Abstract
129I is commonly either the top or among the top risk drivers, along with 99Tc, at radiological waste disposal sites and contaminated groundwater sites where nuclear material fabrication or reprocessing has occurred. The risk stems largely from 129I having a high toxicity, a high bioaccumulation factor (90% of all the body's iodine concentrates in the thyroid), a high inventory at source terms (due to its high fission yield), an extremely long half-life (16M years), and rapid mobility in the subsurface environment. Another important reason that 129I is a key risk driver is that there is uncertainty regarding its biogeochemical fate and transport in the environment. We typically can define 129I mass balance and flux at sites, but cannot predict accurately its response to changes in the environment. As a consequence of some of these characteristics, 129I has a very low drinking water standard, which is set at 1 pCi/L, the lowest of all radionuclides in the Federal Register. Recently, significant advancements have been made in detecting iodine species at ambient groundwater concentrations, defining the nature of the organic matter and iodine bond, and quantifying the role of naturally occurring sediment microbes to promote iodine oxidation and reduction. These recent studies have led to a more mechanistic understanding of radioiodine biogeochemistry. The objective of this review is to describe these advances and to provide a state of the science of radioiodine biogeochemistry relevant to its fate and transport in the terrestrial environment and provide information useful for making decisions regarding the stewardship and remediation of 129I contaminated sites. As part of this review, knowledge gaps were identified that would significantly advance the goals of basic and applied research programs for accelerating 129I environmental remediation and reducing uncertainty associated with disposal of 129I waste. Together the information gained from addressing these knowledge gaps will not alter the observation that 129I is primarily mobile, but it will likely permit demonstration that the entire 129I pool in the source term is not moving at the same rate and some may be tightly bound to the sediment, thereby smearing the modeled 129I peak and reducing maximum calculated risk.
1. INTRODUCTION
The Department of Energy (DOE) is concerned about radioiodine because it is an identified risk in groundwater on the Hanford Site and the Savannah River Site (SRS) and is abundant in nuclear waste currently being processed and disposed at these sites and in the waste that will be eventually disposed at a national repository (e.g., Department of Energy, 2002, 2003, 2012a; Westinghouse Savannah River Company, Citation2008). To illustrate how the properties of 129I magnify its risk, 129I accounts for only 0.00002% of the radiation released offsite from the SRS, but contributes 13% of the population dose, a six orders of magnitude magnification of risk with respect to its radioactivity (Kantelo et al., Citation1990).
The potential nuclear renaissance in response to climate change presents another concern regarding radioiodine. There are 16 license applications to the Nuclear Regulatory Commission to construct 18 new nuclear reactors in the United States, increasing the total number of reactors from 104 to 122 (Parker and Holt, Citation2007). Similarly, India plans to construct another 20–30 reactors by the year 2020 (World Nuclear Association, Citation2009). China presently has 17 reactors under construction and anticipates building another 13 by the year 2020, increasing their nuclear capacity to at least 50 GW (World Nuclear Association, Citation2009). By the year 2030, China anticipates another threefold increase in nuclear power to 120–160 GW. Radioiodine is produced at a rate of 1 Ci (37 giga-Becquerels) per gigawatt of electricity produced by nuclear power (McKay, Citation1984). For example, if all 22 proposed 1-GW U.S. reactors are built, an additional 22 Ci of 129I will be generated annually (reactors are 98% efficient) and within seven years these reactors will generate more 129I waste than currently exists at the Hanford Site (65.5 Ci; Kincaid et al., Citation2006) and SRS (26 Ci; Hiergesell et al., 2008). Thus, if only a fraction of the proposed nuclear growth from the nuclear renaissance is realized, a significant increase in worldwide radioiodine inventory will be created by the nuclear power industry. Understanding the environmental behavior of 129I is critical to assessing the capability and capacity of facilities where this waste will be stored or disposed.
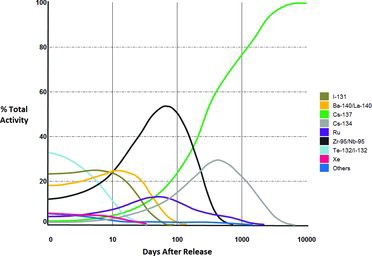
The risk posed by radioiodine can be broadly categorized as being long term and short term. The long-term risks are those resulting from exposure to the long-lived isotope, 129I, with a half-life of 16 million years. The short-term risks are those resulting from exposure to the short-lived isotope, 131I, with a half-life of eight days. By virtue of these differences in radioactivity (or half-lives), 129I is a long-term risk, associated with current groundwater contamination and nuclear waste disposal. 131I decays quickly to stable 131Xe, but its high specific activity makes it an immediate threat to exposed individuals. An example of the short-term risk from 131I is illustrated from the Chernobyl accident (). The majority of the radiation emitted immediately after the Chernobyl accident was from 131I. Studies show a link to thyroid cancer in exposed children that were less than five-years old at the time of the accident (Guiraud-Vitaux et al., Citation2008; Zaichick and Choporov, Citation1996). This was attributed to more than 90% of natural occurring or radioactive iodine in the human body is concentrated in the thyroid (Zaichick and Choporov, Citation1996). Once 131I is taken up by the thyroid, beta particles (primarily 606 keV, 89% abundance) and gamma rays (primarily 364 keV, 81% abundance) bombard nearby tissue, promoting thyroid cancer. Thus, 131I is a problem associated with large-scale accidents or failures at nuclear facilities, whereas 129I is a problem related to environmental remediation and long-term stewardship of disposed nuclear waste. This report focuses on 129I.
The objectives of this review are to (a) provide a current state of the science of radioiodine biogeochemistry relevant to the fate and transport and (b) identify areas of research that will facilitate remediation of 129I contaminated areas and numerically predicting 129I long-term fate and transport in the subsurface environment.
2. RADIOIODINE SOURCES
2.1 Reactor Sources of Radioiodine
The principle mechanism for radioiodine production is neutron-induced fission. Neutron-induced fission is a nuclear reaction in which the nucleus of an atom (e.g., 235U) splits when bombarded with neutrons into lower atomic-weight isotopes (e.g.,129I), often producing free neutrons and energy. Reactions form a variety of fission products, including 19 iodine isotopes (). Typically fission products are by-products and are not the isotopes of interest. At the Hanford Site and SRS, where most 129I exists in the United States, the reactors were fueled with uranium, and part of this uranium was converted into plutonium by the reactor neutron flux.
TABLE 1. Inventory of radioiodine isotopes produced in a representative material irradiated by a production reactor (Kantelo et al., Citation1990)
To provide an idea of the relative contribution of the various iodine isotopes to radiation over time, Kantelo et al. (Citation1990) calculated the decay rate of iodine isotopes from a representative irradiated material (). These calculations demonstrate that radioiodine activity decreases very quickly after reactor shutdown. By letting the irradiated material cool for 200 days, radioiodine activity decreases from 1,000,000,000 Ci to 2.5 Ci. Only two of the 19 radioiodine isotopes remain after 200 days, 129I (t1/2 = 1.6 × 107 years) and 131I (t1/2 = 8 days). shows the radiochemical properties of these isotopes. 129I is the only isotope that can last long enough to be a long-term threat in the environment.
TABLE 2. Radiochemical properties of 129I and 131I
Once the elements have cooled, the targeted isotopes are recovered through chemical separation or reprocessing. It is at this step that most radioiodine is inadvertently introduced into the environment. It has been estimated that radioiodine emissions to the atmosphere from the Hanford reprocessing operations between 1944 and 1972 were 266 kg (Raisbeck and Yiou, Citation1999). Presently, the two largest commercial reprocessing facilities are at Sellafield (England) and La Hague (France) and they have authorized liquid discharge permits for treated waste to the North East Irish Sea and the English Channel, respectively. In 1999, a year of high discharge, their discharge rates were 330 kg/years, with approximately 75% coming from La Hague. The integrated discharges from La Hague (1975–1997) and Sellafield (1961–1997) are estimated to be 1640 kg and 720 kg, respectively (Raisbeck and Yiou, Citation1999). Therefore, the total amount of 129I released over the history of facilities in the United States are less than that recently released in a single year by these two reprocessing facilities ().
TABLE 3. Major sources of 129I in the environment (compiled from Raisbeck and Yiou, Citation1999)
Another way radioiodine can enter the environment is through nuclear power plant accidents, such as the Fukushima Daiichi (Japan) and Chernobyl (Ukraine) reactor accidents. The Fukushima Daiichi accident introduced 1.5 × 107 kg 131I (1017 Bq 131I; note 131I and not 129I as described in ), this is about 10% the 131I mass released from the Chernobyl disaster (MacKenzie, Citation2011; von Hippel, Citation2011).
2.2 Contaminated Sites in the United States
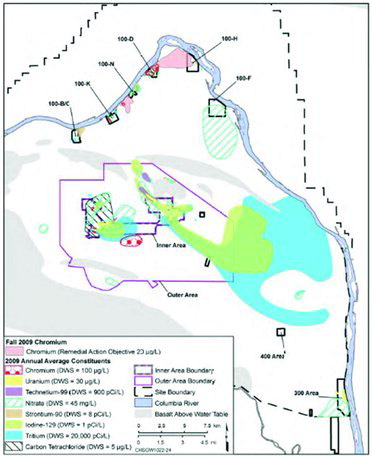
As a product of uranium fission, 129I occurs in the environment at several DOE facilities, but is especially problematic at the Hanford Site (Section 2.2.1) and the SRS (Section 2.2.2; ). Both of these sites have contaminant plumes that contain 129I at concentrations well above the drinking water standard (DWS) of 1 pCi/L. Maximum groundwater concentrations of 2 pCi/L have been reported near nuclear test holes at the Nevada Test Site (Hu et al., Citation2009), but 129I groundwater concentrations in wells away from the immediate nuclear test site have concentrations below 1 pCi/L. Very low concentrations of groundwater 129I have been reported in wells to the south of the Idaho National Laboratory (INL) boundary (Hall, Citation2005). Based on the presence of the co-contaminant 36Cl, the origin of the 129I was the INL waste disposal area. The maximum concentration observed in groundwater at the West Valley Demonstration Project in 2007 was 5.6 pCi/L (West Valley Demonstration Project, Citation2007). Using 129I to 127I ratios, elevated 129I concentrations were detected in soil cores of western New York and were attributed to atmospheric releases from the West Valley reprocessing plant (Rao and Fehn, Citation1999). Because 129I is not commonly analyzed at very low concentrations, it is expected that other sites that processed spent fuels, uranium targets, or other sources of uranium that underwent fission, may have elevated 129I concentrations in their sediments and ground-water.
2.2.1 Hanford Site, Richland, Washington
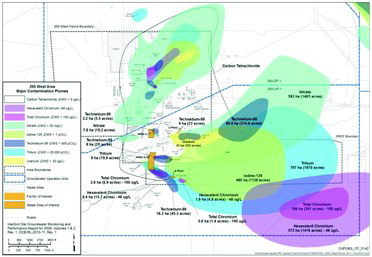
Between 1943 and 1988, operations in the Central Plateau of the Hanford Site, which include 200 East and 200 West Areas, were involved in Pu separation and U recovery operations. In the 200 West Area, the S and U Plant chemical separation and recovery processes generated liquid waste streams, such as process condensate, cooling water, and laboratory waste that were discharged to the ground using ponds, cribs, ditches, and trenches. Mobile radiological and nonradiological contaminants, including 129I, 99Tc, and nitrate, discharged to these facilities have migrated through the vadose zone to the underlying aquifer, some 76-m below grade. Once the contaminants reached the aquifer, they spread relatively rapidly, producing large plumes ( and ). Among these plumes are three 129I plumes covering an area >50 km2. The largest plume extends toward the southeast from the 200 East Area. A smaller arm of the plume is moving toward the northwest between Gable Mountain and Gable Butte. The two smaller plumes emanate from S Plant and U Plant cribs. The 90th percentile 129I concentration is 3.5 pCi/L, meaning 90% of the groundwater 129I concentration data in the plume is below 3.5 pCi/L. There are few recently recorded groundwater concentrations that exceed 10 pCi/L. Therefore, major challenges for treating 200 Area groundwater 129I plumes are that the plumes are extremely extensive and the 129I concentrations are extremely dilute (3.5 pCi/L = 0.00002 mg/L = 1.5 × 10–10 M). A technology evaluation for 129I remediation was completed and concluded that there were no current treatment technologies available that can achieve the federal DWS (Department of Energy, Citation2012a). The present plan for 129I treatment at the site is hydraulic containment that will be performed using withdrawing and injection wells placed at the leading edge of the plume(s) (Department of Energy, Citation2012b). DOE continues to evaluate potential treatment options for 129I (Department of Energy, Citation2012b). 129I is among the three primary radiological risk drivers at the site, along with 99Tc and 3H (Department of Energy, Citation2012b).
A great deal of attention has been directed to airborne emissions of 131I, the short-lived isotope, from the Hanford Site (National Research Council, Citation1995). In 1987, the DOE began the Hanford Environmental Dose Reconstruction Project (HEDR), which analyzed some 19,000 pages of newly released data related to Hanford environmental releases and production (National Research Council, Citation1995). The HEDR estimated the release rate and the exposure risk of nearby populations. For most of those exposed, the greatest part of their total dose came from drinking milk and eating food that was contaminated. According to HEDR's estimates, about 2 million people were exposed through the air or the Columbia River between 1944 and 1972. 131I accounted for >98% of the radiation dose they received (National Research Council, Citation1995; CDC, 2002). The Hanford Thyroid Disease Study reported that thyroid diseases, including thyroid cancer, were not more common among people in the Hanford vicinity (Davis et al., Citation2007). The study screened >3,000 people for thyroid diseases. Presently, essentially all the released 131I that was the subject of the Hanford Study has decayed.
2.2.2 SRS, Aiken, South Carolina
As briefly discussed above, the SRS also has extensive 129I environmental contamination (Denham et al., Citation2009; Kantelo et al., Citation1990). The 129I (and 131I) were produced through similar but not identical operations as existed at the Hanford Site. Targeted radionuclides were extracted from irradiated materials at two locations, F-Area and H-Area, together known as the General Separations Area (). At F-Area and H-Area, low-level radioactive liquid waste was discharged to seepage basins, unlined pits that were ∼4 m deep and several hectares in area. Low mobility radionuclides, such as Pu, Cm, and Pu, have been largely sequestered in the sediments immediately beneath the basins, but more mobile radionuclides have migrated into the groundwater and discharge into Four Mile Branch, a small stream to the south of the General Separations Area that eventually flows into the Savannah River (Westinghouse Savannah River Company, Citation2006). The F- and H-Area Seepage Basins received approximately 3 Ci of 129I from 1955 to 1989. The seepage basins have since been closed and capped with a low permeability engineered cover built to The Resource Conservation and Recovery Act (RCRA) specifications and the contaminant plume is being treated with a funnel and gate system that includes a periodic aqueous base injection into the subsurface.
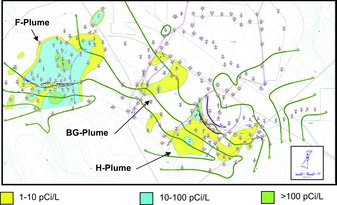
The SRS plumes are about 25 times smaller, <2 km2, than the Hanford plumes. However, 129I concentrations in these plumes are much greater, >20% of the plume has 129I concentrations >50 pCi/L. The F-Area Seepage Basins plume has the highest concentrations of 129I, with numerous wells containing concentrations tens to hundreds of times greater than the DWS (1 pCi/L; ). The highest concentration reported to date, 1060 pCi/L, was in 1996 for a well adjacent to the largest basin. Concentrations of 129I in this well have been rising over the past twenty years, while concentrations of most other radionuclide contaminants have been decreasing (Kaplan et al., Citation2011). The increase likely reflects release of adsorbed 129I from vadose zone and seepage basin sediments as pH has risen with time (Denham and Vangelas, Citation2008; Kaplan et al., Citation2011). The high concentrations within the F-Area Seepage Basins plume make 129I one of the primary risk drivers at this site (along with tritium, uranium, and 90Sr). 129I biogeochemistry in the F-Area plume has been the subject of several recent studies (further discussed in Section 3.5).
The H-Area Seepage Basins plume has lower concentrations of 129I (). The highest concentration observed in the first half of 2009 was 79 pCi/L in a well adjacent to the largest basin. The bulk of the plume has concentrations that are <10 pCi/L. Likewise, the small plume emanating from the Old Radioactive Waste Burial Ground contains relatively low concentrations of 129I. The highest concentration measured in the plume in 2011 was 11 pCi/L. The 129I plumes at both the F- and H-Areas have already travelled from the source to the nearest surface water, Four Mile Branch.
3. GEOCHEMISTRY OF IODINE
3.1 General Iodine Chemistry
Iodine is a complicated element because under environmentally relevant conditions, it can exist in multiple physical (solid, liquid, or gas) and oxidation states (–1, 0, +1, +5, and +7). It readily reacts with organic compounds, further complicating its chemistry in most natural environments. The solubility of elemental iodine in water at 25°C is 340 mg/L with a vapor pressure of 4.1 × 10–4 atm (Lauterbach and Ober, Citation1996). Henry's Law constant at 25°C for I2 is estimated to be 3.8 mol/L·atm (HSC Chemistry® Version 7.1, Outotec, Coeur d’Alene, ID, USA). Iodine hydrolyzes in water by four main reactions (Parsly, Citation1970):
(1)
(2)
(3)
(4)
The reactions for several species are shown in with iodide (I–) as the reactant species. Under conditions prevalent in groundwater and surface water only the –1, 0, and +5 valence states are common. Additional species may occur in natural waters because iodide forms aqueous complexes with various soft metals, but require high iodide concentrations to account for a significant fraction of the metal species.
TABLE 4. Reactions of log K association constants of selected aqueous species of iodine
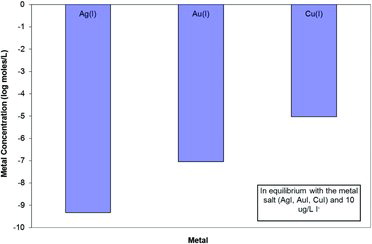
Iodide forms low solubility salts with several metals, notably Ag, Hg, and Cu(I). shows the calculated aqueous concentration of the metal in equilibrium with metal salt and 10 μg/L iodide, assuming ideal behavior and no significant complexing by other constituents. The occurrence of natural iodide minerals containing Ag, Hg, and Cu, though rare, attests to the insolubility of these metal iodides. The solubility of metal iodates is typically much higher than iodides and thus metal iodates are unlikely to exert any control on iodate concentrations in most systems (Fuge and Johnson, Citation1986).
Various forms of iodine react with organic molecules. Elemental iodine reacts in much the same way as chlorine and bromine, to form organo-iodine compounds. The syntheses of polyvalent organo-iodine compounds has been reviewed (Stang, Citation2003; Zhdankin and Stang, Citation2002). Skulski (Citation2000) reviewed 10 years of research at the Medical University of Warsaw, Poland, into aromatic organo-iodine compounds. Another area of research into organic-iodine reactions has been the reactions that might occur with organic substances in nuclear reactors during an accident (e.g., Malinauskas and Bell, Citation1987; Paquette et al., Citation1986; Skulski, Citation2000; Taghipour and Evans, Citation2002; Wren and Ball, Citation2001).
3.2 Aqueous Speciation
3.2.1 Aqueous Iodine–Inorganic Ligand Interactions
Inorganic iodine chemistry under conditions associated with most 129I environmental plumes is somewhat simplified because the only aqueous species that are common are iodide, I2o, and iodate. Based on thermodynamic considerations, shows the relation of these species in Eh-pH space. Iodate is expected to be stable under very oxidized conditions across the range of pH likely to be encountered in most contaminant plumes. Elemental iodine, I2, is stable at moderately oxidized, acidic conditions in which relatively high total iodine concentrations are present, such as in marine environments. Increased total iodine concentration expands the diiodine field as shown by the dotted line in . Iodide is the stable form over much of the range of conditions expected in contaminant plumes.
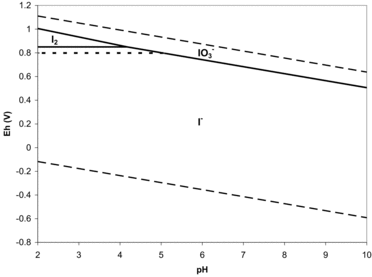
As will be discussed in Section 3.3, important omissions in these calculations are organo-iodine species; these species are omitted because association constants are not available. Because iodine and organic carbon (OC) form extremely strong covalent bonds, the presence of small concentrations of dissolved OC can have significant effects on iodine speciation. Such organo-I species have been shown to account for as much as 40% of the iodine species in estuaries, rivers, and rain (Santschi and Schwehr, Citation2004), or as much as 80% in subsurface aquifer environments (Kaplan et al., Citation2011; Otosaka et al., Citation2011). Low concentrations of organo-I (and iodate) have also been detected recently in Hanford groundwater (Santschi et al., Citation2012; discussed in Section 3.4). In part, iodide was thought to be the thermodynamically favored species because of the way the calculations were done. If calculations were done using oxygen fugacity rather than redox potential (such as ), iodate would be the favored species in all SRS waters examined and presumably in Hanford waters as well (Miles Denham, personal communication, Savannah River National Laboratory, December 11, 2013). Iodine speciation appears to be controlled by complex microbial and organic interactions (Section 4.0). There are likely kinetic controls not captured by thermodynamic analyses. Yet, without thermodynamic constants for all species even this is difficult to say definitively.
3.2.2 Aqueous Iodine–Dissolved Natural Organic Matter Interactions
The coexistence of inorganic and organic iodine species has been reported in various environments (Baker et al., Citation2001; Couture and Seitz, Citation1983; Muramatsu, Citation1989; Schwehr et al., Citation2005a; Schwehr et al., Citation2005b; Xu et al., Citation2012a; Xu et al., Citation2011a; Xu et al., Citation2011b; Xu et al., Citation2011c; Xu et al., Citation2012b; Yuita, Citation1992; Yuita and Kihou, Citation2005). For example, methyl iodide is an important gaseous form of iodine in the marine atmosphere and in releases from nuclear fuel reprocessing facilities, while dissolved organo-I compounds comprise up to 40% of total iodine in aqueous samples from estuaries, rivers, and rain (Santschi and Schwehr, Citation2004). Organic carbon interactions with 129I are discussed in more detail in Section 3.3.
3.3 Uptake to Sediment Minerals and Organic Matter
3.3.1 Iodine Speciation on the Solid Phase
In this review the term sorption is used to describe aqueous iodine partitioning to a solid phase. It is meant to be devoid of implied partitioning mechanism and as such may include adsorption, absorption, complexation, precipitation, coprecipitation, and ionization with organic carbon (the covalent bond between iodine and organic carbon, especially aromatic moieties). When specific partitioning processes were identified, more specific mechanisms are noted.
There have been two different approaches to identifying iodine species associated with soils and sediments: direct spectroscopic methods and indirect extraction methods. Direct spectroscopic methods have been limited to the use of X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure spectra (EXAFS; Fuhrmann et al., 1998; Kodama, 2006; Schlegel et al., Citation2006; Shimamoto and Takahashi, Citation2008). These direct X-ray absorption spectroscopy techniques are limited to analyzing geological samples with elevated iodine concentrations added to the samples. The reason for this is because environmental samples tend to have iodine concentrations that are appreciably lower than this method's detection limits. The detection limit of XANES is in the order of >10 μg/g 129I (Hou et al., Citation2009a). Consequently studies must be conducted at extremely high iodine concentrations, parts per million to percent concentration levels. Shimamoto and Takahashi (Citation2008) conducted XANES studies on soils amended with 55 μg/g I. They reported that most of the iodine in the soil was in an organic form. XANES studies with a pyrite mineral isolate showed the reduction of IO3– to I2 (Fuhrmann et al., Citation1998). This finding is very likely a result of using very high iodine concentrations and likely not a common process occurring in natural terrestrial environments. They also observed that magnetite sorbed iodide from solution but not iodate (this is contrary to popular understanding that iodate sorbs more strongly than iodide to mineral surfaces due to iodate's strong Lewis base characteristic) and that biotite sorbed iodate from solution, but not iodide from solution. XANES was also used to independently determine Kd values of iodate and iodide uptake by a soil (Kodama, 2006).
The second method of assessing the speciation of iodine on the solid phase is via sequential extraction or selective extraction. This technique involves adding extractants designed to remove iodine associated with a given phase (e.g., water soluble, carbonate, Fe-(oxy)hydroxide, OM, residual) or sorbed by a specific process (e.g., exchangeable). Sequential extractions have been carried out to investigate iodine speciation in sediments (Hou et al., Citation2003; Schmitz and Aumann, Citation1995; Stutz et al., Citation1999; Yuita, Citation1992). These are operationally defined parameters and as such do not carry mechanistic interpretations. In practice they suffer a number of pitfalls that are especially important with contaminants that are more prone to complex, change oxidation, or precipitate, such as phosphate or plutonium (Nirel and Morel, Citation1990; Scheckel et al., Citation2003; Tessier and Campbell, Citation1988).
Hou et al. (Citation2003) used a water soluble NH4OAC exchange, carbonate (NH4OAC (pH 5), Fe-oxide (NH2OH, HCl, pH 2, 80–100°C), organic (NaOH 80–100°C), and residue (remaining) fractionation scheme. Schmitz and Aumann (Citation1995) analyzed soils collected from a reprocessing plant in Germany and reported that 39–49% of the 129I was associated with the water soluble fraction, whereas only 4–15% was associated with the organic fraction and 7–13% was associated with the residual fraction. However, a different distribution of stable iodine, 127I, was observed where only <4% occurred in the water soluble fraction. This difference between the two isotopes may be attributed to the different sources of the two isotopes and underscores an important point that the fate of iodine in the environment is controlled by biological as well as geochemical factors. In the coastal and estuarine area of Sellafield, most of the 129I was associated with the oxides (53–66%) and organic (23–43%) fractions, whereas <7.5% was found in the other fractions. A similar result was also obtained from soil samples collected from near the Chernobyl accident: 30–40% of the 129I was associated with the oxides, 40–48% with the organics, and only 6–13% with the water soluble fraction (Hou et al., Citation2003).
3.3.2 Influence of Sediment Organic Matter on Iodine Sorption
Several studies of sorption in shallow soils that contain OM indicate that the OM is a primary control on iodine sorption (Assemi and Erten, Citation1994; Bird and Schwartz, Citation1997; Fukui et al., Citation1996; Kaplan, Citation2003; Neal and Truesdale, Citation1976; Sheppard and Thibault, Citation1991; Whitehead, Citation1974; Yoshida et al., Citation1992; Yu et al., Citation1996). In a survey of 26 soils and sediments samples from across the United States, with natural OM concentrations ranging from 0.046 to 0.5 wt% (except for one peat sample that was 28.1 wt%), Hu et al. (Citation2009) reported that ∼90% of the total iodine in soils was present as organic iodine, while inorganic iodine species became important only in sediments with low OM contents. Similarly, Whitehead (Citation1973) demonstrated that iodine sorption in untreated soils was greater than in soils that were treated to destroy the OM. There was some sorption of iodine in the treated soils, primarily by iron and aluminum oxides (Whitehead, Citation1973). In a single soil profile studied (Bors et al., Citation1988), sorption of iodide correlated with organic carbon content of samples that ranged from 6.2 to 0.06 wt% organic carbon. Similar results were reported in studies involving different heating regimes (Muramatsu et al., Citation1990b; Muramatsu, Citation1990a). When all OM was removed by oxidation at 500°C, sorption of iodine was reduced substantially, but the decrease in sorption was much less when carbon remained as charcoal after heating to 500°C under a nitrogen atmosphere (Muramatsu, Citation1990a).
Another approach to demonstrating the importance of OM to iodine sorption by soils is to compare sorption onto individual components that comprise the soil. Whitehead (Citation1974) took this approach and found that organic compost sorbed iodine more strongly than the mineral components of a soil. He also noted that drying the compost prior to the experiments greatly reduced iodine sorption. Neal and Truesdale (Citation1976) measured sorption of iodide and iodate in riverine sediments and concluded that stronger sorption of these species occurred in peaty sediments than in freshly prepared ferric hydroxide. Sorption of iodine on a soil that was 70% organic carbon was compared to sorption onto various minerals by Assemi and Erten (Citation1994) and sorption was much stronger to the soil. They also found that iodine soil sorption decreased substantially when it was heated to 180°C or irradiated. Yu et al. (Citation1996) demonstrated the importance of OM to iodide sorption by showing that the individual high surface area inorganic phases that make up most of a volcanic soil poorly sorb iodide relative to the bulk soil. A different approach was taken by Fukui et al. (Citation1996) who compared iodine sorption onto a soil to sorption onto pure humic material. Kd values on the humic material were 10 times higher than on the soil.
Sorption of iodine in oxic organic-rich sediments is greater than in anoxic organic-rich sediments (Ashworth and Shaw, Citation2006; Ashworth et al., Citation2003; Maillant et al., Citation2007; Sheppard and Hawkins, Citation1995; Whitehead, Citation1974). This was hinted at by Whitehead (Citation1974) who found that drying organic-rich soils before measuring iodine Kd values substantially reduced the sorption. Unfortunately, the implications of these results on experimental protocol have been overlooked by several subsequent related studies. Sheppard and Hawkins (Citation1995) explicitly noted this difference in iodine sorption between oxic and anoxic organic-rich sediments with experiments on bog soils. Changes in the redox state of an organic-rich soil from oxic to anoxic can also release iodide that was sorbed in the oxic state (Bird and Schwartz, Citation1997). The same relation was observed by Ashworth et al. (Citation2003) and Ashworth and Shaw (Citation2006) in column studies where they noted much lower sorption in saturated anoxic portions of their columns than in unsaturated oxic portions. Likewise, the same observation has been made in the field by Maillant et al. (Citation2007) who returned, after 15 years, to the site of an iodide injection into bog soils documented by Sheppard et al. (Citation1989). Iodine Kd values were approximately seven times higher in the surface bog soils than in the deeper anoxic bog soils. Release of soil-bound iodine has also been observed in two forest soils (9–56-fold increase) under flooded, anaerobic conditions (Yuita, Citation1992). Iodate was the dominant (86% of water-soluble I) form under nonflooded, oxidizing conditions, whereas iodide was the dominant (87% of water-soluble I) form under flooded conditions. In one soil type, the soil solution concentration of organo-iodine increased 2.5-fold under flooded conditions. A very extensive sampling of iodine soil water from a forest, upland field, and rice paddy field in Japan revealed a strong negative relationship between soil Eh and soil water iodine concentrations (Yuita and Kihou, Citation2005). In particular, soil water iodine concentrations increase with decreasing Eh values below 200 mV. In summary, it appears that iodine uptake by sediments is largely controlled by OM concentrations and that iodine binds to surface soils under oxic conditions and is released under strongly reducing conditions. It is not clear from these studies whether the changes in redox alter the iodine speciation or the OM speciation, or both.
Although the above studies demonstrate the importance of OM controlling iodine sorption in sediments, there still remains considerable uncertainty regarding (a) the nature of the organo-iodine bonding mechanism; (b) whether the organo-iodine bond occurs only under abiotic, biotic, or under both soil conditions; (c) why iodine sorption to OM occasionally appears to enhance or have no effect on transport; (d) the potential for abiotic iodide oxidation; and (e) the role of naturally occurring sediment microbes to accumulate iodine and to promote iodide oxidation. Much of this uncertainty arises from the fact that humic substances are inherently heterogeneous and complicated. Natural OM has been described as supramolecular associations of self-assembling heterogeneous and relatively small molecules derived from the decomposition of dead plant and animal residues. These small molecules are held together by weak dispersive forces, such as hydrogen bonding, hydrophobic interactions, and electrostatic interactions.
It is indeed very important to study iodine speciation under ambient conditions (Schwehr et al., Citation2009; Zhang et al., Citation2011). Using a SRS subsurface wetland sediment and ambient iodine (10–8 M) consisting of 29% iodide, 4% iodate, and 67% organo-I, Schwehr et al. (2009) showed that by incrementally adding more total iodide, incrementally less organo-I and more iodide was detected in the aqueous phase of sediment suspensions at steady state. At 1000 μM, only 3% of the iodine existed as organo-I. They attributed this change in detected iodine speciation to the added iodine swamping out the low system OM concentrations. Similarly, Zhang et al. (2012) investigated the sorption, transport, and interconversion of iodine species by comparing their mobility in groundwater at ambient iodine concentrations (10–8 and 10–7 M) to those at artificially elevated concentrations (10–5 M). Iodine mobility greatly depended on iodine concentration, in addition to the type of species. At ambient concentrations, Kd values as high as 49 mL/g were measured, whereas at 10–5 M iodide, the solute traveled along with the water without retardation. Consequently, it is not possible to assess accurately natural iodine speciation using elevated spike loadings (to ease analytical detection).
3.3.3 Influence of Mineralogical and Inorganic Sediment Parameters on Iodine Sorption
Sorption of iodine on organic-poor soils is influenced primarily by mineralogy and pH, but complicated by iodine speciation (Dai et al., Citation2004; Kaplan et al., Citation2000a; Mishra and Srinivasu, Citation1992; Neal and Truesdale, Citation1976; Sazarashi et al., Citation1994; Whitehead, Citation1973; Whitehead, Citation1974; Yoshida et al., Citation1992). Whitehead (Citation1973) showed that when OM was removed from a soil, some iodine still sorbed to the mineral fraction–primarily to iron and aluminum oxides. The magnitude of the sorption was inversely related to pH. In several studies sorption of iodate to the mineral fraction of soils has been greater than sorption of iodide. Neal and Truesdale (Citation1976) observed that there was little sorption of iodide by ferric hydroxide or kaolinite, whereas iodate sorbed strongly to ferric hydroxide. Yoshida et al. (Citation1992) concluded that the difference was that iodide sorption was likely purely electrostatic attraction, while iodate was chemically adsorbed or exchanged by the mineral allophane or sesquioxides. Others have observed the same behavior of iodide versus iodate sorption, with the iodate sorption showing a two-step mechanism–an initial rapid equilibrium sorption, followed by slow non-equilibrium sorption (Nishimaki et al., Citation1994). The conclusions of Fukui et al. (Citation1996) were consistent with Yoshida et al. (Citation1992), except they reported that iodide sorption seemed to be more complicated than pure electrostatic attraction. For 20 different Chinese soils, Dai et al. (Citation2004) observed that the only strong correlation between soil properties and iodate sorption was the content of free iron and aluminum oxides.
Monomineralic studies of iodine sorption show similar results as those using bulk soils, but provide an additional level of uniformity, permitting greater interpretation of the data. In general, iodate sorbs more strongly to individual minerals than iodide and the common soil minerals that most strongly bind iodate are ferric oxides and hydroxides (Couture and Seitz, Citation1983; Neal and Truesdale, Citation1976; Ticknor and Cho, Citation1990). Fuhrmann et al. (Citation1998) observed an exception to the general iodide-iodate sorption relationship, whereby magnetite sorbed iodide more strongly than iodate. They also looked at redox changes in the sorbed iodate using XANES spectra. They observed that no reduction of iodate occurred when sorbed to the ferrous iron containing minerals biotite and magnetite, but reduction to elemental iodine did occur when iodate was sorbed to pyrite. Further reduction of the elemental iodine to iodide was not observed. Ticknor and Cho (Citation1990) reported that iodate sorption to hematite increased with increased total dissolved solids. Their experiments were done in a synthetic groundwater with high concentrations of Na+, Ca+2, Cl–, and SO4–2 (2000, 2170, 6176, and 985 mg/L, respectively), indicating that the presence of one or more of these ions changed the surface chemistry of the hematite to favor iodate sorption. Relatively strong sorption of iodate to freshly precipitated aluminum hydroxide has been measured, but sorption decreased as the aluminum hydroxide aged (Musić et al., Citation1979).
Iodide also sorbs to common soil minerals. Musić et al. (1979) observed weak sorption of iodide to aluminum hydroxide. Sazarashi et al. (Citation1994) measured Kd values of 2–3 mL/g for sorption of iodide onto allophane, but observed no sorption to montmorillonite. Weak sorption of iodide to imogolite and ferrihydrite was observed by Yu et al. (Citation1996), with sorption to ferrihydrite inversely related to pH. However, sorption onto these minerals was much less than onto a bulk soil containing these minerals and OM. Substantial sorption of iodide to common minerals has been observed by some investigators. Kaplan et al. (2000) observed strong sorption of iodide to illite, which was inversely related to pH. Titanium oxides are a common minor component of soils and Mishra and Srinivasu (Citation1992) found that 73% of iodide at a concentration of 10–7 molar sorbed to TiO2 powder. Yet organo-clays show even stronger sorption of iodide (Bors et al., Citation1994).
3.4 Radioiodine Geochemistry at the Hanford Site
A recent review of stable and radioiodine geochemistry at the Hanford Site located in Washington State was recently completed by Kaplan et al. (Citation2012). In a compilation of iodine sorption studies using Hanford sediments (Cantrell et al., Citation2003), it was concluded that the likely range of iodine Kd values was 0 to 2 mL/g and that the most common range of values is between 0 and 0.2 mL/g. The tests he reported were almost exclusively designed to measure the extent that radioiodine sorbs under Hanford subsurface conditions and were not designed to understand iodine speciation or sorption mechanisms (Gee and Campbell, Citation1980; Kaplan et al., Citation1998; Kaplan, Citation1996; Serne et al., Citation1993; Um et al., Citation2004). Among the underlying assumptions in each of these laboratory Kd measurements was that iodide is the primary species in Hanford groundwater and that it sorbs to sediments through the reversible anion exchange process (meaning iodine adsorbs and desorbs at equal rates). Santschi et al. (Citation2012) recently measured iodine speciation in groundwater samples recovered from the Central Plateau region of the Hanford Site and reported the presence of iodide, but also organo-iodine and significant amounts of iodate (). Both stable and radioiodine indicate that iodate is clearly the predominant species in all samples analyzed. Stable 127I, for which there was a sufficient concentration to conduct full iodine speciation, consisted of only 1–2% iodide and 0–14% organo-I and 84–98% iodate.
TABLE 5. Stable 127I and radioactive 129I speciation in groundwater collected in March 2012 from 200 West Area (Santschi et al., Citation2012)
Based on thermodynamic considerations (), iodide was expected to be the dominant species in Hanford groundwater. However, it is quite likely that iodide and iodate, as opposed to only iodide, may have been the dominant species introduced into the cribs that serviced facilities that used hot, strong acids to remove cladding from spent fuels, such as at T and B Plants in the 200 Areas. In a study to provide some insight into these conditions, 10 mg/L iodide was prepared in1 M HNO3 and heated at 80°C for 2 hr (Santschi et al., Citation2012). The following speciation was measured:
4.5 mg/L iodide
2.2 mg/L iodate
Below detection limit concentrations of I2.
These results show a 55% decrease in iodide concentrations and 22% increase in iodate concentrations. Lack of detection of I2 may in part be due to it escaping when the vial was opened for analysis and the analytical sample may have been overly diluted to permit measuring the iodide and iodate.
However, the likely introduction into the subsurface environment of 129I in part as iodate, can only partially explain the observed groundwater speciation () because stable iodine, 127I, also exists predominantly as iodate. The presence of iodine in multiple oxidation states and species within a given aqueous samples is consistent with previous reports of radioiodine and stable iodine speciation measurements at other DOE sites (Denham et al., Citation2009; Kaplan, Citation2011; Kaplan et al., Citation2011; Kaplan et al., Citation2012; Otosaka et al., Citation2011), oligotrophic lake (Gilfedder et al., Citation2008; Gilfedder et al., Citation2009), and marine systems (see references in (Schwehr et al., Citation2005a). To our knowledge there are no iodine speciation studies reported in the literature of arid region groundwater similar to that at the Hanford Site. The 129I plume at the SRS is appreciably more acid (pH 3.1–6.0) and has higher dissolved organic carbon (DOC) levels than the Hanford Site (Section 3.5). All the SRS groundwater samples had varying percentages of iodide, iodate, and organo-iodine that changed as a function of the plume's pH, Eh, and DOC gradients. It was not uncommon to have about equal concentrations of all three species. Iodide concentrations tended to be greater than those reported in . Schwehr et al. (Citation2005a) reported that marine samples, which tend to have a pH of ∼8, closer to that of Hanford groundwater (pH 7–8.5), but relatively higher DOC concentrations, tended to have even distributions of iodide, iodate, and organo-iodine. Gilfedder et al. (Citation2009) reported monthly iodine speciation in an oligotrophic freshwater lake in the Alps. The lake had a pH between 8.0 and 8.5 and the DOC concentrations were very low (<2 mg/L). Organo-iodine, iodide, and iodate concentrations varied greatly during the year, but clearly organo-iodine was the dominant species, generally accounting for >80% of the iodine pool. Iodate concentrations remained fairly constant throughout the year, and iodide concentrations tended to vary. The significance of the data in is that iodate typically sorbs to sediments greater than iodide. No sorption testing has been conducted with organo-iodine, but it too may sorb appreciably more than iodide to sediments. These findings bring into question whether the present Hanford 129I Kd values, albeit conservative, adequately represents the sorption anticipated by the more strongly sorbing iodate and organo-iodine species. The presence of multiple iodine species would not influence the expected extent of a Hanford plume, as the presence of the fastest moving species, iodide, would not change. However, the presence of iodate or organo iodine would be expected to move slower, thereby changing the distribution of the total 129I concentrations within the plume.
Researchers have reported that a large fraction of radioiodine sorbed onto Hanford sediment readily desorbs, consistent with the reversible anion exchange mechanism (or Kd model; Kaplan et al., Citation2000b; Um et al., Citation2004). Yet, there was a more strongly binding radioiodine fraction that did not desorb under extreme chemical conditions conducive to anion exchange. Using a subsurface sediment, (ad)sorption iodide Kd values were <0.3 mL/g (depending on iodide spike concentrations), whereas desorption Kd values using the same sediments were 1.41–4.15 mL/g. A geochemical process that describes strongly sorbing iodine is iodine partitioning to sediment OM (Section 3.3.2). Santschi et al. (Citation2012) recently conducted initial studies related to this issue (). Using three composite sediments recovered from the 200 West Area, iodate Kd values were on average 89% greater than iodide Kd values and both species tended to increase with the amount of OC present in the sediment. It is especially noteworthy that this trend existed at the very low OC concentrations that naturally exist in the Hanford sediments, albeit there were only few samples in this early study. Another observation made by the researchers was that the iodine speciation changed once the spike solution came into contact with the sediments, indicating that is not reporting species-specific-Kd values, but instead represent Kd of a suite of iodine species. The importance of evaluating a species specific approach to describing radioiodine speciation is that it may provide a 129I fraction that is nearly irreversibly bound to the sediment and may provide a more accurate representation of 129I transport.
TABLE 6. Iodide and iodate Kd values after 21 days of composite sediments recovered from 200 West Area borehole cores (Santschi et al., Citation2012)
Um et al. (Citation2004) noted that, although sorption of iodide was very low on Hanford sediments, the iodide was only partially reversible. The sorption Kd was 0.2 mL/g, but the desorption Kd was 1.4 mL/g. Irreversibility of iodide diffusion into shale was observed; a substantial fraction did not diffuse back out (Savoye et al., Citation2006). Hu et al. (Citation2005) made similar observations in studies of iodine sorption to SRS and Hanford sediments. They found that the capacity for reduction of iodate to iodide in SRS sediments was greater in deeper aquifer material than in surface soil or sub-soil. A sample of Hanford sediment had a reduction capacity even greater than the SRS aquifer material. Reduction capacity is caused by the presence of structural Fe(II) in common clay minerals such as illite and smectite (Hu et al., Citation2005).
3.5 Radioiodine Geochemistry at the SRS
One reason that little is known about radioiodine at DOE sites is because very little research funding has been directed at 129I biogeochemistry, certainly not to the extent that has been directed at Cs, Pu, Tc, and U biogeochemistry. Among the reasons for this is that the analytical chemistry at the extremely low concentrations necessary to study ambient 129I (10–7 to 10–11 M) have not been readily available and the analytical procedures were (and still are) arduous (Denham et al., Citation2009). By combining recently developed analytical techniques applicable to stable iodine, 127I, and radioiodine, 129I (Schwehr et al., Citation2005a; Zhang et al., Citation2010) with existing spectroscopic techniques for natural organic matter, it has been possible to address applied and basic geochemistry problems.
Groundwater129I and stable 127I speciation was measured in 14 well samples in the F-Area of the SRS (), which comprises gradients in redox potential (360–654 mV), OC concentrations (5–60 μmol L–1; portion was anthropogenic organic contaminants), and pH (pH 3.2–6.8) in the F-Area plume (Otosaka et al., Citation2011). The 129I in all the groundwater samples were comprised of more than one species, typically all three of the analyzed species: iodide, iodate, and organo-iodine. As was the case at the Hanford Site, these results were not consistent with thermodynamic calculations that predict the presence of almost 100% iodide. Within these 14 groundwater samples, iodate accounted for 7% to 100% of the 129I and 19% to 84% of the 127I. Organo-iodine accounted for 0–70% of the 129I and 8–75% of the 127I. The total groundwater 129I concentration was 232 pCi/L immediately downstream of the seepage basins (source terms) and decreased with distance from the seepage basin. 127I concentration decreased similarly to that of 129I. Near the seepage basins, the majority (55–86%) of iodine existed as iodide for both 129I and 127I. Then as the iodide move down gradient, some of it transformed into iodate and organo-iodine, as indicated by decreased iodide percentages near the source and greater iodate and organo-iodine percentages down gradient. Significant amounts of organo-iodine (30–82% of the total iodine) were also observed at upstream wells, including those outside the mixing waste plume (as stable 127I). Concentrations of groundwater iodide decreased at a faster rate than organo-iodine along the transect from the seepage basin. It was concluded that removal of iodine from the groundwater through the formation of high molecular weight organo-iodine species was complicated by the release of other more mobile organo-iodine groundwater species.
The F-Area plume flows 0.7 km before it is intercepted by a riparian zone. As part of the SRS groundwater monitoring program, it has been documented that 129I concentrates in the riparian zone to extraordinarily high levels, >1000 pCi L–1 (unfiltered samples; Kaplan et al., Citation2011). Studies were undertaken to identify the process/mechanism responsible for the 129I concentrated, and in particular to quantify and understand the role of OM and microbes (Xu et al., Citation2011a; Xu et al., Citation2011c; Xu et al., Citation2012b) The F-Area riparian zone is heavily vegetated and includes an organically rich interface where the aquifer surfaces with the biosphere. Laboratory studies using SRS solid and groundwater materials showed that between 72% and 77% of the newly introduced iodide or iodate were could not be readily exchanged from the organic-rich riparian sediment, while the rest was transformed by the sediment into colloidal and truly dissolved organo-iodine (Xu et al., Citation2011a). Laboratory iodination experiments (the chemical process by which iodine bonds with organic carbon) indicated that iodine was likely covalently bound to aromatic structures of the SOM. Under very acidic conditions, abiotic iodination of SOM was predominant, whereas under less acidic conditions (pH >5), microbial enzymatically assisted iodination of SOM was predominant. It was concluded that although trace amounts of SOM-bound 129I could enhance transport, in generally the SOM in the riparian zone immobilized the 129I.
Iodide added to a subsurface sandy sediment with very low OM concentrations (0.01 wt% OC) did not convert to iodate, but trace levels were converted to organo-iodine (Xu et al., Citation2011a). When iodate was added to the same sediment, it immediately transformed to about 18% iodide and 3% organo-iodine. The iodide (ad)sorption Kd value in the organic-poor sediment was 0.71 mL/g and was 23.89 mL/g for a desorption tests. Similarly for iodate, the (ad)sorption Kd value was 5.16 mL/g and for a desorption test, the Kd values was 9.75 mL/g. By the end of these Kd measurements, portions of the iodide or iodate spikes had transformed to other species, hence these test do not represent species-specific Kd values, but were mixed-species Kd values.
In column studies, Zhang et al. (Citation2011) investigated the sorption, transport, and interconversion of iodine species in an SRS groundwater and subsurface sediment with high OM concentrations, 10.8 mg/kg. At ambient concentrations (10–8 to 10–7 M), iodide and iodate were significantly retarded (Kd values as high as 49 mL g–1), whereas at concentrations three orders of magnitude greater, iodide traveled predominantly at the same rate as water without retardation. Appreciable amounts of iodide were retained in soils during transport due to iodination of OC. When iodate was introduced in SRS subsurface sediment columns, it quickly reduced to iodide and proceeded to migrate through the sediment in a similar manner as the iodide spike system.
Iodine bonding with SOM is greatly influenced by not only the functionality of the SOM, but also the hydrophilic/hydrophobic forces in the SOM (Xu et al., Citation2011a; Xu et al., Citation2011c). Coincident variations in chemical composition, aromaticity, functional groups (e.g., aliphatic), hydrophobicity, and molecular weight indicated that (a) iodine in different humic acids was bound to a small-size aromatic subunit (∼10 kDa), (b) the large-size subunit (∼90 kDa), determined the relative mobility of iodine bound to SOM, and (c) iodine incorporation into the SOM (or iodination) is via covalent aromatic carbon-iodine bonding. The nature of the iodine bond to OM was further evaluated using various proton and 13C nuclear magnetic resonance (NMR) techniques (Xu et al., Citation2012b). Quantitative structure analyses by 13C direct-polarization magic-angle spinning (DPMAS) NMR and solution state 1H NMR of these humic substances indicated that iodine was closely related to the aromatic regions containing esterified products of phenolic and formic acids or other aliphatic carboxylic acids, amide functionalities, quinone-like structures activated by electron-donating groups (e.g., NH2), or hemicellulose-lignin-like complex with phenyl-glycosidic linkages.
The concentration of stable iodine bound to the fulvic acid fraction (519 μg 127I/g-carbon) was approximately an order of magnitude greater than the concentration of stable iodine bound to humic acid fractions (62 μg 127I/g-carbon). However, the contrasting radioiodine contents among the two fractions of SOM (humic acids and fulvic acids) could not be solely explained by the difference in the number of reactive binding sites. This difference may be the result of differences in the micro-molecular environment of the SOM that play additional key roles in the interactions between iodine and organic carbon. These microenvironments can influence the hydrophobic aliphatic periphery hindering the active aromatic cores and the hydrophilic polysaccharides favoring iodine accessibility towards hydrophilic iodine species. Environmental microbial studies involving iodine at the SRS are discussed in Section 4.6.
4. MICROBIAL PROCESSES INFLUENCING 129I FATE AND TRANSPORT
The activity of iodine in biota, the strong chemical bond with OM, the redox sensitivity, and studies of marine iodine cycling have led to the hypothesis that microbial activity is involved in iodine sorption by sediment OM. Many researchers have tested this hypothesis by comparing iodine sorption onto sterilized and unsterilized soils (Behrens, Citation1982; Bird and Schwartz, Citation1997; Bors et al., Citation1991; Sheppard and Hawkins, Citation1995; Sheppard et al., Citation1996; Yoshida et al., Citation1998). These studies have confirmed that microbial activity is involved, but the nature of that involvement is unresolved. Bors et al. (Citation1991) showed that under specific conditions bacteria and fungi cells isolated from a soil took up iodine, though they acknowledged that this did not resolve whether microbes may have exerted an indirect effect on iodine sorption, by their action of degrading OM under oxic conditions. Sheppard et al. (Citation1996) reiterated this conclusion by showing that the organic content of seven soils correlated positively with sorption, but enzymatic activity did not. Some of the iodide in their studies was irreversibly sorbed and they suggested that this iodide had converted to elemental iodine that reacted irreversibly with OM. Results reported by Yamaguchi et al. (Citation2006) and Yamaguchi et al. (Citation2010) using XANES to analyze iodine speciation also demonstrated that elemental iodine was ultimately the species that reacted with OM. In anaerobic soils, iodate (IVO3–) was reduced to elemental iodine (I0) that reacted with OM (Yamaguchi et al., Citation2006). In aerobic soils, iodine reaction with OM involved either reduction of iodate or oxidation of iodide (Yamaguchi et al., Citation2010). Yamaguchi et al. (Citation2010) also concluded that microbes were not necessary for the reduction of iodate by humic substances. Maillant et al. (Citation2007), studying bog samples that were injected with iodide 15 years earlier (Sheppard and Thibault, Citation1989), found considerably more evidence of microbial behavior in the oxic surface layers than in the anoxic layers. They also observed iodine associated with polyphenols in the humified fraction of the soils. This is consistent with the findings of Warner et al. (Citation2000) who observed that sorption rates of elemental iodine onto solid humic material were in the range of sorption rates onto the phenolic compounds (Warner et al., Citation2000). Recently, the microbial enzyme laccase was found to promote the oxidation of iodide and the formation of organically bound iodine in soils (Seki et al., Citation2013). It remains unclear whether laccase contributes significantly to all soil systems, but it is generally agreed that microbes play some role in soil organic matter-iodine interactions.
4.1 Biological Reactions and the Global Iodine Cycle
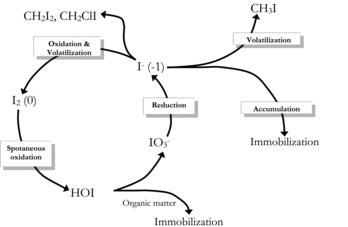
Biological mechanisms are intrinsic to the global iodine cycle. Oceans are considered the primary source of iodine; accordingly, our understanding of biological transformations of iodine is derived largely from those processes that have been studied in marine environments. Based on thermodynamic considerations iodate/iodide levels in seawater are in disequilibrium, with iodide found at levels much higher than would be expected, especially in certain surface waters, coastal and estuary regions, deep oxygenated water, and porewater of marine sediments where iodide concentration up to 40 μg/L have been measured (Amachi, Citation2008; Farrenkopf et al., Citation1997). The conversion of iodate to iodide is thought to be catalyzed primarily via microbial activity, perhaps by the enzyme nitrate reductase or release of iodide from C-I or N-I bonds upon decomposition of OM (Farrenkopf et al., Citation1997; Tsunogai and Sase, Citation1969; Waite and Truesdale, Citation2003; Wong et al., Citation2002; ).
Organic iodine species, including CH3I, CH2ClI, CH2I2, and CH3CH2I, are found at relatively low concentrations (<5% of total I) in the open ocean, but concentrations between 5% and 40% of total dissolved iodine are not uncommon in coastal and estuary systems (Hou et al., Citation2009b; Schwehr and Santschi, Citation2003; Wong and Cheng, Citation1998). Volatilization of iodine (primarily as methyl iodine, CH3I) from the ocean into the atmosphere is a critical feature of the biogeochemical cycling of this element (Fuge and Johnson, Citation1986). Localized concentrations of CH3I can be very high over seaweed beds and this may be the dominant source of gaseous organo-iodine in coastal regions, whereas microalgae, bacteria, and abiotic photochemical processes probably play a larger role in open ocean surface waters (Amachi et al., Citation2004; Baker et al., Citation2001; Moore and Groszko, Citation1988; Richter and Wallace, Citation2004; Smythe-Wright et al., Citation2006).
Importantly, organic-I species are thought to play a key role in the transfer of iodine to the terrestrial environment whereby organic iodine volatilizes from the ocean surface, undergoes various photolytic transformations in the atmosphere, and is transferred to the land surface via wet (1–6 μg/L total I in rainwater) and dry deposition (Fuge et al., Citation2005; Hou et al., Citation2009b; Kolb, Citation2002). Runoff concentration of iodine in fresh waters is typically 1–3 μg/L (Hou et al., Citation2009b). Iodine concentrations in soil ranges from 0.5–40 μg/g with common concentrations of 1–5 μg/g, a value much greater than that of underlying rocks (0.05–0.5 μg/g; Fuge et al., Citation2005; Hou et al., Citation2009b). Within the terrestrial sphere, little research has been devoted to the participation of biological processes in iodine cycling.
4.2 Accumulation and Speciation of Iodine in Biological Samples
Studies examining the accumulation of iodine by microorganisms are exceedingly sparse. A number of bacterial strains capable of iodide accumulation have been isolated from marine sediment (Amachi et al., Citation2005a; Amachi et al., Citation2005b). All strains were classified within the Flavobacteriaceae family of the Bacteroidetes phylum. The maximum iodide content measured in cells was 30 μg/g dry cells, and the maximum concentration factor based on the ratio of iodide in cells to that in the media was 5.5 × 103. By comparison, the brown algae, Laminaria digitata, has exhibited an iodide concentration factor of 1.5 × 105 (Küpper et al., Citation1998).
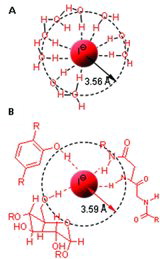
Iodide uptake by bacterial cells was stimulated in the presence of glucose and O2, and the process was saturable (0.073 μM affinity constant; 0.55 pmol/min/mg dry cells maximum velocity; Amachi et al., Citation2005a). Iodate was not accumulated. In a follow up study with one of these iodide-accumulating bacterial strains, it was determined that glucose stimulated iodide uptake through the action of glucose oxidase, which generated extracellular H2O2 (Amachi et al., Citation2007). The authors proposed a model whereby extracellular H2O2, generated by glucose oxidase, is used to oxidize iodide to I2 or hypoiodous acid, HIO, via an unidentified haloperoxidase. HIO is then transported across the cell membrane via a facilitated diffusion-type mechanism. Once inside the cell, HIO would be reduced to iodide or form organo-iodine species. This scenario is strikingly similar to that observed in the brown algae, L. digitata, where iodide is oxidized to hypoiodous acid (HIO) by an extracellular haloperoxidase, which then enters the cell via facilitated diffusion (Küpper et al., Citation1998; ). Inside the cell, iodine is stored in the iodide form and it has been posited that it acts as an inorganic antioxidant, scavenging reactive oxygen species (Kupper et al., Citation2008). Very interestingly, data indicated that while inside L. digitata tissue, iodide is surrounded by organic molecules such as phenols, carbohydrates, and proteins rather than an ordered hydration shell (; Kropper et al., 2008). In the presence of H2O2 and peroxidase activity most of the available iodine is incorporated into aromatic organic molecules that may or may not undergo nucleophilic substitution with Cl–, Br–, or HO– to regenerate iodide.
4.3 Biological Formation of Gaseous Iodine
From early studies it was concluded that volatilization of iodine from surface soils was not a widespread phenomenon. Whitehead (1981) found that among 24 surface soils volatilization was negligible, except in an acid sandy soil where 57% of the iodine was volatilized. It was generally believed that the presence of OM inhibits volatilization in soils by binding available iodine.
Rice paddies have, however, been shown to be a significant source of iodine transfer to the atmosphere. It has been calculated that worldwide methyl iodide volatilization from rice paddies at 16–29 Gg/year or as much as 4% of the atmospheric methyl I balance (Lee-Taylor, 2005; Muramatsu, 1995). In one of the early studies examining iodine volatilization from rice paddies, Muramatsu and Yoshida (Citation1995) determined that iodine volatilized from rice plants was mainly in the form of CH3I. Production of CH3I was greatest when the plants were flooded, and paddy soil incubated with increasing concentrations of iodide (0.1–10 mM) released increasing amounts of CH3I (other alkyl iodides were not detected).
A survey of 53 soil samples collected from rice paddies, forests, upland fields, and wetlands yielded detectable CH3I production from spiked iodide (0.1 μM) in samples from each of these terrestrial environments (Amachi et al., Citation2003). Formation of other alkyl iodides was not observed. The addition of glucose or yeast extract could enhance CH3I production, whereas the addition of antibiotics targeting prokaryotes (streptomycin and tetracycline) was strongly inhibitory (the fungal antibiotic, cyclohexamide had little effect). Very little methyl iodide was produced with autoclaved soils or under anaerobic conditions.
These results suggest that soil bacteria (mainly aerobic soil bacteria) may contribute preferentially to iodine volatilization from these soil environments. Iodide methylation has been observed primarily under aerobic or microaerobic conditions and has been observed under a variety of environments, including freshwater, seawater, and brine waters (Amachi et al., Citation2001; Stephenson, 1995). The extrapolation of iodine methylation rates from laboratory studies with select soils to natural soil environments is, however, difficult because of the large variations in iodine levels among different soil types, the dependence of iodine availability on soil properties, and the concentration-dependent nature of methyl iodine formation (Amachi et al., Citation2001). Of particular interest, it has been suggested that biologically mediated iodine methylation could contribute to localized air concentrations of 129I near nuclear production facilities or reprocessing plants, even after closure (Brauer and Strebin, Citation1982; Muramatsu, 1995).
Wide varieties of terrestrial bacteria are capable of methylating iodine, including multiple taxa within the α-, β-, and γ-Proteobacteria, Bacteroidetes, and Actinobacteria phyla (Amachi et al., Citation2001; Muramatsu et al., Citation2004). Indeed, of 100 bacterial isolates from a variety of sources, including soils, seawater, and marine sediments, approximately 40% were observed to produce CH3I. Rates of CH3I production were directly correlated with iodide concentration over the range of iodide concentrations examined (0.1 μM to 10 mM), thus bacterial CH3I production depends greatly on ambient iodine levels. Of the strains evaluated, iodine methylation may proceed via a S-adenosyl-L-methionine (SAM)–dependent mechanism. An affinity constant for iodide in cell extracts of one strain was 0.26 mM. SAM-dependent iodine methylation activity has been observed in a number of eukaryotes including marine algae, fungi, and higher plants (Amachi et al., Citation2001; Saini et al., Citation1995; Saxena et al., Citation1998; Wuosmaa and Hager, Citation1990). Although halide volatilization has been demonstrated for terrestrial plants, fungi, and bacteria, the relative contribution of each in soil systems has not been established.
4.4 Microbiological Oxidation of Iodine
The initial description of iodide oxidation by a bacterial species was elicited by the death of fish in experimental seawater, presumably due to iodine poisoning. The marine bacterium, P. iodooxidans (the strain is no longer available and the taxonomic assignment is uncertain), was isolated from tank water by observing colonies that produced a blue color on seawater-agar plates containing 4% glycerol, 0.12% starch, and 0.1% potassium iodide (Gozlan, Citation1968). Iodine production could be detected in the presence of 8 mM but not 0.8 mM KI. The microorganism was highly resistant to iodine toxicity. Experimental evidence pointed towards the production of an extracellular haemoprotein peroxidase by P. iodooxidans that could oxidize iodide in the presence of H2O2 (Gozlan and Margalith, Citation1973, 1974).
(7)
Importantly, the physiological status of the bacterial culture needed to be such that H2O2 was produced as a co-substrate for iodide oxidation.
Iodide oxidation has also been confirmed (used 1–5 mM KI) to be catalyzed by two Roseovarius sp. (α-Proteobacteria), one isolated from marine sediment and another from seawater (Fuse et al., Citation2003). Organic iodine species, including CH3I, CH2ClI, CH2I2, and CHI3, were also produced from iodide by these strains, and it was suggested that organic acids in the media or produced by the strains could be the carbon sources for these organo-iodine products. Extracellular peroxidases were implicated in the process.
In the most exhaustive study to date prospecting for iodide-oxidizing bacteria, strains were isolated from iodide-rich (63 μM to 1.2 mM) natural gas brine waters and seawater (Amachi et al. Citation2005). Iodide-oxidizing bacteria were not obtained from the 22 surface soil samples that were tested. Iodine-oxidizing bacteria were isolated directly from brine waters, but were only isolated from seawater after enrichment in the presence of 1 mM iodide (however, it was shown that KI does not influence the levels of iodide-oxidizing activity in the isolates). Why iodide-oxidizing bacteria can be enriched in the presence of high I concentrations (mM) is currently unknown. Amachi et al. (Citation2005) suggested two possibilities. First, iodide oxidation generates energy using oxygen as an electron acceptor, a thermodynamically favorable reaction (2I– + 1/2 O2 + 2H+ → I2 + H2O (ΔG0' = –56 kJ/reaction). However, since the iodide oxidizing enzyme appears to be extracellular, this hypothesis is unlikely. Alternatively, iodide-oxidizing bacteria use oxidized iodine species to attack competing microbes giving them a selective advantage, especially in environments with high levels of I (Amachi et al., Citation2005b; Arakawa et al., Citation2012).
The reaction required O2 but the addition of H2O2 did not stimulate iodide-oxidizing activity, thus the role of a peroxidase in iodide oxidation by these strains was not borne out by the experimental evidence. The authors suggested that an oxidase may be involved. The strains, which were most closely related to Roseovarius and Rhodothalassium spp. (α-Proteobacteria), were also shown to produce organic iodine species (CH2I2 and CH2ClI) in marine broth. It was postulated that I2 reacted with organic molecules in the media to form organo-iodine species. Bromine-oxidizing activity was not detected in these strains.
4.5 Biological Reduction of Iodine
A number of studies have demonstrated that phytoplankton (true algae and cyanobacteria) are involved in transformation of iodate to iodide in seawater (Chance et al., Citation2007; Wong et al., Citation2002). Nitrate-reducing bacteria have also been implicated in biological iodate reduction in marine environments (Tsunogai and Sase, Citation1969). The sulfur- and iron-reducing bacteria, Desulfovibrio desulfuricans and Shewanella oneidensis, have been shown to reduce iodate in pure cultures, but the mechanism and enzymes involved have not been identified (Councell et al., Citation1997; Farrenkopf et al., Citation1997). Iodate was toxic to cells of S. oneidensis at high concentrations (20 mM), and biological iodate reduction was optimal for both organisms at or below a concentration of 250 uM. Importantly, it was observed that two primary products of microbial sulfur and iron reduction, ferrous iron and sulfide, can abiotically catalyze the reduction of iodate to iodide (Councell et al., Citation1997). This abiotic mechanism could contribute to iodate reduction in environments that support microbial iron and sulfate reduction.
Recently, an iodate-reducing bacterium (strain SCT) was isolated from marine sediments (Amachi et al., Citation2007). The microorganism is closely related to the denitrifier Pseudomonas stutzeri and was capable of utilizing iodate (2–4 mM) as the terminal electron acceptor for growth in the presence of organic acids, alcohols, or sugars serving as the electron donor/carbon source. The results indicate that SCT is a dissimilatory iodate reducing bacterium that may couple iodate reduction directly to energy generating electron transport (IO3–/I– ΔG° = 3347 kJ/mol). However, nitrite reductase did not appear to be the primary mechanism behind iodate reduction in this microorganism.
4.6 Influence of Microbiology on Iodine Behavior at the SRS
Studies were undertaken to understand the role of microbes in iodine accumulation and species transformation (Li et al., Citation2012a; Li et al., Citation2011; Li et al., Citation2012b). Measurements of iodide uptake by aerobic bacteria isolates from F-Area sediments indicated that only three of 136 strains could accumulate iodide and they only accumulated between 0.2% and 2.0% of the ambient aqueous iodide (Li et al., Citation2011). Iodide accumulation in these isolates was inversely related to pH levels, significantly increasing at pH levels <6. Spent liquid medium from 27 of 84 bacterial cultures enhanced iodide oxidation 2–10-fold in the presence of H2O2 (Li et al., Citation2012a). Organic acids secreted by the bacteria were found to enhance iodide oxidation by (a) lowering the pH of the spent medium and (b) reacting with H2O2 to form peroxy carboxylic acids, which are extremely strong oxidizing agents. H2O2-dependent iodide oxidation increased exponentially from 8.4 to 825.9 μM with decreasing pH from 9 to 4. As pH decreased (≤5.0), it increased H2O2 hydrolysis, thereby promoting iodide oxidation. However, at pH ≥ 6.0, spontaneous decomposition of peroxy carboxylic acids, generated from H2O2 and organic acids, contributes significantly to iodide oxidation. The results reveal an indirect microbial mechanism, organic acid secretion coupled to H2O2 production that could enhance iodide oxidation and organo-iodine formation in soils and sediments. Most recently, Li et al. (Citation2012b), screened 84 strains for iodide oxidizing activity using a combination of triiodide (I3–) formation, radiography and a recently developed, sensitive iodine speciation assay revealed that 44 of these strains were capable of iodide oxidation. Together these results indicate that readily culturable, aerobic bacteria of the F-area aquifer do not accumulate significant amounts of iodide (Li et al., Citation2011). However, iodide oxidation, albeit at very slow rates, can be supported by a variety of terrestrial bacteria.
4.7 Microbiology of the Hanford Subsurface
A series of recent studies have provided the most in-depth analysis of the microbial community inhabiting subsurface sediments of the Hanford Site. Subsurface cores obtained from the Integrated Field-Scale Subsurface Research Challenge (IFRC) site within the 300 Area were extensively analyzed by biogeochemical measures, high-throughput sequencing and state of the art community analysis tools (Li et al., Citation2012b; Lin et al., Citation2012a; Lin et al., Citation2012b; Lin et al., Citation2012c; Stegen et al., Citation2012). Not unexpectedly, microbial community characteristics of the Hanford sediments were largely delineated based upon the formation from which they were retrieved, Hanford or Ringold. The Hanford formation, which is aerobic and primarily composed of unconsolidated sediments varying in size from silt to sand to large gravel, harbored moderate levels of bacterial biomass (∼5 × 106 to 5 × 107 cells per gram of sediment) and exhibited high diversity representing 12 bacterial divisions (Lin et al., Citation2012a). Transitioning into the anoxic portion of the Ringold, bacterial biomass decreased approximately 50-fold (∼1 × 105 to 1 × 106 cells per gram of sediment), and the bacterial diversity was greatly diminished and was comprised of nearly 90% Proteobacteria.
Because the organic carbon content of sediments from the two formations were similar it was suggested that higher hydrological flux and transmissivity of the Hanford formation enables higher advective flux of nutrients, thus supporting higher levels of biomass than observed in the less porous Ringold formation (Lin et al., Citation2012b); the concentration of DOC in groundwater has been measured <25 μmol/L (<0.3 mg/L C), and sedimentary organic C ranged from <42 μmol/g to 67 μmol/g (<0.05% to 0.08%) in Hanford and upper Ringold sediments though higher organic C contents have been detected deeper in the Ringold, ∼850 μmol/g (1.02% C). Sequencing and culturing results indicated that anaerobic respirers were present in both formations, and it was posited that anoxic microsites or hot spots of electron donors are interspersed throughout the largely aerobic Hanford sediment supporting spaces where anaerobic processes could dominate. Boulder-sized rip-up clasts of Ringold silt and clay found throughout the Hanford formation (Bjornstad et al., Citation2009) could also support anaerobic populations of microorganisms, adding to the complexity of the system and influencing the range of redox reactions possible.
Gaseous products of anaerobic metabolism were detected in both Ringold sediments and lower portions of the Hanford formation (Lin et al., Citation2012b). Indeed, it was suggested that anaerobic activity in the anoxic Ringold formation could support microbial growth in the aerobic Hanford sediments via upward fluxes of H2 and CH4. Yet the extent to which metabolic products of fermentation and methanogenesis support biomass formation in the Hanford formation remains untested.
Microorganisms can catalyze the reduction, oxidation, and alkylation of iodine (Amachi, Citation2008; Denham et al., Citation2009). Within the anoxic portion of the Ringold formation or within anaerobic pockets of the heterogeneous Hanford formation, the potential for iodate reduction to iodide is high since this activity has been readily detected among microorganisms involved in denitrification and iron and sulfur reduction (Amachi et al., Citation2007; Councell et al., Citation1997; Farrenkopf et al., Citation1997). It is important to note that both ferrous iron and sulfide (the two primary products of microbial iron and sulfur reduction) can catalyze the reduction of iodate to iodide (Councell et al., Citation1997). Evidence for denitrification and iron and sulfur reduction within the Ringold formation and at the Hanford/Ringold boundary has been reported, and relatively high proportions of ferrous iron are found even within sediments in the Hanford formation (Lin et al., Citation2012b). Yet, recent analyses of iodine speciation in Hanford groundwater and sediments revealed that the large majority of I exists as iodate (). Therefore, microbial reduction of iodate is either limited within this formation or reduction processes (abiotic and/or biotic) are kinetically hindered.
5. CONCLUSIONS AND TECHNICAL NEEDS
131I poses a very short-term risk (days to weeks) and 129I poses a very long-term risk (years to thousands of years). 129I is commonly among the key proposed risk drivers at disposal facilities (Hanford Intermediate Disposal Site, INL's Remote-Handled Low-Level Waste Disposal Facility, SRS's E-Area Low-Level Waste Disposal Facility; former Yucca Mountain Disposal Facility), and at radionuclide-contaminated plumes emanating from reprocessing facilities. The risk stems largely from 129I having a high toxicity, a high bioaccumulation factor (90% of all the body's iodine concentrates in the thyroid), a high inventory at source terms (due to its high fission yield), an extremely long half-life (16M years), and perceived rapid mobility in the subsurface environment. As U.S. and worldwide inventories of 129I increase, understanding how to minimize its release to the environment and what to do if it is released becomes increasingly important.
129I is a fission product (waste product) from plutonium separation and uranium recovery operations. Within the DOE complex, INL, Nevada Test Site, and the West Valley Demonstration Project have released 129I to the environment, but only low concentrations have been detected. The Hanford Site and the SRS have major plumes containing 129I at concentrations above the drinking water standard; the former are an order of magnitude larger in size, whereas the latter have orders of magnitude greater 129I plume concentrations. The facilities that are generating and introducing the greatest amount of 129I into the environment are in La Havre, France, and Sellafield, England.
Iodine behavior in the environment is complicated by its multiple physical states, multiple redox states, interactions with OM, and microbial transformations. Iodine sorption is mostly reversible, but a small fraction is not. Consequently, measured (ad)sorption Kd values are less than desorption Kd values. Adsorption and interaction with OM are the dominant attenuation mechanisms for 129I in groundwater. Iodate is typically adsorbed more strongly than iodide; adsorption is inversely related to pH. Sediment OM generally decreases iodine mobility, especially when bound to larger organic moieties. To a lesser extent, organo-iodine complexes may increase mobility, especially when iodine is bound to smaller organic moieties. Radioiodine speciation has been measured at the SRS and Hanford Site contaminant plumes. In both cases, iodide was not the dominant species and iodate and organo-iodine aqueous species were also present. This speciation distribution is not predicted by thermodynamic considerations. Iodide was thought to be the thermodynamically favored species because of the way the calculations were done. If calculations were done using oxygen fugacity rather than redox potential, iodate would be the favored species in all SRS waters examined and presumably in Hanford waters as well. Iodine speciation appears to be controlled by complex microbial and organic interactions. There is likely kinetic control not captured by thermodynamic analyses. Yet, without thermodynamic constants for all species even this is difficult to say definitively.
Microorganisms can catalyze iodine oxidation, reduction, accumulation, volatilization, and incorporation into OM. Iodide oxidation, albeit at very slow rates, can be supported by a variety of terrestrial bacteria. Microbial activity is an important factor in iodine biogeochemical cycling, particularly in enhancing iodine binding to high molecular weight OM, which is expected to sorb strongly to sediments. Indirect iodide oxidation mechanisms facilitated by bacteria, such as the production of organic acids and reactive oxygen species, likely influence iodine speciation and transport in the subsurface.
129I has not received the same attention in basic and applied research that other contaminants associated with DOE plumes have received. Other contaminants, such as uranium, plutonium, 90Sr, and 99Tc are more common. Yet, at the Hanford Site and the SRS 129I occurs in groundwater at concentrations significantly above the primary drinking water standard and there is no accepted method for treating it, other than pump-and-treat systems. Furthermore, 129I is likely to continue to be a major risk driver for nuclear disposal facilities and decontamination and decommissioning sites. Given these concerns and the inevitable increase in 129I inventory resulting from nuclear electrical power generation in the United States and world-wide, more rigorous research into 129I behavior in the environment and its remediation is warranted. Gaps in the knowledge of 129I behavior and remediation are presented throughout this review of the state of 129I environmental science. Listed below are lines of scientific inquiry that would significantly advance the goals of basic and applied research programs for accelerating 129I environmental remediation and reducing uncertainty associated with disposal of 129I waste.
Evaluation of amendments or other treatment systems that can be applied in the subsurface to sequester 129I from groundwater. They must be efficient, cost effective, and acceptable to regulators. Because of the long half-life of 129I, 16 million years, a major requirement for the amendment is that it must be effective for a prolong period of time.
Field sampling methods that eliminate the necessity of collecting and shipping large samples of groundwater for 129I analysis. Currently large samples of groundwater must be collected and processed in a laboratory to obtain enough 129I to achieve detection limits near the drinking water standard, 1 pCi/L, by the most widely available radiochemical techniques, gamma spectrometry and liquid-scintillation counting. This represents a significant expense in monitoring for 129I. Flow-through collectors of 129I could be developed that would be easy to use in the field and eliminate much of the laboratory sample preparation. Such devices configured to hang in wells would be useful for long-term monitoring for 129I.
Develop and evaluate ways to manipulate areas with organic-rich soil to maximize 129I sorption or to use natural OM as a remediation amendment. The maximum sorption of 129I is achieved in soils with elevated concentrations of OM, such as wetlands. In general, sorption is greater in these soils if they are oxic rather than anoxic. Thus, measures that minimize fluctuations between redox states of wetland soils or otherwise promote stronger sorption to the soils may minimize periodic releases of sorbed 129I.
Develop analytical techniques that can identify the various 129I species in the subsurface aqueous and solid phases at ambient concentrations and under ambient conditions. 129I speciation in environmental samples has been shown to be concentration dependent (Schwehr et al. Citation2009) and the concentrations of interest are typically very low (5 μg/L background 127I and 1 μg/L 129I [0.006 μg/L 129I]) and close to the detection limits of instruments that are readily available to most scientists, such as ion chromatography, gamma spectrometers, and liquid scintillation counters. Development of analytical techniques for aqueous and solid phase speciation will lead to a better understanding of iodine biogeochemical behavior in the terrestrial environment. Presently, we have a reasonable understanding of where iodine is and how fast it moves from one place to another. Yet, its transitions between iodide, iodate, elemental iodine, and organic forms are poorly understood, in part, because analytical techniques that differentiate these species are difficult and not widely available. Perhaps most importantly, this information is often important to predicting 129I behavior in changing geochemical conditions.
Understand the nature of 129I-organic interactions. Numerous studies have demonstrated the important role that natural OM plays in concentrating 129I, yet the mechanisms and controlling factors by which this is accomplished are poorly understood. It is not clear what role microbes, redox processes, or the composition of the OM play. The 129I-organic interaction is the only process that significantly attenuates 129I in most systems. Further complicating matters, small organic moieties have been shown to complex contaminants and enhance, not attenuate, 129I mobility. A thorough understanding of the processes controlling 129I attenuation by natural OM may lead to a means of using it as an amendment and extending it to areas other than wetlands.
Understand the biological processes that transform iodine species throughout different compartments of subsurface waste sites and the role that these processes have on 129I flux. In particular, the extent that microorganisms influence 129I volatilization and/or incorporation into OM is poorly understood in terrestrial environments. These processes likely play critical roles in 129I fluxes from subsurface aquifers to the surface environment, where bioconcentration mechanisms could impact human health.
Additional information
Funding
REFERENCES
- Amachi, S. (2008). Microbial contribution to global iodine cycling: Volatilization, accumulation, reduction, oxidation, and sorption of iodine. Microbes and Environments 23, 269–276.
- Amachi, S., Kamagata, Y., Kanagawa, T., and Muramatsu, Y. (2001). Bacteria mediate methylation of iodine in marine and terrestrial environments. Applied and Environmental Microbiology 67, 2718–2722.
- Amachi, S., Kasahara, M., Fujii, T., Shinoyama, H., Hanada, S., Kamagata, Y., Ban-nai, T., and Muramatsu, Y. (2004). Radiotracer experiments on biological volatilization of organic iodine from coastal seawaters. Geomicrobiology Journal. 21, 481–488.
- Amachi, S., Kasahara, M., Hanada, S., Kamagata, Y., Shinoyama, H., Fujii, T., and Muramatsu, Y. (2003). Microbial participation in iodine volatilization from soils. Environmental Science & Technology 37, 3885–3890.
- Amachi, S., Kimura, K., Muramatsu, Y., Shinoyama, H., and Fujii, T. (2007). Hydrogen peroxide-dependent uptake of iodine by marine Flavobactefiaceae bacterium strain C-21. Applied and Environmental Microbiology 73, 7536–7541.
- Amachi, S., Mishima, Y., Shinoyama, H., Muramatsu, Y., and Fujii, T. (2005a). Active transport and accumulation of iodide by newly isolated marine bacteria. Applied and Environmental Microbiology 71, 741–745.
- Amachi, S., Muramatsu, Y., Akiyama, Y., Miyazaki, K., Yoshiki, S., Hanada, S., Kamagata, Y., Ban-nai, T., Shinoyama, H., and Fujii, T. (2005b). Isolation of iodide-oxidizing bacteria from iodide-rich natural gas brines and seawaters. Microbial Ecology 49, 547–557.
- Arakawa, Y., Akiyama, Y., Furukawa, H., Suda, W., and Amachi, S. (2012). Growth stimulation of iodide-oxidizing a-Proteobacteria in iodide-rich environments. Microbial Ecology 63, 522–531.
- Ashworth, D.J., and Shaw, G. (2006). Effects of moisture content and redox potential on in situ Kd values for radioiodine in soil. Science of the Total Environment 359, 244–254.
- Ashworth, D.J., Shaw, G., Butler, A.P., and Ciciani, L. (2003). Soil transport and plant uptake of radio-iodine from near-surface groundwater. Journal of Environmental Radioactivity 70, 99–114.
- Assemi, S., and Erten, H.N. (1994). Sorption of radioiodine on organic rich soil, clay minerals, and alumina. Journal of Radioanalytical and Nuclear Chemistry 178, 193–204.
- Baker, J.M., Sturges, W.T., Sugier, J., Sunnenberg, G., Lovett, A.A., Nightingale, P.D., and Penkett, S.A. (2001). Emissions of CH3Br, organochlorines, and organoiodines from temperate macroalgae. Chemosphere Global Change Science 3, 93–106.
- Behrens, H. (1982). New insights into the chemical behavior of radioiodine in aquatic environments In Environmental Migration of Long-Lived Radionuclides (H. Seidler, ed.), International Atomic Energy Agency, Vienna, Austria.
- Bird, G.A., and Schwartz, W. (1997). Distribution coefficients, Kds, for iodide in Canadian Shield lake sediments under oxic and anoxic conditions. Journal of Environmental Radioactivity 35, 261–279.
- Bjornstad, B.N., Homer, J.A., vermeal, V.R., Lanigan, D.C., and Thorne, P.D. (2009). Borehold completion and conceptual hydrogeologic model for the IFRC Well Field, 300 Area, Hanford Site. Rep. No. PNNL-18340, Pacific Northwest National Laboratory, Richland, WA.
- Bors, J., Erten, H., and Martens, R. (1991). Sorption studies of radioiodine on soils with special references to soil microbial biomass. Radiochimica Acta 52/53, 317–325.
- Bors, J., Gorny, A., and Dultz, D. (1994). Some factors affecting the interactions of organophilic clay minerals with radioiodine. Radiochimica Acta 66/67, 309–313.
- Bors, J., Martens, R., and Kuhn, W. (1988). Studies on the role of natural and anthropogenic organic substances in the mobility of radioiodine in soils. Radiochimica Acta 44/45, 201–206.
- Brauer, F., and Strebin, R.J. (1982). Environmental migration of long-lived radionuclides. Rep. No. IAEA-SM-257, International Atomic Energy Agency, Vienna, Austria.
- Cantrell, K.J., Serne, R.J., and Last, G.V. (2003). Hanford contaminant distribution coefficient database and users’ guide. Rep. No. PNNL-13895, Pacific Northwest National Laboratory, Richland, WA.
- CDC (Centers for Disease Control and Prevention). (2002). Summary of the Hanford Thyroid Disease Study, Final Report. Centers for Disease Control and Prevention (CDC), Atlanta, GA.
- Chance, R., Malin, G., Jickells, T., and Baker, A.R. (2007). Reduction of iodate to iodide by cold water diatom cultures Marine Chemistry 105, 169–180.
- Councell, T.B., Landa, E.R., and Lovley, D.R. (1997). Microbial reduction of iodate. Water Air and Soil Pollution 100, 99–106.
- Couture, R.A., and Seitz, M.G. (1983). Sorption of anions of iodine by iron oxides and kaolinite. Nuclear Chemical Waste Management 4, 301–306.
- Dai, J.L., Zhang, M., and Zhu, Y.G. (2004). Adsorption and desorption of iodine by various Chinese soils - I. Iodate. Environment International 30, 525–530.
- Davis, S., Kopecky, K.J., and Hamilton, T.E. (2007). Hanford thyroid disease study final report. Rep. No. CDC 200–89–0716, Fred Hutchinson Cancer Research Center, Seattle, WA.
- Denham, M.E., Kaplan, D.I., and Yeager, C.M. (2009). Groundwater radioiodine: prevalence, biogeochemistry, and potential remedial approaches. Savannah River National Laboratory, Aiken, SC.
- Denham, M.E., and Vangelas, K.M. (2008). Biogeochemical gradients as a framework for understanding waste-site evolution. Remediation 19, 5–17.
- Department of Energy. (2002). Yucca Mountain site suitability evaluation. Rep. No. DOE/RW-0549, U.S. Department of Energy–Office of Civilian Radioactive Waste Management.
- Department of Energy. (2003). Preliminary Performance Assessment for Waste Management Area C at the Hanford Site, Rep. No. DOE/ORP-2003–11, Department of Energy, Richland Operations Office, Richland, WA.
- Department of Energy. (2012a). Proposed plan for remediation of the 200-UP-1 groundwater operable unit, Rep. No. DOE/RL-2010–05, Rev. 0, Deaprtment of Energy, Richland Operations Office, Richland, WA.
- Department of Energy. (2012b). Remedial investigation/feasibility study for the 200-UP-1 groundwater operable unit. Rep. No. DOE/RL-2009–122, U.S. Department of Energy, Richland Operations Office, Richland, WA.
- Farrenkopf, A.M., Dollhopf, M.E., Chadhain, S.N., Luther, G.W., and Nealson, K.H. (1997). Reduction of iodate in seawater during Arabian Sea shipboard incubations and in laboratory cultures of the marine bacterium Shewanella putrefaciens strain MR-4. Marine Chemistry 57, 347–354.
- Fuge, R., and Johnson, C.C. (1986). The geochemistry of iodine–a review. Environmental Geochemistry and Health 8, 31–54.
- Fuge, R., Selinus, O., Alloway, B., Centeno, J., Finkelman, R., Lindh, U., and Smedley, P. (2005). Soils and iodine deficiency. In Essentials of Medical Geology (O. Selinus, ed.), pp. 417–433. Elsevier Academic Press, Amsterdam.
- Fuhrmann, M., Bajt, S., and Schoonen, M.A. (1998). A sorption of iodine on minerals investigated by X-ray absorption near edge structure (XANES) and 125I tracer sorption experiments. Applied Geochemistry 13, 127–141.
- Fukui, M., Fujikawa, Y., and Satta, N. (1996). Factors affecting interaction of radioiodide and iodate species with soil. Journal of Environmental Radioactivity 31, 199–216.
- Fuse, H., Inoue, H., Murakami, K., Takimura, O., and Yamaoka, Y. (2003). Production of free and organic iodine by Roseovarius spp. Fems Microbiology Letters 229, 189–194.
- Gee, G.W., and Campbell, A.C. (1980). Monitoring and physical characterization of unsaturated zone transport–Laboratory analysis. Rep. No. PNL-3304, Pacific Northwest Laboratory, Richland, WA.
- Gilfedder, B.S., Petri, M., and Biester, H. (2008). Iodine speciation and cycling in limnic systems: Observations from a humic rich headwater lake (Mummelsee). Biogeosciences Discussions 5, 25–64.
- Gilfedder, B.S., Petri, M., and Biester, H. (2009). Iodine speciation and cycling in fresh waters: A case study from a humic rich headwater lake (Mummelsee). Journal of Limnology 68, 396–408.
- Gozlan, R.S. (1968). Isolation of iodine-producing bacteria from aquaria. Antonie van Leeuwenhoek 34, 226.
- Gozlan, R.S., and Margalith, P. (1973). Iodide oxidation by a marine bacterium. Journal of Applied Microbiology 36, 407–417.
- Gozlan, R.S., and Margalith, P. (1974). Iodide oxidation by Pseudomonas iodooxidans. Journal of Applied Microbiology 37, 493–499.
- Guiraud-Vitaux, F., Elbast, M., and Colas-Linhart, N. (2008). Thyroid cancer after Chernobyl: Is iodine-131 the only culprit? Impact on clinical practice. Bulletin du Cancer 95, 191–195.
- Hall, F. (2005). Concentrations of selected trace metals, common ions, nutrients and radiological analytes in ground water for selected sites, Eastern Snake River Plain Aquifer, South of Idaho National Laboratory. Idaho, Rep. No. OP-06.03, Idaho National Laboratory, Idaho Falls, ID.
- Hou, X.L., Fogh, C.L., Kucera, J., Andersson, K.G., Dahlgaard, H., and Nielsen, S.P. (2003). Iodine-129 and Caesium-137 in Chernobyl contaminated soil and their chemical fractionation. The Science of the Total Environment 308, 97–109.
- Hou, X.L., Hansen, V., Aldahan, A., Possnert, G., Lind, O.C., and Lujaniene, G. (2009b). A review on speciation of iodine-129 in the environmental and biological samples. Analytica Chimica Acta 632, 181–196.
- Hou, X.L., Hansen, V., Aldahan, A., Possnert, G., Lind, O.C., and Lujaniene, G. (2009a). A review on speciation of iodine-129 in the environmental and biological samples. Analytica Chimica Acta 632, 181–196.
- Hu, Q.H., Moran, J.E., and Blackwood, V. (2009). Geochemical cycling of iodine species in soils. In Comprehensive Handbook of Iodine (V.R. Preedy, G.N. Burrow and R. Watson, eds.), pp. 93–107. Oxford Academic Press, Oxford.
- Hu, Q.H., Zhao, P.H., Moran, J.E., and Seaman, J.C. (2005). Sorption and transport of iodine species in sediments from the Savannah River and Hanford Sites. Journal of Contaminant Hydrology 78, 185–205.
- International Atomic Energy Agency. (2006). Chernobyl legacy: Health, environmental and socio-economic impacts, and recommendations to the governments of Belarus, the Russian Federation and Ukraine. The Chernobyl Forum: 2003–2005. (Second revised version). International Atomic Energy Agency, Vienna, Austria.
- Kantelo, M.V., Bauer, L.R., Marter, W.L., Murphy, C.E. Jr., and Zeigler, C.C. (1990). Radioiodine in the Savannah River site environment. Rep. No. WSRC-RP-90–424–2, Westinghouse Savannah River Company, Aiken, SC.
- Kaplan, D.I. (2003). Influence of surface charge of an Fe-oxide and an organic matter dominated soil on iodide and pertechnetate sorption. Radiochimica Acta 91, 173–178.
- Kaplan, D.I. (2011). Estimated neptunium sediment sorption values as a function of pH and measured barium and radium kd values. Rep. No. SRNL-STI-2011–00011, Savannah River National Laboratory, Aiken, SC.
- Kaplan, D.I., Mattigod, S.V., Parker, K.E., and Iverson, G. (2000a). Experimental work in support of the 129I-disposal special analysis, Westinghouse Savannah River Company, Aiken, SC.
- Kaplan, D.I., Mattigod, S.V., Parker, K., and Iverson, G. (2000b). I-129 test and research to support disposal decisions. Rep. No. WSRC-TR-2000-00283, Savannah River National Laboratory, Aiken, SC.
- Kaplan, D.I., Parker, K.E., and Orr, R.D. (1998). Effects of high-pH and high-ionic-strength groundwater on iodide, pertechnetate, and selenate sorption to Hanford sediments: Final report for subtask 3a, Rep. No. PNNL-11964, Pacific Northwest National Laboratory, Richland, WA.
- Kaplan, D.I., Roberts, K.A., Schwehr, K.A., Lilley, M.S., Brinkmeyer, R., Denham, M.E., Diprete, D., Li, H.P., Powell, B.A., Xu, C., Yeager, C.M., Zhang, S.J., and Santschi, P.H. (2011). Evaluation of a radioiodine plume increasing in concentration at the Savannah River Site. Environmental Science & Technology 45, 489–495.
- Kaplan, D.I., Serne, R.J., Owen, A.T., Conca, J.A., Wietsma, T.W., and Gervais, T.L. (1996). Radionuclide adsorption distribution coefficients measured in hanford sediments for the low-level waste performance assessment project. Pacific Northwest National Laboratory, Richland, WA.
- Kaplan, D.I., Yeager, C., Denham, M.E., Zhang, S., Xu, C., Schwehr, K.A., Li, H.-P., Ho, Y.-F., Brinkmeyer, R., and Santschi, P.H. (2012). Biogeochemical considerations related to the remediation of 129I plumes. Rep. No. SRNL-STI-2012–00425, Savannah River National Laboratory, Aiken, SC.
- Kincaid, C.T., Eslinger, P.W., Aaberg, R.L., Miley, T.B., Nelson, I.C., Strenge, D.L., and Evans, J.C. (2006). Inventory data package for Hanford Assessments. Pacific Northwest National Laboratory, Richland, WA.
- Kodama, S., Takahashi, Y, Okumura, K. and Uruga, T. (2006). Speciation of iodine in solid environmental samples by iodine K-edge XANES: Application to soils and ferromanganese oxides. Science of the Total Environment 363, 275–284.
- Kolb, C.E. (2002). Atmospheric chemistry: Iodine's air of importance. Nature 417, 597–598.
- Küpper, F.C., Carpenter, L.J., McFiggans, G.B., Palmer, C.J., Waite, T.J., Boneberg, E.-M., Woitsch, S., Weiller, M., Abela, R., Grolimund, D., Potin, P., Butler, A., Luther, G.W., Kroneck, P.M. H., Meyer-Klaucke, W., and Feiters, M.C. (2008). Iodide accumulation provides kelp with an inorganic antioxidant impacting atmospheric chemistry. Proceedings of the National Academy of Sciences 105, 6954–6958.
- Küpper, F.C., Schweigert, N., Gall, E.A., Legendre, J.M., Vilter, H., and Kloareg, B. (1998). Iodine uptake in Laminariales involves extracellular, haloperoxidase-mediated oxidation of iodide. Planta 207, 163–171.
- Lauterbach, A., and Ober, G. (1996). Iodine and iodine compounds. In Encyclopedia of Chemical Technology, Vol. 14 (Kirk-Othmer, ed.), pp. 709–737. Wiley, Boston.
- Lee-Taylor, J., and Redeker, K.R. (2005). Reevaluation of global emissions from rice paddies of methyl iodide and other species. Geophysical Research Letters 15, 1–5.
- Li, H.-P., Brinkmeyer, R., Jones, W.L., Zhang, S., Xu, C., Ho, Y.-F., Schwehr, K.A., Kaplan, D.I., Santschi, P.H., and Yeager, C.M. (2012a). Iodide oxidizing activity of bacteria from subsurface sediments of the Savannah River Site. In Interdisciplinary Studies on Environmental Chemistry Vol. 6 - Environmental Pollution and Ecotoxicology (K.M. M. Kawaguchi, H. Sato, T. Yokokawa, T. Itai, T.M. Nguyen, J. Ono, S. Tanabe, ed.), Vol. 6, pp. 89–97, Terra Scientific Publishing Company, Tokyo.
- Li, H.-P., Brinkmeyer, R., Jones, W.L., Zhang, S., Xu, C., Schwehr, K.A., Santschi, P.H., Kaplan, D.I., and Yeager, C.M. (2011). Iodide accumulation by aerobic bacteria isolated from subsurface sediments of a I-129-contaminated aquifer at the Savannah River Site, South Carolina. Applied and Environmental Microbiology 77, 2153–2160.
- Li, H.-P., Yeager, C.M., Brinkmeyer, R., Zhang, S., Ho, Y.-F., Xu, C., Jones, W.L., Schwehr, K.A., Otosaka, S., Roberts, K.A., Kaplan, D.I., and Santschi, P.H. (2012b). Organic acids produced by subsurface bacteria enhance iodide oxidation in the presence of hydrogen peroxide. Environmental Science & Technology 46, 4837–4844.
- Lin, X., Kennedy, D., Fredrickson, J., Bjornstad, B., and Konopka, A. (2012a). Vertical stratification of subsurface microbial community composition across geological formations at the Hanford Site. Environmental Microbiology 14, 414–425.
- Lin, X., Kennedy, D., Peacock, A., McKinley, J., Resch, C.T., and Fredrickson, J. (2012b). Distribution of microbial biomass and potential for anaerobic respiration in Hanford Site 300 Area subsurface sediment. Applied and Environmental Microbiology 78, 759–767.
- Lin, X., McKinley, J., Resch, C.T., Kaluzny, R., Lauber, C.L., Fredrickson, J., Knight, R., and Konopka, A. (2012c). Spatial and temporal dynamics of the microbial community in the Hanford unconfined aquifer. ISME J 6, 1665–1676.
- MacKenzie, D. (2011). Fukushima radioactive fallout nears Chernobyl levels. New Scientist 17, 14.
- Maillant, S., Sheppard, M.I., Echevarria, G., Denys, S., Villemin, G., Tekely, P., Leclerc-Cessac, E., and Morel, J.L. (2007). Aged anthropogenic iodine in a boreal peat bog. Applied Geochemistry 22, 873–887.
- Malinauskas, A.P., and Bell, J.T. (1987). The chemistry of fission-product iodine under nuclear reactor accident conditions. Nuclear Safety 28, 505–514.
- McKay, A. (1984). The making of the atomic age. Oxford University Press, New York.
- Mishra, S.P., and Srinivasu, N. (1992). Radiotracer technique in adsorption study–IX. Adsorption of iodide ions on titanium(IV) oxide powder. Applied Radiation and Isotopes 43, 789–793.
- Moore, R.M., and Groszko, W. (1988). Methyl iodide distribution in the ocean and fluxes to the atmosphere. Journal of Geophysical Research 11, 163–171.
- Muramatsu, Y., Uchida, S. and Ohmomo, Y. (1990a). Determination of I-129 and I-127 in soil and tracer experiments on the adsorption of iodine on soil. Journal of Radioanalytical and Nuclear Chemistry 138, 377–384.
- Muramatsu, Y., Uchida, S., Sriyotha, P., and Sriyotha, K. (1990b). Some consideration on the sorption and desorption phenomena of iodide and iodate on soil. Water Air and Soil Pollution 49, 125–138.
- Muramatsu, Y., Uchida, S., Sumiya, M., Ohmomo, Y., and Obata, H. (1989). Tracer experiments on transfer of radio-iodine in the soil—rice plant system. Water, Air, & Soil Pollution 45, 157–171.
- Muramatsu, Y., and Yoshida, S. (1995). Volatilization of methyl iodide from the soil-plant system. Atmospheric Environment 29, 21–25.
- Muramatsu, Y., Yoshida, S., Fehn, U., Amachi, S., and Ohmomo, Y. (2004). Studies with natural and anthropogenic iodine isotopes: iodine distribution and cycling in the global environment. Journal of Environmental Radioactivity 74, 221–232.
- Musić, S., Šipalo-Žuljerić, J., and Wolf, R.H. H. (1979). Radiochemical study of the sorption of iodide and iodate on aluminum(III) hydroxide precipitate. Isotopenpraxis 15, 93–96.
- NCRP (National Council Radiation Protection). (1983). Iodine-129: Evaluation of releases from nuclear power generation, NCRP Report 75, National Council on Radiation Protection and Measurements, Bethesda, MD.
- National Research Council. (1995). A review of two Hanford Environmental Dose Reconstruction Project (HEDR) dosimetry reports: Columbia River pathway and atmospheric pathway. National Academy Press, Washington, DC.
- Neal, C., and Truesdale, V.W. (1976). Sorption of iodate and iodide by riverine sediments - Its implications to dilution gauging and hydrochemistry of iodine. Journal of Hydrology 31, 281–291.
- Nirel, P.M. V., and Morel, F.M. M. (1990). Pitfalls of sequential extractions. Water Research 24, 1055–1056.
- Nishimaki, K., Satta, N., and Maeda, M. (1994). Sorption and migration of radioiodine in saturated sandy soil. Journal of Nuclear Science and Technology 31, 828–838.
- Otosaka, S., Schwehr, K.A., Kaplan, D.I., Roberts, D.A., Zhang, S., Xu, C., Li, H.-P., Ho, Y.-F., Brinkmeyer, R., Yeager, C.M., and Santschi, P.H. (2011). Transformation and transport processes of 127I and 129I species in an acidic groundwater plume at the Savannah River Site. The Science of the Total Environment 409, 3857–3865.
- Paquette, J., Wren, D.J., Ford, B.L., and Toth, L.M. (1986). Iodine chemistry. In The Three Mile Island Accident–Diagnosis and Prognosis, ACS Symposium Series, Vol. 293. American Chemical Society, Washington, DC.
- Parker, L., and Holt, M. (2007). CRS report for congress. Nuclear power: Outlook for new U.S. Reactors, Congress Research Service, Washington, DC.
- Parsly, L.F. (1970). Design considerations of reactor containment spray systems–Part IV. Calculation of Iodine-water partition coefficients, Oak Ridge National Laboratory, Oak Ridge, TN.
- Raisbeck, G.M., and Yiou, F. (1999). 129I in the oceans: Origins and applications. The Science of the Total Environment 237/238, 31–41.
- Rao, U., and Fehn, U. (1999). Sources and reservoirs of anthropogenic iodine-129 in western New York. Geochimica et Cosmochimica Acta 63, 1927–1938.
- Richter, U., and Wallace, D.W. R. (2004). Production of methyl iodide in the tropical Atlantic Ocean. Geophysical Research Letters 31, 1–4.
- Saini, H., Attieh, J., and Hanson, A. (1995). Biosynthesis of halomethanes and methanethiol by higher plants via a novel methyl-transferase system. Plant Cell and Environment 18, 1027–1033.
- Santschi, P.H., and Schwehr, K.A. (2004). I-129/I-127 as a new environmental tracer or geochronometer for biogeochemical or hydrodynamic processes in the hydrosphere and geosphere: The central role of organo-iodine. Science of the Total Environment, 257–271.
- Santschi, P.H., Xu, C., Zhang, S., Ho, Y.F., Li, H.P., Schwehr, K.A., and Kaplan, D.I. (2012). Laboratory results on iodine (129I and 127I) speciation, transformation and mobility in Hanford groundwater, suspended particles and sediments. Rep. No. SRNL-STI-2012–00592. Savannah River National Laboratory, Aiken, SC.
- Savoye, S., Michelot, J.L., and Wittebroodt, C. (2006). Evaluation of the reversibility of iodide uptake by argillaceous rocks by the radial diffusion method. Radiochimica Acta 94, 699–704.
- Saxena, D., Aouad, J., Attieh, J., and Saini, H. (1998). Biochemical characterization of chloromethane emission from the wood-rotting fungus Phellinus pomaceus. Applied and Environmental Microbiology 64, 2831–2835.
- Sazarashi, M., Ikeda, Y., Seki, R., and Yoshikawa, H. (1994). Adsorption of I– ions on minerals for 129I waste management. Journal of Nuclear Science and Technology 31, 620–622.
- Scheckel, K.G., Impellitteri, C.A., Ryan, J.A., and McEvoy, T. (2003). Assessment of a sequential extraction procedure for perturbed lead-contaminated samples with and without phosphorus amendments Eniron. Sci. Technol. 37, 1892–1898.
- Schlegel, M.L., Reiller, P., Mercier-Bion, F., Barre, N., and Moulin, V. (2006). Molecuylar environment of iodine in naturally iodinated humic substances: Insight from X-ray absorption spectroscopy. Geochimica 70, 5536–5551.
- Schmitz, K., and Aumann, D.C. (1995). A study on the association of two iodine isotopes of natural I-127 and of the fission product I-129, with soil components using a sequential extraction procedure. Journal of Radioanalytical Nuclear Chemistry 198, 229–236.
- Schwehr, K.A., and Santschi, P.H. (2003). Sensitive determination of iodine species, including organo-iodine, for freshwater and seawater samples using high performance liquid chromatography and spectrophotometric detection. Analytica Chimica Acta 482, 59–71.
- Schwehr, K.A., Santschi, P.H., and Elmore, D. (2005a). The dissolved organic iodine species of the isotopic ratio of (129)I/(127)I: A novel tool for tracing terrestrial organic carbon in the estuarine surface waters of Galveston Bay, Texas. Limnology and Oceanography-Methods 3, 326–337.
- Schwehr, K.A., Santschi, P.H., Kaplan, D.I., Yeager, C.M., and Brinkmeyer, R. (2009). Organo-iodine formation in soils and aquifer sediments at ambient concentrations. Environmental Science & Technology 43, 7258–7264.
- Schwehr, K.A., Santschi, P.H., Moran, J.E., and Elmore, D. (2005b). Near-conservative behavior of 129I in the Orange County aquifer system, California. Applied Geochemistry 20, 1461–1472.
- Seki, M., Oikawa, J., Taichi, T., Ohnuki, T., Muramatsu, Y., Sakamoto, K., and Amachi, S. (2013). Laccase-catalyzed oxidation of iodide and formation of organically bound iodine in soils. Environmental Science & Technology 47, 390–397.
- Serne, R.J., Conca, J.L., LeGore, V.L., Cantrell, K.J., Lindenmeier, C.W., Campbell, J.A., Amonette, J.E., and Wood, M.I. (1993). Solid-waste leach characteristics and contaminant-sediment interactions. Volume 1: Batch leach and adsorption tests and sediment characterization. Rep. No. PNL-8889, Pacific Northwest National Laboratory, Richland, WA.
- Sheppard, M.I., and Hawkins, J.L. (1995). Iodine and microbial interactions in an organic soil. Radioactivity 29, 91–109.
- Sheppard, M.I., and Thibault, D.H. (1989). Iodine dispersion and effects on groundwater chemistry following a release to a peat bog, Manitoba, Canada. Applied Geochemistry 4, 423–432.
- Sheppard, M.I., and Thibault, D.H. (1991). A 4-Year mobility study of selected trace-elements and heavy-metals. Journal of Environmental Quality 20, 101–114.
- Sheppard, M.I., Hawkins, J.L., and Smith, P.A. (1996). Linearity of iodine sorption and sorption capacities for seven soils. Journal of Environmental Quality 25, 1261–1267.
- Shimamoto, Y.S., and Takahashi, Y. (2008). Superiority of K-edge XANES over L-III-edge XANES in the speciation of iodine in natural soils. Analytical Sciences 24, 405–409.
- Skulski, L. (2000). Organic iodine (I, III, and V) chemistry: 10 years of development at the Medical University of Warsaw, Poland. Molecules 5, 1331–1371.
- Smythe-Wright, D., Boswell, S.M., Breithaupt, P., Davidson, R.D., Dimmer, C.H., and Diaz, L.B. (2006). Methyl iodide production in the ocean: Implications for climate change. Global Biogeochemical Cycles Vol. 20, pp. . BG 3003.
- Stang, P.J. (2003). Polyvalent iodine organic chemistry. Journal of Organic Chemistry 68, 2997–3008.
- Stegen, J.C., Lin, X., Konopka, A., McKinley, J., and Fredrickson, J. (2012). Stochastic and deterministic assembly processes in subsurface microbial communities. ISME Journal 6, 1653–1664.
- Stephenson, M., and Motycka, M (1995). Volatility of 125I in fresh water. Journal of Environmental Radioactivity 28, 295–311.
- Stutz, J., Hebestreit, K., Alicke, B., and Platt, U. (1999). Chemistry of halogen oxides in the troposphere: Comparison of model calculations with recent field data. Journal of Atmospheric Chemistry 34, 65–85.
- Taghipour, F., and Evans, G.J. (2002). Modeling of iodine radiation chemistry in the presence of organic compounds. Radiation Physics and Chemistry 64, 203–213.
- Tessier, A., and Campbell, P.G. C. (1988). Partitioning of trace metals in sediments. In Metal Speciation: Theory, Analysis and Application (J.R. Kramer and H.E. Allen, eds.), pp. 183–217. LewiV, Chelsea, MI.
- Ticknor, K.V., and Cho, Y.H. (1990). Interaction of iodide and iodate with granitic fracture-filling minerals. Journal of Radioanalytical and Nuclear Chemistry 140, 75–90.
- Tsunogai, S., and Sase, T. (1969). Formation of iodide-iodine in the ocean. Deep-Sea Research 16, 489–496.
- Um, W., Serne, R.J., and Krupka, K.M. (2004). Linearity and reversibility of iodide adsorption on sediments from Hanford, Washington under water saturated conditions. Water Research 38, 2009–2016.
- Von Hippel, F.N. (2011). The radiological and psychological consequences of the Fukushima Daiishi accidnet. Bulletin of the Atomic Scientists 67, 27–36.
- Waite, T.J., and Truesdale, V.W. (2003). Iodate reduction by Isochrysis galbana is relatively insensitive to de-activation of nitrate reductase activity: Are phytoplankton really responsible for iodate reduction in seawater. Marine Chemistry 81, 137–148.
- Warner, J.A., Casey, W.H., and Dahlgren, R.A. (2000). Interaction kinetics of I−2(aq) with substituted phenols and humic substances. Environmental Science & Technology 34, 3180–3185.
- Washington Savannah River Company. (2008). E-area low-level waste facility DOE 435.1 performance assessment. Rep. No. WSRC-STI-2007–00306, REVISION 0, Washington Savannah River Company, Aiken, SC.
- West Valley Demonstration Project. (2007). West Valley Demonstration Project annual site environmental report: Calendar year 2007. Rep. No. DE-AC3007CC30000
- Westinghouse Savannah River Company. (2006). Annual corrective action report for the F-Area hazardous waste management facility, the H-area hazardous waste management facility, and the mixed waste management facility. Rep. No. WSRC-RP-2006–4011, Westinghouse Savannah River Company, . LLC, Aiken, SC.
- Whitehead, D.C. (1973). The sorption of iodide by soils as influenced by equilibrium conditions and soil properties. Journal of the Science of Food and Agriculture 24, 547–556.
- Whitehead, D.C. (1974). The sorption of iodide by soil components. Journal of the Science of Food and Agriculture 25, 73–79.
- Wong, G.T. F., and Cheng, X.H. (1998). Dissolved organic iodine in marine waters: Determination, occurrence and analytical implications. Marine Chemistry 59, 271–281.
- Wong, G.T. F., Piumsomboon, A., and Dunstan, W. (2002). The transformation of iodate to iodide in marine phytoplankton cultures. Marine Ecology Progress Series 237, 27–39.
- World Nuclear Association. (2009). Nuclear power in India. Retrieved from http://www.world-nuclear.org/info/Country-Profiles/Countries-G-N/India/
- Wren, J.C., and Ball, J.M. (2001). LIRIC 3.2: An updated model for iodine behavior in the presence of organic impurities. Radiation Physics and Chemistry 60, 577–596.
- Wuosmaa, A., and Hager, L. (1990). Methyl chloride transferase: A carbocation route for biosynthesis of halometabolites. Science of the Total Environment 249, 160–162.
- Xu, C., Chen, H., Sugiyama, Y., Zhang, S., Li, H.P., Ho, Y., Chuang, C.Y., Schwehr, K.A., Kaplan, D., Yeager, C.M., Roberts, K.A., Brinkmeyer, R., Hatcher, P.G., and Santschi, P.H. (2012a). Novel molecular-level evidence of iodine binding to natural organic matter from Fourier transform ion cyclotron resonance mass spectrometry. Environmental Science & Technology (submitted).
- Xu, C., Miller, E.J., Zhang, S., Li, H.P., Ho, Y.F., Schwehr, K.A., Kaplan, D.I., Otosaka, S., Roberts, K.A., Brinkmeyer, R., Yeager, C.M., and Santschi, P.H. (2011a). Sequestration and remobilization of radioiodine (I-129) by soil organic matter and possible consequences of the remedial action at Savannah River Site. Environmental Science & Technology 45, 9975–9983.
- Xu, C., Santschi, P.H., Hung, C.C., Zhang, S.J., Schwehr, K.A., Roberts, K.A., Guo, L.D., Gong, G.C., Quigg, A., Long, R.A., Pinckney, J.L., Duan, S.W., Amon, R., and Wei, C.L. (2011b). Controls of (234)Th removal from the oligotrophic ocean by polyuronic acids and modification by microbial activity. Marine Chemistry 123, 111–126.
- Xu, C., Zhang, S., Ho, Y.-F., Miller, E.J., Roberts, K.A., Li, H.-P., Schwehr, K.A., Otosaka, S., Kaplan, D.I., Brinkmeyer, R., Yeager, C.M., and Santschi, P.H. (2011c). Is soil natural organic matter a sink or source for mobile radioiodine (129I) at the Savannah River Site? Geochimica et Cosmochimica Acta 75, 5716–5735.
- Xu, C., Zhong, J.Y., Hatcher, P.G., Zhang, S., Li, H.P., Ho, Y., Schwehr, K.A., Kaplan, D.I., Roberts, K.A., Brinkmeyer, R., Yeager, C.M., and Santschi, P.H. (2012b). Molecular environment of stable iodine and radioiodine (129I) in natural organic matter: Evidence inferred from NMR and binding experiments at environmentally relevant concentrations. Geochimica et Cosmochimica Acta 97, 166–182.
- Yamaguchi, N., Nakano, M., Takamatsu, R., and Tanida, H. (2010). Inorganic iodine incorporation into soil organic matter: Evidence from iodine K-edge absorption near-edge structure. Journal of Environmental Radioactivity 101, 451–457.
- Yamaguchi, N., Nakano, M., Tanida, H., and Fujiwara, H. (2006). Redox reaction of iodine in paddy soil investigated by field observation and I K-edge XANES spectroscopy. Journal of Environmental Radioactivity 86, 212–226.
- Yoshida, S., Muramatsu, Y., and Uchida, S. (1992). Studies on the sorption of I– (iodide) and IO3– (iodate) onto Andosols. Water Air and Soil Pollution 63, 321–329.
- Yoshida, S., Muramatsu, Y., and Uchida, S. (1998). Soil-solution distribution coefficients, Kds, of I– and IO3– for 68 Japanese soils. Radiochimica Acta 82, 293–297.
- Yu, Z., Warner, J.A., Dahlgren, R.A., and Casey, W.H. (1996). Reactivity of iodine in volcanic soils and noncrystalline soil constituents. Geochim. Cosmochim. Acta 60, 4945–4956.
- Yuita, K. (1992). Dynamics of iodine, bromine, and chlorine in soil. II. Chemical forms of iodine in soil solutions Soil Science and Plant Nutrition 38, 281–287.
- Yuita, K., and Kihou, N. (2005). Behavior of iodine in a forest plot, an upland field, and a paddy field in the upland area of Tsukuba, Japan: Vertical distribution of iodine in soil horizons and layers to a depth of 50 m. Soil Science and Plant Nutrition 51, 455–467.
- Zaichick, V.Y., and Choporov, Y.Y. (1996). Determination of the natural level of human intra-thyroid iodine by instrumental neutron activation analysis. Journal of Radioanalytical Nuclear Chemistry 207, 153–161.
- Zhang, S., Du, J., Xu, C., Schwehr, K.A., Ho, Y.F., Li, H.P., Roberts, K.A., Kaplan, D.I., Brinkmeyer, R., Yeager, C.M., Chang, H.S., and Santschi, P.H. (2011). Concentration-dependent mobility, retardation, and speciation of iodine in surface sediment from the Savannah River Site. Environmental Science & Technology 45, 5543–5549.
- Zhang, S., Schwehr, K.A., Ho, Y.-F., Xu, C., Roberts, K., Kaplan, D.I., Brinkmeyer, R., Yeager, C.M., and Santschi, P.H. (2010). Determination of 127I and 129I speciation in environmental waters using a novel gas chromatography-mass spectrometry method. Environmental Science & Technology 44, 9042–9048.
- Zhdankin, V.V., and Stang, P.J. (2002). Recent developments in the chemistry of polyvalent iodine compounds. Chemical Reviews 102, 2523–2584.