
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
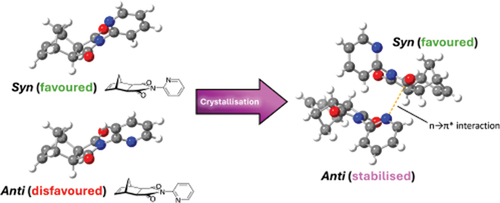
In recent studies, n→π* interactions featuring nucleophilic pyridine have been investigated using Fourier transform microwave spectroscopy and high-level theoretical computations. Examples of n→π* of this type in the solid state, however, are limited. The norbornene-based 2-pyridyl imide 1 is a compound that can adopt two conformers, in which the pyridyl nitrogen is either syn or anti to the norbornane bridge. It would be expected that the syn conformation of imide 1 would be more favourable, as indicated in a previous computational study. Here, tandem crystallographic and additional computational studies provide evidence that the otherwise unfavourable anti conformer of imide 1 becomes favoured upon stabilisation by an n→π* interaction in the solid state.
GRAPHICAL ABSTRACT
Introduction
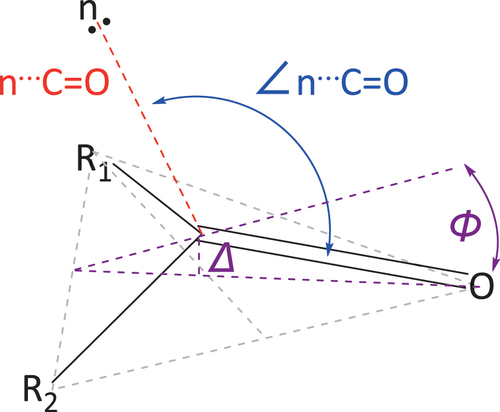
In supramolecular n→π* interactions, electron density is donated from the lone pair of a ‘nucleophile’ into the empty π* antibonding orbital of a carbonyl or aryl ‘electrophile’ [Citation1–3]. When the π* orbital belongs to a carbonyl group, the interactions typically follow the Bürgi – Dunitz trajectory for nucleophilic attack on a carbonyl, exhibiting an ∠n⋯C=O angle of 109 ± 10° within an n⋯C=O distance of ≤3.2 Å () [Citation4–6]. These interactions can also cause the carbonyl carbon to distort out of the expected trigonal planar geometry towards the nucleophile, a phenomenon known as pyramidalisation [Citation1–3]. The distance that the carbonyl carbon atom moves out of the plane of the carbonyl moiety (Δ) and the angle the carbonyl oxygen has with the plane of the other atoms in the moiety (Φ) are parameters used to describe the degree of pyramidalization () [Citation1,Citation2].
Figure 1. n→π* interaction in a carbonyl electrophile.
The n→π* interactions are weak, ranging from approximately 1.3 kJmol−1 to 2.9 kJmol−1, however the prevalence of carbonyl groups in both natural and synthetic chemistry makes these interactions particularly interesting [Citation2]. Extensive study has identified that they (i) contribute significantly to protein stability [Citation7–11] and (ii) stabilise the most favoured conformers of biologically relevant small molecules including N-acyl homoserine [Citation9], aspirin [Citation12], γ-aminobutyric acid [Citation13], 4(S)- and 4(R)-hydroxyproline [Citation14], and HGlyProOH [Citation15].
Recent studies by Blanco and López et al. have confirmed the action of pyridine as a nucleophile in n→π* interactions [Citation16–18]. These studies made use of Fourier transform microwave spectroscopy in tandem with high-level theoretical computations to characterise complexes of pyridine with simple ketones and aldehydes [Citation16–18]. Examples of this pyridine⋯C=O interaction in the solid state, however, are limited.
The 2-pyridyl-functionalised imide 1 is a known compound that has been reported to adopt two conformers, one in which the lone pair of the pyridyl nitrogen is syn to the norbornane bridge, and one in which it is anti () [Citation19]. The preference of the molecule, however, is somewhat uncertain, with computational evidence [Citation19] suggesting the syn conformer is more favoured, while an earlier NMR study [Citation20] instead infers a preference for the anti conformer.
Figure 2. Syn and anti conformers of 2-pyridyl imide 1.
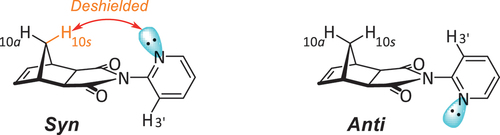
The 1H NMR chemical shift of this 10s proton of the norbornyl methylene bridge has been used to assess conformation, with computational evidence asserting that the proximity of the lone pair causes deshielding () [Citation19]. Deciphering changes in this shielding, however, is a somewhat challenging process as the peaks for the 10s and 10a bridge protons in 2-pyridyl imide 1 overlap at ambient temperature [Citation19].
As the similarity of the 10s and 10a protons in the methylene bridge of imide 1 makes tracking changes in the 10s bridge proton difficult, a new molecule in which the two bridge protons experience different environments was devised in [5]polynorbornane 2 (). In similar polynorbornane systems, steric compression by the adjacent oxanorbornane bridges causes significant deshielding of the 31a and 33a bridge protons [Citation21–26], and with the 31a and 33a protons being distinguishable from 31s and 33s, the influence of pyridyl ring rotation should be more readily identified.
Figure 3. Structure of 2-pyridyl [5]polynorbornane 2. The left 2-pyridyl heterocycle is shown in the syn conformation, while the right is shown in the anti conformation.
![Figure 3. Structure of 2-pyridyl [5]polynorbornane 2. The left 2-pyridyl heterocycle is shown in the syn conformation, while the right is shown in the anti conformation.](/cms/asset/3508dac0-8f9a-4602-935d-bfdd012247f2/gsch_a_2384908_f0003_oc.jpg)
Cyclical N-arylimides [Citation27–31] and, more specifically norbornene imides [Citation32–34], have seen extensive use in the study of rotamers, using the carbonyl of the rigid imide ring to restrict the rotation of the rotors. Imide 1, however, would not be expected to exhibit strongly restricted rotation and has been reported to rotate freely at ambient temperatures [Citation19].
In this study, crystallographic and computational studies are used to provide evidence for the stabilisation of the less favoured conformer in imide 1 by n→π* interactions. This is further evidenced by studies of related [5]polynorbornane 2, related computational studies, and VT 1H NMR analysis.
Results and discussion
Synthesis
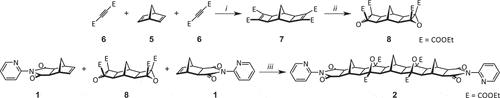
Using a method adapted from the literature [Citation35] norbornene-exo-anhydride 3 and 2-aminopyridine (2-AP) 4 were reacted at 130°C in DMF to give pyridyl imide 1 ().
Scheme 1. Reagents and conditions: (i) 130°C, DMF, 16 h, 72%.
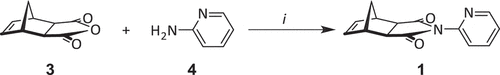
The combination of exo-functionalised norbornene imides and bis(epoxide) building blocks has previously been reported as a means to access [5]polynorbornane frameworks with a linear overall shape [Citation24]. The exo stereochemistry and extended length of this framework also ensures clear separation of the terminal imide moieties, eliminating the risk of an extraneous variable in intramolecular interaction of the two pyridyl moieties.
The synthesis of polynorbornane scaffolds can be achieved through a 1,3-dipolar alkene cyclobutane epoxide (ACE) cycloaddition [Citation36,Citation37]. To produce the bis(2-pyridyl) [5]polynorbornane framework 2, pyridyl-functionalised norbornene imide 1 was reacted with the previously reported bis(epoxide) 8 () [Citation38] which in this work was synthesised in two steps from 2,5-norbornadiene 5. First, a twin [2 + 2] Ru-catalysed Mitsudo cycloaddition between norbornadiene and diethyl acetylenedicarboxylate (DEtAD) 6 afforded tetraester 7 [Citation39]. Weitz – Scheffer epoxidation of this di-ene tetraester 6 using t-BuOOH and t-BuOK gave bis(epoxide) 8 [Citation40]. The microwave assisted [Citation37] ACE cycloaddition of imide 1 and bis(epoxide) 8 building blocks gave the novel [5]polynorbornane 2.
Scheme 2. Synthesis of the bis(2-pyridyl) framework 2. Reagents and conditions: (i) RuH2(CO)(PPh3)3, 100°C, DMF, 16 h, 46%; (ii) t-BuOOH, t-BuOK, −10°C – RT, THF, 16 h, 43%; (iii) 140°C, DMF, microwave, 30 min, 66%.
Computational studies
The geometries of the conformers and transition states of imide 1 were optimised using density functional calculations at the B3LYP/6-311 G* level (298.15 K) using Spartan ’14 [Citation41] (). NMR chemical shifts, vibrational energies, and thermodynamic properties were calculated using these simulated structures ().
Figure 4. Calculated (a) transition state, (b) syn conformer and (c) anti conformer of imide 1, with annotated C3-N4-C2’-C3’ dihedral angles.
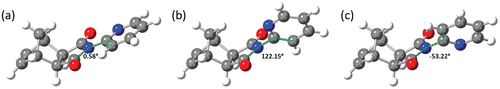
Table 1. Calculated (B3LYP/6-311 G* (298.15 K)) properties of conformers of imide 1.
The syn conformer was found to be 2.46 kJmol−1 lower in energy than the anti conformer, while in the previous study by Gómez-Sandoval et al. the syn conformer was found to be 1.55 kJmol−1 lower in energy [Citation19]. While this is not an insignificant discrepancy, both studies indicate the syn conformer to be more favourable. The energy of activation (ΔG‡) for the rotation of the pyridyl ring of imide 1 was calculated using the average free energy of both conformers and found to be 16.11 kJmol−1.
Given this relatively low energy barrier and the calculated1H NMR chemical shift of the syn proton in both conformers (), it was estimated (using the modified Eyring equation for calculating ΔG‡ from coalescence temperature) [Citation42] that slow exchange would not be visible until well below −150°C.
VT NMR
VT NMR was used to assess the rotation of the 2-pyridyl moiety of imide 1 about the C-N bond. At lower temperatures (< −20°C), the singlet associated with the 10a and 10s bridge protons began to form a distinct AB pair, with complete coalescence occurring as temperatures increased to 19°C and higher (). All four peaks of the AB pair could only be identified at temperatures well under −20°C. The available equipment was limited to the lowest stable temperature of −37.5°C, yet even at this temperature, the AB pair attributed to the 10s (1.63 ppm [ 2JAB= 10.2 Hz) and 10a (1.60 ppm 2JAB = 10.1 Hz) protons of the methylene bridge (assigned by inference using the simulated spectra) were readily distinguished.
Figure 5. VT 1H NMR stackplot of 2-pyridyl imide 1 (1.670–1.565 ppm), CDCl3, 500 MHz.
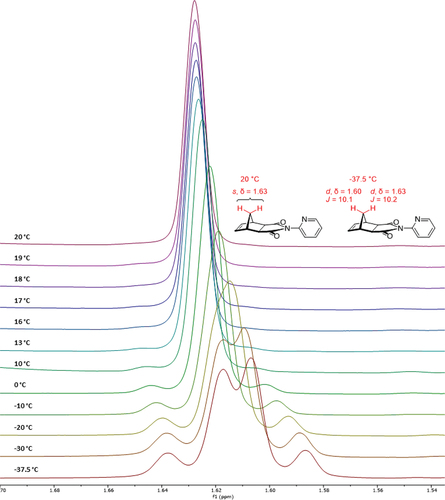
At a temperature of −37.5°C, there was no significant broadening or additional splitting seen in the aromatic region of the spectrum (Figure S2). If the additional complexity in the peak(s) attributed to the bridgehead were due to restricted rotation, it would be expected that this complexity would also be seen in the aromatic region. As this phenomenon is not observed, it was assumed that the appearance of the AB pair at lower temperatures is not caused by the freezing out of distinct conformers.
In 2-pyridyl [5]polynorbornane 2, due to the steric compression of the anti protons by the oxanorbornane bridges, the peaks caused by the terminal bridgeheads are clearly separated at ambient temperature, forming an AX pair with the anti proton at 2.40 ppm 2JAX = 11.8 Hz) and the syn proton at 1.06 ppm 2JAX = 11.8 Hz). This enhanced clarity allows 2-pyridyl [5]polynorbornane 2 to be used to study the possibility of restricted rotation around the C-N bond of the imide. VT NMR was used to investigate this rotation, with special attention paid to the aromatic region of the spectra as well as the doublet attributed to the 31s and 33s protons. From these experiments, it could be seen that at a temperature of −37.5°C, there was no additional complexity in the signals attributed to the aromatic region or terminal bridges of the [5]polynorbornane (). This lack of significant change between spectra further cements the notion that there is no significant restriction on rotation at ambient temperatures. Given the similarity of the imide system in imide 1, it can be inferred that the pyridine similarly rotates freely at ambient temperature.
Figure 6. VT 1H NMR stackplot of 2-pyridyl [5]polynorbornane 2 (9.00–0.75 ppm), CDCl3, 500 MHz. Insets display the doublet assigned to the 31s and 33s protons.
![Figure 6. VT 1H NMR stackplot of 2-pyridyl [5]polynorbornane 2 (9.00–0.75 ppm), CDCl3, 500 MHz. Insets display the doublet assigned to the 31s and 33s protons.](/cms/asset/71a6dbb8-5c7d-482f-9637-95d7ee7f2e6f/gsch_a_2384908_f0006_oc.jpg)
Crystallography
Recrystallisation of imide 1 by evaporation of DCM from a 1:9 n-heptane:DCM yielded colourless block crystals. Subsequent SC-XRD analysis and refinement gave a crystal structure of 2-pyridyl imide 1 () of high quality solved in the triclinic space group P (Table S1).
Figure 7. (a) Asymmetric unit of 2-pyridyl imide 1 highlighting the n→π* interaction (orange) and (b) annotated n→π* interaction of 2-pyridyl imide 1.
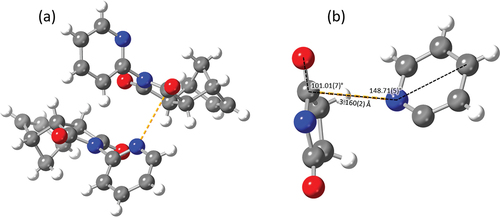
Of particular interest, it can be seen that the crystal structure of 2-pyridyl imide 1 includes both conformers of the molecule. While crystal packing effects cannot be discounted, the presence of both conformers in a 1:1 ratio in the crystal structure suggests the conformers are similar in energy in solution [Citation43].
The presence of the anti conformer in the crystal structure was particularly interesting, as it would be expected that the molecule would adopt the lower energy syn conformation. Examination of the crystal structure reveals the presence of an n→π* interaction between the pyridyl lone pair of the anti conformer and a carbonyl π* orbital of the syn conformer.
Examples where the carbonyl components of an imide act as electrophiles in n→π* interactions in the solid state are limited [Citation44,Citation45]. By comparison, interactions featuring pyridyl nucleophiles have been studied extensively and as such are an excellent basis for comparison [Citation16–18].
The noncovalent bonding distance and angles of the interaction in imide 1 largely agree with the experimental and theoretical values reported for pyridyl⋯carbonyl complexes in literature [Citation16–18]. The value of ∠n⋯C=O was found to be 101.01(7)° (), a value clearly within the observed literature range of 94.7(6) to 104.5° [Citation16–18]. The n⋯C=O distance in the structure of imide 1 was found to be 3.160(2) Å and the angle between the pyridyl ring and the noncovalent bond (∠C4’·n⋯C) was 148.71(5)° (), each of these falling just outside the literature ranges of 2.855(4) to 3.118 Å and 152.2° to 157.4°. The increased length of this interaction is potentially due to steric hindrance of the imide ring, as more hindered carbonyl electrophiles result in increased interatomic distances in similar n→π* interactions [Citation18]. The carbonyl carbon displays subtle pyramidalization of approximately 0.005 Å towards the pyridyl lone pair. Nevertheless, as the other carbonyl carbons in the structure are between 0.003 Å and 0.014 Å out of plane, pyramidalization of 0.005 Å does not of itself guarantee an n→π* interaction
Natural Bond orbital (NBO) [Citation46] analysis was performed using Gaussian 09 [Citation47] using the RHF method at the 6-31 G(d,p) level. From this analysis, it was found that the n→π* interaction was the strongest stabilising charge transfer between the two molecules in the asymmetric unit. This interaction had a calculated stabilisation energy (from second-order perturbation theory analysis) of 4.0 kJmol−1. This stabilisation energy is slightly lower than that of the weakest known pyridyl nucleophile n→π* interaction (4.9 kJmol−1, pyridyl⋯acetone) [Citation18], likely as the imide 1 is more hindered than the previously studied ketones.
As the magnitude of this stabilisation energy still exceeds the energy difference between the syn and anti conformers of imide 1, it is a reasonable assertion that the n→π* interaction assists significantly in the stability of the anti conformer in the solid state.
Conclusions
Computational and VT NMR studies have been used to determine that rotation of the 2-pyridyl moiety in imide 1 is likely not significantly restricted at ambient temperatures and that the syn conformer is likely favoured in solution. NBO analysis and energy calculations support the notion that n→π* interactions stabilise the anti conformer in the solid state.
Experimental
General experimental
Norbornene anhydride 3 was purchased from Combi-Blocks, 2-aminopyridine 4 and 2,5-norbornadiene 5 was purchased from AK Scientific, and all other chemicals were purchased from Sigma-Aldrich. All chemicals were used without further purification, with the exception of anhydrous tert-butyl hydroperoxide (t-BuOOH), which was prepared in toluene according to the method described by Sharpless [Citation48]. The RuH2(CO)(PPh3)3 catalyst was prepared according to the literature method. [Citation49] Tetrahydrofuran (THF) was dried through use of a silica gel 60 (230–400 mesh) column and stored over 4 Å molecular sieves [Citation50].
1H NMR and1C NMR were recorded on a Bruker Ascend 400 or 500 MHz FT-NMR spectrometer. Sample temperature control for VT NMR was performed using a stream of chilled nitrogen gas from a Bruker BCU II Smart Cooler. HRMS data were recorded using a Shimadzu LCMS-9030 QTOF mass spectrometer. Melting points of compounds were determined by use of a Stuart SMP30 melting point apparatus.
Crystallographic data were collected using the MX2 beamline at the Australian Synchrotron [Citation51]. Crystals were mounted from their respective mother liquors with the assistance of paratone oil before crystallographic data was collected under a stream of nitrogen at 100 K. Crystal structures were solved using the program SHELXT [Citation52] and refined using SHELXL [Citation53] within Olex2 graphic user interface [Citation54].
Computational details
Conformers of imide 1 were generated using Spartan ’14 [Citation41]. Structures of imide 1 given 20 points of rotation around the imide C-N bond were calculated using Hartree Fock 6-31 G*, energy maxima and minima from these calculations were used as a starting point to optimise the geometries of the conformers and transition state using density functional calculations at the B3LYP/6-311 G* level (298.15 K). NMR chemical shifts, vibrational energies, and thermodynamic properties were calculated with these models.
Synthesis
Tetraethyl(1α,2β,5β,6α,7β,10β)-tetracyclo[4.4.1.02,5.07,10]undeca-3,8-diene-3,4,8,9-tetracarboxylate (7)
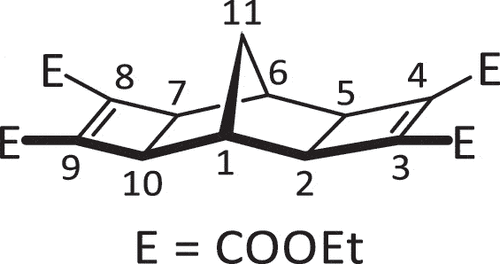
2,5-Norbornadiene 5 (2.20 mL, 1.99 g, 21.6 mmol), DEtAD 6 (8.80 mL, 9.36 g, 55.0 mmol, 2.54 equiv.) and RuH2(CO)(PPh3)3 (570 mg, 0.621 mmol, 0.29 equiv.) were combined in DMF (6.0 mL). The reaction vessel was equipped with an air condenser before heating to 100°C for 16 h and stirred in a sand bath behind a blast shield (HAZARD! Potential exotherm). After cooling on ice, water was added dropwise to the mixture, causing the precipitation of crude product as a dark brown powder which was collected using vacuum filtration. The resultant dark brown powder was recrystallised in EtOH (by heat, adding a drop of water while boiling) to produce needle-like brown crystals. The crystals were collected using vacuum filtration and washed with MeOH (10 mL) to yield the desired pure product (4.28 g, 46%). mp: 98.9–99.7°C. 1H NMR (400 MHz, CDCl3) δ 4.23 (q, J = 7.1 Hz, 8 H, 4 × Et CH2), 2.75 (s, 4 H, H2,5,7,10), 2.41 (s, 2 H, H1,6), 1.39 (s, 2 H, H11), 1.32 (t, J = 7.1 Hz, 12 H, 4 × Et CH3). 13C NMR (100 MHz, CDCl3) δ 161.2, 142.5, 61.1, 46.1, 31.8, 23.8, 14.3. HRMS calcd. for C23H28O8 [M + Na]+ 455.16764; found 455.16976.
Tetraethyl 4,10-dioxahexacyclo [5.5.1.02,6.03,5.08,12.09,11]trideca-3,5,9,11-tetracarboxylate (8)
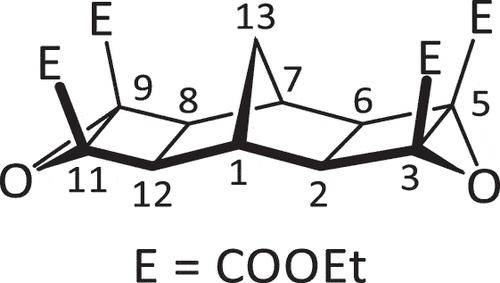
Under an inert N2 atmosphere, a solution of tetraester 7 (1.000 g, 2.312 mmol) in anhydrous THF (150 mL) was cooled on an ice bath before a solution of t-BuOOH in toluene (1.52 mL, 3.824 M, 5.81 mmol, 2.5 equiv.) was added dropwise. The mixture was stirred vigorously for 10 min before the addition of t-BuOK (140 mg, 1.25 mmol, 0.54 equiv.) in two portions, the second portion being added 30 min after the first. The mixture was then stirred for further 10 min before being allowed to warm to room temperature and left to stir for 16 h. Saturated sodium sulphite was used to quench the reaction, with the biphasic mixture stirred for an additional 20 min. The mixture was then reduced to approximately one-third of its original volume under reduced pressure, and the aqueous phase was extracted with CHCl3 (4 × 60 mL). The organic phases were then combined and dried (MgSO4) and the solvent was removed under reduced pressure.
The resultant yellow-tinged liquid solidified upon standing at room temperature in air. The solid was then triturated (i-PrOH). Collection using vacuum filtration and washing with i-PrOH (10 mL) yielded the pure product as a white powder (461 mg, 43%). mp: 121.4–123.8°C1H NMR (400 MHz, CDCl3) δ 4.27 (ABX3, JAB = 10.8 Hz, JAX = JBX = 7.1 Hz, 8 H, 4 × Et CH2), 3.29 (s, 2 H, H1,7), 2.26 (s, 4 H, H2,6,8,12), 1.94 (s, 2 H, H11), 1.31 (t, J = 7.1 Hz, 12 H, 4 × Et CH3). 13C NMR (100 MHz, CDCl3) δ 163.9, 64.4, 62.1, 48.9, 36.7, 28.7, 14.3. HRMS calcd. for C23H28O10 [M + Na]+ 487.15745; found 487.15987. Compound reported previously (as oil) without characterisation data [Citation38].
Tetraethyl (1α,2β,3α,4β,5α,6β,10β,11α,12β,13α,14β,15α,16β,17α,18β,19α,20β,24β,25α,26β,27α,28β)-7,9,21,23-tetraoxo-8,22-bis(pyridin-2-yl)-30,32-dioxa-8,22-diazadodecacyclo[13.13.11,15.13,13.15,11.117,27.119,25.02,14.04,12.06,10.016,28.018,26.020,24]tritriaconta-3,13,17,27-tetracarboxylate (2)
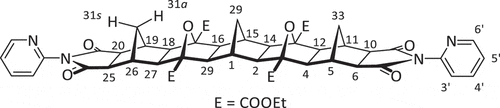
Bis(epoxide) 8 (100 mg, 0.215 mmol) and pyridine imide 1 (109 mg, 0.454 mmol, 2.1 equiv.) were combined in DMF (0.8 mL) in a 10 mL microwave vial before being heated to 140°C for 30 min with microwave radiation (200W). The mixture was allowed to cool before water was added to precipitate an off-white powder. The crude solid was collected using vacuum filtration and washed with i-PrOH, drying under vacuum to yield the final product as a white powder (135 mg, 66%). mp: > 330°C dec. 1H NMR (500 MHz, CDCl3) δ 8.63 (dd, J = 1.9 Hz, J = 4.9 Hz, 2 H, H6’), 7.85 (td, J’ = 7.8 Hz, J = 1.9 Hz, 2 H, H4’), 7.36 (dd, J = 7.6 Hz, J = 4.8 Hz, 2 H, H5’), 7.21 (d, J = 8.0 Hz, 1 H, H3’), 4.32 (ABX3, JAB = 10.9 Hz, JAX = JBX = 7.1 Hz, 8 H, 4 × Et CH2), 2.73 (s, 4 H, H6,10,20,24), 2.72 (s, 4 H, H5,11,19,25), 2.40 (d, J = 11.8 Hz, 2 H, H31a,33a), 2.21 (s, 4 H, H4,12,18,26), 2.10 (s, 2 H, H1,15), 2.00 (s, 4 H, H2,14,16,28), 1.92 (s, 2 H, H29), 1.33 (t, J = 7.1 Hz, 12 H, 4 × Et CH3), 1.06 (d, J = 11.8 Hz, 2 H, H31s, 33s). 13C NMR (100 MHz, CDCl3) δ 176.3, 168.2, 150.0, 146.0, 138.7, 124.4, 122.2, 89.8, 61.7, 55.0, 54.8, 48.5, 42.2, 40.7, 29.4, 28.9, 14.6. HRMS calcd. for C51H52N4O14 [M + H]+ 945.35528; found 945.35484.
File not for review.docx
Download MS Word (762.2 KB)ESI.pdf
Download PDF (1.4 MB)Response to Reviewer Comments.docx
Download MS Word (181.6 KB)Acknowledgments
W. M. gratefully acknowledges Deakin University for a Deakin University Postgraduate Research Scholarship (DUPR, including scholarship extension) and the Deakin Centre for Sustainable Bioproducts for additional funding. W.M. is also thankful for A/Prof. Luke O’Dell of Deakin University for assistance in performing VT NMR. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Deposition number 2356985 in the Cambridge Crystallographic Data Centre (CCDC) contains the supplementary crystallographic data for this paper.
Supplemental material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/10610278.2024.2384908
References
- Singh SK, Das A. The n→π* interaction: a rapidly emerging non-covalent interaction. Phys Chem Chem Phys. 2015;17(15):9596–9612. doi: 10.1039/C4CP05536E
- Newberry RW, Raines RT. The n→π* interaction. Acc Chem Res. 2017;50(8):1838–1846. doi: 10.1021/acs.accounts.7b00121
- Wang X-D, Zhu J, Wang D-X. Intermolecular n→π* interactions in supramolecular chemistry and catalysis. ChemPluschem. 2023;88(9):e202300288. doi: 10.1002/cplu.202300288
- Bürgi HB, Dunitz JD, Shefter E. Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J Am Chem Soc. 1973;95(15):5065–5067. doi: 10.1021/ja00796a058
- Bürgi HB, Dunitz JD, Shefter E. Chemical reaction paths. IV. Aspects of O…C = O interactions in crystals. Acta Crystallogr Sect A. 1974;30(6):1517–1527. doi: 10.1107/S0567740874005188
- Bürgi HB, Lehn JM, Wipff G. Ab initio study of nucleophilic addition to a carbonyl group. J Am Chem Soc. 1974;96(6):1956–1957. doi: 10.1021/ja00813a062
- Hinderaker MP, Raines RT. An electronic effect on protein structure. Protein Sci. 2003;12(6):1188–1194. doi: 10.1110/ps.0241903
- Bartlett GJ, Choudhary A, Raines RT, et al. n→π* interactions in proteins. Nat Chem Biol. 2010;6(8):615–620. doi: 10.1038/nchembio.406
- Newberry RW, VanVeller B, Guzei IA, et al. n→π* interactions of amides and thioamides: implications for protein stability. J Am Chem Soc. 2013;135(21):7843–7846. doi: 10.1021/ja4033583
- Wenzell NA, Ganguly HK, Pandey AK, et al. Electronic and steric control of n→π* interactions: stabilization of the α-helix conformation without a hydrogen bond. Chembiochem. 2019;20(7):963–967. doi: 10.1002/cbic.201800785
- Khatri B, Majumder P, Nagesh J, et al. Increasing protein stability by engineering the n → π* interaction at the β-turn. Chem Sci. 2020;11(35):9480–9487. doi: 10.1039/d0sc03060k
- Choudhary A, Kamer KJ, Raines RT. An n→π* interaction in aspirin: implications for structure and reactivity. J Org Chem. 2011;76(19):7933–7937. doi: 10.1021/jo201389d
- Blanco S, López JC, Mata S, et al. Conformations of γ-Aminobutyric acid (GABA): the role of the n→π* interaction. Angew Chem Int Ed. 2010;49(48):9187–9192. doi: 10.1002/anie.201002535
- Lesarri A, Cocinero EJ, López JC, et al. Shape of 4(S)- and 4(R)-hydroxyproline in gas phase. J Am Chem Soc. 2005;127(8):2572–2579. doi: 10.1021/ja045955m
- León I, Alonso ER, Cabezas C, et al. Unveiling the n→π* interactions in dipeptides. Commun Chem. 2019;2(1):3. doi: 10.1038/s42004-018-0103-2
- Blanco S, López JC. Rotational characterization of an n→π* interaction in a pyridine–formaldehyde adduct. J Phys Chem Lett. 2018;9(16):4632–4637. doi: 10.1021/acs.jpclett.8b01719
- Blanco S, Macario A, López JC. Pyridine–acetaldehyde, a molecular balance to explore the n→π* interaction. Phys Chem Chem Phys. 2019;21(37):20566–20570. doi: 10.1039/C9CP04088A
- López JC, Alkorta I, Macario A, et al. Characterizing the n→π* interaction of pyridine with small ketones: a rotational study of pyridine⋯acetone and pyridine⋯2-butanone. Phys Chem Chem Phys. 2022;24(25):15484–15493. doi: 10.1039/D2CP01611G
- Hernández-Madrigal J, Pineda-Contreras A, Vázquez-Vuelvas O, et al. Synthesis of novel pyridinium betaine precursors from exo-norbornene dicarboximides. Lett Org Chem. 2011;8:249–257. doi: 10.2174/157017811795371458
- Mahanti S, Verma SM. Steric requirements of sp2-hybridized lone electron pair: a PMR study of restricted rotation about a pyridyl carbon-nitrogen bond. Indian J Chem Sect B. 1982;21B(12):1098.
- Warrener RN, Margetić D, Amarasekara AS, et al. Synthesis of functionalized cavity structures via 1,3-dipolar cycloaddition of angle-shaped alkenes to curved Norbornene-framed dipoles. Org Lett. 1999;1(2):203–206. doi: 10.1021/ol990538d
- Margetić D, Butler D, Warrener R. The synthesis of rigid chromophore–spacer–chromophore dyads and three-armed triads by the 1,3-dipolar reaction of cyclobutene epoxides with aromatic dipolarophiles. Synthesis. 2013;45(24):3413–3425. doi: 10.1055/s-0033-1340305
- Golić M, Johnston MR, Margetić D, et al. Use of a 9,10-dihydrofulvalene pincer cycloadduct as a cornerstone for molecular architecture. Aust J Chem. 2006;59(12):899–914. doi: 10.1071/CH06286
- Shang M, Warrener RN, Butler DN, et al. Synthesis of bis-peptides attached on poly[n]norbornene molecular scaffolds with well-defined relative positions and distances. Mol Divers. 2011;15(2):541–560. doi: 10.1007/s11030-010-9279-9
- Johnstone MD, Schwarze EK, Clever GH, et al. Modular synthesis of linear bis- and tris-monodentate fused [6]Polynorbornane-based ligands and their assembly into coordination cages. Chem Eur J. 2015;21(10):3948–3955. doi: 10.1002/chem.201405037
- Troselj P, Briš A, Murata Y, et al. Structural evidence for the arc-shaped topology of hetero[5]polynorbornanes. Struct Chem. 2012;23(3):791–799. doi: 10.1007/s11224-011-9925-6
- Dial BE, Pellechia PJ, Smith MD, et al. Proton grease: an acid accelerated molecular rotor. J Am Chem Soc. 2012;134(8):3675–3678. doi: 10.1021/ja2120184
- Han S, Wu Y, Duan R, et al. Fluoride-controlled molecular brake systems. Asian J Org Chem. 2019;8(1):83–87. doi: 10.1002/ajoc.201800614
- Dial BE, Rasberry RD, Bullock BN, et al. Guest-accelerated molecular rotor. Org Lett. 2011;13(2):244–247. doi: 10.1021/ol102659n
- Li P, Vik EC, Maier JM, et al. Electrostatically driven CO−π aromatic interactions. J Am Chem Soc. 2019;141(32):12513–12517. doi: 10.1021/jacs.9b06363
- Wu Y, Wang G, Li Q, et al. A multistage rotational speed changing molecular rotor regulated by pH and metal cations. Nat Commun. 2018;9(1):1953. doi: 10.1038/s41467-018-04323-4
- Vik EC, Li P, Pellechia PJ, et al. Transition-state stabilization by n→π* interactions measured using molecular rotors. J Am Chem Soc. 2019;141(42):16579–16583. doi: 10.1021/jacs.9b08542
- Vik EC, Li P, Madukwe DO, et al. Analysis of the orbital and electrostatic contributions to the lone pair–aromatic interaction using molecular rotors. Org Lett. 2021;23(21):8179–8182. doi: 10.1021/acs.orglett.1c02878
- Lin B, Liu H, Karki I, et al. Pnictogen interactions with nitrogen acceptors. Angew Chem. 2023;135(28):e202304960. doi: 10.1002/ange.202304960
- Kong P, Drechsler S, Balog S, et al. Synthesis and properties of poly(norbornene)s with lateral aramid groups. Polym Chem. 2019;10(16):2057–2063. doi: 10.1039/C9PY00187E
- Warrener NR, Schultz CA, Butler N, et al. A new building block technique based on cycloaddition chemistry for the regiospecific linking of alicyclic sub-units as a route to large, custom-functionalised structures. Chem Commun. 1997;1997(11):1023–1024. doi: 10.1039/A700010C
- Foitzik R, Lowe A, Pfeffer F. Microwave-accelerated 1,3-dipolar cycloaddition for the formation of fused [n]polynorbornanes. Tetrahedron Lett. 2009;50(21):2583–2584. doi: 10.1016/j.tetlet.2009.03.079
- Warrener RN, Butler DN, Margetić D, et al. New and improved ‘LEGO’ BLOCK protocols for the direct synthesis of hydrophilic ribbon molecules with acid, ester or peptide functionality. Tetrahedron Lett. 2000;41(23):4671–4675. doi: 10.1016/S0040-4039(00)00685-7
- Johnstone M, Lowe A, Henderson L, et al. Rapid synthesis of cyclobutene diesters using a microwave-accelerated ruthenium-catalysed [2+2] cycloaddition. Tetrahedron Lett. 2010;51(45):5889–5891. doi: 10.1016/j.tetlet.2010.08.119
- Johnstone MD, Frank M, Clever GH, et al. Rapid solvent-free synthesis of pyridyl-Functionalised [5]Polynorbornane-based ligands for metal–organic rings and cages. Eur J Org Chem. 2013;2013(26):5848–5853. doi: 10.1002/ejoc.201300647
- Spartan ’14. Irvine: Wavefunction Inc.; 2014.
- Huggins MT, Kesharwani T, Buttrick J, et al. Variable temperature NMR experiment studying restricted bond rotation. J Chem Educ. 2020;97(5):1425–1429. doi: 10.1021/acs.jchemed.0c00057
- Li P, Hwang J, Maier JM, et al. Correlation between solid-state and solution conformational ratios in a series of N-(o-Tolyl)Succinimide molecular rotors. Cryst Growth Des. 2015;15(8):3561–3564. doi: 10.1021/acs.cgd.5b00906
- Hendi Z, Jamali S, Chabok SMJ, et al. Bis-N-Heterocyclic carbene complexes of coinage metals containing four naphthalimide units: a structure–emission properties relationship study. Inorg Chem. 2021;60(17):12924–12933. doi: 10.1021/acs.inorgchem.1c01302
- Howe ENW, Bhadbhade M, Thordarson P. Highly sheared anti-parallel dipolar Carbonyl···Carbonyl interaction in the crystal packing of strapped crown-3-pyromellitimide. Aust J Chem. 2012;65(10):1384–1389. doi: 10.1071/CH12085
- Glendening ED, Landis CR, Weinhold F. NBO 7.0: new vistas in localized and delocalized chemical bonding theory. J Comput Chem. 2019;40(25):2234–2241. doi: 10.1002/jcc.25873
- Frisch M, Trucks G, Schlegel H, et al. Gaussian 09 (Revision A02). Wallingford (CT): Gaussian Inc; 2009.
- Hill JG, Rossiter BE, Sharpless KB. Anhydrous tert-butyl hydroperoxide in toluene: the preferred reagent for applications requiring dry TBHP. J Org Chem. 1983;48(20):3607–3608. doi: 10.1021/jo00168a063
- Ahmad N, Levison JJ, Robinson S, et al. Complexes of Ruthenium, Osmium, Rhodium, and Iridium Containing Hydride, Carbonyl, or Nitrosyl Ligands. In: Parshall GW, editor. Inorganic Syntheses McGraw-Hill.1974;15:45–64.
- Williams DBG, Lawton M. Drying of organic solvents: quantitative evaluation of the efficiency of several desiccants. J Org Chem. 2010;75(24):8351–8354. doi: 10.1021/jo101589h
- Aragão D, Aishima J, Cherukuvada H, et al. MX2: a high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian synchrotron. J Synchrotron Radiat. 2018;25(3):885–891. doi: 10.1107/S1600577518003120
- Sheldrick G. SHELXT – integrated space-group and crystal-structure determination. Acta Crystallogr Sect A. 2015;71(1):3–8. doi: 10.1107/S2053273314026370
- Sheldrick GM. Crystal structure refinement with SHELXL. Acta Crystallogr Sect C. 2015;71(Pt 1):3–8. doi: 10.1107/s2053229614024218
- Dolomanov OV, Bourhis LJ, Gildea RJ, et al. OLEX2: a complete structure solution, refinement and analysis program. J Appl Crystallogr. 2009;42(2):339–341. doi: 10.1107/S0021889808042726