 164.25 (C-OH), 165.83 (CO2Et) ppm. IR (ATR-diamond, ν): 705, 739, 789, 820, 862, 891, 918, 944, 998, 1020, 1044, 1105, 1156, 1227, 1332, 1366, 1394, 1404, 1458, 1568, 1603, 1721, 1737, 1980, 2114, 2166, 2480, 2582, 2684, 2797, 2907, 2941, 2985 cm–1. ESI-MS: m/z calculated for C11H14NO5 [M+H]+ 240.086649; found: 240.086014.
Diethyl 4-iodopyridine-2,6-dicarboxylate (4)
Tert-butyl ammonium iodide (1.85 g, 5.02 mmol, 1.20 equiv.) and phosphorus pentoxide (1.66 g, 5.85 mmol, 1.40 equiv.) were dissolved in dry toluene (100 mL) under argon atmosphere and the solution was heated for 1 hour at 80 °C. Then, the ester (3, 1.00 g, 4.18 mmol, 1.00 equiv.) was added to the reaction mixture. The reaction was refluxed for 2 days at 110 °C. After cooling to room temperature, the supernatant was taken aside, and the oily residue was heated up with toluene (50 mL) for 1 hour. The supernatant and the solution of the residue were combined, washed with water (100 mL), potassium thiosulphate solution (100 mL) and brine (100 mL). The organic layer was dried over magnesium sulphate and the solvents were evaporated to yield iodide (4, 1.46 g, 4.18 mmol, quant.) as white solid. Mp: 77 °C-79 °C. Rf = 0.59 (dichloromethane/methanol 50:1). 1H NMR (400 MHz, DMSO-d6, δH): 1.34 (t, 6H, J = 7.11 Hz,-CH3), 4.38 (q, 4 H, J = 7.11 Hz, -CH2CH3), 8.56 (s, 2 H, arom.) ppm. 13C NMR (100 MHz, DMSO-d6, δC): 14.1 (-CH3), 61.9 (-CH2CH3), 108.7 (-C-I), 136.2 (2C, C1), 148.2 (2C, C2), 163.0 (CO2Et) ppm. IR (ATR-diamond; ν): 661, 689, 708, 733, 779, 865, 872, 899, 936, 943, 1018, 1110, 1150, 1239, 1260, 1318, 1367, 1407, 1425, 1445, 1472, 1554, 1714, 1880, 2099, 2167, 2325, 2872, 2906, 2936, 2977, 3065, 3408 cm–1. ESI-MS: m/z calculated for C11H12INO4 [M+Na]+ 371.970327; found: 371.970612.
N1,N3,N5-Tris(4-ethynylphenyl)benzene-1,3,5-tricarboxamide (7)
The 1,3,5-benzenetricarbonyl trichloride (5, 500 mg, 1.88 mmol, 1.00 equiv.) and 4-dimethylaminopyridine (690 mg, 5.65 mmol, 3.00 equiv.) were dissolved in dry THF (200 mL) at 0 °C. 4-Ethynyl aniline (6, 662 mg, 5.65 mmol, 3.00 equiv.) was added as a solution in THF (20 mL) and the solution was allowed to warm to room temperature. After 4 hours, the white precipitate was filtered off, taken up in dichloromethane (100 mL) and washed with hydrochloric acid (0.1 M, 3 × 50 mL) and brine (50 mL). The combined organic layers were dried over magnesium sulphate and evaporation of the solvents gave N1,N3,N5-tris(4-ethynylphenyl)benzene-1,3,5-tricarboxamide (7, 469 mg, 0.92 mmol, 49%) as white-beige solid. Mp: 196 °C-198 °C. Rf = 0.20 (hexane/ethyl acetate 1:1). 1H NMR (400 MHz, DMSO-d6, δH): 4.13 (s, 3 H, H9), 7.51 (d, 6H, J = 8.7 Hz, H6), 7.87 (d, 6H, J = 8.8 Hz, H5), 8.71 (s, 3 H, H1), 10.77 (s, 3 H, NH) ppm. 13C NMR (100 MHz, DMSO-d6, δC): 80.1 (3C, C9), 83.5 (3C, C8), 116.1 (3C, C7), 120.1 (6C, C5), 130.1 (3C, C1), 132.3 (6C, C6), 135.1 (3C, C2), 139.5 (3C, C4), 164.6 (3C, C3) ppm. IR (ATR-diamond; ν): 687, 764, 833, 911, 953, 999, 1018, 1048, 1109, 1175, 1244, 1294, 1313, 1401, 1507, 1587, 1648, 1917, 2102, 2324, 3099, 3244, 3285 cm–1. ESI-MS: m/z calculated for C33H21N3O3 [M+Na]+ 530.147512; found: 530.147508.
Hexaethyl-4,4’,4”-((((benzene-1,3,5-tricarbonyl)tris(azanediyl))tris(benzene-4,1-diyl))tris(ethyne-2,1-diyl))tris(pyridine-2,6-dicarboxylate) (1)
Diethyl 4-iodopyridine-2,6-dicarboxylate 4 (124 mg, 0.36 mmol, 3.00 equiv.), tetrakis(triphenylphosphine)palladium(0) (41.0 mg, 0.04 mmol, 0.30 equiv.) and copper iodide (13.5 mg, 0.07 mmol, 0.60 equiv.) were stirred at room temperature in a degassed THF/triethylamine (15 mL/15 mL) solvent mixture. After 30 minutes, a degassed solution of N1,N3,N5-tris(4-ethynylphenyl)benzene-1,3,5-tricarboxamide 7 (60.0 mg, 0.12 mmol, 1.00 equiv.) in THF (5 mL) was added dropwise. The solution was stirred overnight at room temperature under argon atmosphere. Then, the solvents were evaporated, and the resulting residue was taken up in dichloromethane (50 mL), washed with water (30 mL) and brine (30 mL) and dried over magnesium sulphate. The solvents were evaporated, and the crude product was purified by column chromatography (dichloromethane/methanol 20:1, then 10:1). The protected ligand 1 (16.0 mg, 0.01 mmol, 12%) could be obtained as pale yellow solid. Mp: 238 °C-240 °C. Rf = 0.42 (dichloromethane/methanol 20:1). 1H NMR (400 MHz, DMSO-d6, δH): 1.37 (t, 18 H, J = 7.1 Hz, -CH3), 4.42 (q, 12 H, J = 7.1 Hz, -CH2CH3), 7.74 (d, 6H, J = 8.7 Hz, H6), 8.09 (d, 6H, J = 8.8 Hz, H5), 8.29 (s, 6H, H11), 8.86 (s, 3H, H1), 11.15 (s, 3H, NH) ppm. 13C NMR (100 MHz, CDCl3: DMSO-d6, δC): 14.2 (6C, -CH3), 62.5 (6C, -CH2CH3), 85.4 (3C, C9), 97.0 (3C, C8), 116.9 (3C, C7), 120.2 (6C, C5), 129.4 (6C, C6), 133.0 (6C, C11), 134.5 (3C, C2), 135.5 (3C, C10), 140.1 (3C, C4), 148.8 (6C, C12), 164.3 (6C, CO2Et), 164.6 (3C, CONH) ppm. IR (ATR-diamond, ν): 658, 682, 700, 728, 761, 779, 824, 900, 948, 1004, 1022, 1048, 1104, 1155, 1174, 1234, 1296, 1313, 1350, 1376, 1404, 1443, 1512, 1584, 1672, 1717, 1738, 2127, 2205, 2257, 2903, 2983, 3086, 3391 cm–1. ESI-MS: m/z calculated for C66H54N6O15 [M+Na]+ 1193.3545; found: 1193.3555.
Associated content
Supporting Information contains 1H and 13C NMR spectra, IR and ESI-MS for all the synthesised compounds along with absorption, fluorescence and excitation spectra recorded for 1 in various organic solvents. Part of the spectroscopic titration data of 1 vs Eu(CF3SO3)3⋅6H2O, its non-linear regression analysis and Eu(III)-centred luminescence decay data is also represented.
Accession Code CCDC 2231908 (7) contains the supplementary crystallographic data.
Supplemental Material
Download MS Word (2.9 MB)Acknowledgments
We thank Dr. Gary Hessman for mass spectrometry measurements, Drs. Manuel Ruether and John O’Brien for the help with NMR analysis. Authors would like to thank the Science Foundation Ireland (PI awards 13/IA/1865 to T.G.), SFI AMBER Center (12/RC/2278_P2), the School of Chemistry, and Trinity College Dublin for the provided infrastructure and financial support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/10610278.2023.2177162
Additional information
Funding
References
- a) Lehn J.-M. Toward self-organization and complex matter. Science. 2002;295(5564):2400–2403. b) Philp, D.; Stoddart, J. F. Angew. Chem. Int. Ed. 1996, 35, 1155-1196; c) Amabilino, D. B.; Smith, D. K.; Steed. J. W. Chem. Soc. Rev. 2017, 46, 2404-2420; d) Antipin, I. S. et al. Russ. Chem. Rev. 2021, 90, 895-1107.
- a) Kulkarni C., Meijer E. W., Palmans A. R. A. Cooperativity scale: a structure–mechanism correlation in the self-assembly of benzene-1,3,5-tricarboxamides. Acc. Chem. Res. 2017;50(8):1928–1936. b) Su, L.; Mosquera, J.; Mabesoone, M. F. J.; Schoenmakers, S. M. C.; Muller, C.; Vleugels, M. E. J.; Dhiman, S.; Wijker, S.; Palmans, A. R. A.; Meijer, E. W. Science. 2022, 377, 213-218; c) Schnitzer, T.; Preuss, M. D.; van Basten, J.; Schoenmakers, S. M. C.; Spiering, A. J. H.; Vantomme, G. and Meijer, E. W. Angew. Chem. Int. Ed. 2022, 61, e202206738; d) Schoenmakers, S. M. C.; Spiering, A. J. H.; Herziger, S.; Böttcher, C.; Haag, R.; Palmans, A. R. A. and Meijer, E. W. ACS Macro Letters. 2022, 11(5), 711-715.
- a) Beseniusa P., Portaleb G., Bomansc P. H. H., et al. Controlling the growth and shape of chiral supramolecular polymers in water. PNAS. 2010;107(42):17888–17893. b) Kotova, O.; Daly, R.; dos Santos, C. M. G.; Boese, M.; Kruger, P. E.; Boland, J. J. and Gunnlaugsson, T. Angew. Chem. Int. Ed. 2012, 51, 7208-7212; c) Lynes, A. D.; Hawes, C. S.; Byrne, K.; Schmitt, W. and Gunnlaugsson, T. Dalton Trans. 2018, 47, 5259-5268; d) Lynes, A. D.; Hawes, C. S.; Ward, E. N.; Haffner, B.; Möbius, M. E.; Byrne, K.; Schmitt, W.; Pal, R. and Gunnlaugsson, T. Cryst. Eng. Comm. 2017, 19, 1427-1438; e) Kotova, O.; Bradberry, S. J.; Savyasachi, A. J. and Gunnlaugsson, T. Dalton Trans. 2018, 47, 16377-16387.
- a) Eliseeva S. V., Bünzli J.-C. G. Rare earths: jewels for functional materials of the future. New J. Chem. 2011;35(6):1165–1176. b) Li, X.-Z.; Tian, C.-B.; Sun, Q.-F. Chem. Rev. 2022, 122(6),6374-6458; c) Heffern, M. C.; Matosziuk, L. M.; Meade, T. J. Chem. Rev. 2014, 114, 4496-4539; d) Bell, D. J.; Natrajan, L. S.; Riddell, I. A. Coord. Chem. Rev. 2022, 472, 214786.
- a) Eliseeva S. V., Bünzli J.-C. G.Lanthanide luminescence for functional materials and bio-sciences. Chem. Soc. Rev. 2010;39(1):189–227. b) Binnemans, K. Coord. Chem. Rev. 2015, 295, 1-45.
- a) Bradberry S. J., Savyasachi A. J., Martínez-Calvo M., et al. Development of responsive visibly and NIR luminescent and supramolecular coordination self-assemblies using lanthanide ion directed synthesis. Coord. Chem. Rev. 2014;273-274:225–241. b) Barry, D. E.; Caffrey, D. F.; Gunnlaugsson, T. Coord. Chem. Rev. 2016, 45, 3244-3274; c) Xian, T.; Meng, Q.; Gao, F.; Hu, M.; Wang, X. Coord. Chem. Rev. 2023, 474, 214866.
- Chauvin A.-S., Gumy F., Imbert D., et al. Europium and terbium tris (dipicolinates) as secondary standards for quantum yield determination. Spectrosc. Lett. 2004;37(5):517–532. erratum: Chauvin, A.-S.; Gumy, F.; Imbert, D.; Bünzli, J.-C. G. Spectrosc. Lett. 2006, 40, 193.
- a) Nozary H., Piguet C., Tissot P., et al. Bent tridentate receptors in calamitic mesophases with predetermined photophysical properties: new luminescent lanthanide-containing materials, J. Am. Chem. Soc. 1998;120: 12274–12288. b) Shavaleev, N. M.; Gumy, F.; Scopelliti, R. and Bünzli, J.-C. G. Inorg. Chem. 2009, 48, 5611-5613; c) Di Pietro, S.; Gautier, N.; Imbert, D.; Pécauta, J. and Mazzanti, M. Dalton Trans. 2016, 45, 3429-3442.
- a) Leonard J. P., Jensen P., McCabe T., et al. Self-assembly of chiral luminescent lanthanide coordination bundles. J. Am. Chem. Soc. 2007;129(36):10986–10987. b) Kotova, O.; Kitchen, J. A.; Lincheneau, C.; Peacock, R. D. and Gunnlaugsson, T. Chem. Eur. J. 2013, 19, 16181-16186.
- a) Starck M., MacKenzie L. E., Batsanov A. S., et al. Excitation modulation of Eu:BPEPC based complexes as low-energy reference standards for circularly polarised luminescence (CPL). Chem. Commun. 2019;55(94):14115–14118. b) Andres, J.; Chauvin, A.-S. Eur. J. Inorg. Chem. 2010, 2700-2713; c) Caffrey, D.; Gunnlaugsson, T. Dalton Trans. 2014, 43, 17964-17970; c) Johnson, K. R.; de Bettencourt-Dias, A. Inorg. Chim Acta. 2021, 514, 120003; d) Monteiro, J. H. S. K.; Fetto, N. R.; Tucker, M. J.; Sigoli, F. A.; de Bettencourt-Dias, A. J. Lumin. 2022, 245, 118768; e) Rajah, D.; Pfrunder, M. C.; Chong, B. S. K.; Ireland, A. R.; Etchells, I. M. and Moore, E. G. Dalton Trans. 2021, 50, 7400-7408; f) Kotova, O.; O’Reilly, C.; Barwich, S. T.; Mackenzie, L. E.; Lynes, A. D.; Savyasachi, A. J.; Ruether, M.; Pal, R.; Möbius, M.; Gunnlaugsson, T. Chem. 2022, 8(5), 1395-1414.
- D’Aléo A., Picot A., Beeby A., et al. Efficient sensitization of europium, ytterbium, and neodymium functionalized tris-dipicolinate lanthanide complexes through tunable charge-transfer excited states. Inorg. Chem. 2008;47(22):10258–10268.
- a) Picot A., D’Aléo A., Baldeck P. L., et al. Long-lived two-photon excited luminescence of water-soluble europium complex: applications in biological imaging using two-photon scanning microscopy. J. Am. Chem. Soc. 2008;130(5):1532–1533. b) D’Aléo, A.; Pointillart, F.; Ouahabb, L.; Andraudc, C.; Maury, O. Coord. Chem. Rev. 2012, 256, 1604-1620.
- a) Barsu C., Fortrie R., Nowika K., et al., Synthesis of chromophores combining second harmonic generation and two photon induced fluorescence properties. Chem. Commun. 2006;45:4744–4746. b) Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874-922.
- a) Gampp H., Maeder M., Meyer C. J., et al. Calculation of equilibrium constants from multiwavelength spectroscopic data—IV model-free least-squares refinement by use of evolving factor analysis. Talanta. 1986;33(12):943–951. b) Gampp, H.; Maeder, M.; Meyer, C. J.; Zuberbìhler, A. D. Talanta 1985, 32, 1133-1139.
- Picot A., Feuvrie C., Barsu C., et al. Synthesis, structures, optical properties, and TD-DFT studies of donor-π-conjugated dipicolinic acid/ester/amide ligands. Tetrahedron. 2008;64(2):399–411.
- Abergel R. J., Kozimor S. A. Innovative f-element chelating strategies. Inorg. Chem. 2020;59(1):4–7.
- Lakowicz J. R. Principles of fluorescent spectroscopy. 3rd ed. New York NY: Springer; 2006. p. 954.
- a) Gampp H., Maeder M., Meyer C. J., et al. Calculation of equilibrium constants from multiwavelength spectroscopic data—IV model-free least-squares refinement by use of evolving factor analysis. Talanta. 1986;33(12):943–951. b) Gampp, H., Maeder, M., Meyer, C. J., Zuberbìhler, A. D., Talanta 1985, 32, 1133-1139.
- Bruker-AXS. Bruker APEX-3, Bruker-AXS Inc. Madison, WI; 2016.
- Bruker-AXS. SADABS 2016/2, Bruker-AXS Inc. Madison, WI; 2016.
- Sheldrick G. M. SHELXT – integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A. 2015;71(1):3–8.
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015;71(1):3–8
- Dolomanov O. V., Bourhis L. J., Gildea R. J., et al. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009;42(2):339–341.