
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Interleukin 7 (IL-7) is an essential cytokine that acts as a potent growth factor of T-cells and supports the growth of B-cell precursors. IL-7 binds to a heterodimeric receptor consisting of an IL-7 receptor alpha (IL-7Rα) and the common gamma chain receptor (γc) which is shared with IL-2, IL-4, IL-9, IL-15 and IL-21. The discovery of small-molecule agonists of cytokines would be of great pharmaceutical interest with the increasing scientific rationale. In this study, a series of molecular modelling methods, including field-based pharmacophore virtual screening, protein-protein docking and molecular dynamics simulations, led to the identification of two compounds (i.e. 1 and 2) of different classes that exhibit enhanced agonistic effects by activating the IL-7 signalling cascade. One of these compounds was selected as a hit and represents the first small-molecule agonist of IL-7Rα with single-digit micromolar activity. Moreover, the prediction model of the active compound to the IL-7Rα/γc interaction complex provides insight into the binding of a small-molecule agonist to its receptor.
Introduction
Interleukin 7 (IL-7) is a haematopoietic growth factor protein that is secreted by stromal cells in the thymus and bone marrow [Citation1–3]. IL-7 is one of the essential cytokines for the homoeostatic maintenance and development of B and T lymphocytes [Citation4,Citation5]. Its signalling cascade starts with IL-7 binding to the IL-7 receptor, which is composed of common gamma chain receptor (γc) and IL-7 receptor alpha (IL-7Rα) [Citation6,Citation7]. Binding results in a cascade of signals, including the expression of multiple transcription factors (Jak3, STAT, PI3K and SRC) which are crucial for T-cell proliferation and activation [Citation8,Citation9]. IL-7 is considered one of the most important immunotherapy agents for treating various malignancies and viral infections including human immunodeficiency virus (HIV) [Citation10–12]. Previous studies have shown that IL-7 signalling promotes osteoclastogenesis and fibroblast activation, which are involved in tissue damage in chronic inflammation [Citation13]. IL-7 signalling also regulates antigen-reactive T cells, which suggests that abnormal IL-7 signalling can be related to the pathogenesis of autoimmune diseases, such as type 1 diabetes, rheumatoid arthritis, multiple sclerosis and ulcerative colitis [Citation14]. IL-7 signalling also induces the production of TNF-α, IL-1β and IL-1α by monocytes, and these cytokines inhibit melanoma growth [Citation5]. In addition, IL-7 signalling induces an increase in CD8 + T lymphocytes, which can attack cancerous cells.
Recent studies have suggested that the role of IL-7 in immunotherapy is not limited to treating viruses but can also be applied to treating cancers [Citation15,Citation16]. A human clinical study with cancer patients suggested that administration of IL-7 can transiently lead to an expansion of CD8+ and CD4 + T cell populations along with a decrease in CD4+ regulatory T cell populations [Citation17]. This modulation of T cell homoeostasis can exert potential antitumor effects and has been intensely studied recently [Citation18,Citation19].
However, due to the high cost of manufacturing recombinant IL-7 proteins [Citation20] and the limitations on delivery route (mainly intravenous) [Citation21,Citation22] searching for small-molecule agonists showing similar immunotherapeutic effects via activation of the IL-7 signalling cascade is worth undertaking. Here, we present our efforts in a small-molecule search for potential IL-7 agonists via intensive virtual screening and molecular assays.
Materials and methods
In silico molecular modelling studies
Shape- and electrostatic field- based virtual screening
IL-7Rα in complex with IL-7 was available, and the X-ray crystal structure of the IL-7Rα/IL-7 complex was obtained from the Protein Data Bank (PDB code 3DI3) [Citation23]. The IL-7 protein was used to generate a pharmacophore model using ligand-based virtual screening within OpenEye Scientific software [Citation24]. A pharmacophore query model was created from the binding site of IL-7 with IL-7Rα based on the binding motif and consists of a helix; its residues are amino acids (aa) 72–91. The 557,360-membered KCB library database was prepared using OMEGA v3.0.1, thereby generating a maximum number of 200 low-energy conformers for each molecule. The 3D shape similarity search of the helix as a query was performed using ROCS v3.2.2, and the top 10,000 hits were selected according to the ROCS_TanimotoCombo score. We then performed a 3D electrostatic potential field map similarity search for the screened 10,000 compounds using EON v2.2.0, and the top 1,000 hits were selected by the EON_ET_Combo score. A total of 182 compounds were ultimately selected by eye selection for the in vitro assay.
Similarity-based virtual screening using two-dimensional fingerprints
To identify more analogues of active compound 1, we performed a second round of virtual screening using the fingerprint similarity method. The similarity between single pairs of compounds was calculated using Pipeline Pilot 2020 [Citation25]. The KCB library database was screened using a Tanimoto coefficient ≥ 0.8 compared with active compound 1, and 9 analogues were ultimately selected by eye selection for the in vitro assay.
Protein-protein docking
To understand the IL-7Rα/γc heterodimeric complex in the presence of compound 1, we performed a rigid-body protein-protein docking simulation using the ZDOCK protocol in Discovery Studio 2020 [Citation26]. The structure of γc extracted from the structure of the IL-2Rβ/γc complex (PDB code 5M5E) [Citation27] was used. The receptor-ligand complex model for IL-7Rα bound to compound 1 was retrieved by superimposition of compound 1 onto IL-7, which was a pharmacophore query model for ligand-based virtual screening. The IL-7Rα/compound 1 and γc protein structures were assigned the CHARMM force field for protein-protein docking procedures. The top poses of docking models between IL-7Rα/compound 1 and γc were chosen by visual inspection with the ZDOCK score from the top 500 poses. Five protein-protein docking models were selected by high scoring and eye selection.
Molecular dynamics simulation
For further refinement of IL-7Rα/γc docking models and the molecular interaction analysis of compound 1, molecular dynamics (MD) simulations were performed using Desmond v6.4 with the OPLS3e force field in Schrodinger Suite 2020–4 [Citation28]. System Builder was used for solvation, employing predefined TIP3P water molecules in an orthorhombic box with dimensions of 15 Å x 15 Å x 15 Å, and the overall complex was neutralized by adding Cl- counterions at least 25 Å from ligand compound 1. The NaCl concentration was 0.15 mol/L. MD simulations 500 ns in length were produced under periodic boundary conditions in the NPT ensemble at normal temperature (300 K) and pressure (1.01325 bar). Recording intervals of 1.2 and 500 ps were used for energy calculations and trajectory analyses. The final proposed IL-7Rα (bound to compound 1)/γc docking model for the equilibrium state of the MD simulations was represented using Discovery Studio 2020.
Biological assays
Materials
The PathHunter IL-7 dimerization assay kit was purchased from Eurofins DiscoveRX (Luxembourg). Human interleukin 7 protein (IL-7) was obtained from Sino Biological Inc. (Beijing, China).
IL-7 agonism assay
PathHunter-expressing dimerized cells (DLD-1-based) were prepared as described in the manufacturer’s protocol. In brief, cells were thawed and seeded on 96-well plates at a density of 10,000 cells/well and maintained at 37°C in a humidified incubator with 5% CO2 for 48 hours in 100 µL of cell culture medium. Cells were then treated with 10 µL of compounds at the designated concentrations (1, 10, 100 and 1000 μM) and IL-7 in an incubator at 37°C. After 24 hours of incubation, 110 µL of detection reagents was added, and the plates were further incubated at room temperature. After 1 hour of incubation, chemiluminescence signals were monitored with a spectrophotometer [Citation29].
Statistical analysis
Statistical analysis was performed using OriginPro 8.5 [Citation30]. The half maximal effective concentration (EC50) of compound 1 and IL-7 was estimated using a sigmoidal logistic function, type 1, in Origin 8.5.
Each experiment was performed in triplicate, and values are expressed as the mean ± standard deviation (SD).
Results and discussion
Ligand-based virtual screening by shape and electrostatic pharmacophore
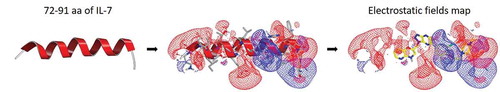
To discover novel small-molecule agonists that promote IL-7Rα/γc heteodimerization, we explored the IL-7 helix domain region where IL-7Rα interacts to the three-dimensional X-ray crystal structure of the IL-7Rα/IL-7 complex (PDB code 3DI3) was utilized to generate a shape- and field-based pharmacophore model based on aa 72–91 that are involved in the binding interface of IL-7 to IL-7Rα () for the similarity search. Ligand-based virtual screening was performed using OpenEye Scientific software. The Korea Chemical Bank (KCB) library, which consists of 557,360 compounds, was prepared for the 3D similarity search using OMEGA v3.0.1, which was used to generate up to 200 low-energy conformers for each molecule. For virtual screening using a large chemical database, two computational strategies were employed. First, a 3D shape similarity search of peptide molecules consisting of aa 72–91 in IL-7 as a query was performed using ROCS v3.2.2, and the top 10,000 hits for query molecules were selected based on the ROCS_TanimotoCombo score. Second, a 3D electrostatic property similarity search was executed for the 10,000 selected compounds using EON v2.2.0. After the positive and negative field contour maps with EON_ET_Combo scores were analysed, the top 500 hits were selected. An additional visual inspection was carried out to remove the PAINS [Citation31]. As a result, 182 compounds were ultimately selected to examine their IL-7R agonist effects in vitro.
Figure 1. Ligand-based virtual screening using 3D shape and electrostatic fields. The IL-7Rα binding motif, a helix consisting of aa 72–91 from the IL-7 crystal structure (PDB code 3DI3), was utilized as a template for 3D similarity screening. Representation of electrostatic potential maps for the helix consisting of aa 72–91. The positive contours are coloured blue, and the negative contours are coloured red. A small-molecule compound obtained from EON is coloured yellow
In vitro assay for IL-7 agonistic activity
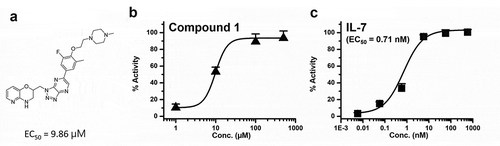
The IL-7 agonistic activities of the 182 selected compounds were measured and compound 1 was most potent and promoted for further biological activity study. We used a commercially available IL-7Rα/γc dimerization assay kit and detected compound/ligand-induced dimerization of two subunits of IL-7Rα and γc. Engineered DLD-1 cells coexpress one receptor subunit fused to an enzyme donor and a dimerizing partner receptor fused to an enzyme acceptor, and binding of an agonist to the receptor subunit induces dimerization with the partner receptor, forcing complementation of the two enzyme fragments. This complementation results in the formation of a functional enzyme, which can generate a chemiluminescent signal that can be monitored. The EC50 of compound 1 () was 9.86 μM (), while the EC50 of IL-7 was 0.71 nM (). The other hit (compound 2 in Figure S1) was derived from virtual screening; this compound from a structurally different class than that of compound 1 also induced dimerization of IL-7Rα and γc, although it showed less potency (EC50 = 48.8 μM) than that of compound 1. The detailed structure and biochemical activity of compound 2 are presented in Figure S1. To this end, compound 1 from the primary virtual screening was selected for further studies.
Figure 2. (a) Compound 1 structure, (b) the biochemical activity of compound 1 and (c) the biochemical activity of IL-7
Similarity-based virtual screening using 2D fingerprints
To validate compound 1 as a primary hit, we performed a second round of virtual screening based on two-dimensional fingerprint similarity. The KCB library was screened based on a Tanimoto coefficient ≥ 0.8 compared with active compound 1 using Pipeline Pilot. Nine analogues were chosen by eye selection for further IL-7 agonistic activity measurements. However, none of the newly selected compounds 183–191 were as effective as compound 1, and their agonist activities are summarized in .
Table 1. Structure-activity relationships of IL-7 agonist activities
3D shape and electrostatic similarity analysis of compound 1
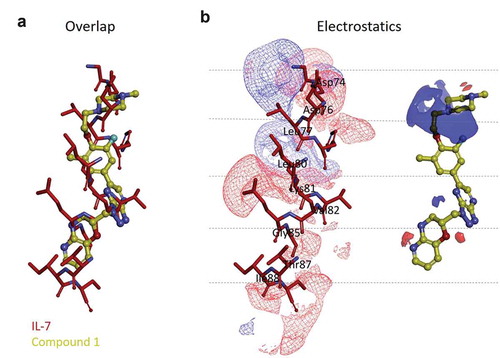
The Electrostatic Tanimoto combo (ET_combo) scores for the 182 virtual hits ranged from 0.158 to 0.278, while the value for compound 1 was 0.240 (the electrostatic field and shape similarity scores were 0.092 and 0.148, respectively), and the compound ranked in the top 10 among the 182 hits. The electrostatic potential maps of aa 72–91 in IL-7 and compound 1 are depicted in . The potential maps revealed distinct differences in the electrostatic surfaces between the two molecules. Regarding the similarity to the α-helix consisting of aa 72–91 in IL-7 as a query, compound 1 is radically different with respect to its 2D and 3D structures but similar in the 3D shape to a Shape Tanimoto (ST) similarity value of 0.148. Although no part in compound 1 was aligned to the contour map with aa 72–91 of IL-7, the sequence adjacent to the piperazine group was comparable (). The other three parts were not electrostatically similar to aa 72–91 of IL-7 but could overlap well by shape (). Compound 1 had an ST value of 0.148, which was higher than 0.092 with the Poisson-Boltzmann Electrostatic Tanimoto (ET_pb) value. Overall, our field-based pharmacophore virtual screening results suggest that compound 1 and the helix consisting of aa 72–91 of IL-7 may be structurally dissimilar and therefore bind to different binding sites. Full details for 182 ligand-based virtually screened compounds, including their structures, EON ranks, ET_combo scores, ST scores, ET_pb scores and primary screening activity data, are provided in Supplementary Table 1.
Figure 3. Three-dimensional shape and electrostatic contour map comparison between the α-helix consisting of aa 72–91 of IL-7 crystal structure and compound 1. (a) overlay of the aa 72–91 helix (red stick model) and compound 1 (yellow ball and stick model) and (b) structures of the aa 72–91 helix and compound 1 with electrostatic contour maps
Protein-protein docking and molecular dynamics simulation for the prediction of the IL-7Rα/compound 1/γc heterodimer
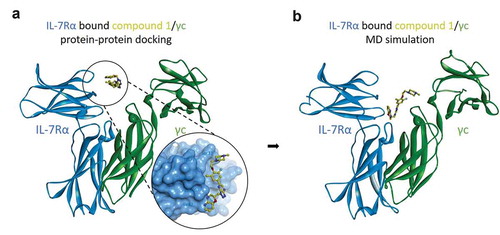
To explore the structural basis of IL-7Rα/γc heterodimerization, we performed protein-protein docking and MD simulations. The X-ray crystal structure of the IL-7Rα/γc complex was not yet available at the time of this work. As a surrogate model, we used the γc protein structure from the IL-2Rβ/γc complex structure (PDB code 5M5E). According to our previous ligand-based pharmacophore model and the release of the IL-7Rα/IL-7 complex (PDB code 3DI3), we hypothesized that compound 1 binds to the IL-7Rα/IL-7 binding interface. The initial binding mode of compound 1 to IL-7Rα was retrieved by superimposition of compound 1 onto IL-7. Then, a protein-protein docking simulation was run to predict the binding model of the γc protein with IL-7Rα/compound 1 by employing the ZDOCK protocol in Discovery Studio. As shown in , γc binds to IL-7Rα complexed with compound 1 in our protein-protein docking study based on high scoring selection. This was supported by relaxation of the docking poses conducted with MD simulation using Desmond in Schrodinger 2020–4. The binding mode of IL-7Rα with γc remained similar in 500 ns MD simulations relative to the initial structure. Interestingly, compound 1 binds deeper into the IL-7Rα/γc binding interface and closer to γc (). Further analysis of the structural changes of IL-7Rα/γc in the presence of bound IL-7 revealed no significant changes in the dimeric conformations between the X-ray crystal structure and the structure determined by MD simulation. The helix consisting of aa 72–91 (Asp74, Leu77, Leu80, Lys81 and Ile88) of IL-7 forms hydrophobic interactions with Val58, Leu80 and Ile82 and hydrogen bonds with Ser31 and Lys77 in the binding site of IL-7Rα (Figure S2b).
Figure 4. Molecular dynamics simulations of IL-7Rα/compound 1/γc complex models. (a) the protein-protein docking model of IL-7Rα bound compound 1, the pose of which is the superimposition of the 3D conformation by ligand-based virtual screening and γc, and (b) the equilibrium state complex model after 500 ns MD simulations. IL-7Rα subunit: blue ribbon model; compound 1: yellow ball and stick model; γc subunit: green ribbon model
Molecular interaction analysis of compound 1
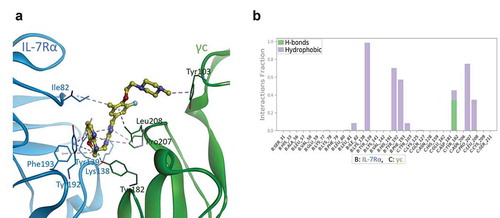
Through the application of MD simulations, the insight gained through protein-protein docking was further extended [Citation32]. We performed an in-depth analysis of the binding interactions formed between compound 1 and the IL-7Rα/γc complex for molecular analysis of ligand recognition, in which the binding mode of compound 1 was also obtained from the corresponding MD model. As shown in , the 3,4‐dihydro‐1,3‐pyridooxazine ring of compound 1 forms significant π-π stacking interactions with IL-7Rα around protein residue Tyr192 and alkyl hydrophobic interactions with IL-7Rα around protein residue Phe193. Notably, the N4-nitrogen atom of oxazine forms hydrogen bonds with the hydroxy group of the Tyr182 residue from γc. The triazolopyrazine ring of compound 1, located between the two proteins, shows very strong π-π stacking interactions with Tyr139 of IL-7Rα, in addition to other alkyl hydrophobic interactions with Pro207 of γc (see the protein-ligand interaction diagram in ). Moreover, the methyl group of the 1-fluoro-3-methylbenzene ring in compound 1 also forms alkyl hydrophobic interactions with Ile82 of IL-7Rα, and the 1-fluoro-3-methylbenzene ring makes ca-π interactions with Leu208 of γc. The terminal methylpiperazine substituent is oriented towards γc by forming ca-π interactions with Tyr103. Taken together, our molecular modelling studies suggest that the important residues for small-molecule agonist binding promote IL-7Rα/γc dimerization throughout MD simulation.
Figure 5. Proposed binding model and interaction profile analysis of compound 1 for IL-7Rα/γc proteins. (a) predicted binding mode of compound 1 (yellow ball and stick model) after 500 ns MD simulations. The IL-7Rα and γc proteins are displayed as blue and green ribbon models, respectively. The hydrogen bonds are shown as green dashed lines, and hydrophobic interactions are represented by pink dashed lines. (b) contact histograms of IL-7Rα (B chain)/γc (C chain) proteins with compound 1 are represented as a coloured stacked bar plot between 200 and 500 ns MD simulations. Hydrogen bonding is displayed by yellow-green coloured bars, and hydrophobic interactions are displayed by lavender coloured bars for clarity
Conclusion
To date, to the best of our knowledge, no IL-7Rα agonist with low molecular weight has been described in the literature. In this study, we report the results of virtual screening with the aim of identifying agonists capable of promoting the IL-7Rα/γc complex. This is the first attempt to target IL-7Rα by an agonist approach, and this study proposes an in silico approach for the discovery of new chemical entities for IL-7. A combined computational approach, including virtual screening and protein-protein docking and MD simulations, has been applied to identify small-molecule agonists other than IL-7 from KCB chemical library databases. A total set of 191 compounds (182 first-round virtual hits by ligand-based virtual screening and 9 second-round virtual hits by a 2D fingerprint similarity search with compound 1) were tested to validate our results in vitro, of which two compounds were shown to have agonistic effects, promoting IL-7Rα/γc dimerization. Compound 1, as a first-round virtual screening hit, exerts single-digit micromolar efficiency on heterodimerization by binding to IL-7Rα/γc. Following an in silico characterization of the IL-7Rα/γc complex interface, a protein-protein docking simulation was performed. In addition, the use of an MD simulation method enabled the discovery of a small-molecule agonist and its binding-determining structural features against the IL-7Rα/γc complex, which will aid in the further development of agonists. In the present research work, a series of compounds with similar functional groups were tested in vitro, and the effects of substituents on compound 1 were determined. The strategy described herein offers a feasible and effective approach to discover other bioactive compounds using in silico virtual screening. In this study, we identified the first class of small-molecule agonists of the IL-7Rα/γc complex, and we are currently searching for more analogues capable of higher-level IL-7Rα/γc dimerization.
Supplemental Material
Download PDF (1.3 MB)Acknowledgement
This work was supported by Korea Research Institute of Chemical Technology intramural funding (grant numbers KK2062–132 and SI2131–50). The chemical library used in this study was provided by Korea Chemical Bank and by Hee Jung Jung Ph.D. of KRICT for donating the compound.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed at: https://doi.org/10.1080/1062936X.2021.1969684.
Additional information
Funding
References
- N. ElKassar and R.E. Gress, An overview of IL-7 biology and its use in immunotherapy, J. Immunotoxicol. 7 (2010), pp. 1–7. doi:https://doi.org/10.3109/15476910903453296.
- J. Lorenzo, Chapter 9 - The effects of immune cell products (cytokines and hematopoietic cell growth factors) on bone cells, in Osteoimmunology, 2nd, J. Lorenzo, M.C. Horowitz, Y. Choi, H. Takayanagi, and G. Schett, eds., Academic Press, Cambridge, 2016, pp. 143–167.
- H. Spits, In interleukin-7, in Encyclopedia of Hormones, H.L. Henry and A.W. Norman, eds., Academic Press, Cambridge, 2003, pp. 439–446.
- N. Bosco, F. Agenès, and R. Ceredig, Effects of increasing IL-7 availability on lymphocytes during and after lymphopenia-induced proliferation, J. Immunol. 175 (2005), pp. 162–170. doi:https://doi.org/10.4049/jimmunol.175.1.162.
- J. Lin, Z. Zhu, H. Xiao, M.R. Wakefield, V.A. Ding, Q. Bai, and Y. Fang, The role of IL-7 in immunity and cancer, Anticancer Res. 37 (2017), pp. 963–967.
- R. Mazzucchelli and S.K. Durum, Interleukin-7 receptor expression: Intelligent design, Nat. Rev. Immunol. 7 (2007), pp. 144–154. doi:https://doi.org/10.1038/nri2023.
- A. Sheikh and N. Abraham, Interleukin-7 receptor alpha in innate lymphoid cells: More than a marker, Front. Immunol. 10 (2019), pp. a2897. doi:https://doi.org/10.3389/fimmu.2019.02897.
- B.R. Lawson, R. Gonzalez-Quintial, T. Eleftheriadis, M.A. Farrar, S.D. Miller, K. Sauer, D.B. McGavern, D.H. Kono, R. Baccala, and A.N. Theofilopoulos, Interleukin-7 is required for CD4+ T cell activation and autoimmune neuroinflammation, Clin. Immunol. 161 (2015), pp. 260–269. doi:https://doi.org/10.1016/j.clim.2015.08.007.
- S.L. Silva, A.S. Albuquerque, P. Matoso, B. Charmeteau-de-muylder, R. Cheynier, D. Ligeiro, M. Abecasis, R. Anjos, J.T. Barata, and R.M. Victorino, IL-7-induced proliferation of human naive CD4 T-cells relies on continued thymic activity, Front. Immunol. 8 (2017), pp. a20. doi:https://doi.org/10.3389/fimmu.2017.00020.
- A. Chahroudi and G. Silvestri, Interleukin-7 in HIV pathogenesis and therapy, Eur. Cytokine Netw. 21 (2010), pp. 202–207.
- Y. Levy, C. Lacabaratz, L. Weiss, J.-P. Viard, C. Goujard, J.-D. Lelièvre, F. Boué, J.-M. Molina, C. Rouzioux, and V. Avettand-Fénoêl, Enhanced T cell recovery in HIV-1–infected adults through IL-7 treatment, J. Clin. Invest. 119 (2009), pp. 997–1007.
- C. Sportès and R.E. Gress, Interleukin-7 immunotherapy, in Immune-Mediated Diseases. Advances in Experimental Medicine and Biology, M.R. Shurin and Y.S. Smolkin, eds., Springer, New York, 2007, pp. 321–333.
- A. Bikker, C.E. Hack, F.P.J.G. Lafeber, and J.A.G. van Roon, Interleukin-7: A key mediator in T cell-driven autoimmunity, inflammation, and tissue destruction, Curr. Pharm. Res. 18 (2012), pp. 1–10. doi:https://doi.org/10.2174/138161212800165979.
- M. Yasunaga, S. Manabe, and Y. Matsumura, Immunoregulation by IL-7R-targeting antibody-drug conjugates: Overcoming steroid-resistance in cancer and autoimmune disease, Sci. Rep. 7 (2017), pp. 1–14. doi:https://doi.org/10.1038/s41598-017-11255-4.
- J. Gao, L. Zhao, Y.Y. Wan, and B. Zhu, Mechanism of action of IL-7 and its potential applications and limitations in cancer immunotherapy, Int. J. Mol. Sci. 16 (2015), pp. 10267–10280. doi:https://doi.org/10.3390/ijms160510267.
- C.-H. Yuan, X.-Q. Yang, C.-L. Zhu, S.-P. Liu, B.-C. Wang, and F.-B. Wang, Interleukin-7 enhances the in vivo anti-tumor activity of tumor-reactive CD8+ T cells with induction of IFN-gamma in a murine breast cancer model, Asian Pac. J. Cancer Prev. 15 (2014), pp. 265–271. doi:https://doi.org/10.7314/APJCP.2014.15.1.265.
- S.A. Rosenberg, C. Sportès, M. Ahmadzadeh, T.J. Fry, L.T. Ngo, S.L. Schwarz, M. Stetler-Stevenson, K.E. Morton, S.A. Mavroukakis, and M. Morre, IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells, J. Immunother. 29 (2006), pp. 313–319. doi:https://doi.org/10.1097/01.cji.0000210386.55951.c2.
- W. Dummer, A.G. Niethammer, R. Baccala, B.R. Lawson, N. Wagner, R.A. Reisfeld, and A.N. Theofilopoulos, T cell homeostatic proliferation elicits effective antitumor autoimmunity, J. Clin. Invest. 110 (2002), pp. 185–192. doi:https://doi.org/10.1172/JCI0215175.
- J.E. Bower, P.A. Ganz, N. Aziz, J.L. Fahey, and S.W. Cole, T-cell homeostasis in breast cancer survivors with persistent fatigue, J. Natl. Cancer Inst. 95 (2003), pp. 1165–1168. doi:https://doi.org/10.1093/jnci/djg0019.
- I. Marković and S.N. Savvides, Modulation of signaling mediated by TSLP and IL-7 in inflammation, autoimmune diseases, and cancer, Front. Immunol. 11 (2020), pp. a1557. doi:https://doi.org/10.3389/fimmu.2020.01557.
- Y. Song, Y. Liu, R. Hu, M. Su, D. Rood, and L. Lai, In vivo antitumor activity of a recombinant IL7/IL15 hybrid cytokine in mice, Mol. Cancer Ther. 15 (2016), pp. 2413–2421. doi:https://doi.org/10.1158/1535-7163.MCT-16-0111.
- L. Belarif, C. Mary, L. Jacquemont, H. Le Mai, R. Danger, J. Hervouet, D. Minault, V. Thepenier, V. Nerrière-Daguin, and E. Nguyen, IL-7 receptor blockade blunts antigen-specific memory T cell responses and chronic inflammation in primates, Nat. Commun. 9 (2018), pp. 1–13. doi:https://doi.org/10.1038/s41467-018-06804-y.
- C.A. McElroy, J.A. Dohm, and S.T. Walsh, Structural and biophysical studies of the human IL-7/IL-7Rα complex, Structure 17 (2009), pp. 54–65. doi:https://doi.org/10.1016/j.str.2008.10.019.
- OpenEye Scientific Software, Santa Fe, NM, USA, 2018.
- BIOVIA, Dassault systèmes, Pipeline Pilot, San Diego, CA, USA, 2020.
- BIOVIA, Dassault systèmes, Discovery Studio, San Diego, CA, USA, 2020.
- C. Klein, I. Waldhauer, V.G. Nicolini, A. Freimoser-Grundschober, R. Nayak, D.J. Vugts, C. Dunn, M. Bolijn, J. Benz, and M. Stihle, Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines, Oncoimmunology 6 (2017), pp. e1277306. doi:https://doi.org/10.1080/2162402X.2016.1277306.
- Desmond Molecular Dynamics System, D. E. shaw research, New York, NY, USA, 2020. Maestro Desmond Interoperability Tools, Schrödinger, New York, NY, USA, 2020.
- SpectraMax M5e, Molecular devices, San Jose, CA, USA, 2010.
- OriginLab Corporation, Northampton, MA, USA, 2016.
- J. Baell and M.A. Walters, Chemistry: Chemical con artists foil drug discovery, Nature 513 (2014), pp. 481–483. doi:https://doi.org/10.1038/513481a.
- J. Mortier, C. Rakers, M. Bermudez, M.S. Murgueitio, S. Riniker, and G. Wolber, The impact of molecular dynamics on drug design: Applications for the characterization of ligand–macromolecule complexes, Drug Discov. Today 20 (2015), pp. 686–702. doi:https://doi.org/10.1016/j.drudis.2015.01.003.