Abstract
A comprehensive review of quantitative structure-activity relationships (QSAR) allowing the prediction of the fate of organic compounds in the environment from their molecular properties was done. The considered processes were water dissolution, dissociation, volatilization, retention on soils and sediments (mainly adsorption and desorption), degradation (biotic and abiotic), and absorption by plants. A total of 790 equations involving 686 structural molecular descriptors are reported to estimate 90 environmental parameters related to these processes. A significant number of equations was found for dissociation process (pKa), water dissolution or hydrophobic behavior (especially through the KOW parameter), adsorption to soils and biodegradation. A lack of QSAR was observed to estimate desorption or potential of transfer to water. Among the 686 molecular descriptors, five were found to be dominant in the 790 collected equations and the most generic ones: four quantum-chemical descriptors, the energy of the highest occupied molecular orbital (EHOMO) and the energy of the lowest unoccupied molecular orbital (ELUMO), polarizability (α) and dipole moment (μ), and one constitutional descriptor, the molecular weight. Keeping in mind that the combination of descriptors belonging to different categories (constitutional, topological, quantum-chemical) led to improve QSAR performances, these descriptors should be considered for the development of new QSAR, for further predictions of environmental parameters. This review also allows finding of the relevant QSAR equations to predict the fate of a wide diversity of compounds in the environment.
1. INTRODUCTION
The high number and the wide diversity of manmade organic compounds (e.g., pesticides, pharmaceuticals, polycyclic aromatic hydrocarbons (PAH), polychlorinated biphenyls (PCB)) that have been or will be released in the environment constitute the most important challenge for research on the fate and effects of these contaminants. About 100,000 substances have been registered for use in United States or Europe over the past 30 years (Hansen et al., Citation1999b; Muir and Howard, Citation2006). However, they cannot be studied on a case-by-case basis, in particular because experimental studies are time-consuming and/or cost prohibitive (Reddy and Locke, Citation1994a; Russom et al., Citation2003; Sabljic, Citation1989; Türker Saçan and Balcioğlu, 1996). Therefore, the vast majority of existing and new chemical substances are not monitored in environmental media, and their fate and effects remain unknown, so that regulators face the task of reviewing the potential risk for chemicals having little or no empirical data (Muir and Howard, Citation2006; Russom et al., Citation2003).
Reliable environmental fate and risk assessment procedures strongly rely on the ability to accurately measure or estimate various environmental parameters and molecular properties of chemicals (Sabljic, Citation2001). Therefore, the development of in silico methods of prediction based on quantitative structure activity relationships (QSAR) or quantitative structure property relationships (QSPR) has received an increasing interest for many years (Cronin et al., Citation2003; Hermens et al., Citation1995; Mackay et al., Citation2001; Sabljic, Citation1991; Walker et al., Citation2002). The QSAR approach is based on the assumptions that the structure of a molecule contains the features responsible for its physical, chemical, and biological properties, and that variations in the fate within a series of similar structures can be correlated with changes in descriptors that reflect their molecular properties (Reddy and Locke, Citation1994a; Sabljic, Citation2001; Walker et al., Citation2003). The QSAR have the potential to estimate the risks of chemicals for environment and human health, for example, while reducing time, monetary cost, and animal testing currently needed for ecological risk assessment of chemicals (Organization for Economic Cooperation and Development, 2013; Reddy and Locke, Citation1994a).
The QSAR can be based on (a) physicochemical properties that can be determined experimentally (e.g., water solubility, octanol-water partition coefficient) or (2) structural molecular descriptors that include constitutional (number of atoms, atom types), geometric (e.g., surface, volume), topological (connectivity indices), and quantum-chemical (dipole moment, polarizability, energies) properties (Doucette, Citation2003; Sabljic, Citation2001; Tao et al., Citation1999; Todeschini et al., Citation1996). However, approaches based on experimental properties, such as water solubility or octanol-water distribution coefficient, are prone to experimental errors in the input variables, which may result in some severe statistical problems (Lohninger, Citation1994; Nguyen et al., Citation2005; Sabljic, Citation1991; Sabljic and Piver, Citation1992). Therefore, some of the advantages of the exclusive use of structural molecular descriptors are that they are free of the uncertainty of experimental measurements and that they can be calculated for organic compounds under development, not yet synthesized (Gramatica and Di Guardo, 2002; Karelson et al., Citation1996).
The objective of this work was thus to do the first comprehensive review of QSAR allowing the prediction of the fate of organic compounds in the environment from their structural molecular properties. The major processes that were considered are water dissolution, dissociation, volatilization, retention, degradation, and absorption by higher plants. The reviewed QSAR were analyzed according to two criteria: the frequency of the use of one descriptor in all reviewed equations, and the generic character of one descriptor (i.e., if it is involved in the assessment of a high diversity of processes). In addition, the physical meaning of the structural molecular descriptors was considered. This allowed the identification of the most relevant descriptors for the assessment of the environmental parameters related to the considered processes.
The different types and categories of molecular descriptors are first presented followed by the review of the QSAR equations for each selected process, and by the synthesis and discussion of the results.
2. STRUCTURAL MOLECULAR DESCRIPTORS USED FOR ENVIRONMENTAL CONCERNS
The central axiom of QSAR is that the activity of molecules is reflected in their structures (Organization for Economic Cooperation and Development, 2013). The structure of a molecule (e.g., its geometric or quantum properties) can be represented by several structural molecular descriptors, and the information contained in the descriptors reflects the nature of the molecular representation used. For example, a graph theoretical representation describes the molecule as a set of vertices (atoms) and edges (bonds). This allows the estimation of topological descriptors. A more sophisticated representation views a molecule as a collection of nuclei bound together by overlapping electron orbitals. Such representation can be used to derive descriptors such as atomic charge and dipole moment. Another type of structural representation characterizes the molecule as a set of hard spheres connected by bonds possessing specific stretching, bending, and torsion energies. The shape of the molecule is determined by the strain placed on the bonds as the spheres are allowed to interact. Information that is related to the geometry of the molecule (size and shape) can be obtained from such representation. A realistic representation of a molecule must lie in the combination of these representations (Stanton and Jurs, Citation1990). A lot of structural molecular descriptors that take into account different aspects of chemical information have been proposed and reviewed by Todeschini and Consonni (Citation2000). This section is focused on concise presentation of the 686 descriptors (Table S1) that were found in the 790 equations reported in this work allowing the assessment of 90 environmental parameters (Table S2). The descriptors were classified in seven categories: constitutional, geometric, geometric-topological, geometric-electronic, topological, electro-topological, and quantum-chemical. The review of the different QSAR will be then organized and discussed according to these seven categories. All structural molecular descriptors can be calculated with different softwares such as ChemOffice (Citation2009), comprehensive descriptors for structural and statistical analysis PRO (CODESSA PRO) (Katritzky et al., 2005), Dragon (2007), Gaussian 09 (Frisch et al., Citation2009), HyperChem (Citation2007) or parameter estimation for the treatment of reactivity applications (PETRA) (TORVS Research Team, 1999).
2.1 Constitutional Descriptors
Constitutional descriptors reflect the molecular composition of a compound without any information about its molecular geometry (Ma et al., Citation2010). One hundred and forty one descriptors were used in the reviewed equations. The simplest constitutional descriptors are the molecular weight or the number of atoms, bonds, functional groups, and rings (Table S1).
Several other constitutional descriptors were found such as indicators of the presence of different chemical groups (e.g., ester, epoxide, nitro group), H attached to heteroatom (H-050), the hydrophilic factor (HY), or the gravitation index (IG; Table S1). H-050 is one of the atom-centered fragment descriptors that describe each atom by its own atom type, and the bond types and atom types of its first neighbors. It represents the first neighbor (hydrogen) of heteroatom (Habibi-Yangjeh et al., Citation2009). The HY is based on atom and group counting (e.g., number of hydrophilic groups [‒OH, ‒NH, ‒SH], number of carbon atoms, and number of non-hydrogen atoms). It is related to the presence of hydroxyl groups in the molecule (Gramatica and Di Guardo, 2002; Gramatica et al., Citation1999b). The gravitation index IG reflects the effective mass distribution within the molecule and depicts the molecular dispersion forces in a bulk liquid media (Estrada et al., Citation2004; Katritzky et al., Citation1998).
In the fragment approaches, a molecular structure is divided into fragments (atom or larger functional groups), and values of each atom or group are summed together to give the estimate of one environmental parameter. The polarity of organic compounds can be taken into account through polarity correction factors (Meylan et al., Citation1992; Müller and Kördel, Citation1996; Sabljic, Citation1987; Tao and Lu, Citation1999). There are two types of fragment approaches: the constructionist approach (Hansch and Leo, 1979; Meylan and Howard, Citation1995) and the reductionist approach. The constructionist approach consists of determining the environmental parameter values of a set of small molecules very accurately and then calculating fundamental chemical fragments from these values: single fundamental fragments consist of (a) isolated carbons or (b) a hydrogen or heteroatom plus multiple atom (e.g., ‒CN) with correction factors. The reductionist approach assumes the deduction of coefficients for individual fragments derived by statistical relationships between the molecular properties and individual constitutive fragments. However, there are two main limits of the fragment approaches: first, they require a large data set to obtain a contribution of each functional group or fragment; second, there can be missing fragment, which means that if a compound contains a missing fragment, the parameter cannot be precisely predicted (Hou et al., Citation2004; Leo, 1975; Meylan and Howard, Citation1995; Schüürmann et al., Citation2006; Sun et al., Citation1996; Tao et al., Citation1999).
2.2 Geometric Descriptors
Geometric descriptors give information about molecular size and shape, thus require accurate three-dimensional coordinates of the optimized geometry of the compounds (McElroy and Jurs, 2001). In this review, 77 different geometric descriptors were inventoried (Table S1).
The simplest geometric descriptors are related to the dimensions of atoms or molecules: radius, diameter, length, perimeter, ovality, thickness, surface, and volume (Table S1). Among the 15 descriptors related to the surface, the FOSA (hydrophobic component of the total solvent accessible surface area) is a measure of the hydrophobic property of a molecule and as it increases, the polarity of the molecule will decrease. The FISA is the hydrophilic component of the total solvent accessible surface area, and the PSA is the Van der Waals surface area of polar nitrogen and oxygen atoms. Both PSA and FISA give measures of hydrophilic properties. As they increase, the polarity of the molecules will rise (Cao et al., Citation2009). For the molecular volume, 13 descriptors were found. Among them, the parachor (P) relates the surface tension to the molecular volume, allowing the comparison of molecular volumes under conditions such that surface tensions are equivalent (Zhao et al., Citation2003). The McGowan volume (Vx) is derived from the parachor and is calculated by a method of group contribution (Abraham and McGowan, Citation1987). For a molecule, the van der Waals volume (VdW) is the volume enclosed by the van der Waals surface, it is usually calculated with software through the estimation of the van der Waals radius. It was shown that the McGowan volumes are equivalent to computer-calculated van der Waals volumes (Reddy and Locke, Citation1994a; Zhao et al., Citation2003). The Le Bas molar volume (VLB) is based on the summation of atomic volumes with adjustment for the volume decrease arising from ring formation (Cousins and Mackay, Citation2000).
VolSurf is a computational program that generates 2D molecular descriptors from 3D molecular interaction energy grid maps (Cruciani et al., Citation2000a; Cruciani et al., Citation2000b). The base of VolSurf is to compress the information present in 3D maps into a few 2D numerical descriptors that are simple to understand and to interpret. These descriptors quantitatively characterize the size, shape, polarity, and hydrophobicity of molecules as well as the balance between them. Molecular shape, which affects packing and solvent interactions, can be described through geometry dependent descriptors such as SHDW, GEOM, and GRAV (McElroy and Jurs, 2001; Table S1). The VolSurf BV31OH2, as for it, is the volume descriptor representing one of the best hydrophilic volumes generated by a water probe calculated at –1 kcal mol−1 energy level (Bordás et al., Citation2011; Cruciani et al., Citation2000a; Cruciani et al., Citation2000b).
Weighted holistic invariant molecular (WHIM) descriptors form another group of geometric descriptors (Todeschini and Gramatica, Citation1997a, 1997b; Todeschini et al., Citation1996). They are built to capture the relevant molecular 3D information regarding the molecular size, shape, symmetry, and atom distribution with respect to some invariant reference frames. WHIM descriptors are obtained from the molecular coordinates of the 3D structure of the molecule (i.e., from its spatial conformation). The algorithm consists in performing a principal component analysis on the centered molecular coordinates by using six different weighting schemes: unweighted (u), weighted by the atomic mass (m), by the van der Waals volume (v), by the Mulliken atomic electronegativity (e), by the atomic polarizabilities (p), or by the electro-topological index of Hall et al. (1991; s). For each weighting scheme, a set of statistical indices is calculated on the atoms projected onto each principal component (1, 2, and 3). The WHIM approach can be viewed as a generalization searching for the principal axes with respect to a defined atomic property (the weighting scheme). Unlike topological descriptors (see section 2.5), the WHIM descriptors are able to distinguish different conformations of the same molecule and different geometric isomers. There are a total of 66 directional WHIM descriptors, and 33 global WHIM descriptors: directional descriptors related to size (λ), shape (θ), symmetry (γ), and atom distribution and density around the origin (κ); and nondirectional descriptors related to the total dimension of the molecule (T(λ) and A(λ) related to linear and quadratic contribution to the total molecular size, and V(λ) being the complete expression), its shape (K(λ)), the total molecular symmetry (G(γ)), and its total density (D(η), with η being related to the quantity of unfilled space per projected atom; Todeschini and Gramatica, Citation1997a, 1997b; Todeschini et al., Citation1996).
3D molecule representation of structures based on electron diffraction (3D-MoRSE) descriptors and 2D autocorrelation descriptors were also involved in some QSAR (Tables S3, S4, S6, S7, and S9). The 3D-MoRSE descriptors are derived from infrared spectra simulation using a generalized scattering function (Habibi-Yangjeh et al., Citation2009; Todeschini and Consonni, Citation2000). The Mor(12p) and Mor(31)v descriptors relate to polarizabilities and van der Waals volumes of the atoms, respectively (Habibi-Yangjeh et al., Citation2009). The 2D autocorrelation descriptors are calculated from molecular graph by summing the products of atom weights of the terminal atoms of all the paths of the considered path length (the lag). For example, the GATS1p, Geary autocorrelation lag 1, weighted by atomic polarizabilities (Table S1), is one of the 2D autocorrelation descriptors. The Geary coefficient is a distance-type function, a function being any physicochemical property calculated for each atom of the molecule, such as atomic mass and polarizability. For GATS1p, the function is the polarizability. Therefore, the molecule atoms represent the set of discrete points in space, and the atomic property the function evaluated at those points (Habibi-Yangjeh et al., Citation2009).
Finally, some other miscellaneous geometric descriptors were used: the summation of the steric factors of the additional substituents (Es; Peijnenburg et al., Citation1992); the molecular refraction (MR), which affords information about the molecular volume and polarizability (Kim et al., Citation2007); the excess molar refraction (R2; Abraham, Citation1993); steric parameters, ΣD and ΣS, that reflect the overall dimension of the molecule (Chaumat et al., Citation1992); and the sum of the core count for non-hydrogen vertex (ΣαH) that may be taken as a measurement of the molecular bulk (Roy et al., Citation2007).
2.3 Geometric-Topological Descriptors
The geometry, topology, and atom-weights assembly (GETAWAY) descriptors include the geometric information given by the influence molecular matrix, and the topological information given by the molecular graph, weighted by chemical information encoded in selected atomic weightings (Consonni et al., Citation2002). Two sets of molecular descriptors have been devised: H-GETAWAY descriptors have been calculated from the molecular influence matrix H, while R-GETAWAY descriptors are from the influence/distance matrix R where the elements of the molecular influence matrix are combined with those of the geometry matrix. The molecular influence matrix H contains some useful information on the molecular geometry, and especially the diagonal elements (leverages) of the matrix allow discrimination among the atoms according to their position in the 3D molecular space with respect to the molecule center (Consonni et al., Citation2002). In the equations reviewed in this work, five GETAWAY descriptors were found: H4p, H5e, HATS7p, HTp, and R3e (Bordás et al., Citation2011; Gramatica et al., Citation2003; Table S1). The number of donatable hydrogens, count of all donatable hydrogens (CTDH), and the accessibility of the acidic oxygen atom in a molecule (Aaccess,O (2D)) can also be classified as combined geometric-topological descriptors (McElroy and Jurs, 2001; Zhang et al., Citation2006; Table S1).
2.4 Geometric-Electronic Descriptors
The geometric-electronic descriptors mainly belong to the charged partial surface area (CPSA) descriptors, which combine molecular surface area and partial atomic charge information. They encode features responsible for polar interactions between molecules. The molecular representation views a molecule as having a surface defined by the overlap of hard spheres, defined by the van der Waals radii of the atoms, which is traced by a sphere representing a solvent molecule (water by default). The surface traced out by the center of the solvent sphere has been termed the solvent-accessible surface. The molecule is further defined as having a specific electron distribution, thus yielding a representation of a charged contact surface where polar intermolecular interactions can take place. Depending on the method used to combine the surface area and the charge information, there are different kinds of descriptors such as three partial positive surface area descriptors (PPSA-1, PPSA-2, PPSA-3), and an equal number of the partial negative surface area descriptors (PNSA-1, PNSA-2, PNSA-3). Also, there is a set of three differences in partial surface area descriptors (DPSA-1, DPSA-2, DPSA-3), six fractional charged surface area descriptors (three positive: FPSA-1, FPSA-2, FPSA-3, and three negative: FNSA-1, FNSA-2, FNSA-3), and a similar set of six total surface weighted partial positively (WPSA-1, WPSA-2, WPSA-3) or negatively (WNSA-1, WNSA-2, WNSA-3) charged surface area descriptors (Stanton and Jurs, Citation1990). In addition to the charged surface area descriptors, the relative influence of the most highly charged (positive and negative) atom on the overall charge of the molecule can be taken into account. This information is combined with the accessible surface area of the most highly charged atoms to obtain the relative positive and the relative negative charged surface area descriptors (RPCS and RNCS, respectively; Stanton and Jurs, Citation1990). From these CPSA descriptors, new descriptors were created to account for hydrogen bonding effects: the SAAA-i (i = 1 – 3), which is the summation of the surface area of atoms that are capable of accepting hydrogen bonding interactions; and the CHAA-i (i = 1 – 3), which is the sum of charges on acceptor atoms, which encodes similar information as SAAA-i (Bakken and Jurs, Citation1999; Sutter and Jurs, Citation1996; Table S1).
Two VolSurf descriptors can also be classified as geometric-electronic (Table S1): the H-bonding capacity derived with the CO probe (HB5O), a hydrogen bond descriptor calculated with carboxyl oxygen probe; and the high values of the hydrophilic region of the hydrogen bond acceptor probe (W4O), which accounts for polarizability (Bordás et al., Citation2011). The last geometric-electronic descriptor is the hydrogen bonding parameter, HB1, which was used to estimate the vapor pressure (Basak et al., Citation1997; Table S6).
2.5 Topological Descriptors
Topological descriptors encode information about the atom types, bond types, and connectivity of the molecule without the need for optimized geometry (McElroy and Jurs, 2001; Sabljic, Citation1991; Sabljic and Trinajstic, Citation1981). They describe both the size and shape of molecules (Organization for Economic Cooperation and Development, 1993). One hundred and forty one topological descriptors were found in the different equations (Table S1).
The best known topological descriptors are the molecular connectivity indices (MCI); (symbol: χ), which characterize the degree of the molecular branching (Randic, Citation1975). The molecule is considered to be a sum of parts, namely, the bonds connecting pairs of atoms. Each atom in a molecule is represented by a cardinal number, δ, the count of all bonded atoms other than hydrogen. The molecule is dissected into fragments or bonds, each retaining the δ values assigned in the original graph. This decomposition produces a set of fragments encoded by the two δ values of the atoms comprising each bond (Kier and Hall, Citation2000). The MCI encode, in the various indices, information on molecular size, branching, cyclization, unsaturation, and heteroatom content. Four types of MCI exist: path (χ), cluster (χc), path-cluster (χpc), and chains (χch; Gerstl and Helling, Citation1987; Sabljic, Citation1991). Zero-order path-type index (0χ) defines individual atom (vertices), first-order (1χ) deals with single-bond length, and so on. The first-order MCI (1χ) correlates extremely well with the molecular surface area (Sabljic, Citation1991; Sabljic and Horvatic, Citation1993; Sabljic and Piver, Citation1992). In addition, Kier and Hall (Citation2002) demonstrated that the 1χ is the contribution of one molecule to the bimolecular interactions arising from encounters of all bonds among two molecules. From order 3, as the order of path index goes higher, MCI describe some local structural properties and possibly long-range interactions. The main characteristic of cluster-type indices is that all bonds are connected to the common, central atom (star-type structure). The third-order cluster molecular connectivity index (3χc) is the first, simplest member of the cluster-type indices where three bonds are joined to the common central atom. For this kind of index, orders higher than four do not have much chemical and structural sense for organic chemicals (Sabljic, Citation1991). The fourth-order path-cluster molecular connectivity index (4χpc) is the first, simplest member of the path-cluster type indices. It refers to subgraphs consisting of four adjacent bonds between non-hydrogen atoms, three of which are joined to the same non-hydrogen atom. Orders higher than six do not have much chemical and structural sense either. The cluster and path-cluster indices describe local structural properties, mainly the extent or degree of branching in a molecule. They are very useful as steric descriptors (Sabljic, Citation1991; Sabljic and Piver, Citation1992). The chain type molecular connectivity indices (χch) describe the type of rings that are present in a molecule as well as the substitution patterns on those rings. Thus, chain type indices also describe more local-type properties. The lowest order for the chain type index is third order, and increases up to the largest ring in any particular molecule (Sabljic, Citation1991). In the valence approximation, non-hydrogen atoms are described by their atomic valence values calculated from their electron configuration (Kier and Hall, 1986, in Sabljic and Piver, Citation1992). For example, the zero-order valence MCI, 0χv, is a simple and good approximation for the molecular volume (Sabljic, Citation1991; Sabljic and Piver, Citation1992).
Based on MCI, several descriptors were then developed: the polarity index (1Fχv), which is the 1χv normalized to the number of discrete functional groups (Sekusak and Sabljic, Citation1992), and the ith order (i = 0 or 1) valence nondispersive factor (Δiχv), which is equal to the difference between the MCI for the nonpolar molecular structure and the same-order MCI (Bahnick and Doucette, Citation1988). To account for negative and positive contribution of individual atoms to the modeled property within the same molecule, Pompe and Randic (Citation2007) developed modified variable connectivity indices (1χf and 1χfλ). As classical MCI were shown to be not applicable to organometallic compound, Sun et al. (1996) introduced the radius-corrected MCI (1χr) and the bond-length-corrected MCI (iχb, i = 1 – 6; Table S1).
A second important group of topological descriptors is related to the information indices such as complementary information content (CIC), information content (IC), or structural information content (SIC), which quantify the degree of heterogeneity and redundancy of topological neighborhoods of atoms in a molecule (Basak, Citation1999; Basak et al., Citation1996, 1997; Estrada et al., Citation2004; Gramatica and Di Guardo, 2002; Gramatica et al., Citation2001; Gramatica et al., Citation1999a; Gramatica et al., Citation2000; Huibers and Katritzky, Citation1998; Katritzky et al., Citation1998; Ma et al., Citation2011; Niemi et al., Citation1992; Table S1). These descriptors view the molecular graph as a source of different probability distributions to which the Shannon's entropy and related expressions can be applied. They are insensitive to molecular geometry, accounting for structural characteristics such as size, branching patterns, bonding types, and cyclicity (Gramatica et al., Citation2000).
A set of weighted path descriptors (WTPT) was also used (Bakken and Jurs, Citation1999; Mitchell and Jurs, Citation1998; Randic, Citation1984; Sutter and Jurs, Citation1996; Table S1). They are based on the molecular identification number (ID) that combines features of connectivity indices and path counts, and characterize molecular branching (Randic, Citation1984). Each contiguous path in the molecule can be assigned a weight based on the number of atoms adjacent to the atoms in the path. The molecular ID is the summation of all paths of the compound.
Four Moran autocorrelation descriptors (MATS6e, MATS7e, MATS4p, and MATS1v) were found in the equations (Bhhatarai and Gramatica, Citation2011; Goudarzi et al., Citation2009; Gramatica et al., Citation2003). The structural variables introduced by Moran correspond to bidimensional autocorrelations between pairs of atoms in the molecule, and are defined to quantify the contribution of a considered atomic property to the analyzed property. These can be readily calculated by summing products of terms including the atomic weights for the terminal atoms in all of the paths of a prescribed length. For example, for MATS6e, the path connecting a pair of atoms has a length of 6 and involves the atomic Sanderson electronegativities as weighting scheme (Goudarzi et al., Citation2009).
Liu et al. (Citation1998) developed a molecular distance-edge between carbon atoms vector (MDE) based on two fundamental structural variables: one for distance between atoms in the molecular graph, and one for edges of the adjacency in the graph. In these descriptors, carbon atoms are divided into four types: (a) primary (‒CH3), (b) secondary (>CH2), (c) ternary (>CH-), and (d) quaternary (>C<). A distance edge term is computed for all pairwise combinations of carbon types, for a total of 10 descriptors. For example, MDE-13 represents the distance edge descriptor between primary and ternary carbons. In addition to describing carbon bonding, these descriptors include information regarding distance between atoms (Bakken and Jurs, Citation1999).
Several other miscellaneous topological descriptors were found in the equations listed in Tables S3–S10. Among BCUT descriptors (Burden - CAS - University of Texas eigen-values; Burden, 1989), BEHe7 brings 2D information which takes into account the weight of different atoms in the structure (Burden matrix) and their electronegativities (Papa et al., Citation2009). The DELS could be a measure of total charge transfer in the molecule (Gramatica et al., Citation2000). The group philicity (ωg+) is a descriptor of reactivity that allows a quantitative classification of the global electrophilic nature (electron accepting capacity) of a molecule within a relative scale (Parthasarathi et al., Citation2006). The Lu index is interpreted as a parameter characterizing molecular size, and the DAI characterizes the degree of branching on aromatic ring (Lu et al., Citation2006). The sum of topological distances between oxygen and bromine atoms (T(O…Br)) gives a double structural information: its values increases according to both the number and the distance of bromine substituents, thus T(O…Br) also takes into account the information related to the position of the bromine atoms on the phenyl rings (Papa et al., Citation2009). The eccentric connectivity index (ξC) is a topological index accounting for both size and branching of compounds (Sharma et al., Citation1997), and the bond connectivity index (ϵ) can be understood as molecular size corrected for geometric accessibility with respect to van der Waals contact (Schüürmann et al., Citation2006). The Kapa index (3κ), which is based on path lengths, is a shape index (Bakken and Jurs, Citation1999; Dunnivant et al., Citation1992; Kier, Citation1986). The numbers of different sp hybridized orbitals between carbon atoms were also used: 1SP2, 2SP2, 3SP2. They encode information concerning attack sites for the radical (Bakken and Jurs, Citation1999). The Wiener index (W) is defined as the number of bonds between all pairs of atoms in an acyclic molecule. It measures the compactness of the molecule (Bogdanov et al., Citation1989). The calculation of the 3D Wiener index for the hydrogen-suppressed geometric distance matrix (3DW) consists in summing the entries in the upper triangular submatrix of the topographic Euclidean distance matrix for a molecule (Basak et al., Citation1996). This index is considered as a measure of molecular shape (Consonni and Todeschini, Citation2010). The characteristic root index (CRI) is the sum of the positive characteristic roots obtained from the characteristic polynomial of the matrix with the entries calculated from the electronic input information. It was shown that it is correlated to the molecular surface area (Türker Saçan and Balcioğlu, 1996; Türker Saçan and Inel, 1995). MAXDN represents the maximum negative intrinsic state difference in the molecule and can be related to the nucleophilicity of the molecule, while MAXDP represents the maximum positive intrinsic state difference and can be related to the electrophilicity of the molecule (Gramatica et al., Citation2000). The superpendentic index (PND) can be calculated from the pendent matrix, a submatrix of distance matrix. This index takes into consideration all pendent vertexes, and its value changes significantly with a small change in the branching of a molecule (Gupta et al., Citation1999). Finally, the Kier symmetry index (S0K) is used to encode the shape contribution due to symmetry (Todeschini and Consonni, Citation2000). The lower the S0K of a compound, the greater the topological symmetry, and thus the lower the change in molecular freedom (Ding et al., Citation2006).
2.6 Electro-Topological Descriptors
Electro-topological state (E-state) is a method for describing and encoding molecular structure at the atom level. In the E-state formalism, each atom is viewed as having an intrinsic state which is perturbed by every other atom in the molecule. The intrinsic state combines valence state electronegativity with the local topology of the atom. Perturbation is dependent on the difference between intrinsic state values, and diminishes as the square of the graph distance between atoms. The result is that the E-state index, S, for an atom represents electron accessibility at that site. There are several extensions of the E-state concept. E-states indices may be computed separately for hydrogen atoms, the hydrogen E-state indices. Also E-state indices may be summed for all atoms of a given type in a molecule. These atom type E-state indices encode electron accessibility, presence or absence of groups, and count of groups (Hall and Story, Citation1996; Hall et al., Citation1991; Huuskonen et al., Citation1999; Kier and Hall, Citation1999).
The E-state index for a given atom (or atom type) varies from molecule to molecule, and depends on the detailed structure of the molecule. In the different atom-type E-state index, the set of bonds to a skeletal atom is given by a chain of lowercase letters: s (single), d (double), t (triple), and a (aromatic). The element is given by its symbol together with the number of hydrogen atoms. For example, SdCH2 represents the S values for a terminal CH2 group on a double bond, while SssCH2 represents the methylene group with two single bonds; SaasC stands for an aromatic carbon to which a substituent is bonded (Hall and Story, Citation1996; Huuskonen, Citation2001a, 2001b; Huuskonen et al., Citation1999). As indicated previously, E-state index can also be calculated for group or molecule by adding the contributions of the single atoms present in the group or the molecule: to each atom is ascribed an index encoding the intrinsic electronic and topological state of the atom as well as the effect of the molecular environment in which the atom under study resides (Gombar and Enslein, Citation1996; Thomsen et al., Citation1999).
Finally, the mean E-state (Ms), the average E-state value over all heteroatoms (EAVE-2), and the sum of E-state values over all heteroatoms (ESUM-2) can also be calculated (Habibi-Yangjeh et al., Citation2009; McElroy and Jurs, 2001; Table S1).
2.7 Quantum-Chemical Descriptors
Two hundred and forty eight quantum-chemical descriptors were inventoried in this review, representing the highest number of descriptors for one category (Tables S1 and S3–S10).
The most well-known quantum-chemical molecular descriptors are related to energies, and in particular to the energies of the highest occupied molecular orbital (EHOMO) and lowest unoccupied molecular orbital (ELUMO). Orbitals play a major role in most of chemical reactions, they are particularly involved in the formation of covalent bonds and thus of many charge-transfer complexes. The EHOMO is directly related to the ionization potential and characterizes the susceptibility of the molecule toward attack by electrophiles (Karelson et al., Citation1996). The ELUMO measures the ability of a molecule to accept electrons in intermolecular interactions (Chen et al., Citation2002a). EHOMO represents the proton acceptance ability in forming hydrogen bond, while ELUMO represents the proton donation ability in formation of hydrogen bond (Zhou et al., Citation2005). Two descriptors are derived from EHOMO and ELUMO (Table S1): the absolute electronegativity (EN), which provides insight into the energetics of the reactant molecule (Bakken and Jurs, Citation1999; Hu et al., Citation2000; Müller and Klein, Citation1991), and the hardness (Hard; Bakken and Jurs, Citation1999). The HOMO and LUMO are generally the most important orbitals, but in cases where lower-energy occupied orbitals are close in energy to the HOMO, and higher-energy orbitals are close in energy to the LUMO, other orbitals may also play a role (Brown and Mora-Diez, Citation2006b; Table S1).
Klamt (Citation1993) defined a new set of descriptors called local frontier orbital descriptors: the charge-limited effective HOMO energy at H atom (ECHH (q)), which is the weighted average of the orbital energies starting with the HOMO and extending to lower orbitals until the amount of charge taken into account reaches q; the energy-weighted effective HOMO energy (EEHH (ϵ)), which is similar to an effective EHOMO of the electrons of atom H calculated with an energetic penetration length ϵ; and the energy limited effective frontier orbital charges QLA (E), where QLA (E) is the amount of electronic charge available at atom A in the lower unoccupied orbitals down to an energy limit E.
In contrast to conventional molecular orbital based descriptors such as the EHOMO and ELUMO, local quantum-chemical molecular descriptors are designed to extract, from the delocalized molecular orbital wavefunctions and energies, energy and charge information that reflects the local characteristics of a given atomic site in the molecular environment. The energy-weighted donor energy (EEocc) describes the electron donor ability of a molecule at an atomic site, and is constructed through a sum of occupied molecular orbital energies Ei, weighted by exponential terms involving reference energy Eref. EEocc ranges between the EHOMO as delocalized limit (for Eref close to 0) and the sum of the orbital energies weighted only by pi (for Eref close to ∞). The energy-weighted acceptor energy (EEvac) is defined accordingly through unoccupied molecular orbitals. It characterizes the capability of the molecule to accept additional electron charge at an atomic site, and thus represents a localized generalization of the ELUMO. Another local reactivity parameter is the charge-limited acceptor energy (EQvac (q, r)). It characterizes the energy gain upon accepting charge q at atomic site r, and can be understood as a further local generalization of the ELUMO. As a general trend, EQvac (q, r) becomes increasingly local with increasing amount of the charge penetration depth q. A complementary approach is to evaluate, for a given energy loss or gain, the associated amount of charge released from or taken up at site r. Therefore, an energy-limited donor charge QEocc (ϵ, r) can be defined as amount of charge being removed from center r when offering the energy ϵ. Atomic sites with high electron donor ability are characterized by large values for QEocc. The defined energy-limited acceptor charge QEvac involves unoccupied molecular orbitals and quantifies the amount of accepted electron charge that is associated with a predefined energy gain ϵ (Yu et al., Citation2011). The electron affinity (EA) represents the energy difference associated with the gain of an electron, which should correlate with the ease or difficulty of the reduction of a compound (Colón et al., Citation2006).
In addition to EHOMO and ELUMO and the derived energies, the number of descriptors related to energy is very large (Table S1). As most of these energy descriptors are well-known—total energy (TE, TE2), torsional energy (TOE), electronic energy (EE), attraction (EN1, EN1c, EN1x) or repulsion (CCR, EE1, EE1c, NN2, NRE) energies, resonance energy (J, MinOH), Gibbs energy (G, ΔGaq, ΔGdiss)—no detailed description is given here.
The interaction process between an acid and a base can be dissected into two steps: a charge transfer process resulting in a common chemical potential describing the strengths of the acid and the base, at a fixed external potential, followed by a reshuffling process at a fixed chemical potential. The fractional number of electron transfer, ΔN, and the associated energy change in the charge transfer, ΔEe, depend on the interplay between electronegativity and hardness of the acid and the base. The ΔEe is the energy lowering due to this electron transfer from a species of higher chemical potential (base) to that of a lower chemical potential (acid; Gupta et al., Citation2007). The inclusion of pi-electronegativity of the α carbon atom, ENπ,αC, can represent the different hybridization states (i.e., sp, sp2, and sp3) of the α carbon atom in an acid, and ENσ,O is the σ-electronegativity for the oxygen atom in the acidic hydroxyl group (Zhang et al., Citation2006).
Several descriptors related to the electronegativity were also used, such as the molecular electronegativity distance vector (MEDV). The properties of a molecule mainly depend on various interactions between its atoms. These interactions vary with the electronegativity of the atoms and the distances of chemical bonds formed between atoms. To describe an organic molecule and to construct its MEDV, the atomic types of all non-hydrogen atoms in the molecule have to be specified. If an atom is linked to k non-hydrogen atoms through chemical bonds, then the atom belongs to the k atomic type. There are at best four atomic types (considering the non-hydrogen atoms are often carbon, oxygen, nitrogen, or halogen atoms) and, therefore, to express the approximate interactions, MEDV include a maximum of 10 elements (e.g., M11, M12, M13, M14, M22, M23, M24, M33, M34, M44). These ten elements combine atomic attributes (both the chemical element type and the chemical bond type of each atom), bond length, relative distance between atoms, and their relative electronegativities. The relative electronegativity of a non-hydrogen atom is defined as the ratio of Pauling's electronegativity of the non-hydrogen atom to Pauling's electronegativity of a carbon atom, and the relative distance or relative bond length of a chemical bond is defined as the ratio of the length of this bond to the bond length of the C-C bond (Liu et al., Citation2002; Sun et al., Citation2007).
Sixty-four of the 248 quantum-chemical descriptors that were found in the equations are related to the charges of atoms or molecules. Others are related to the dipole moment, polarizability, superdelocalizability, electrostatic potential, moments, and bond order (Table S1).
In addition to the dipole moment μ, two descriptors derived from μ were found useful: the Z-component of the dipole moment (Bordás et al., Citation2011), and the total local dipole, μtot. The latter is defined as the difference in charges of each atom in the bond, divided by the length of the bond, and summed over all bonds in the molecule. The resulting μtot describes the dipolarity as a single number independent of direction, and alleviates some of the problems encountered in using the dipole moment μ (Famini and Wilson, Citation1997).
The polarizability α was involved in many equations (Tables S3, S4, S6 to S9), but the atom self-polarizability (ALPij; Berger et al., 2001; Berger et al., Citation2002; Tehan et al., Citation2002b; Von Oepen et al., 1991), the second (α2) and third (α3) principal polarizabilities (Dunnivant et al., Citation1992), and the polarizability of the hydroxyl oxygen atom in an acid (αO) were also used. Other polarizability terms are specially used in linear solvation energy relationships (LSER) equations (see section 2.8).
The superdelocalizability of an atom is a measure of its available electron density. It is calculated by the sum over all orbitals of the ratio between orbital densities and the corresponding orbital energies. The superdelocalizability of the HOMO (SHOMO) and of the LUMO (SLUMO), and the electrophilic (SE) and nucleophilic superdelocalizabilities (SN) can also be calculated (Reddy and Locke, Citation1996). The SE has been designed to quantify the susceptibility of a molecule for an electrophilic attack (Tehan et al., Citation2002b).
Concerning the electrostatic potential, the most used descriptors are the molecular electrostatic potential minima (Vmin), the surface molecular electrostatic potential maxima (VS,max) and minima (VS,min), and the sum of the surface maxima (ΣV+s) or minima (ΣV−s) values of the electrostatic potential. The ΣV−s should not be viewed as a hydrogen bond basicity descriptor but one reflecting nonspecific intermolecular interactions despite the fact that similarities exist between each other. Vmin measures the hydrogen bond-accepting tendency or hydrogen bond basicity of a molecule, whereas VS,max measures the hydrogen bond-donating tendency or hydrogen bond acidity of a molecule. The electrostatic potential V(r) is created in the space surrounding a molecule by its nuclei and electrons. The electrostatic potential surrounding spherically symmetric, neutral atoms is positive everywhere. However, when atoms combine to form molecules, regions of negative potential develop. These are usually in regions surrounding electronegative atoms, above and below multiple carbon-carbon bonds and aromatic rings, and along the outer edges of strained carbon-carbon bonds. Each such negative region has one or more spatial minima, Vmin, associated with it. These Vmin, as well as surface minima, VS,min, have served as a mean for ranking sites for susceptibility toward electrophilic attack (Gross et al., Citation2001; Ma et al., Citation2004; Xu et al., Citation2007; Zou et al., Citation2002). Other descriptors related to the electrostatic potential are indicated in Table S1, such as molecular electrostatic potential (MEP) on the acidic atom N, O, or S (Liu and Pedersen, 2009); highest hydrogen bond acceptor potential (VHHA); and highest hydrogen bond donor potential (VHHD; Yan and Gasteiger, 2003).
Another group of quantum-chemical descriptors is related to the moments (Table S1): σ-moments are derived from quantum chemical density functional calculations combined with the continuum solvation model (COSMO). The zero-moment M0 is identical with the molecular surface, the second moment M2 is a measure of the overall electrostatic polarity of the solute, the third moment M3 is a measure of the asymmetry of the polarization charge density profile, and the hydrogen-bond moments Macc and Mdon are quantitative measures of the acceptor and donor capacities of the compound, respectively (Klamt et al., Citation2002). The second principal moment of inertia (SMI) is derived from 3D representations of the molecules (Dunnivant et al., Citation1992). The magnitude of the principal moments of inertia of a molecule (PMI) encodes information about spatial distribution of mass and its rotational properties. It also expresses the role of molecular size and volume in occupying the space between water molecules (Dimitriou-Christidis et al., Citation2008). The quadrupole moment (in particular Qzz) was also found in some equations (Staikova et al., Citation2004; Zeng et al., Citation2012; Table S6).
Several descriptors related to bond order were useful for some environmental parameters (Tables S4, S5, and S10): the bond order of the carbon-halogen bonds (BO; Chen et al., Citation1998b; Chen et al., Citation2001c; Zhao et al., Citation2001); the OH bond order (BOOH), which is a measure of the strength of OH bond (Citra, Citation1999; Hollingsworth et al., Citation2002); the average bond order (ABO(N)), which is a term to correct the deficiency of the electrostatic or hydrogen-bonding parameter for the N-atom containing compounds (Katritzky et al., Citation1998); and the minimum bond order of an atom C (MinC; Pompe and Veber, Citation2001). The bond strength (BS) of the carbon-halogen bond to be broken enables the differentiation between the reactivity of the various halogen atoms (F, Br, Cl, and I; Peijnenburg et al., Citation1992).
Some descriptors are defined by using a combination of topological invariants, such as interatomic connectivity, and quantum-chemical information, such as atomic charges and bond orders: the atom quantum-connectivity index of path type of the order 2 defined on the basis of graphs weighted by charge density (2ΩpC(q)), and the bond quantum-connectivity index of chain type of the order 6 based on graph weighted by bond orders (6ϵRg(ρ); Table S1). The 2ΩpC(q) accounts for the topological structural features of the sequence of three consecutive atoms (order 2), and includes quantum chemical information through the use of atomic charge densities. It controls the influence of the number of substitutions at different sites in the molecule. The 6ϵRg(ρ), as for it, accounts for the influence of bond order weighted cyclic fragments of six bonds, that is, six-atom rings (Estrada et al., Citation2004).
Several other miscellaneous descriptors were found (Table S1) such as the hydrogen bonding donor charged surface area, (HDCA(2)), which is connected with the hydrogen-bonding ability of compounds (Katritzky et al., Citation1998); and the hydrogen acceptor dependent hydrogen donors surface area-2 (HDSA(2)), based on quantum chemical partial charge, which is also directly related to hydrogen-bond acceptor capability of a molecule (Modarresi et al., Citation2007). The D3DRY and D6DRY, that are VolSurf descriptors, represent the hydrophobic energy calculated with the hydrophobic probes (Bordás et al., Citation2011). The spin density (SD) is a measure of free spin concentrated on the benzylic carbon after hydrogen atom abstraction (Beasley et al., Citation2009).
Finally, some quantum-chemical descriptors were specially used with the LSER approach, their description is given in the next section.
2.8 Descriptors Related to the Solvation Energy
Linear solvation energy relationships (LSER) are a part of the wider field of linear free energy relationships (LFER; Platts et al., 1999). LSER use a mechanistic understanding of the partition process, which considers the interaction energies that contribute to the overall free energy of the transfer process (Abraham and McGowan, Citation1987; Goss and Schwarzenbach, Citation2001; Kamlet et al., Citation1988). It takes into account the energy term for cavity formation (proportional to the size of the molecule which can be related to the geometric descriptors Vi or Vx), and the interaction terms that can be decomposed in four terms as follows: (a) the induction of dipoles within the solutes (London dispersive forces, Debye forces) represented by the excess molar refraction (R2), (b) electrostatic interactions represented by dipolarity/polarizability term (π*), (c) overall hydrogen bond donor acidity Σα2H (H-donor or electron-acceptor), and (d) overall hydrogen bond acceptor basicity Σβ2H (H acceptor or electron donor; Gawlik et al., Citation1997; Nguyen et al., Citation2005; Platts et al., Citation2000; Van Noort et al., 2010; Wauchope et al., Citation2002). For transfer between water and wet solvents, such as wet octanol or ethyl acetate, the Σα2H and Σβ2H are replaced by Σα2O and Σβ2O for certain functional groups (e.g., pyridines, sulfoxides), whose basicity is found to change substantially between wet and dry solvents (Platts et al., Citation1999 and 2000).
In theoretical LSER (TLSER), the energy term for cavity formation is the molar volume (Vm). The hydrogen bond acceptor basicity is represented by two terms: the covalent basicity (ϵβ), based on EHOMO, and the solute electrostatic basicity (largest negative net atomic charge on an atom q−). The hydrogen bond acidity is similarly divided into a covalent acidity (ϵα), which is a function of the ELUMO, and the solute electrostatic acidity (most positive atomic net charge on a hydrogen atom QH+; Cramer et al., Citation1993; Famini and Wilson, Citation1997; Wilson and Famini, Citation1991)
To simplify the TLSER equation, Chen et al. (Citation1996a) developed a modified TLSER (MTLSER): the equation only depends on the polarizability (α), the dipole moment (μ), the EHOMO and ELUMO, QH+, and q−. These descriptors were presented in the previous section (2.7).
3. PARAMETERS FOR PROCESSES GOVERNING THE FATE OF ORGANIC COMPOUNDS IN THE ENVIRONMENT
The fate of organic compounds in the environment is mainly regulated by their behavior in the soils on which they might be applied directly (i.e., after application of pesticides) or indirectly through rain and water leaching, atmospheric deposition or organic waste amendments. In soils, organic compounds are affected by various physicochemical and biological processes conditioning their biotic and abiotic degradation, their retention (adsorption, desorption); their transfer toward groundwater, surface water, plant, and atmosphere; and consequently their bioavailability and potential side effects on the organisms living in the contaminated environment. The organic compounds also undergo adsorption phenomena in sediments, and degradation in water, sediments, and atmosphere (Katayama et al., Citation2010).
From QSAR that are reviewed in this work, we propose to classify the processes governing the fate of organic compounds in the environment in six main categories (Table S2): water dissolution, dissociation, volatilization, retention, degradation, and absorption by higher plants. These processes are represented by 90 different environmental parameters. Their description and their relationships to the structural molecular descriptors are included in the following sections.
3.1 Water Dissolution Process
The physicochemical properties of substances related to their dissolution in water control their partitioning between air, water, soils, sediments, and biota, and thus the accumulation and the rates of transfer between these different compartments. The key parameters include water solubility (SW) and octanol-water partition coefficient (KOW; Shiu et al., 1988; Table S2). A review of the QSAR allowing, in particular, the prediction of SW and KOW was written by Katritzky et al. (Citation2000). In the following paragraphs, we have completed and updated this review by considering the results that have been published since then.
3.1.1 Water Solubility
Water solubility, SW, plays an important role in the fate of organic compounds in the environment. One of the routes of transport of contaminants in the environment is water, therefore SW affects the ability of a compound to be transported as well as its rates of transfer between water and other environmental compartments (Kühne et al., Citation1995). Table S3 summarizes the 65 QSAR found for the prediction of SW. They are classified in three parts: relationships with one descriptor, with one category of descriptors, and with several descriptors and categories. The equations involve from 1 to 30 descriptors (excluding the equations based on the fragment approach), but most of them involve 1–3 descriptors. The QSAR were developed for a wide diversity of organic compounds (Table S3).
Several of the one-descriptor relationships involved descriptors related to the molecular surface and volume (Cao et al., Citation2009; Huibers and Katritzky, Citation1998; Katritzky et al., Citation2000; Puzyn et al., Citation2009; Shiu et al., Citation1988; Table S3). For 13 androgens, the SW were moderately correlated with the hydrophobic component of the total solvent accessible surface area (FOSA), the hydrophilic component of the total solvent accessible surface area (FISA) or the van der Waals surface area of polar nitrogen and oxygen atoms PSA (r2 < 0.55; Table S3). As FISA and PSA increase, the polarity of the molecules rises and the aqueous solubility should rise (Cao et al., Citation2009). For 10 chloronaphthalenes, a relationship between SW and the solvent accessible molecular volume (SAVw) in the water SAVw was proposed. The correlation coefficient is good (r2 = 0.950; Table S3), however, the number of compounds used to develop the QSAR is low, therefore giving uncertainty on its robustness. The SAVw is directly related to the cavitation, and formation of cavitation in the solvent plays a critical role in dissolving highly hydrophobic compounds. In the case of chloronaphthalenes, the SAVw mainly depends on the chlorination degree: it increases from mono- to octa-chloronaphthalenes. The influence of the substitution pattern on SW is less pronounced. The electrostatic and dispersive interactions occurring between the solvent and solute after formation of the caves are less important for the dissolving process. However, for chloronaphthalenes, those factors become significant when comparing each other congeners with the same number of chlorine substituents (Puzyn et al., Citation2009). The molecular volume Vm was a good descriptor to estimate the SW of 241 hydrocarbons and halogenated hydrocarbons (r2 = 0.904; Table S3; Huibers and Katritzky, Citation1998), but also of 15 polychlorinated dibenzo-p-dioxins (PCDD; Shiu et al., 1988). Water as a solvent would much prefer to interact with itself or other hydrogen bonding or ionic species than with a nonpolar solute, so there is a lower SW for larger hydrocarbon solutes. However, the major problem with Vm as the sole descriptor for SW is that it does not take into account steric interactions or conformational effects (Huibers and Katritzky, Citation1998). For 209 polybrominated diphenyl ethers (PBDE) and hexabromobenzenes (HBB), there was a good correlation between SW and the 3D-MoRSE-signal 23 weighted by atomic masses, Mor23m (r2 = 0.918; Table S3). Mor23m brings complex 3D information related to the weights of the atoms in the structure, as viewed by an angular scattering function (Papa et al., Citation2009). Topological descriptors were involved in several relationships: the Lu index (Lu, Citation2009), and four different MCI: 0χv, 1χv, 3χv or 5χv. The best correlations were obtained with 1χv and 3χv: r2 > 0.933, but the number of compounds considered is low (Table S3; Gerstl and Helling, Citation1987). Using constitutional descriptors, relationships were found between SW and the molecular weight (MW) for only six PBDE (Wania and Dugani, Citation2003), or with the number of chlorine atoms nCl for 15 PCDD (Shiu et al., Citation1988). For each chlorine added, there is a 5× drop in solubility. Finally, the polarizability α allowed good estimate of the SW of 20 substituted phenols (r2 = 0.951; Table S3; Xie et al., Citation2008), and 75 PCDD (r2 = 0.978; Table S3; Yang et al., Citation2007). SW is inversely proportional to α mainly because α is correlated with the molecular volume. Indeed, the molecular volume can reflect the absorbed energy during the formation of cavity in the solvent (Yang et al., Citation2007).
Table S3 reports 21 QSAR developed with descriptors belonging to the same category. Chen et al. (Citation2007) used the positions of Cl substitution method to predict the SW of 107 polychlorinated diphenyl ethers (PCDE; Table S3). The best model involved four descriptors relative to the positions of chlorine atoms in the compound—N2(6), N3(5), N4, Nm—but the effect of N3(5) on SW was the most important one. For 53 miscellaneous compounds, the quantum-chemical descriptors atom quantum-connectivity index of path type of the order 2 defined on the basis of graphs weighted by charge density (2ΩpC(q)), and bond quantum-connectivity index of chain type of the order 6 based on graph weighted by bond orders (6ϵRg(ρ)) were better descriptors to predict SW than constitutional (nC, and gravitation index (IG)), geometric (molecular surface S, molecular volume divided by xyz box Vxyz), or topological ones (3χ, average structural information content index of order two I2av), and better descriptors than the quantum-chemical descriptors minimal net atomic charge Qmin, and α (Estrada et al., Citation2004). Three equations were based on electro-topological descriptors (Huuskonen, Citation2001a; Thomsen et al., Citation1999; Table S3). Two of them were obtained by Huuskonen (Citation2001a) for 674 compounds: one only with atom-type E-state index, and one with E-state index and three simple indicator variables that are applied, for example, to compounds containing only aliphatic C and H, or to pyridines and their alkyl derivatives. Then, the SW of up to six carbon atoms phthalates in the alkyl chain was moderately reproduced with electro-topological descriptors related to polar/hydrophilic (Sester) and polar/hydrophobic (Salkyl) characters (r2 = 0.574; Table S3). For high molecular weight phthalates, other processes such as the formation of micro-droplets prevail, leading to unexpected high apparent water solubility (Thomsen et al., Citation1999). Several MCI (0χv, 1χ,1χv, 3χ, 3χv, 3χvpc, 5χv, 6χvc, or 6χvpc) were used to predict the SW of 50 alcohols, 14 derivates of benzanilides, and miscellaneous compounds (Dai et al., Citation1998; Gerstl and Helling, Citation1987; Nirmalakhandan and Speece, Citation1988a): the best correlations were obtained for alcohols (r2 = 0.961) and the worst for pesticides (r2 = 0.389; Table S3). The SW of 107 PCDE was correlated to six quantum molecular electronegativity-distance vectors (M11, M12, M13, M22, M23, M33; Sun et al., Citation2007). The combination of the polarizability α and the total energy TE was found useful to estimate the SW of 20 substituted phenols (r2 = 0.980; Xie et al., Citation2008), and of 107 PCDE (r2 = 0.956; Yang et al., Citation2003). The addition of the most positive atomic net charge on a hydrogen atom (QH+) to the equation allowed to improve the correlation for the substituted phenols (r2 = 0.985; Xie et al., Citation2008; Table S3). For these phenols, SW increases with the increase in TE, because TE can be related to molecular volume. The molecule tends to be more hydrophobic and the substituted phenol has thus difficulty in entering water phase, resulting in low SW (Xie et al., Citation2008). Similar results were found for the 107 PCDE (Yang et al., Citation2003). For 27 halogenated anisoles, SW increases with ELUMO and Qyy, and conversely with the decrease in QH+. The effect of Qyy was the most remarkable (r2 = 0.980; Table S3; Zeng et al., Citation2012). Some relations based on the fragment approach were successfully developed for high number of compounds (Clark, Citation2005; Hou et al., Citation2004; Kühne et al., Citation1995; Table S3).
The number of descriptors involved in the several descriptors and categories equations ranged from 2 to 30 (Table S3). The combination of different categories of descriptors improved the estimation of SW (increase in r2; Huibers and Katritzky, Citation1998), probably because it allows considering simultaneously different representations and properties of the molecule. Constitutional descriptors (C/H, MW, number of double bonds (NBD), number of single bonds (NSB), nC, nCl, nH, nO, NCl) were used in more than half of the equations, combined either with topological (mainly MCI but also MDE, WTPT, or CIC), quantum-chemical (α, number of independent points of the positive electrostatic potentials on molecular surface N+v, charges) and/or geometric (GEOM, GRAV) descriptors (Bhhatarai and Gramatica, Citation2011; Huuskonen, Citation2000; McElroy and Jurs, 2001; Müller and Klein, Citation1992; Nirmalakhandan and Speece, Citation1988a, 1989, 1990; Patil, Citation1994; Sutter and Jurs, Citation1996; Xu et al., Citation2010; Table S3). For 107 PCDE, SW depended on the NCl because the larger PCDE molecules would yield stronger dispersion-type interactions between them and tend to be excluded from water (i.e., the SW value becomes smaller). PCDE congener with smaller N+v will likely have stronger interaction with the water, and thus produce higher SW. The introduction of σ2tot in the equation indicates that the uniformity of electrostatic potential distribution has effect on aqueous solubility of PCDE (Xu et al., Citation2010). For 52 pesticides, acceptable correlation between SW and C/H, 0χ, 0χv and α was found (r2 = 0.810; Table S3), but this relationship cannot be suitably used for O-analogues and compounds with C/H ratio higher or equal to 2 (Patil, Citation1994). The QSAR developed by Sutter and Jurs (Citation1996) to estimate the SW of 123 organic compounds relied on nine descriptors: two constitutional (nC, nO), one geometric (GEOH), three geometric-electronic (SAAA-1, SAAA-2, FNSA-3), two topological (WTPT-1, WTPT-2), and one quantum-chemical (QSUM). The nO, QSUM, and FNSA-3 descriptors could encode dipole interactions; nC, GEOH, WTPT-1, and WTPT-2 could have been responsible for London dispersion forces; and SAAA-1 and SAAA-2 were probably encoding the hydrogen bonding interactions between the molecules and solvent (Sutter and Jurs, Citation1996).
Combination of quantum-chemical descriptors (bond order of nitrogen atom ABO(N), EHOMO, ELUMO, fractional area-weighted surface charge of hydrogen bonding donor atoms FHDSA(2), number of electron Nel, minimal net atomic charge Qmin, QH+, electronic spatial extent Re, TE or solvation free energy ΔGs) with geometric (molecular contact surface area CSA, largest bond length between two carbon atoms Lcc), geometric-electronic relative negative charged surface area (RNCS), or topological structural information content of zeroth order (0SIC) descriptors allowed good estimate of the SW of various compounds (Katritzky et al., Citation1998; Lu et al., Citation2008; Schüürmann, Citation1995; Table S3). For 411 organic compounds, the two most important descriptors were the number of electrons (Nel) and the most negative partial charge in the molecule (Qmin). The Nel can be related to cavity-size effects (dispersion and cavity formation), and the Qmin can be related to one specific type of solute-solvent interaction, as solute-solvent interaction is a major determining factor for the SW of compounds. Among the other descriptors involved in the relationship, the structural information content of a graph based on zero-order neighborhood of vertices (0SIC) describes the atomic connectivity in the molecule and encodes the size and the degree of branching in the compound. The size and the shape of the molecule also directly affect the intermolecular interaction. The average bond order of nitrogen atom (ABO(N)) seems to correct the deficiency of the electrostatic or hydrogen-bonding parameter for the N-atom containing compounds (Katritzky et al., Citation1998).
Some equations were found combining geometric (magnitude of the third geometric moment GEOM-3, shadow area SHDW-i, Vm), topological (total weighted number of paths in the molecule divided by the total number of atoms ALLP-4, structural information content of zero-order 0SIC, average structural information content index of order zero I0av, MDE-i, WTPT-2, 1χvrc), electro-topological (average E-state value over all heteroatoms (EAVE-2), sum of E-state values over all heteroatoms (ESUM-2)), quantum-chemical (sum of charges on all donatable hydrogens (CHDH), electrostatic hydrogen bonding basicity (EHBB), maximum partial charge for a hydrogen atom Qmax(H), average surface area times charge on donatable hydrogen (SCDH-2)), and/or several geometric-electronic (sum of charges on acceptor atoms (CHAA-2), difference in partial surface areas (DPSA-i), fractional positively charged partial surface areas (FPSA-1), partial negative surface area (PNSA-i), partial positive surface area (PPSA-1), sum of the surface area of acceptor atoms (SAAA-3), surface weighted negatively charged partial surface area (WNSA-1), surface weighted positively charged partial surface area (WPSA-2)) descriptors (Estrada et al., Citation2004; Huibers and Katritzky, Citation1998; McElroy and Jurs, 2001; Mitchell and Jurs, Citation1998; Table S3). For 241 hydrocarbons and halogenated hydrocarbons, the molecular volume Vm was nevertheless the most performing descriptor (high t test value; Huibers and Katritzky, Citation1998). As indicated previously, in order for a solute to enter into aqueous solution, a cavity must be formed in the solvent for the solute molecule. For 265 miscellaneous compounds, Mitchell and Jurs (Citation1998) developed a nonlinear model involving nine descriptors: SHDW-3 and GRAV, which are geometry-based descriptors, three geometric-electronic descriptors (PPSA-1, FPSA-3, WPSA-3), three topological descriptors (2SP3, ALLP-3, WTPT-4) that contained information about weighted paths and carbon types, and one quantum-chemical descriptor (q−). This nonlinear model provided better estimate of SW (r2 = 0.974) than the linear one (r2 = 0.931; Table S3).
Atom type E-state index and topological descriptors (essentially MCI) were correlated to the SW of pharmaceuticals and miscellaneous compounds (Huuskonen, Citation2000; Huuskonen et al., Citation1997, 1998). However, the model gave poor predictions for the subgroup of pesticides containing phosphate or thiophosphate group and polychlorinated hydrocarbons (Huuskonen et al., Citation1998).
The LSER and TLSER approaches were used successfully to estimate the SW of numerous compounds (Famini and Wilson, Citation1997; Feng et al., Citation1996; Hickey and Passino-Reader, Citation1991; Xie et al., Citation2008; Table S3). Finally, for 797 miscellaneous compounds, the development of a relationship based on the 3D structure of the molecules and eight descriptors: five constitutional (AROM, ALIF, nH, nO, nHG), and three quantum-chemical (highest hydrogen bond acceptor potential VHHA, highest hydrogen bond donor potential VHHD, and α) allowed good estimate of the SW (Yan and Gasteiger, 2003).
As a conclusion, the descriptors related to the surface (CSA, FISA, FOSA, PSA, S) and volume (SAVw, Vi, Vm, Vxyz) of molecules seem to be the most appropriate ones to estimate the SW of organic compounds, but descriptors such as 0χ, 0χv, the number of chlorine atoms nCl, and the polarizability α (which can be related to the volume) also play an important role. All categories of descriptors were used in the equations.
3.1.2 Octanol-Water Partition Coefficient
The octanol-water partition coefficient, KOW, is the most frequently used parameter to characterize the hydrophobicity (or lipophilicity) of chemicals, which is a very important property in environmental sciences (Katritzky et al., Citation2000). Therefore, a high number of QSAR were developed to predict the KOW. The 115 equations that are reported in Table S4 contain from 1 to 19 molecular descriptors, most of them relying on one, three, or four descriptors, and they were developed for a wide diversity of organic compounds.
As for SW, the prediction of KOW with only one descriptor mainly involved geometric descriptors related to the molecular surface or volume (Table S4). Indeed, the size of the molecule is a major factor in determining its solubility and partition behavior (Doucette and Andren, Citation1988). The FOSA, FISA, or the PSA (Table S1) of polar nitrogen and oxygen atoms allowed good estimates of the KOW of several hormones (r2 > 0.782; Table S4; Cao et al., Citation2009). The FPSA-3 was not well correlated to the KOW of 133 PCB (r2 = 0.251; Table S4; Lü et al., Citation2007), but the total surface area (TSA) was very well correlated with the KOW of PCB and several miscellaneous compounds (r2 > 0.870; Table S4; Doucette and Andren, Citation1988; Hansen et al., Citation1999a; Hawker and Connell, Citation1988; Lü et al., Citation2007), and the SAS with the KOW of 139 PCB (r2 = 0.898; Table S4; Makino, Citation1998). The volume, as for it, was used to predict the KOW of 15 PCDD (Shiu et al., Citation1988), and of 142 compounds including haloalkanes, aromatics, haloaromatics, and alkenes (Bodor and Buchwald, Citation1997). Then, some relationships were found with MW for 139 PCB, only six PBDE or 64 aromatic compounds (Doucette and Andren, Citation1988; Makino, Citation1998; Wania and Dugani, Citation2003), the maximum valency of C atom (MVC) for 133 PCB (Lü et al., Citation2007), nBr for only nine PBDE (Braekevelt et al., Citation2003), and nCl for 15 PCDD (Shiu et al., Citation1988). The addition of chlorine substituents results in an increase in the KOW. In general, topological descriptors such as MCI (Dai et al., Citation1999; Doucette and Andren, Citation1988; Gerstl and Helling, Citation1987; Güsten et al., Citation1991; Sabljic, Citation2001), CRI (Türker Saçan and Inel, 1995), the Lu index (Lu, Citation2009), and T(O…Br) (Papa et al., Citation2009) allowed good estimate of the KOW of various compounds (HBB, PAH, and their alkyl derivatives, PBDE, PCB, polychlorinated organic compounds (PCOC), and phthalates; Table S4). In particular, 0χv, which is a simple and acceptable approximation for the molecular volume, was found to be a good descriptor for the KOW of PAH and their alkyl derivatives (Güsten et al., Citation1991; Sabljic, Citation1991, 2001; Sabljic and Piver, Citation1992) but not for that of nonacid pesticides and miscellaneous organic compounds (Gertsl and Helling, 1987). Among the quantum-chemical descriptors involved in one-descriptor relationships, the ionization potential (IP) and the dipole moment (μ) were used to estimate the KOW of 139 PCB, however, the correlations were very bad (r2 < 0.340; Makino, Citation1998). Yang et al. (Citation2007) modeled KOW using the polarizability α of 75 PCDD and dibenzo-p-dioxins: the greater the α is, the larger the KOW is, suggesting that PCDD molecule with large α possesses great dispersion force and can easily enter the octanol phase. Similarly, the KOW of 133 PCB was correlated with α (Lü et al., Citation2007). Finally, the electron affinity (EA) and the final heat of formation (HOF) were satisfactorily correlated with the KOW of 139 PCB (r2 = 0.743 and 0.870, respectively; Makino, Citation1998), and the total energy (TE) with the KOW of 20 substituted phenols (r2 = 0.843; Xie et al., Citation2008; Table S4).
Using several descriptors of the same category, it was shown that constitutional descriptors based on the numbers of chlorine atoms (N2(6), N3(5), N4) allowed good prediction of the KOW of 107 PCDE (r2 = 0.983) and of 209 PCB (r2 = 0.949; Chen et al., Citation2007; Han et al., Citation2006). As observed before for PCDD (Shiu et al., Citation1988), the higher the number of substituted chlorine atom is, the larger the KOW value of PCDE is. For 22 polychlorinated diphenyl sulfides (PCDPS), the pairwise of Cl atoms at meta position Nm was added to N2(6), N3(5), and N4, and KOW was shown to increase with Nm for compounds with the same number of chlorine substituents (Shi et al., Citation2012). But, these equations were only developed for organochlorines, therefore their applicability domain remains limited. Atom/fragment contribution approaches were used successfully for high number of compounds (Clark, Citation2005; Katritzky et al., Citation2000; Meylan and Howard, Citation1995; Table S4). However, the fragment constant approach leads to oversimplification of steric and conformational effects of complex structures. In addition, there is a need for correctional factors, and it is not possible to estimate KOW for uncorrelated or unknown fragments. Some equations were based on several topological descriptors: MCI, the number of path length (P7, P10), the Balaban index based on distance (Jb), the Wiener index (W), and information indices (CIC, IC, SIC) led to good estimate of KOW of various organic compounds (r2 > 0.652; Table S4; Basak et al., Citation1996; Dai et al., Citation1998; Niemi et al., Citation1992). However, for pesticides and miscellaneous compounds, the KOW were not well correlated to MCI (r2 < 0.425; Table S4; Gerstl and Helling, Citation1987). For 50 aromatic hydrocarbons, 300 pharmaceutical compounds and 14 phthalates, the KOW were correlated to E-state indices (Gombar and Enslein, Citation1996; Huuskonen et al., Citation1999; Thomsen et al., Citation1999), but these indices cannot account for 3D and conformational effects, which may play a major role for solubility properties of chemical compounds. The polarizability α was combined with μ to predict the KOW of 22 PCDPS (Shi et al., Citation2012); with μ and the superdelocalizability of the highest unoccupied molecular orbital (SHOMO) to predict the KOW of 17 ureas (Reddy and Locke, Citation1996); with μ, ELUMO and the largest negative net atomic charge on an atom (q−) for 28 alkyl (1-phenylsulfonyl) cycloalkane-carboxylates (Chen et al., Citation1996a); with EHOMO and ELUMO for 209 PCB (Zhou et al., Citation2005); and with the partial atomic charge on nitrogen (q2N) and the partial atomic charge on oxygen (q2O) to predict the KOW of 592 miscellaneous compounds (Xing and Glen, Citation2002). As indicated before, the bigger the α, the more hydrophobic the molecule is predicted to be: molecules which require a bigger cavity are predisposed to move into the octanol layer. The μ relating with the intermolecular dipole-dipole and dipole-induced dipole interactions also plays a role in the variations of KOW because molecules with larger μ also tend to transfer from octanol phase to water phase (Shi et al., Citation2012). Finally, the atomic charges q2N and q2O are derived from computed charge densities of nitrogen and oxygen atoms of the molecule, which is a measure of their ability to form hydrogen bonds with the solvent molecules (Xing and Glen, Citation2002). For the 209 PCB, EHOMO had the most important influence on KOW. Large values of EHOMO and ELUMO would result in small values of KOW for PCB because EHOMO represents the proton acceptance ability in forming hydrogen bond, while ELUMO represents the proton donation ability in formation of hydrogen bond. Therefore, the compounds with large values of EHOMO and ELUMO tend to donate or accept protons easily (Zhou et al., Citation2005). The KOW of 49 halogenated anisoles was correlated to ELUMO, the most positive atomic net charge on a hydrogen atom (QH+), and the quadrupole moment (Qzz). The KOW increases with increasing QH+, which suggests intermolecular electrostatic interactions between halogenated anisoles and octanol molecules, with the carbon atoms in halogenated anisoles to accept electrons and the oxygen atoms in octanol molecules to donate electrons. On the contrary, KOW increases with decreasing ELUMO (see previous) and Qzz (Zeng et al., Citation2012). Finally, quantum-chemical (EHOMO, q−, QH+, TE, μ) descriptors allowed the estimate of the KOW of 70 PCOC (r2 = 0.931; Table S4; Dai et al., Citation1999), and for 107 PCDE, the equation was based on the interactions between non-hydrogen atoms M11, M13, M22, and M33 (r2 = 0.984; Table S4; Sun et al., Citation2007).
In general, as for SW, the combination of descriptors of several categories improved the quality of the prediction of the KOW (Dai et al., Citation1998; Güsten et al., Citation1991; Lü et al., Citation2007; Makino, Citation1998; Xie et al., Citation2008; Table S4). Numerous relationships were based on the molecular volume (V, VdW, VdWA, Vi, Vm) and/or surface (CSA, PSA, S, SAS, TSA), associated with several constitutional (Ialkane, Iv, MW, Np), topological (MCI, CICi, ICi, P10, 3DW), or quantum-chemical (EA, EHOMO, ELUMO, HOF, IP, QN, qO, QO, QON, SHOMO, SN, TE, VS,max, Vmin, ΔGs, μ, μtot, ΣV−s) descriptors (Basak et al., Citation1996; Bodor and Buchwald, Citation1997; Bodor et al., Citation1989; Edward, Citation1998; Famini and Wilson, Citation1997; Li et al., Citation2008; Makino, Citation1998; Nandihalli et al., Citation1993; Reddy and Locke, Citation1994b, 1996; Schüürmann, Citation1995; Xie et al., Citation2008; Zou et al., Citation2002; Table S4). The molecular volume was a key descriptor because molecules with larger size would tend to distribute into octanol: it is much easier to open a cavity in octanol than in water (fewer hydrogen bonds) therefore larger molecules will preferentially solvate in the octanol layer (Famini and Wilson, Citation1997). For 118 compounds, including basic heterocycles, halogenated compounds, multiple substituted benzenes derivatives, and pharmaceuticals, the geometric descriptors, surface S (indirectly, the volume), and ovality O, but also the MW, which is also a volume-related descriptor, were the most significant descriptors. The need to include Ialkane (classifier indicator for alkane) in the relationship may arise from the different nature of partition of alkanes: they cannot participate in any special interaction, such as hydrogen bonding and electrostatic effect with the surrounding solvent molecules; therefore, the alkanes cannot account for a quasi-structured hydrate environment. All the remaining descriptors are derived from computed charge densities of nitrogen and oxygen atoms of the molecules, these elements being capable of forming hydrogen bonding with the solvent molecule (Bodor et al., Citation1989).
As indicated previously, the polarizability α plays an important role in the prediction of KOW (the greater the α is, the larger the KOW is) and it was involved in several equations (but never in equations containing descriptors related to surface or volume; Table S4). The polarizability α with FPSA-3 and/or MVC, allowed good prediction of the KOW of 133 PCB (r2 = 0.928; Table S4; Lü et al., Citation2007), and with MW and ΔHf it provided satisfactory estimate of the KOW of 106 PCDE (r2 = 0.976; Table S4; Yang et al., Citation2003). The relationship obtained by Patil (Citation1994) for 55 pesticides, and based on carbon to hydrogen ratio C/H, α, and two MCI (0χ and 0χv), cannot be used acceptably for O-analogues and compounds with C/H ratio higher or equal to 2.
Constitutional descriptors, such as the number of the chlorine atoms on phenyl rings NCl, the number of ortho-chlorine substituents normalized to the molecular weight of the compound NrCl0, the number of meta-chlorine substituents ClMETA, the number of meta/para pairs of chlorine substituents ClMP-PAIR or the number of branching points in the carbon skeleton NoB3 were combined to MCI to estimate the KOW of chlorinated compounds and of PAH and their alkyl derivatives (Güsten et al., Citation1991; Sabljic et al., Citation1993), or to quantum-chemical descriptors (the number of independent points of the positive electrostatic potentials on molecular surface N+v, and the average of the sum of the surface minima values of the electrostatic potential V−s,av) to estimate the KOW of 107 PCDE (Xu et al., Citation2010). For PCDE, the NCl term was introduced in the equations because the larger PCDE molecule would yield stronger dispersion-type interaction between the octanol molecule (i.e., the KOW value becomes higher), and tend to be excluded from water. As V−s,av can be viewed as a descriptor reflecting nonspecific intermolecular interactions, the introduction of this descriptor in the equation reveals that nonspecific intermolecular interaction is statistically significant to KOW (Xu et al., Citation2010). For 64 benzotriazoles, the geometric descriptor Geary autocorrelations-lag 3 weighted by atomic masses (GATS3m) was combined with two topological descriptors (2D binary fingerprint that takes into account the presence of C-C (C-C single bond) at a topological distance 8B08[C-C], and Moran autocorrelations lag (1) weighted by atomic van der Waals volume MATS1v), and nN to give correct estimate of the KOW (r2 = 0.886; Table S4; Bhhatarai and Gramatica, Citation2011). With global descriptors, constituted by 19 constitutional and quantum-chemical descriptors, Cheu et al. (1996) modeled the KOW of 30 phenylthio, phenylsufinyl, and phenylsulfonyl acetates. E-state indices associated to MW or to several topological descriptors were used successfully to estimate the KOW of miscellaneous organic compounds (Gombar and Enslein, Citation1996; Tetko et al., Citation2001) but the results were not good for drug compounds (Huuskonen et al., Citation1999; Table S4). Finally, for 122 nonionic organic compounds, the use of several functionality index [η‘F] allowed good estimate of the KOW (r2 = 0.960; Table S4; Roy et al., Citation2007).
Some relationships were developed using LSER, TLSER, and MTLSER approaches (Chen et al., Citation1996a; Dai et al., Citation1998; Kamlet et al., Citation1988; Katritzky et al., Citation2000; Xu et al., Citation2002). In general, the molecular volume was again the most significant descriptor (Famini and Wilson, Citation1997; Feng et al., Citation1996; He et al., Citation1995; Xie et al., Citation2008; Xu et al., Citation2002). The coefficient was positive, indicating an endoergic effect consistent with most cavity/bulk effects. As indicated previously, cavity effects are critical, and therefore larger molecules will preferentially solvate in the octanol layer (Famini and Wilson, Citation1997). For 28 alkyl(1-phenylsulfonyl)cycloalkane-carboxylates, the polarizability α was the most significant descriptor, but it is in direct proportion to the intrinsic molecular volume (Chen et al., Citation1996a). TLSER equation was also combined with a fragment approach to estimate the KOW of 148 various organic compounds (Platts et al., Citation1999 and 2000).
Considering all the results that are summarized in this section, it appeared that the molecular descriptors related to the volume (V, VdW, VdWA, Vi, Vm, Vx) were the most relevant ones to assess the KOW of organic compounds, but MW, 0χv, 1χv, α, μ, EHOMO and ELUMO were also frequently involved in the equations and found to allow good estimate of the KOW. All categories of descriptors were used in the equations, except geometric-topological descriptors.
3.2 Dissociation Process
The dissociation constant, pKa, which describes the extent to which a compound dissociates in solution, is a fundamental physical property of a chemical. It is a key feature, which governs the chemical reactivity of the substances with other compounds in any solvent, and also the interaction with the solvent itself, and thus its hydrophobicity and water solubility (Citra, Citation1999; Jover et al., Citation2008). Lee and Crippen (2009) focused on the description of the methods used to predict the pKa (e.g., quantum chemical and continuum electrostatic methods or artificial neural networks), rather than to review the different descriptors used in the relationships. This section is therefore different and complementary to the work of Lee and Crippen.
A very high number (145) of QSAR were developed to estimate the pKa of organic compounds (Table S5). Most of these relationships use only one molecular descriptor but they can use up to eight descriptors, and most of the descriptors are quantum-chemical descriptors (the pKa values themselves reflect electronic properties in a direct manner). Few constitutional, geometric, and topological descriptors have been also involved (Table S5). The QSAR were developed for several specific classes of organic compounds but not, for example, for pesticides (Table S5).
The estimation of the pKa using one descriptor mostly involves the electrophilic superdelocalizability (SE) of the atoms N, C or O. The best correlations were found for anilines and amines, and ortho phenols (r2 > 0.900), and the worst for heterocycles, pyridines and tertiary amines (r2 < 0.570; Tehan et al., Citation2002a; Tehan et al., Citation2002b; Yu et al., Citation2010; Table S5). SE quantifies the susceptibility of a molecule for an electrophilic attack. The negative value of SE indicates that increasing SE correlates with increasing pKa and thus decreasing acidity (Yu et al., Citation2010; Table S5). The charges of atoms or groups were also involved in numerous equations: the atomic partial charges of carbon connected to hydroxyl or carboxyl groups (apc(C)), the atomic partial charges of hydrogen of the hydroxyl group (apc(H)), the atomic partial charges of oxygen of the hydroxyl group (apc(O)) (Hanai, Citation2003), the natural charge on the amino nitrogen (Qn(N)) (Gross and Seybold, Citation2000; Gross et al., Citation2001), the natural charge on the neutral amino group (Q(NH2)), the natural charge on the cationic ammonium group (Q(NH3+)) (Seybold, Citation2008), the natural charge on the phenoxide oxygen (Qn(O−)) and the natural charge on the phenolic hydrogen (Qn(H)) (Gross and Seybold, Citation2001), the Mulliken charge of COO− group (QM(COO−)), the Löwdin charge of hydrogen (QL(H)) and of COOH (QL(COOH)), the AIM charge of hydrogen (QA(H)), the natural population analysis charge of COOH group (Qn(COOH)) (Hollingsworth et al., Citation2002), the charge on the acidic hydrogen (QacidH) and the charge on the basic nitrogen (QbaseN) (Brown and Mora-Diez, Citation2006b), and the charge on phenolic O or phenolate O atoms, (dOphenolic or dOphenolate) (Grüber and Buß, Citation1989). In general, the correlation coefficients were high (r2 > 0.810), except for protonated benzimidazoles (r2 = 0.644), but the number of compounds used to develop the QSAR were sometimes very low, in particular for fluorinated ethylamines (Table S5). For phenols, the significant correlation between pKa and dOphenolic (r2 = 0.810; Table S5) can be explained by the heterocyclic dissociation of the OH bond that should be facilitated by a positive charge on O, both by polarizing the bond and by accommodating the developing negative charge. The correlation with dOphenolate was nevertheless better: the charge distribution of the anion, especially at the acidic center, is an indicator of the ability of the system to accommodate excess negative charge and as such should correlate with acid strength (Grüber and Buß, Citation1989). The Qn(H) and the Qn(O−) should serve as good measures of acidity, more acidic hydrogens having lower electron densities. In the same manner, delocalization of the phenoxide oxygen negative charge is expected to impart stability to the phenoxide, favoring its formation and lowering the pKa (Gross and Seybold, Citation2001). The QacidH is related to the delocalization of charge over the molecule and in turn to the acidity: a lower value of QacidH indicates good delocalization and should produce a higher pKa. The higher pKa could also be explained by stating that the lower the positive charge on the acidic hydrogen, the less polarized the N-H bond and the less acidic the compound. The values of QbaseN become more negative with increasing pKa which indicates that the larger the negative charge on the basic nitrogen of the neutral molecular form, the stronger it will be as a base, hence a weaker acid once protonated; that is well illustrated in the case of the benzimidazole (Brown and Mora-Diez, Citation2006b). Hanai (Citation2003) developed another approach to estimate the pKa: the pKa is the sum of the pKa (base compounds) derived from the atomic partial charge (apc) of basic compounds, and of ΔpKa (substitute effect) derived from the difference in the atomic partial charge between derivatized and base compounds. Good correlations between the partial charges apc(H), and apc(O) were obtained, but again the number of data is very low (r2 > 0.864; Table S5). The substitute effect on atomic partial charge Δpc was balanced between the atomic partial charge of the substituted phenol and phenol, and was well correlated with ΔpKa (r2 > 0.900; Table S5; Hanai, Citation2003).
Equations were also developed with the molecular electrostatic potential minima (Vmin), the surface molecular electrostatic potential minima (VS,min) or the surface molecular electrostatic potential maxima (VS,max) for phenols, phenolates, benzoic acids, benzoates and anilines. In any case, the correlation coefficients were very good (r2 > 0.868; Table S5). Vmin and VS,min are related to the initial attraction that brings the proton into the vicinity of the amino group. When the VS,min values are more negative, the anionic conjugate base is more attractive to the approach of an electrophile, and the acidity is lower. VS,min indicates how negative is the electrostatic potential of the oxygen, and VS,max how positive is the electrostatic potential of the hydrogen. The VS,min values of the conjugate bases reflect the tendencies of electrophiles to approach the anions to reform the acids; the VS,max values of the acids are good indicators of the ease of proton loss (Gross et al., Citation2001; Ma et al., Citation2004). Some equations were found with the molecular electrostatic potential on the acidic atom (MEP) for amines, anilines, carboxylic acids, alcohols, sulfonic acids, and thiols (r2 > 0.878; Table S5). The pKa of these compounds were also correlated to ΔMEP (i.e., the MEP evaluated for the isolated neutral acidic atom subtracted from the MEP value on the acidic nucleus for each category of compounds; Liu and Pedersen, 2009).
Several QSAR were developed using the energies of orbitals (EHOMO, EacidHOMO−1, EacidHOMO, EbaseHOMO-2, EbaseHOMO-1, EbaseHOMO, ELUMO, EacidLUMO, EacidLUMO+2, EbaseLUMO; Brown and Mora-Diez, Citation2006b; Gross and Seybold, Citation2001; Grüber and Buß, Citation1989; Soscún Machado and Hinchliffe, 1995; Tehan et al., Citation2002a; Yu et al., Citation2010; Table S5). For 18 protonated benzimidazoles, the EacidLUMO+2 showed the strongest correlation to the experimental data (r2 = 0.843; Table S5), followed by EbaseHOMO-2 (r2 = 0.768; Table S5): an empty orbital of an acid will be more involved in its deprotonation process, while an occupied orbital of the base will be more involved in its protonation process. The energy of the LUMO of the protonated species is related to the formation of hydrogen bonds with the solvent molecules and the subsequent deprotonation. A lower value for EacidLUMO means that hydrogen bonds will form with greater ease, and allows for easier deprotonation and a lower pKa (Brown and Mora-Diez, Citation2006b). Among the other quantum-chemical descriptors, the sum of the valence p natural atomic orbitals of the atom (NAO) was correlated to the pKa of amines, anilines, carboxylic acids, alcohols, sulfonic acids, and thiols (r2 > 0.905; Table S5; Liu and Pedersen, 2009). For 17 benzoic acids and benzoates, 19 phenols and phenolates, and 36 anilines, pKa values increase as the minimum molecular surface local ionization energy (IS,min) values decrease. The IS,min is related to the subsequent charge sharing or charge transfer. When the IS,min values are low, there is a greater tendency for a proton to transfer back to form the neutral acid, leading to a lower acidity and a higher pKa (Gross et al., Citation2001; Ma et al., Citation2004). Finally, some relationships were found using (a) the OH bond order (BOOH): the pKa of 17 substituted benzoic acids are proportional to the strength of the bond between hydrogen and oxygen in the carboxylic acid group (Hollingsworth et al., Citation2002); (b) the energy difference between acid and conjugated base (Hf) for phenols (Grüber and Buß, Citation1989); (c) the energy difference in aqueous phase between the neutral amines and their cationic forms (ΔEaq) for 26 amines (Seybold, Citation2008); (d) the energy difference in gas phase between the neutral amines and their cationic forms (ΔEd) for four fluorinated ethylamines and 28 amines (Seybold, Citation2008); (e) the relative proton transfer energy (ΔEprot) for 17 substituted benzoic acids or 19 phenols derivatives, or the relative proton transfer enthalpy (ΔHprot) for 36 anilines (Gross and Seybold, Citation2001; Gross et al., Citation2001; Hollingsworth et al., Citation2002): a positive value of ΔEprot indicates that the substituted phenol is less acidic than phenol itself, and a negative ΔEprot suggests the substituted phenol is more acidic; (f) the aqueous Gibbs free energy (ΔGaq) for benzimidazoles (Brown and Mora-Diez, Citation2006a, 2006b); (g) the Gibbs free energy of dissociation (ΔGdiss) for 64 organic and inorganic acids (Klamt et al., Citation2003); (h) the standard free energy (ΔGO) for 12 aliphatic, alicyclic, and aromatic amines (Kallies and Mitzner, Citation1997); (i) the fractional number of electron transfer (ΔN), or the associated energy change (ΔEe) for 58 acids (Gupta et al., Citation2007): a larger ΔEe value denotes a stronger Lewis acid and that corresponds to a smaller pKa value implying a stronger Brønsted acid. A larger value of ΔN indicates a greater amount of electron transfer and hence a better Lewis acid-base pair. Since the base remains the same for all the acid-base pairs studied, a larger ΔN would indicate a stronger acid and a smaller pKa value (stronger Brønsted acid).
With topological descriptors, relationships were developed with the group philicity (ωg+) for small number of compounds: nine substituted phenols, nine alcohols or 14 substituted anilines and phosphoric acids, but also for 31 substituted carboxylic acids (r2 > 0.722; Table S5; Parthasarathi et al., Citation2006), and with two modified MCI, 1χf and 1χfλ, for 31 carboxylic and halogenated carboxylic acids (Pompe and Randic, Citation2007; Table S5). For 18 benzimidazoles, Brown and Mora-Diez (2006b) tried to develop a relationship with the change in the volume of the solvent cavity going from the protonated to the neutral species (ΔVSC), but the correlation was poor (r2 = 0.269; Table S5).
The QSAR based on several descriptors of the same category only involve quantum-chemical descriptors. Ten equations combine self-polarizability (e.g., ALPC1, ALPC2, ALPC4, ALPO3, and ALPN1), partial atomic charges of atom N or O at position 1 (e.g., AQN1, AQO1, AQO2, AQO3), superdelocalizability (e.g., SEC1, SEC2, SEC4, SEN, SEN1, SEO, and SEO3), charge descriptors (e.g., Coulson net atomic charge of the nitrogen atom QC(N), net atomic charge at the carbonyl O of the carboxylic group Q = O), the nucleophilic frontier electron density of atom N at position 1 (FNN1), the electrophilic frontier electron density of atom O at position 1 or 3 (e.g., FEO1, FEO3), and ELUMO (r2 > 0.690; Table S5; Tehan et al., Citation2002a; Tehan et al., Citation2002b; Yu et al., Citation2010). One relationship was based on four charges of carbon at different positions (dC4, dC12, dC16, dC18), and one charge of oxygen (dO11) for 48 benzoic acids (r2 = 0.880; Table S5; Grüber and Buß, Citation1989). For phenols, and aromatic and aliphatic carboxylic acids, EHOMO, Hf and charges play an important role in the prediction of pKa. The other descriptors, relying on atomic charges, enter the regression equations with negative signs showing that the acid strength increases as excess negative charge decreases. For phenols, the high regression coefficient of the meta-carbon (dC15) seems to indicate that resonance effects which are primarily related to ortho- and para-positions do not play a dominant role in differentiating acid strength (Grüber and Buß, Citation1989). Three relationships contained different combinations of two of the following descriptors (Yu et al., Citation2011): the energy-weighted acceptor energy (EEvac), the energy-weighted donor energy (EEocc), the charge-limited acceptor energy (EQvac), the energy-limited acceptor charge (QEvac), and the energy-limited donor charge (QEocc). The correlations were good (r2 > 0.820; Table S5). For 29 phenols with intramolecular H bonding, the pKa was correlated to the EEvac evaluated at the acidic H atom, and EEocc evaluated at the oxygen atom bonded to this H atom. The EEvac increases with increasing electron acceptor strength of H, indicating a larger energy demand for ionizing H to become H+. Thus, EEvac increases with decreasing acidity and accordingly increasing pKa, which is reflected by its positive regression coefficient. The EEocc evaluated at the oxygen bonded to H increases with increasing oxygen donor strength, which in turn reflects an increasing OH bond strength and thus a lower tendency for bond fission. Accordingly, EEocc correlates with decreasing acidity and thus with increasing pKa. EEvac evaluated at H is also used as local reactivity parameter of the pKa prediction models calibrated for the 190 aliphatic carboxylic acids. For this compound class, the QEocc is used as second molecular descriptor. QEocc increases with increasing amount of loose electron charge ready for donation, which stabilizes the carboxylic O-H bond. QEocc increases with decreasing acidity (decreasing O-H bond fission tendency) and thus with increasing pKa (Yu et al., Citation2011). The pKa of 19 phenols and phenolates was well correlated to IS,min and VS,max (r2 = 0.908; Table S5; Ma et al., Citation2004), and that of 13 benzimidazoles was found to depend on the difference in molar energies of the ground states of the product and reactants at 0 K (ΔEo), molecular partition functions (qfi), and the stoichiometric coefficient (νi) of each species (Brown and Mora-Diez, Citation2006a; Table S5). Other equations involve the gas-phase free energy of the proton abstraction (ΔGg) and the solvation free energy (ΔGs) for only eight weak acids (Topol et al., Citation2000); or the charge on hydrogen atom (qH) and the net atomic charges on the oxygen atom (qO) combined with BOOH for various compounds (Citra, Citation1999; Table S5).
There is no result allowing the comparison of equations using one category of descriptors and several categories of descriptors (Table S5). Using constitutional descriptors such as Iortho associated with EEvac, Yu et al. (Citation2011) found good pKa predictions for 150 aromatic carboxylic acids (r2 = 0.820; Table S5). The Iortho has a negative regression coefficient, indicating an increased acidity for ortho-substituted compounds. Ortho substitution might destabilize the molecular ground state as compared to meta and para substitution, and the associated steric repulsion decreases upon dissociation, thus supporting the cleavage of H+. For 190 phenols without intramolecular H bonding, the pKa was correlated to EQvac, QEvac, and Iortho (r2 = 0.900; Table S5). The H atoms with a large EQvac prefer more strongly to retain electron charge and accordingly provide more resistance in donating charge to their bonding partner in order to become dissociated. Larger QEvac values reflect larger amounts of electron charge per unit energy transferred to H and thus a larger polarizability of H in its bonding situation. Because increasing polarizability indicates a decreasing resistance to changing local electron density, QEvac increases with increasing readiness of the acidic H atom to become ionized upon dissociation. Accordingly, QEvac increases with increasing acidity and thus with decreasing pKa as indicated by the negative sign of its regression coefficient. For 288 alcohols, the pKa was correlated to the inductive descriptor of the acidic oxygen atom in an acid (Qσ,O), the pi-electronegativity for the α carbon atom in an acid (ENπ,αC), Icarboxy, and Iamino (Zhang et al., Citation2006). The negative coefficient sign for the atomic inductive descriptor Qσ,O is consistent with the physical meaning of this descriptor, as a large Qσ,O value means a large inductive effect of the substituent. For 1122 aliphatic carboxylic acids, the best multiple linear-regression equation is obtained with five molecular descriptors: the accessibility of the acidic oxygen atom in an acid (Aaccess,O (2D)), ENπ,αC, Iamino, Qσ,O, and αO (Zhang et al., Citation2006). As for alcohols, there is a small pKa value if there is a large Qσ,O value. The positive sign for Aaccess,O (2D) means that a large steric hindrance will increase the pKa and therefore decrease the acidity of the acid, as access of water to take up the proton is hindered (Zhang et al., Citation2006). The geometric descriptor ΔVSC combined with EacidLUMO+2 and QacidH provided excellent results to estimate the pKa of benzimidazoles (r2 = 0.990; Table S5) however the number of data used for the correlation was not indicated in the study (Brown and Mora-Diez, Citation2006b). For 15 imidazol-1-yl alcanoic compounds, there was a good correlation between pKa and ELUMO, nester, and QN3 (r2 = 0.978; Table S5). For the protonated species, LUMO is located over the azole ring. A low ELUMO would suggest an easy formation of a hydrogen bond with the water molecule and donation of a proton by overlapping with the HOMO of water, namely, a low pKa (Soriano et al., Citation2004). Finally, the charges on hydrogen (qHδ+) and on oxygen (qOδ-) atoms combined with the change in O-H bond length (bl(OH)) provided good assessment of the pKa of 74 aromatic acids derivatives (r2 = 0.988; Table S5). Increasing qOδ- on the hydroxyl oxygen induces more hard characteristics to oxygen and consequently lowers the tendency for dissociation: this causes the molecule to be a weaker acid (Ghasemi et al., Citation2007).
Using artificial neural network methodology, Jover et al. (2007, 2008) introduced, in addition to five descriptors relative to the solute, two descriptors relative to the solvent. For 136 benzoic acids, they found good correlations between the pKa and the FPSA-2, the maximum electron-electron repulsion for an O atom (MaxeeO), the minimum resonance energy for O-H bond (MinOH) the maximum valency of C atom (MVC) and the maximum partial charge for a hydrogen atom (Qmax(H)) (in all cases, the involved hydrogen atom corresponds to the carboxylic group) for the solute, and the hydrogen bond donor acidity (αm) and the standard internal energy of vaporization (ΔvapUO) for the solvent (Jover et al., Citation2008). Thus, four solute descriptors of this model are related to the stability, reactivity, or the tendency to dissociate the carboxylic group. The Qmax(H) reflects the polarity of the OH bond that is cleaved in the dissociation process. Also, the MinOH descriptor is related to the energy of the bond that dissociates, and MaxeeO and MVC account for the reactivity of the atoms, respectively. On the other hand, the fifth solute descriptor, FPSA-2, represents a density of charge of the solute and explains the intermolecular interactions, in this case those between the benzoic acid and the solvent. All these solute descriptors can be associated to the nonspecific solute/solvent interactions. Of the two solvent descriptors, the ΔvapUO indicates the energy involved in the solute cavity formation in the bulk of the solvent in the dissolution process and can also be associated to the nonspecific solute/solvent interactions. The αm is associated to the specific hydrogen-bonding interactions and stands for the hydrogen-bond donor properties of the solvents. Similarly, for 199 phenols, the pKa was correlated to ELUMO+1, the maximum electron-neutron attraction for a C-O bond (MaxenC-O), the maximum partial charge for a hydrogen atom (Qmax(H)), the relative positive charged surface area (RPCS), and the polarizability α for the solute; and αm and μ for the solvent. The dipole moment μ of the solvent, and the solute descriptors MaxenC-O, RPCS, and α contain information related to the nonspecific solute/solvent interactions. On the other hand, the ELUMO+1 and αm encode information related to the specific interactions (Jover et al., Citation2007).
Methods such as principal component-genetic algorithm-multiparameter linear regression (PC-GA-MLR) and principal component-genetic algorithm-artificial neural network (PC-GA-ANN) models were employed to predict the pKa of miscellaneous organic compounds (Habibi-Yangjeh et al., Citation2009; Lee and Crippen, 2009). For 282 nitrogen-containing compounds, 15 principal components were first selected by PC-GA-MLR, then descriptors having the highest correlations with the principal components were retained, leading to a selection of eight descriptors: two constitutional (H attached to heteroatom H-050, number of total primary C(sp3) nCp), three geometric (Geary autocorrelation-lag 1 weighted by atomic polarizabilities GATS1p; 3D-MoRSE-signal 12 weighted by atomic polarizabilities Mor(12)p; 3D-MoRSE-signal 31 weighted by atomic van der Waals volumes Mor(31)v), two topological (mean information content index based on the zero-order neighborhood of vertices in a graph IC0, structural information content of a graph based on one-order neighborhood of vertices SIC1) and one electro-topological (constitutional descriptor of the mean E-state of the molecule related to the polarizability, Ms). The Mor(12p) and Mor(31)v descriptors relate to polarizabilities and van der Waals volumes of the atoms, respectively, whereas IC0 and SIC1 are related to information content, which is a measure of the degree of diversity of the elements in the set. The Ms gives information related to the electronic and topological state of the atom in the molecule. Therefore, it is concluded that polarizabilities and van der Waals volumes of the atoms, diversity of the elements in compounds, electronic state of the atoms in the molecule, and the number of first neighbor (hydrogen) of heteroatom play main roles in the pKa of the compounds. With the same 15 principal components, the PC-GA-ANN method gave higher correlations (Habibi-Yangjeh et al., Citation2009; Table S5). Finally, some relationships can take into account the temperature (Brown and Mora-Diez, Citation2006a; Topol et al., Citation2000).
In summary, the most efficient descriptors to predict the pKa were related to the superdelocalizability, the charges and to EHOMO. No geometric-topological and no geometric-electronic descriptors were used in the equations, and contrary to SW and KOW, no MCI was involved.
3.3 Volatilization Process
Volatilization from soil and volatilization from leaf surfaces are distinguished as the factors that are involved may be different. Indeed, trying to correlate observed volatilization fluxes with physicochemical properties of the compounds, the vapor pressure alone (liquid vapor pressure PL, solid vapor pressure PS) and the Henry's law constant (KH) divided by the adsorption coefficient are often found to give the best fit when considering volatilization from leaves and from soil, respectively (e.g., Woodrow et al., Citation1997). The octanol-air partition coefficient (KOA) is also thought to be a key descriptor of the distribution of semi-volatile compounds between the atmosphere and terrestrial organic phase (Zhao et al., Citation2005). Finally, a volatility index (VIN) can also be used to differentiate between volatile and nonvolatile compounds (Gramatica and Di Guardo, 2002).
3.3.1 Vapor Pressure
The 40 equations allowing the estimate of vapor pressure involve from 1 to 12 descriptors, and more than half of them are only based on one descriptor. The QSAR were developed for a wide diversity of organic compounds, but not for pesticides (Table S6).
In contrast to other environmental relevant compounds properties such as water solubility or lipophilicity, vapor pressure is highly dependent on temperature. Therefore the temperature was introduced in a few number of QSAR (Ding et al., Citation2006; Kühne et al., Citation1997).
The prediction of the liquid vapor pressure (PL) of organic compounds with one descriptor mainly involved the polarizability α, and correlations were good (r2 > 0.731; Table S6; Liang and Gallagher, Citation1998; Staikova et al., Citation2004). In any case, an increase in α led to a decrease in PL. Indeed, α is related to dispersion forces or induced dipole-induced dipole interactions, which are the main component of the intermolecular forces in nonpolar compounds. On the other hand, polarizability α showed lower correlations for relatively polar compounds, such as the alcohols, amines, and halogenated ketones. This may be due to the potential for hydrogen bonding and/or dipole interactions which are not adequately accounted for by α (Liang and Gallagher, Citation1998; Table S6). Other equations involve nCl, MW, Vm, or T(O…Br) as single descriptor (Papa et al., Citation2009; Shiu et al., Citation1988; Wania and Dugani, Citation2003). PL tends to fall by a factor of eight per chlorine atom added (Shiu et al., Citation1988).
Using several topological descriptors, Basak et al. (1997) developed four relationships to estimate the PL of 476 various organic compounds (Table S6). The efficiency of topological descriptors to estimate PL (r2 ranges from 0.515 to 0.804; Table S6) showed that adjacency and distance in chemical graphs, being general descriptors of molecular size, shape, and branching, are important in predicting this parameter. For 23 PBDE or 72 PCDE, PL was very well related to three quantum-chemical descriptors: the largest negative net atomic charge on an atom (q−), α and μ (r2 > 0.988; Table S6; Wang et al., Citation2008; Zeng et al., Citation2007). PCDE molecules with great absolute values of q− tend to have great intermolecular electrostatic interactions and limited volatilization. The larger the value of μ is, the smaller the value of PL is, because intermolecular interactions are in direct proportion to μ2, therefore PCDE molecules with larger μ values tend to volatilize less. As indicated previously, increasing α value of the PCDE leads to decreasing PL (Zeng et al., Citation2007). For 15 PCDD, PL was also very well correlated to α and to the most positive atomic net charge on a hydrogen atom (QH+; r2 = 0.985; Table S6). Again, the PCDD with larger α values tend to have lower PL values. The more chlorine atoms present in the parent structures, the greater α is, and thus the lower the PL value is. Increasing QH+ values leads to a decrease in PL values because PCDD molecules with great QH+ values also tend to have great intermolecular interactions that reduce volatilization (Zeng et al., Citation2013). The PL of 11 chlorinated compounds was again well correlated to α, and Qzz (r2 = 0.952; Table S6) but it has to be noticed that the number of data used to develop the equation is low (Staikova et al., Citation2004). For 72 PCDE, the relationship involved the number of chlorine atoms at different positions: the more the number of substituted chlorine atoms is, the lower the PL value of PCDE is. Furthermore, PL decreases with the different chlorine positions: N2(6) < N3(5) < N4. This indicates that congeners with Cl substitutions in the ortho positions to the etherlink, that is, the (2, 6, 2, 6′) positions, have higher vapor pressures compared to those with Cl substitutions in the nonortho positions to the ether-link, that is, the (3, 5, 3, 5’) and (4, 4’) positions. Furthermore, for compounds with the same number of chlorine substitution, the values of PL increases with the increase in pairwise of Cl atoms at meta position (Nm; Zeng et al., Citation2007). Finally, PL of 107 PCDE was correlated to five quantum-chemical molecular electronegativity-distance vectors (M11, M12, M13, M23, M33): M11, M13, and M33 increase with the degree of chlorination, but M22 decreases with increasing degree of chlorination. PCDE congeners with higher chlorination have lower PL (Sun et al., Citation2007).
As observed in previous sections for SW and KOW, the combination of descriptors of different categories allows improvement in the assessment of PL (Basak et al., Citation1997; Liang and Gallagher, Citation1998; Table S6). Quantum-chemical descriptors were involved in almost all equations, combined with constitutional and/or geometric descriptors (Table S6). For 22 PBDE, PL was well correlated to the molecular volume V, and to the sum of the surface maxima values of the electrostatic potential ΣVs+ (r2 = 0.981; Table S6). Indeed, the larger PBDE molecule would yield stronger dispersion-type interaction between each other (lowering the volatility and liquid vapor pressure; Xu et al., Citation2007). For 107 PCDE, PL can be estimated with NCl, ΣVs+, and σ2tot (Xu et al., Citation2010), but also with MW, TE, α, and ΔHf (Yang et al., Citation2003). Based on these descriptors, it can be concluded that PL of PCDE congeners are mainly governed by the intermolecular dispersive interactions (Yang et al., Citation2003). The dispersion forces are a function of the molecule's polarizability, while hydrogen bonding can be facilitated by the presence of ‒OH, ‒NH, or ‒SH groups. Indeed, the addition of the number of polar functional groups ‒OH (nOH), ‒C = O (nC = O), ‒NH (nNH), ‒COOH (nCOOH), ‒NO2 (nNO2), and ‒C≡N (nC≡N) to the polarizability α allowed improvement of the estimate of PL (r2 = 0.960; Table S6; Liang and Gallagher, Citation1998). The PL of 17 benzenes was correlated to the molecular contact surface area (CSA) and to the solvation free energy (ΔGs): positive ΔGs coefficient and negative CSA coefficient agree with theory because increasing (negative) electrostatic interactions and increasing (positive) dispersion interactions both lead to decreasing PL and thus decreasing volatilization (Schüürmann, Citation1995). For 411 compounds, a five-descriptor performing relationship (r2 = 0.949; Table S6) was developed: the most important descriptor was the gravitation index (IG), and the second most important descriptor was the hydrogen-bonding donor charged surface area (HDCA(2)). The combination of IG and HDCA(2) adequately represents the forces of intermolecular attraction: IG is connected with the dispersion and cavity-formation effects in liquid, and HDCA(2) is connected with the hydrogen-bonding ability of compounds. The three additional descriptors were the maximum net atomic charge for a chlorine atom (MNAC(Cl)), the sum of the surface area of fluorine atoms (SA-2(F)), and the surface area of nitrogen atoms (SA(N)). The inclusion of these three descriptors in the model shows that the IG and HDCA(2) could not describe adequately the intermolecular interactions with solute molecules containing fluorine, chlorine, or nitrogen atoms (Katritzky et al., Citation1998). Finally, for 33 benzotriazoles, Bhhatarai and Gramatica (Citation2011) developed a relationship based on combination of one constitutional number of rotatable bonds (RBN), and two topological descriptors (BCUT 2D descriptor encoding the lowest eigenvalue number 2 of the Burden matrix weighted by atomic polarizabilities, BELp2; and 2D binary fingerprint that corresponds to the presence of a N-Cl bond at topological distance 9, B09[N-Cl]). They found good results (r2 = 0.809; Table S6).
Only two relationships were reported to estimate the solid vapor pressure PS of 257 PCDD and PCDF, they involve the temperature (Ding et al., Citation2006; Table S6). The main factors governing PS values, from important to less important, are temperature, intermolecular dispersive interactions (through α), entropic factor (through the Kier index S0K), and intermolecular dipole-dipole and dipole-induced dipole interactions (through μ).
The synthesis of these results shows that the polarizability α is a fundamental descriptor to explain the vapor pressure range of organic compounds and to estimate this parameter. No geometric-topological and no electro-topological descriptors were used in the equations.
3.3.2 Henry's Law Constant
Henry's law constant, KH, is a physical property of a chemical that is a measure of its partitioning between two phases, air and water. Chemicals with low KH will tend to stay in the aqueous phase, while those with high KH will partition more into the gas phase. As air and water are the major compartments of the environment, and water is considered to act as a vector among air, soil, sediment, and biota, the knowledge of KH is very important in assessing environmental risks associated with a chemical (Nirmalakhandan and Speece, Citation1988b). Dearden and Schüürmann (Citation2003) reviewed some of the methods developed for the estimation of KH. This review is completed and updated in this section. The 17 equations reported in Table S6 involve from 1 to 10 descriptors. As for vapor pressure, the QSAR were developed for a wide diversity of organic compounds but not for pesticides.
Seven one-descriptor equations are reported in Table S6 to estimate KH. For 15 PCDD, KH was correlated to nCl: KH tend to fall by a factor of 1.6 per chlorine added (Shiu et al., Citation1988). KH was also correlated to geometric descriptors such as the molecular volume Vm for 15 PCDD (Shiu et al., Citation1988), or the TSA for 58 PCB (Brunner et al., Citation1990). With topological descriptors, QSAR were based on 4χpc for 58 PCB (Brunner et al., Citation1990), and on the highest eigenvalue number 7 of Burden matrix, weighted by Sanderson electronegativities (BEHe7) for 209 PBDE and HBB (Papa et al., Citation2009). BEHe7 brings 2D information which takes into account the weight of different atoms in the structure (Burden matrix) and their electronegativities (Papa et al., Citation2009). The quantum-chemical descriptor average of the sum of the surface minima values of the electrostatic potential (V−s,av) provided correct prediction of the KH of 7 PBDE but based on a low number of compounds (r2 = 0.929; Table S6; Xu et al., Citation2007). Finally, for 17 benzenes, a relationship was developed using the solvation free energy (ΔGs). An incomplete account of the cavity formation energy in water in the ΔGs term could have lowered the regression coefficient, which is nevertheless good (r2 = 0.830; Table S6; Schüürmann, Citation1995).
QSAR with several constitutional descriptors (nCl and northo Cl) or with several MCI (4χ, 4χpc, or 6χpc) allowed good estimate of KH of PCB (r2 > 0.908; Table S6; Brunner et al., Citation1990; Sabljic and Güsten, Citation1989). Sabljic and Güsten showed that the degree of ortho-substitution was the major factor that governs the magnitude of KH. In their model, this structural feature is described by 4χ whose size is proportional to the number of ortho-chlorine atoms. Its positive regression coefficient indicates that the PCB with the greatest ortho-substitution have higher KH and show tendency to stay in the air. The second structural property that controls the KH is the relative position of chlorine substituents (i.e., their distribution between two phenyl rings and their relative position within each ring). The accumulation of chlorine atoms on one phenyl ring in di- and trichlorinated biphenyls tends to increase the value of corresponding KH. This structural feature is in part described by the 4χc whose size is proportional to the extent of adjacent substitution. Congeners which have chlorine substituents together have smaller KH. The third structural feature identified to influence the KH is the degree of branching. This feature is also described by 4χc, which is highly sensitive to changes in branching. The negative regression coefficient indicates that branching lowers the magnitude of KH. Unfortunately, branching overlaps, to a certain degree, some of the structural features described previously (i.e., the extent of adjacent substitution; Sabljic, Citation1991; Table S6). For seven PBDE, KH was correlated to two quantum-chemical descriptors: V−s,av and the equilibrium parameter of electrostatic potentials on molecular surface (ν). The correlation coefficient is high (r2 = 0.998; Table S6), but the number of data points submitted to the regression is low. The larger ν value is, the better the balance between positive and negative electrostatic potentials is, and the smaller KH is (Xu et al., Citation2007).
Meylan and Howard (Citation1991) developed a bond contribution method based on 345 compounds to estimate KH. The advantage of the bond contribution method is its ability to estimate KH for many types of chemical structures. It is relatively accurate for predicting KH for hydrocarbons, monofunctional compounds, and many multifunctional compounds. The major disadvantage is that inaccuracy is introduced by the occurrence of multiple polarizable groups. As chemical structures become more complex, the bond contribution method is more likely to become inaccurate. Comparing several methods of KH estimation (experimental, bond contribution, MCI, and LSER), Brennan et al. (Citation1998) found that the bond contribution method of Meylan and Howard (Citation1991), and the use of MCI as done by Nirmalakhandan and Speece (1988b; see next paragraph), were the most performing ones.
As for vapor pressure, the combination of descriptors of different categories improves the estimate of KH (Schüürmann, Citation1995; Table S6); and quantum-chemical descriptors were involved in almost all equations. Modarresi et al. (Citation2007) developed a performing 10-descriptor QSAR for a wide set of 770 organic compounds (r2 = 0.925; Table S6): the model involved four constitutional, one geometric, one geometric-electronic, and four quantum-chemical descriptors. The four constitutional descriptors were nF, nNO2, nOH, and nR6. The nNO2 most probably represents hydrogen-bond acceptor ability, and the nOH reflects molecular capability for hydrogen-bond acceptance or donation. The quantum-chemical descriptors, sum of absolute Ca and Cd values (hydrogen-bond free energy acceptor and donor factors, respectively) for all H-bond donor and acceptor atoms in molecule (ΣCad(o)), and largest Ca (hydrogen-bond free energy acceptor) factor value in molecule (Max(Ca(o))), demonstrated marked influences on the performance of the QSAR model. Inclusion of hydrogen bonding descriptors reveals that hydrogen bonding is the most important molecular feature of solvent-solute interaction in governing KH of organic compounds in the air-water system. These results are consistent with the nature of water molecules as solvent, as they are very good hydrogen-bond acceptors and donors. Electrostatic intermolecular forces between solute and solvent molecules are characterized by the Geary autocorrelation-lag 1 weighted by atomic Sanderson electronegativities (GATS1e), the partial negative surface area (PNSA-1), and the relative positive charge based on quantum chemical partial charge (RPCG; Modarresi et al., 2007). For 31 PCB, Dunnivant et al. (1992) related KH to two topological descriptors (path-three κ index 3κ, and 4χ) and three quantum-chemical descriptors (second moment of inertia SMI, second- and third-principal polarizability α2 and α3; r2 = 0.899; Table S6). For 17 benzenes, KH was found to be correlated to ΔGs and CSA (r2 = 0.870; Table S6; Schüürmann, Citation1995), and for 180 miscellaneous organic compounds KH was correlated to α and MCI (0χ and/or 1χv; r2 > 0.932; Table S6; Nirmalakhandan and Speece, Citation1988b). Finally, Goss (Citation2006) used the TLSER equation to estimate the KH of 408 miscellaneous organic compounds and obtained very good results (r2 = 0.998; Table S6).
The temperature dependency of KH was only taken into account in one model, developed from 456 miscellaneous organic compounds. This model is based on 45 parameters: 18 fragments, 26 correction factors, and 1 indicator (presence of halogen in 2-position to ring O as in polychlorinated dibenzodioxins or -furans). The correlation coefficient was found to be good (r2 = 0.810; Kühne et al., Citation2005).
It has to be noticed that, after reviewing a large number of calculation schemes focusing on the KH, Dearden and Schüürmann (Citation2003) concluded that the prediction capability of the tested methods was found inferior to the expected one.
As a conclusion, no particular descriptor appeared to be more relevant than another one to estimate KH. No geometric-topological and no electro-topological descriptors were used in the equations.
3.3.3 Octanol-Air Partition Coefficient
As indicated previously, the environmental fate of semivolatile organic compounds depends strongly on their distribution between different environmental compartments. Thus, accurate estimation and/or prediction of environmental distribution coefficients are essential. The octanol-air partition coefficient, KOA, is a key descriptor of the distribution of semi-volatile organic compounds between the atmosphere and terrestrial organic phase (Zhao et al., Citation2005). Twenty-one QSAR allowing the assessment of KOA are reported in Table S6, they involve from 1 to 16 descriptors. As for vapor pressures and Henry's constant, the QSAR were developed for a wide diversity of organic compounds but not for pesticides.
Four of the eight one-descriptor relationships involved topological descriptors (Table S6): MCI (1χv, 2χ, 2χv), but the numbers of compounds used to develop the QSAR were very low (Zhao et al., Citation2005), or sum of topological distances between oxygen and bromine atoms (T(O…Br)) for 209 PBDE and HBB (Papa et al., Citation2009). The T(O…Br) descriptor gives a double structural information: its value increases according to both the number and the distance of bromine substituents, on each phenyl ring, from the oxygen ether. Thus, T(O…Br) also takes into account the information related to the position of the bromine atoms on the phenyl rings. For only six PBDE, KOA was correlated to MW (Wania and Dugani, Citation2003), and for 22 phthalates, KOA was correlated to the Le Bas molar volume (VLB; Cousins and Mackay, Citation2000; Table S6). Then, for 82 chlorinated organic compounds, good estimate of KOA was found using the polarizability α (r2 = 0.979; Table S6; Staikova et al., Citation2004); and similarly good correlation was found with α for a low number of PCDD (r2 = 0.983; Table S6; Zeng et al., Citation2013). Increasing α value of PCDD molecule leads to increasing the KOA values. Molecules with great α values may have great intermolecular dispersive forces with octanol molecules thus favoring partitioning into the octanol phase (Zeng et al., Citation2013). This is consistent with the previous results showing that PL decreases when α increases (Liang and Gallagher, Citation1998; Table S6).
Combination of several topological (1χv, 2χ, 3χv) or several quantum-chemical (largest negative net atomic charge on an atom q−, quadrupole moment Qzz, α, μ) descriptors led to good estimate of KOA of PBDE, polychlorinated naphthalenes and chlorinated organic compounds (r2 > 0.927; Table S6; Staikova et al., Citation2004; Wang et al., Citation2008; Zhao et al., Citation2005).
All the equations based on descriptors of different categories involved quantum-chemical descriptors (Table S6). It is not possible to determine if the combination of several descriptors of several categories improved the prediction of KH as no result has been published to allow a rigorous comparison. For 22 PBDE, KOA was very well correlated to the molecular volume V and to the sum of the surface maxima values of the electrostatic potential (ΣVs+; r2 = 0.976; Table S6). The V term means that the larger PBDE molecule would yield stronger dispersion-type interactions with the octanol molecule (i.e., the KOA value becomes larger; Xu et al., Citation2007). For 16 hydroxylated polybrominated diphenyl ethers (OH-PBDE) and eight methoxylated polybrominated diphenyl ethers (MeO-PBDE), KOA was estimated with six descriptors: electronic energy (EE), ELUMO, MW, the largest positive atomic charge on a bromine atom (QBr+), the most positive atomic net charge on a hydrogen atom (QH+), and μ. The descriptors MW and ELUMO were the two most important factors governing KOA values. Increasing MW, QBr+, QH+, and μ of OH-PBDE and MeO-PBDE led to the increase in KOA. On the contrary, increasing ELUMO and EE can lead to the decrease in KOA (Chen et al., Citation2001d; Chen et al., Citation2003a; Zhao et al., Citation2010). Similarly, for 209 PCB, increasing CCR, MW, most positive net atomic charge on a chlorine atom (qCl), and α values of the PCB led to increasing KOA, whereas increasing EE, total energy (TE), and standard heat of formation (ΔHf) values of the PCB led to decreasing KOA. In this case, the α was the most significant descriptor. The more chlorines in PCB molecules, the larger the size of PCB molecules, the greater the MW, and the greater the α and KOA values. ELUMO was also a significant descriptor, and increasing ELUMO of the PCB leads to decreasing KOA. ELUMO measures the ability of a molecule to accept electrons in intermolecular interactions: molecules with a low ELUMO tend to easily accept electrons. So the lower the ELUMO values, the greater the tendency of PCB molecules to accept electrons in intermolecular interactions, the greater the intermolecular interactions between PCB and octanol molecules, and thus the greater the KOA values (Chen et al., Citation2002a).
Some relationships were developed taking into account the temperature (Chen et al., Citation2002b; Chen et al., Citation2003b; Chen et al., Citation2003c; Chen et al., Citation2004; Table S6). Very good results were obtained for both correlations done with a small and a high number of compounds (r2 > 0.918; Table S6). The most significant descriptors were still ELUMO, MW, TE, and α. As observed previously, increasing α and MW leads to increasing KOA values, while increasing TE, and ELUMO leads to decreasing KOA.
As for the vapor pressure, the polarizability α is the descriptor that explains the best the variation of KOA. The molecular weight MW, the dipole moment μ, and the ELUMO and EE energies also play an important role. No geometric-topological, no geometric-electronic, and no electro-topological descriptors were used in the equations.
3.3.4 Potential of Transfer to the Atmosphere
Only one equation was found to estimate the potential of transfer of organic compounds to the atmosphere (Table S6). For 135 pesticides, Gramatica and Di Guardo (2002) developed a VIN allowing a preliminary ranking of the pesticides according to their tendency to distribute in the atmosphere. The VIN was well correlated (r2 = 0.771) to three constitutional descriptors (HY, number of multiple bonds (nBM), number of rings NoRING), two topological descriptors (0χv, mean information content index on vertex degree equality IEdeg) and one geometric descriptor asphericity (ASP) (Table S6). The most important descriptor is HY, which is related to the presence of hydroxyl groups in the molecule. Then, the next most important descriptors are 0χv and nBM, whereas the least important one is IEdeg.
3.4 Retention Processes
The mobility of organic compounds in the environment mainly depends on their retention on soils and sediments, which is essentially determined by adsorption. Therefore, retention is one of the most important processes that control the fate of organic compounds in the environment because it regulates their availability for degradation, absorption by plants and for transfer toward ground and surface water, and air. In this section, only the adsorption of organic compounds at the liquid-solid interface, the most important retention process, is considered (Katayama et al., Citation2010).
A total of 102 QSAR are inventoried for adsorption on soils, and 38 QSAR for the adsorption on sediments. The nonlinearity and nonequilibrium of adsorption, and the desorption were rarely studied by QSAR approaches: only 18 equations were found for these phenomena (Table S7). Finally, a small number of equations (6) has been published to estimate the potential of transfer of organic compounds to groundwater, and no equation has been published concerning the potential of transfer to surface water (Table S7).
3.4.1 Adsorption Processes
3.4.1.1 Adsorption on Soils
To estimate the adsorption of organic compounds on soils, the QSAR were generally developed for Koc (adsorption coefficient normalized to soil organic carbon content; e.g., Doucette, Citation2003; Gawlik et al., Citation1997; Wauchope et al., Citation2002), and few of them were developed for Kom (adsorption coefficient normalized to soil organic matter content; Briggs, Citation1981; Sabljic, Citation1987 and 1989; Sabljic and Piver, Citation1992), Kf (Freundlich adsorption coefficient; Hance, Citation1969; Jin et al., Citation1997), Kd (linear adsorption coefficient; Hansen et al., Citation1999a; Hu et al., Citation1995), or KL (Langmuir adsorption coefficient; Mon et al., Citation2006; Table S2). The reviews of Doucette (Citation2003), Gawlik et al. (Citation1997), and Wauchope et al. (Citation2002) are completed and updated in this section. In addition, the present review is focused on QSAR only based on structural molecular descriptors.
The number of descriptors used for the estimation of adsorption coefficients ranged from 1 to 11, excluding QSAR based on the fragment approach (Table S7). The increase in the number of descriptors does not improve the significance of the QSAR but the degree of correlation rather decreases with increasing heterogeneity of the training set (Reddy and Locke, Citation1994a, 1994b; Wauchope et al., Citation2002). The QSAR equations that are reported in Table S7 were developed for a wide diversity of organic compounds.
The prediction of the adsorption with only one descriptor mainly involved the MCI (Table S7), and in particular a lot of equations involved 1χ and 1χv to estimate the Koc, Kom, or Kd of many organic chemicals (Bahnick and Doucette, Citation1988; Baker et al., Citation1997; Baker et al., Citation2001; Dai et al., Citation1999; Doucette, Citation2003; Gawlik et al., Citation1997; Gerstl and Helling, Citation1987; Gerstl, Citation1990; Hu et al., Citation1995; Liu and Yu, Citation2005; Meylan et al., Citation1992; Müller and Kördel, Citation1996; Sabljic, Citation1984, 1987, 1989, 2001; Sabljic et al., Citation1995; Sabljic and Piver, Citation1992; Thomsen et al., Citation1999; Von Oepen et al., 1991; Wauchope et al., Citation2002). The r2 are highly variable depending on the classes and on the number of compounds used to develop the relationship (r2 ranges from 0.006 to 0.973; Table S7), and it seems that the inclusion of polar compounds in the dataset led to a decrease in r2. Some correlations were also found between the Koc and 0χ, 0χv, 2χ, 2χv, 3χv, or 5χv for different classes of organic compounds (pesticides, alcohols; Baker et al., 1997; Gerstl and Helling, Citation1987; Müller and Kördel, Citation1996), and between the KL and 9χ for only nine dye tracers (Mon et al., Citation2006). Indeed, MCI encode intermolecular accessibility, and, for example, 1χ correlates very well with the molecular surface, and also represents the contribution of one molecule to the bimolecular interactions arising from encounters of bonds among two molecules (Kier and Hall, Citation2000; Sabljic and Piver, Citation1992). Other topological descriptors were used successfully to estimate the Koc: the CRI for 36 various compounds (r2 = 0.964; Türker Saçan and Balcioğlu, 1996), and the Lu index for only 11 phthalates (r2 = 0.788; Lu, Citation2009; Table S7). The Koc could also be estimated using MW (Kanazawa, Citation1989; Liu and Yu, Citation2005), the TSA (Doucette, Citation2003; Hansen et al., Citation1999a), and the volume of the molecules: molar volume Vm (Von Oepen et al., 1991); parachor P (Hance, Citation1969; Briggs, Citation1981) or van der Waals volume VdW (Hu et al., Citation1995). A larger molecular volume is unfavorable for partitioning to the aqueous phase where strong hydrogen bonds have to be broken to create room for the solute molecule (Baker et al., Citation1997; Briggs, Citation1981). However, the correlations with Vm were not good in some soils for acids and amines (r2 < 0.360; Von Oepen et al., 1991). Finally, Baker et al. (1997), Liu and Yu (Citation2005), and Von Oepen et al. (1991) tried to establish correlations with only one topological (second- (Δ2χ) or third-order (Δ3χ) simple nondispersive force factor), or one quantum-chemical descriptor (self-polarizability ALP, probability of nucleophilic attack DN, EHOMO, ELUMO, MR, net negative atomic charges on atom N in anilines or atom O in phenols q, average charge of molecule Qave, total charge of molecule Qtot, α, and μ) but the correlation coefficients were sometimes very low, and depended on soil characteristics (Table S7).
The combination of several MCI allowed acceptable estimates of Koc (r2 ranges from 0.575 to 0.905; Table S7; Baker et al., Citation2001; Gerstl, Citation1990; Gerstl and Helling, Citation1987; Tao and Lu, Citation1999; Uddameri and Kuchanur, Citation2004). Nevertheless, the QSAR only based on MCI were not suitable for polar organic chemicals, and polarity correction factors have to be introduced in the equations to improve the prediction (Meylan et al., Citation1992; Müller and Kördel, Citation1996; Sabljic, Citation1987; Tao and Lu, Citation1999). The results may be erroneous if an estimate is desired for a compound that has a polar group for which there was not enough data to develop a polarity correction factor (Baker et al., Citation1997). Good correlations were also found between the Koc and several topological indices (1χ and sum over all atoms of the intrinsic state differences DELS, Lu index, and different distance-based atom-type topological index DAI; r2 > 0.900; Table S7; Gramatica et al., Citation2000; Lu et al. Citation2006). To take into account the nonhydrophobic contribution to Koc, Bahnick and Doucette (Citation1988) included, in addition to 1χ, a first-order valence nondispersive force factor (Δ1χv). The two descriptors relate to intermolecular interactions due to molecular size and nondispersive effects, and are important in predicting Koc for molecules exhibiting substantial hydrophilicity. These nondispersive factors were also used by Baker et al. (Citation1997) for 14 miscellaneous compounds (Table S7). A relationship based on constitutional descriptors, 24 structural correction factors (e.g., number of triple bond or number of quaternary carbon) and 74 group fragments, allowed a robust assessment of the Koc of a wide variety of organic compounds (r2 = 0.969; Table S7). These results confirmed the ability of the fragment approach to predict Koc of untested chemicals (Tao et al., Citation1999). Thomsen et al. (Citation1999) developed a concept of grouped E-state index, and in particular, they showed that the Koc of eight phthalates is correlated to Sester and Salkyl that consider the polar/hydrophilic and nonpolar/hydrophobic effects, respectively (r2 = 0.822; Table S7). However, the number of data used to develop the relationship was low. Finally, for 387 miscellaneous compounds, the Koc was estimated using several moment descriptors (M0, M2, M3, Macc, Mdon; Klamt et al., 2002), and for 65 PCOC, the relationship based on several quantum-chemical descriptors such as EHOMO, TE or μ showed good results (r2 = 0.854; Table S7; Dai et al., Citation1999; Table S7).
As observed for SW, KOW, PL, and KH in previous sections, when several descriptors of different categories are used together, the assessment of the adsorption coefficients is improved (Reddy and Locke, Citation1994a). Reddy and Locke (Citation1994a) obtained better correlations between Koc and EHOMO, VdW, α and μ for 71 herbicides than with the volume alone (Table S7). The combination of some constitutional descriptors (MW, structural fragments) and topological (MCI, bond connectivity index) and/or electro-topological descriptors led to correct estimations of Koc for a lot of organic compounds (r2 > 0.772; Table S7; Huuskonen, Citation2003; Lohninger, Citation1994; Schüürmann et al., Citation2006). Sekusak and Sabljic (Citation1992) introduced the number of polar (NoPP) and nonpolar parts (NoNP) in a molecule, the number of rings (NoRING), and the polarity index (1Fχv), and found good correlations for 11 amides, 15 triazole, 16 dinitroanilines, and 21 acetalinides (r2 > 0.846; Table S7). For 44 substituted phenylureas, the estimation of Koc based on VdW, μ and ELUMO was correct (r2 = 0.700; Table S7) but the range of experimental data and/or structural differences among substituted phenylureas in terms of substitution on N3 atom was narrow (Reddy and Locke, Citation1994b).
Gramatica et al. (Citation2000) developed several equations based on a representation of molecular structure by different types of descriptors, such as count descriptors, topological indices, information indices, fragment-based descriptors, and WHIM descriptors. For example, the Koc of 29 carbamates was correlated to the presence of electronegative atoms in the molecules: the greater the number of electronegative atoms in the molecule, the higher the probability of H-bonding with water, leading to a decrease in soil adsorption; and to the eccentric connectivity index (ξC): the size increase leads to increased hydrophobic effects and compound tendency to bind with the soil organic matter. The Koc of 43 phenylureas were well correlated to the MW, nCl, and NoRING, and to two directional WHIM descriptors (r2 = 0.911; Table S7). Most of these descriptors indicated that an increase in phenylurea size favors their adsorption.
Several other relationships were established with various descriptors: scanning a very large number of molecular descriptors (1,457), Goudarzi et al. (Citation2009) identified seven descriptors leading to accurate prediction of the Koc of 62 pesticides: three 2D descriptors (one BCUT index: BEHm2, and two Moran autocorrelation descriptors: MATS6e and MATS4p) and four 3D descriptors (one GETAWAY: HTp, one MoRSE: Mor(05)m, and two WHIM: G1m and G3v; Table S7). Winget et al. (Citation2000) developed a set of effective solvent descriptors that characterize the organic carbon component of soil, and combined these descriptors with solute atomic surface tension parameters to predict the Koc of any solutes composed of H, C, N, O, F, P, S, Cl, Br, and I. However, the errors in predicting experimental data were greater than those resulting from other methods. Nevertheless, the developed models do not use different parameter sets for different classes of solutes, and thus are applicable to totally new classes of molecules. In addition, no experimental data are needed for a new compound once the molecular structure is known, and since the resulting solvent descriptors have reasonable values, it is possible to understand the sources of different partitioning phenomena in cases where the results exhibit significant fragment interactions.
Finally, the LSER and TLSER approaches, based on a mechanistic understanding of the partition process (section 2.8), were also efficient for the estimation of the Koc (r2 > 0.720; Table S7; Baker et al., Citation1997; Famini and Wilson, Citation1997; Feng et al., Citation1996; Poole and Poole, Citation1999; Xu et al., Citation2002). In any case, the leading terms in the equations were those that measure cavity formation (the intrinsic volume Vi or the McGowan characteristic molecular volume Vx), and the overall or summation solute hydrogen bond basicity (Σβ2H or Σβ2O). The correlation coefficient seems to decrease with the heterogeneity of the compounds in the dataset (Table S7).
As a conclusion, it was shown that the prediction of adsorption of organic compounds in soils mainly depends on MCI and especially on 1χ, 1χv, and 3χv, but also on MW and on descriptors related to the volume (P, Vi, Vm, VdW). Indeed, compounds with high branching properties and high volume are less susceptible to partition in the aqueous phase. No geometric-electronic descriptor was used in the equations.
3.4.1.2 Adsorption on Sediments
The 38 QSAR equations reported in Table S7 allow the assessment of seven parameters related to the measurement of the adsorption on sediments: the association coefficient between PCB and humic marine substances (Kh), the Freundlich adsorption coefficient (Kfs), the linear adsorption coefficient (Kds), the adsorption coefficient normalized to sediment organic carbon content (Kocs; or total organic carbon normalized sediment-porewater distribution coefficient KTOC), the sediment water partition coefficient (KS/W), and the maximum concentration of the compound that can be adsorbed (Csm; Tables S2 and S7). The equations involve from one to four descriptors, the majority of them involve one or two descriptors. Contrary to adsorption on soils, there is notably no QSAR for pesticides (Table S7). In general, the number of compounds used for the QSAR development was very low.
The 28 one-descriptor equations developed to estimate the adsorption on sediments involve constitutional, geometric, topological, or quantum-chemical descriptors (Table S7). For chlorobenzenes, PCDD and PCDF, Arp et al. (2009) found correlations between the KTOC and nCl, and for PAH, they found a relation between the KTOC and naromatic-C. However, the number of compounds used in these QSAR was not indicated in their study. The Kocs and Csm of 5 chlorobenzenes on sediment was very well correlated to the molar volume Vm (r2 > 0.890; Table S7), but the number of chemicals used to develop this relationship was low (Von Oepen et al., 1991). Similarly, Vm was well correlated to the Kocs of esters, acids, and amides (not with that of amines), but the number of data was also very low (Djohan et al., Citation2005). For 26 PCB, Lara and Ernst (Citation1989) found better correlation between the Kh and the TSA calculated for the planar configuration (r2 = 0.954) of the PCB than with the nonplanar configuration (r2 = 0.919; with phenyl rings perpendicular to each other; Table S7). However, a disadvantage of using molecular surface areas as structural descriptors is that the molecular geometry has to be well known (Sabljic et al., Citation1989) and, indeed, bad correlations were found between the Kfs of eight organotin (organometallic species) and the TSA (r2 = 0.095; Table S7; Sun et al., Citation1996). Bad correlations were also obtained with several MCI or the Leo fragment constant (π; r2 < 0.135; Table S7; Sun et al., Citation1996), and, similarly, there was no good correlation between MCI and the Kds of 11 naphthoic acids and five quinoline compounds (r2 < 0.220; Table S7; Burgos and Pisutpaisal, Citation2006). The authors rather observed that the adsorption of naphthoic acids increased with the addition of ortho-substituent groups and with increasing chain length of the acid group, and that the adsorption of quinoline decreased with substituent group addition (except for nitro group) and with additional heterocyclic N atoms, but they did not develop any QSAR. On the contrary, for several esters, acids, amides and amines, Von Oepen et al. (1991) found some satisfactory correlations between Kocs and 1χ, 1χv, self-polarizability (ALP), probability of nucleophilic attack (DN), average charge of molecule (Qave), or total charge of molecule (Qtot), but it depended on the soil and the number of data was low (Table S7).
The seven equations developed with several descriptors of the same category involved constitutional, topological, or quantum-chemical descriptors (Table S7). For PCB, the KTOC was correlated to nCl and to northo Cl. Orthochlorine atoms cause the planar conformation of PCB to be energetically unfavorable, which in turn causes lower adsorption (Arp et al., Citation2009; Hawthorne et al., Citation2011). Using MCI, Sabljic et al. (1989) found good correlations between the Kh of 26 PCB and 1χ, following a quadratic function (r2 = 0.948; Table S7). These results show that the adsorption of PCB on marine humic substances is primarily influenced by the size of molecule which is described by 1χ: the larger PCB molecules showed a higher affinity for humic Qtot substances than smaller ones. For 11 alcohol ethoxylates (Kiewiet et al., Citation1996) and for 31 alcohol ethoxylates and four other alcohols (Van Compernolle et al., 2006), the Kds was found to be correlated to the ethoxylate chain length (#EO), and to the alkyl chain length (#C). The dominant influence of the alkyl chain suggests a hydrophobic adsorption mechanism (Table S7).
For 26 PCB, the addition of the number of orthochlorine substituents (noCl) to MCI improved the prediction of Kh (r2 = 0.990; Table S7; Sabljic et al., Citation1989). The negative regression coefficient of noCl indicates that the adsorption decreases with the degree of ortho substitution: noCl provides an estimate for the extent of nonplanarity of PCB. Thus, it may be viewed as third dimension corrections for the 1χ index that is unable to completely handle 3D situations (stereochemistry). However, this term limits the value in modeling the adsorption for other classes of nonionic chemicals with dissolved humic substances, as the noCl is specific for PCB. The quadratic relationship also indicates that each new chlorine substituent increases the extent of adsorption less than the previous one (Sabljic, Citation1991, 2001). Combining topological and quantum-chemical descriptors, Dai et al. (2000) showed that the Kocs of 14 substituted benzaldehydes was well predicted with QH+, μ, 2χv and 3χpc (r2 = 0.922; Table S7). The dipole moment μ was the most significant descriptor.
He et al. (Citation1995) used the LSER approach to estimate the Kocs of 28 phenylsulfonyl acetates, and found excellent results (r2 = 0.976; Table S7). The intrinsic volume Vi was a leading term in the adsorption of these compounds on sediments. Finally, with the MTLSER approach, Chen et al. (Citation1996a) showed, for 22 alkyl(1-phenylsulfonyl)cycloalkane-carboxylates, that α and μ were leading terms influencing the Kocs. The polarizability α is in direct proportion to the intrinsic molecular volume, and therefore it increases the Kocs since the larger molecules would tend to be excluded from the water and be adsorbed on the sediments. The dipole term decreases the Kocs probably because greater dipole would increase interactions between the solutes and more polar water, increasing solubility in water.
As for the adsorption on soils, the variations of the adsorption of organic compounds on sediments are mainly represented by MCI (mainly 1χ, and 1χv). No geometric-topological, no geometric-electronic, and no electro-topological descriptors were used in the equations.
3.4.1.3 Nonlinearity of Adsorption
The nonlinearity of adsorption is indirectly taken into account in the QSAR designed to assess the Kf or KL. However, there is no estimation of the parameters such as nf, the Freundlich exponent.
Only Droge et al. (Citation2009) tried to develop QSAR describing the nonlinearity of adsorption. They proposed to consider the nonlinear adsorption of nine alcohol ethoxylates into the illite clays and sediments using a dual-model combining a Langmuir (KLs) and a linear (KII) adsorption term. They showed that both adsorption coefficients are correlated to the ethoxylate chain length #EO and to the alkyl chain length #C (r2 > 0.910; Table S7). The KLs increases with both chain lengths, on the contrary, the KII slightly decreases with #EO. This probably results from the increased solubility with longer ethoxylate chains and because sorbate-sorbate interactions mainly occur via the alkyl chain, leaving the ethoxylate chains solvated with water. For illite, the increase in KLs with both chain lengths is consistent with adsorption mechanism that depends on both hydrophobic properties and polar interactions with the surface. The enhanced nonlinearity of isotherms with longer ethoxylate chains is explained by both an increasing adsorption coefficient and a decreasing bilayer formation affinity with additional ethoxylate units (Droge et al., Citation2009).
3.4.1.4 Nonequilibrium Adsorption and Desorption
Very few results have been published concerning the nonequilibrium adsorption and the desorption of organic compounds, and only 14 equations were found: 13 for nonequilibrium adsorption and one for desorption (Table S7).
Brusseau (Citation1993) and Hu et al. (Citation1995) demonstrated that the nonequilibrium adsorption in soils of several classes of compounds (PAH, pesticides), measured through a mass transfer coefficient (MT), was correlated to 1χv (r2 > 0.830), and to a lesser extent to the VdW (r2 > 0.700). Indeed, most of the rate-limited adsorption behavior can be explained by accounting for the size and structure of the solute molecule. However, it has to be noticed that few compounds were used to develop the QSAR (Table S7).
Colón et al. (Citation2002) studied the adsorption kinetics in sediment slurries of 21 anilines with substituents in the ortho, meta or para positions. They introduced fast and slow rates of adsorption (kfast, kslow) because the adsorption kinetics of the substituted anilines were characterized by an initial, rapid adsorption process followed by a much slower adsorption process. The rates of adsorption were correlated to the EHOMO or to the one electron oxidation potentials (E1), however the correlation coefficients were not very high (r2 < 0.598; Table S7).
Only one equation was found for the desorption (Table S7): assuming a reversible adsorption, Hsieh and Mukherjee (Citation2003) showed that the desorption of six halogenated aliphatic hydrocarbons from biosolids was very well related to 1χ (r2 = 0.979; Table S7), but the number of data is low. There is no result concerning the estimation of the desorption when the adsorption is partially irreversible.
3.4.2 Potential of Transfer to Ground and Surface Waters
Transfer of organic compounds to ground and surface waters first depends on their availability to be leached through the soil profile or to be mobilized by runoff water. This availability for water extraction changes with time since the compound has reached the soils either directly or indirectly, due to retention and degradation processes, and to climatic and soil conditions that enhance or inhibit these two processes (Louchart and Voltz, Citation2007; Sharer et al., Citation2003; Walker et al., Citation2005). It is therefore difficult to derive organic compound transfer properties on the basis of time-variable environmental parameters such as Koc and half-life (DT50). A few authors defined time-varying environmental parameters equivalent to Koc, taking into account a reference Koc, and either the time (Beigel et al., Citation1997; Renaud et al., Citation2004) or the cumulative rainfall (Louchart and Voltz, Citation2007), as organic compound application. Nevertheless, a few indicator or parameter of the potential for organic compound to be transferred toward water bodies have been developed on the basis of constant Koc and DT50 values (Gustafson, Citation1989; Rao et al., Citation1985).
The occurrence of organic compound in both ground and surface waters also depends on the water fluxes through the soil and at the soil surface, respectively. In fact, water fluxes are driven by meteorological conditions, soil hydrodynamics and initial soil water profile, and soil surface characteristics. This occurrence is classically estimated by means of complex mathematical fate models that take into account these water fluxes, and the environmental parameters such as DT50 and Koc, that can be derived from QSAR models. But the use of such model is time consuming and do not permit to simulate the occurrence of each organic compound in water bodies easily. Another approach is to use QSAR to estimate the occurrence of organic compounds in groundwater (Worrall, Citation2001; Worrall and Thomsen, Citation2004). However, the number of QSAR that have been developed is very low: only six equations were found to estimate the potential of transfer to groundwater (Table S7), and no relationship was found to estimate the potential of transfer to surface water. In addition, these QSAR were only built with pesticides data, no equation has been developed for other organic compounds that are susceptible to reach groundwater (e.g., pharmaceuticals, PCB).
To assess the potential transfer of pesticides to groundwater, the leaching index (LIN) was developed by Gramatica and Di Guardo (2002) in order to give a preliminary ranking of these compounds according to their tendency to distribute in different environmental media. For 135 pesticides, LIN was correlated to three constitutional (nX, nNO2, nS), one topological radial centric information index (ICR), and one electro-topological (constitutional descriptor of the mean E-state of the molecule related to the polarizability, Ms) descriptors with satisfactory model performance (r2 = 0.870; Table S7).
To distinguish polluting from nonpolluting compounds, only on the basis on molecular topology and/or quantum-chemical descriptors, Worrall (Citation2001) and Worrall and Thomsen (Citation2004) used a dataset of 56 pesticides monitored in 303 boreholes across 12 states in the midwestern United States during 1991–1992. With logistic regression, they estimated the probability of finding these pesticides at a level concentration > 0.1μg L−1, θ, using 6χv and 7χvpc, the hydration energy ΔHhyd, and the dipole moment μ, which gave good results (r2 = 0.910; Table S7). Furthermore, they obtained more than 85% of variance explanation by considering the rule that a compound can be found in groundwater if 6χv < 0.55 (Worrall, Citation2001) or if 0.28 μ < 6χv (Worrall and Thomsen, Citation2004). They concluded that the dependence of leaching potential on the descriptors that control solubility (μ and ΔHhyd) indicates that predictions of environmental fate based on this approach may represent a strong alternative to the use of adsorption and degradation parameters.
3.5 Degradation Processes
Degradation is one of the key processes governing the fate of organic compounds in the environment as it determines their removal and/or transformation to metabolites, conditioning their persistence and transfer to different components of the environment (soil, water, sediment, plant, or air). The degradation can be biotic (following degradation by living organisms including microorganisms) or abiotic (hydrolysis, photolysis). The QSAR that have been found to assess both types of degradation processes are reviewed here.
3.5.1 Biodegradation
The structure of this section is slightly different from the structure of the others because, contrary to other processes, the biodegradation of organic compounds can be estimated using a wide range of environmental parameters (Table S2), and nonetheless few QSAR equations were developed for each parameter (Table S8). Indeed, as explained in the following paragraphs, the measurement of the biodegradation of organic compounds can be done according to different methods and in different experimental conditions relying on the use of artificial media to test degrading activity of pure bacterial cultures under laboratory conditions or of more complex natural matrices such as soils, sediments, or water, or man-made matrices such as wastewater or activated sludge incubated under controlled conditions; leading to the determination of different kinds of QSAR. A total of 75 equations is reported in Table S8 for a wide diversity of compounds.
Microbial biodegradation is often considered as the major driving force of the fate of organic compounds in the environment. It is the result of aerobic or anaerobic enzymatic activities of microorganisms. Biodegradation acts on the transformation of organic compounds (primary degradation with production of metabolites), and on their removal (mineralization or ultimate degradation refers to the complete degradation of an organic species to stable inorganic species) from the environment as well. Consequently, biodegradation determines the persistence of organic compounds. Persistence is defined by the length of time a compound remains in an environmental compartment before it is transported to another one or is chemically or biologically transformed. As persistence is a usual criteria used in regulatory bodies, the estimation of the biodegradability (ability of a compound to be biodegraded), which is one way to define such criteria (a substance that is easily degraded is considered non persistent) is fully developed particularly with in silico models such as QSAR. The biodegradation ability can be experimentally determined by measuring half-lives of organic compounds in experimental tests conducted under environmental conditions. The main guidelines are given by Organization for Economic Cooperation and Development (e.g., Organization for Economic Cooperation and Development, 1992, 2009). These experimental results are highly dependent on the environmental conditions found in the matrix (temperature, water content, pH, redox potential, oxygen, substrates, salinity, trace element type, and concentration), on the nature of the microbial community (diversity, density, activity), and on the tested chemicals (structure, concentration). All these aspects are fully discussed in Howard (Citation2000). Some of the available sources of biodegradation databases are given by Pavan and Worth (Citation2008). The two most famous databases used in recent QSAR development are the MITI-I database made of the information on more than 1000 chemicals from one uniform biodegradation test, and the BIODEG database developed by the U.S. Environmental Protection Agency (EPA) and Syracuse Research Corporation (SRC) containing biodegradation information on 815 chemicals.
From these databases, the training sets used in QSAR development are more qualitative data than those issued from the standardized OECD tests. These tests measure either oxygen consumption, or CO2 production, or disappearance of dissolved carbon by comparison to that resulting from easily mineralized compound such as sodium acetate. This type of test results in the classification of chemicals into two main categories readily biodegradable and not readily biodegradable. These categories can be divided to give biodegrades fast, biodegrades fast after acclimation, biodegrades slowly, biodegrades slowly even after acclimation or biodegrades sometimes (Rücker and Kümmerer, Citation2012). Each classification is rated according to the quality of the summarized data on one chemical, with rate 1 (three consistent results), 2 (at least two consistent results), and 3 (one result only or severely conflicting results). This results in two types of data used in QSAR development: either quantitative data such as half-lives (DT50), biodegradation rates, or kinetic constants (BS, k, K, kb, kx/kh, RC), theoretical oxygen demand (%ThOD), biological oxygen demand (BOD), or qualitative data using Boolean-type logic (BM; 1 for readily biodegradable and 0 for not readily biodegradable; Table S2).
The first QSAR and quantitative structure biodegradability relationship (QSBR) models were developed in the 1980s on chemicals with similar structure (homologous models). They were based on octanol/water partition coefficients, Hammett constants or alkaline hydrolysis rate constants not considered there (Peijnenburg, Citation1994; Yonezawa and Urushigawa, Citation1979), or on molecular descriptors such as MW (Boethling, Citation1986), van der Waals radius (YVdW; Paris and Wolfe, Citation1987; Paris et al., Citation1983), MCI (2χ, 2χv, 3χ, 3χc, 3χv, 3χvc, 4χ, 4χc; Boethling, Citation1986), and charge difference in the modulus charges on the atom of specified bonds (Δδx−y; Dearden and Nicholson, Citation1986, 1987) that were well correlated to biodegradability endpoints (r2 > 0.722; Table S8). Some equations were also found using EHOMO, ELUMO, IP, μ, and charges (Peijnenburg, Citation1994). However, these models were often limited in terms of applicability, prediction (class specific compounds and few numbers of compounds) and quality of datasets.
The construction of the two high-quality biodegradation datasets in the 1990s (BIODEG and MITI databases) led to the development of more reliable and accurate biodegradation prediction models. This was also facilitated by the application of new and advanced modeling approaches such as multivariate techniques such as principal component analysis (PCA) and partial least square analysis (PLS), nonlinear neural network, artificial intelligence, computer-automated structure evaluation (CASE), or empirical knowledge. These allowed the development of heterologous models describing the degradability of compounds displaying varying chemical structures.
The majority of these models are based on one category of descriptors which are functional groups or structural composition of chemicals combined or not with the molecular weight MW: indeed the so-called group contribution models (OECD hierarchical approach, Biodegradation Probability Program-Biowin 1–7 models) are based on the counts of structural/substructural fragments (number of carboxyl, hydroxyl, unsubstituted aromatic, phosphate ester). The 40-year work of Howard, Boethling, and collaborators (Table S8) contributed to the development of the most used models in predicting aerobic degradation in water, the Biowin models, that are based on constitutional descriptors: MW and 36 or 42 defined chemical substructures (Boethling, Citation1986; Boethling et al., Citation1994; Howard, Citation2000, 2008; Howard et al., Citation1991; Howard et al., Citation1992; Howard et al., Citation2005; Meylan et al., Citation2007; Sabljic and Peijnenburg, Citation2001; Tunkel et al., Citation2000). Loonen et al. (1996, 1999) proposed a similar ready/nonready aerobic categorization but using a PLS model based on a bigger training set (894 substances) and 127 predefined substructures. All these group contribution models present an acceptably accurate prediction with only a single set of chemical substructures (for models performances comparison, see Pavan and Worth, Citation2008; Raymond et al., Citation2001; Rorije et al., Citation1999; Rücker and Kümmerer, Citation2012). As indicated in the previous sections, the main drawback of this method is the nonprediction for compounds that did not contain any of the substructures. In order to overcome this drawback, models (MultiCASE approach) were developed based on the random generation of all fragments (all linear and terminally branched substructures), and the most significant ones were statistically correlated to the endpoint (Klopman, Citation1992). The fragments that activate the aerobic degradation were called biophores (aliphatic, carboxyl substructures) and the ones that inactivate biophobes (aromatic substructures). It was successfully applied on N-heterocycles compounds by Philipp et al. (Citation2007) with 99% of correct classification.
Various statistical techniques were used to correlate descriptors to biodegradation data, including linear, multilinear, and nonlinear (e.g., neural networks) regressions; classification tools; and expert system based on if-then-else rules and artificial intelligence (Baker et al., Citation2004; Blockeel et al., Citation2004; Cuissart et al., Citation2002; Gamberger et al., Citation1996; Tabak and Govind, Citation1993). If the most used endpoints are the data from MITI-I and BIODEG databases (qualitative), other quantitative endpoints were also used, such as the percentage of theoretical biological oxygen demand achieved in five days (BOD; Sedykh and Klopman, Citation2007), first-order biodegradation constants (k; Desai et al., Citation1990; Tabak and Govind, Citation1993), or Monod constants (Tabak et al., Citation1992). To go further than predicting the extent of biodegradation, other models (META/MultiCASE and CATABOL/CATALOGIC) were developed to predict possible degradation products and pathways based on either expert system or machine-learning systems. The META/MultiCASE approach is an expert system that can predict the metabolic transformation thanks to hierarchized metabolic rules defined by constitutional descriptors. Indeed the 70 general rules are based on a set of MultiCASE biophores (fragments that activate biodegradation), the weights of the fragments being used to define the hierarchy of transformation rules for a given chemical structure (Klopman et al., Citation1994; Klopman et al., Citation1995). CATABOL is a hybrid system predicting the extent of degradation through BOD evaluation, and biotransformation pathways through an expert knowledge-based system (Jaworska et al., Citation2002). The BOD of one compound is there considered as a complex sequence of reaction steps occurring spontaneously (abiotic) or being catalyzed by enzymes: BOD is thus modeled by a sum of terms, each term being the product of the BOD of one step multiplied by the probability of the step to occur. The set of reactions (550) were extracted from the literature and were based on constitutional descriptors of the parent and daughter molecule taking into account the presence/absence of inhibiting fragments for each transformation reaction.
All these models, from the first ones of Geating (Citation1981) and Boethling and Howard's (Boethling et al., Citation1994; Howard et al., Citation1991; Howard et al., Citation1992; Howard et al., Citation2005) to the following ones, are based on the common assumption that attack by microorganisms takes place at a specific functional group of the molecular skeleton and that the presence of a particular fragment may enhance or delay the degradation. This assumption of similar structure-similar microbial metabolism is a strong hypothesis, not always verified. For example, EHOMO-ELUMO gap and ionization potential (IP) may be useful descriptors to describe redox behavior (Rücker and Kümmerer, Citation2012). But, consequently to this assumption, other descriptors, such as constitutional or topological descriptors, that take into account the global structure of chemicals, were assumed not to describe this microbial ability, particularly if the models are used for compound sets of highly diverse structure. Nevertheless, some models were developed using these descriptors: constitutional such as MW or number of rotatable bonds RBN, geometric such as solvent accessible surface area SAS, or topological such as MCI or Wiener index W, alone (Boethling, Citation1986; Boethling and Sabljic, Citation1989; Dearden and Nicholson, Citation1986; Kim et al., Citation2007; Li and Xi, Citation2007; Lindner et al., Citation2003), or in combination with other constitutional descriptors such as the number of chlorine atoms (nCl), geometric descriptors such as the MR, quantum-chemical descriptors such as the charge on the substituted carbon (C12), EHOMO, the sum of charges on all carbons on the substituted benzene ring C8-C12 (SUMC8:C12) and μ (Boethling and Sabljic, Citation1989; Gombar and Enslein, Citation1991; Kim et al., Citation2007; Li and Xi, Citation2007; Lindner et al., Citation2003) for a series of similar compounds (Greaves et al., Citation2001; Kompare, Citation1998), but also for compounds with varying structures (Delisle and Dixon, Citation2004; Jaworska et al., Citation2003; Okey and Stensel, Citation1996; Table S8). Boethling and Sabljic (Citation1989) observed that the structural features found to have a major influence on ultimate biodegradation of chemicals were size and shape, degree of chlorination, and degree of branching. The size and shape of chemicals accounted for the major part of the variation in the biodegradation data. In the model, this structural feature is described by 2χv, the magnitude of which is directly proportional to the size and shape of the chemical. Its positive regression coefficient indicates that microbial biodegradation will be slower for the bigger and more extended chemicals than for the smaller and more compact ones. The second structural property that controls the biodegradation rate of chemicals was the degree of chlorination (halogenation). The relative importance of this structural feature decreases with the size of the chemical. The third structural feature influencing the biodegradation is the degree of branching. It is described by 4χpc, which is highly sensitive to changes in branching, and its value rapidly increases with the degree of branching. The positive regression coefficient of 4χpc indicates that the degree of branching increases the period required for ultimate biodegradation. The relative importance of this structural feature decreases with the size of the chemical. It can be concluded that the ultimate biodegradation rate of a chemical will be a balance between the global structural features and some local structural features, the presence of particular functional groups (Sabljic, Citation1991).
Some models were developed without constitutional descriptors but based on the combined use of geometric (molecular diameter D, solvent accessible surface area SAS; Kim et al., 2007; Yang et al., Citation2004), topological MCI (Huuskonen, Citation2001b, Yang et al., Citation2004, 2006), and/or quantum-chemical descriptors (EHOMO, ELUMO, repulsion energy NRE, q−, natural charge on the NH group Q(NH), spin density SD, α, ΔGs, μ; Beasley et al., Citation2009; Berger et al., Citation2002; Kim et al., Citation2007; Yang et al., Citation2004; Table S8). They were, however, developed on a small set of similar compounds. Finally, for 18 PAH, the biotransformation affinity coefficient (Ks) and the maximal specific biotransformation rate (qmax) were well correlated to geometric (radius of gyration (RadOfGyration), fraction of the area of the projection of a molecule on the y,z-plane divided by the area of the rectangle enclosing the projection of the molecule SHDW-Yzfrac, length of the projection of the molecule on the y axis SHDW-Ylength), topological (Kier flexibility index ø), geometric-electronic (PNSA-1) and quantum-chemical (final heat of formation (HOF), magnitude of the principal moment of inertia [PMI]) descriptors (r2 > 0.821; Table S8; Dimitriou-Christidis et al., Citation2008). Descriptors related to 3D shape of the molecules were found essential. Indeed, PMI and RadOfGyration encode information about spatial distribution of mass and rotational properties of a molecule. The SHDW-Yfrac and SHDW-Ylength encode information about size, shape and orientation, and ø expresses the conformational flexibility of a molecule (Dimitriou-Christidis et al., Citation2008). Wammer et al. (2005) did not find any reliable QSAR to estimate the first-order biomass-normalized rate coefficients of 22 PAH, however their study did not include a comprehensive analysis of all possible molecular descriptors.
As for other environmental parameters (see previous subsections), it was observed that the combination of several categories of descriptors improves the prediction of B, BOD, COD, and AERUD (Boethling and Sabljic, Citation1989; Kim et al., Citation2007; Li and Xi, Citation2007; Tables S2 and S8). We finally ended up with the concept that the combination of descriptors may be the best option: group contribution models may be good for initial screening of the chemicals, but adding other descriptors than fragments may be good to better understand the biodegrading mechanisms.
As a conclusion, although the existence of an important number of studies, it remains difficult to identify descriptors explaining the huge variations in biodegradation of organic compounds. However, MCI were involved in almost half of the reviewed equations (33 over 75), especially 2χv and 4χc, and they provided satisfactory results. MW was also involved in 11 equations, and EHOMO and ELUMO in eight equations. No geometric-topological and no geometric-electronic descriptors were used.
3.5.2 Abiotic Degradation
Among the abiotic degradation processes, the chemical degradation occurring in soil, water, and sediment is the most studied for the development of QSAR (84 equations), followed by the degradation in atmosphere (35 equations; Table S9). Few QSAR were developed to estimate the degradation of organic compounds at the surface of the leaves of plants (2 equations). Most of the equations involve one or two descriptors to estimate the different abiotic degradation processes (i.e., hydrolysis, photolysis, reduction, and oxidation), but the number of descriptors can reach a maximum of 26 (photolysis, reduction; Table S9). The environmental parameters that are considered are essentially the degradation rates and the half-lives of the compounds, but several relationships were also developed using the quantum yield of the reactions Φ, the one-electron reduction potential EoH, and the activation energy Ea (Tables S2 and S9).
A number of reviews were written concerning the photooxidation processes (Mill, Citation1989), the tropospheric degradation (Güsten et al., Citation1995), the radical reactions of benzene derivatives (Hansch and Gao, Citation1997), the photoreaction rates in surface waters (Mill, Citation1999), and the atmospheric oxidation of chemicals (Meylan and Howard, Citation2003). However, almost all the reviewed QSAR are not based on structural molecular descriptors.
3.5.2.1 Abiotic Degradation in Soils, Water, and Sediments
Among the fate processes essentially occurring in natural surface, ground and interstitial water, and sediment, hydrolysis is one of the most important; however, only 12 QSAR equations have been found (Table S9). These equations only involved just one or two quantum-chemical descriptors, and were only developed for a small number of phenylurea and sulfonylurea herbicides. There was no QSAR for other types of organic compounds.
The pseudo-first-order reaction rate constant, khy, of six sulfonylureas herbicides decreased significantly with higher ELUMO (r2 > 0.702; Table S9). This is related to electron affinity and thereby reflects the energy for the uptake of electrons (Berger and Wolfe, Citation1996). In general, the self-polarizability of the carbonyl carbon (ALPCO) and self-polarizability at the heterocycle atom 4 (ALPHeterocycle atom, 4), or the superdelocalizability of the carbonyl carbon (SE(CO)) and at the heterocycle atom 4 (SE(4)) allowed correct prediction of the khy of 11 sulfonylureas in buffer, sterile soil and sterile sediment (Berger et al., Citation2002). The carbonyl group drives the hydrolysis at the sulfonylurea bridge, and the carbon 4 of the heterocycle part of the molecule is the atom where the substitution reaction takes place (hydrolysis of the methoxy group). The ALPCO describes the reactivity of the pi electron system, reflecting the higher reactivity at the carbonyl-carbon of methylmethoxy-substituted compared with dimethyl-substituted phenylureas.
The transformation rates T of 10 phyenylureas in sterile soil and water can be well estimated with ALPij (r2 > 0.745; Table S9; Berger et al., Citation2001).
The 39 QSAR that are summarized in Table S9 to estimate the photolysis of organic compounds were mainly developed for the quantum yield φ, and more rarely for the degradation rates kp or for the half-life T1/2ph (Table S9). The equations are mostly based on one descriptor, but they can involve up to 27 descriptors. They were developed for several compounds but neither for PCB or pesticides, for example.
Twenty-four one-descriptor QSAR are reported to estimate the quantum yield φ of various aromatic halides. The equations mostly involved quantum-chemical descriptors (bond order for the carbon-halogen bonds BO, bond strength of the carbon-halogen bond to be broken BS, electronic energy EE, EHOMO, ELUMO, electron-nuclear attraction energy of the one-center term for the halogen atoms EN1, electron-nuclear attraction energy of the two-center term of the weakest carbon-halogen bond EN2, nuclear-nuclear repulsion energy of the two-center term of the weakest carbon-halogen bond NN2, net atomic charges on the carbon atoms in the benzene ring that are connected with the halogen atoms qc, net atomic charges on the halogen atoms qx, total of electronic and nuclear energy of the two-center term of the weakest carbon-halogen bond TE2, α, μ), but also constitutional (MW) or geometric (summation of the steric factors of the additional substituents Es) descriptors (Chen et al., Citation1998b; Peijnenburg et al., Citation1992; Table S9). The best correlations were found with qx for 15 substituted chlorobenzenes (r2 = 0.829) and Es for 12 substituted aromatic halides (r2 = 0.810), and the worst with BS for 12 substituted aromatic halides (r2 = 0.000), μ for 17 substituted bromo- and iodobenzenes (r2 = 0.242), and EN2 for 15 substituted chlorobenzenes (r2 = 0.267; Table S9). Es was one of the most efficient descriptor showing that steric effects seem to play a dominant role during the rate-limiting step of photolysis. However, the number of data used to develop the QSAR was low (Peijnenburg et al., Citation1992).
All equations based on descriptors of the same categories used quantum-chemical descriptors, and in particular EHOMO and ELUMO. The absolute electronegativity was a common descriptor able to predict the quantum yields of different substituted aromatic halides (bromo-, iodo-, chloro-, and fluorobenzenes) showing φ are dependent on the overall character of the halides, the character of carbon-halogen bond to be broken, and/or the nature of halogen atoms to be replaced. Considering each group of substituted aromatic halides independently, EN1 was the best descriptor for chlorobenzenes; and EHOMO for fluorobenzenes (Chen et al., Citation1998b). Similarly, the quantum yield of 41 substituted halides was correlated to EN2 and ELUMO (r2 = 0.807; Table S9; Chen et al., Citation1998a). For 11 PBDE, the quantum yields (in methanol/water) increased with EHOMO, ELUMO, qC− and QH+, and decrease with CCR, QBr+, and QO− (r2 = 0.982; Table S9; Niu et al., Citation2006).
Using a number of molecular descriptors of different categories, Chen et al. (1998a) developed some relationships to predict the quantum yields of several classes of organic compounds (Table S9). The quantum yields of 41 substituted aromatic halides can be well predicted from EE2, ELUMO, and MW (r2 = 0.848; Table S9). Compounds with higher ELUMO and EE2 could have a higher probability of intersystem crossing, a higher probability of formation of excited triplet state, and thus a higher probability of photochemical reaction. A relationship was also developed using four factors condensing different types of information: (a) the strength of the carbon-halogen bonds, (b) the most positive or negative net atomic charges on an atom, (c) molecular ability to be oxidized or reduced, and (d) structural information related to polarizability α. This may imply that the weaker the carbon-halogen bonds are, the higher the quantum yields are. Thus, the compounds with higher polarizability tend to have smaller quantum yields. Because the electrons in the molecules of the compounds with higher polarizability can relatively move easily, both excited singlet and triplet states of the molecules of such compounds may be unstable (i.e., they may easily undergo processes such as internal conversion and fluorescence) resulting in smaller quantum yield (Chen et al., Citation1998a). The quantum yields in water/acetonitrile of PCDD, as for it, mainly depended on core-core repulsion energy (CCR), EE, electron-electron repulsion energy of the one-center term for the oxygen atoms (EE1-O), EHOMO, ELUMO, electron-nuclear attraction energy of the one-center term for the oxygen atoms (EN1-O), HOF, MW, largest negative atomic charge on a carbon atom (qC-), largest positive atomic charge on a chlorine atom (qCl), net atomic charges on the oxygen atom (qO), TE, and α. The correlation coefficient was good (r2 = 0.972; Table S9), but the number of data used for the regression is small. Increasing bulkiness and polarity of PCDD led to decrease in quantum yield values; increasing ELUMO, EHOMO, and HOF values led to increase in quantum yield. EE1-O, EN1-O, and qO describe the character of the oxygen atoms and play an important role in the relation. This supports the suggestion that fission of the ether bond in the dioxin ring is the most likely route for direct photolysis (Chen et al., Citation2001c). The combination of MW and bond order of the carbon-halogen bonds (BO) led to correct estimate of φ of 17 substituted bromo and iodobenzenes (r2 = 0.789; Table S9; Chen et al., Citation1998b), and the quantum yield of 12 substituted aromatic halides was best correlated with BS and Es (r2 = 0.940; Table S9; Peijnenburg et al., Citation1992). Finally, PAH with large average polarizability α, HOF and MW values tend to have smaller quantum yield, and PAH with great ELUMO and ELUMO/EHOMO values, and small EHOMO values tend to have great φ (Chen et al., Citation2000).
For 11 PBDE, CCR, MW, TE, and ELUMO or EHOMO were common descriptors to estimate their photolysis rate constants kp in different solutions. As all PBDE congeners have a same parent diphenyl ether, it can be concluded that the more bromine atoms in the parent molecule, the higher the photolysis rate. Because a nucleophile reacts by means of its EHOMO values, the compound with higher EHOMO values will be a better nucleophile and will generate more stable. Similarly, PBDE with big absolute EHOMO – ELUMO values tend to be more stable. This result implies that photolysis rates of PBDE are also affected by the characteristics of solution in which they take place (Niu et al., Citation2006). EHOMO and ELUMO were also good descriptors for the assessment of kp of 17 PAH (r2 = 0.848; Table S9; Chen et al., Citation1996b).
The direct photolysis (in water) half-lives T1/2ph of 13 PAH under irradiation of sunlight was mainly correlated to ELUMO + EHOMO and ELUMO – EHOMO, but also to α (r2 = 0.912; Table S9; Chen et al., Citation2001e): the half-lives decrease when EHOMO and α increase, but they increase with ELUMO – EHOMO gap and ELUMO. As indicated before, chemical structures tend to be more stable at larger values of the ELUMO – EHOMO gap. In sunlight, ELUMO – EHOMO was presumed to be an indicator of the wavelengths absorbed, and therefore of the energy of the intermediate excited state. The half-lives also decreased with MW therefore it can be concluded that the larger the PAH molecules, the faster the degradation rate. This is probably mainly caused by enhanced spectral overlap of the absorption spectra of the high molecular weight PAH with solar radiation. In methanol/water solution, it was shown that PAH with larger ELUMO – EHOMO gap absorbs light with small wavelengths and may exhibit greater photolysis rate (Chen et al., Citation1996b).
The most relevant descriptors to explain the variability of the photolysis of organic compounds were found to be EHOMO and ELUMO. The molecular weight MW, and the quantum-chemical descriptors α and μ were also relevant. Only constitutional, geometric, and quantum-chemical descriptors were used in the equations.
Reductive transformation is the dominant reaction pathway for many organic compounds in anoxic environments, and reducing environments abound in nature (e.g., subsurface waters and soils, aquatic sediments, sewage sludge, oxygen-free segments of eutrophic rivers). Most of the research in reductive transformations of chemicals focuses on dehalogenation of chlorinated aliphatic or aromatic contaminants, and on the reduction of nitroaromatic compounds (Tratnyek et al., Citation2003). Only nine QSAR were found to estimate three parameters related to reduction processes: rate constants kred, k for dechlorination, and one-electron reduction potential EOH, which quantifies the tendency for a reduction reaction to occur (Tables S2 and S9). The diversity and number of organic compounds from which the equations were developed is low: only nitroaromatics, chlorinated aliphatics and halogenated aliphatic hydrocarbons.
The ELUMO was the best descriptor to explain the variability in the reactivity data for the reduction of six nitroaromatics (r2 = 0.990; Table S9) and 12 chlorinated aliphatics (r2 = 0.832; Table S9; Colón et al., Citation2006; Scherer et al., Citation1998). ELUMO characterizes the tendency of a compound to accept electrons or to be reduced: the greater the ELUMO values are, the lower the tendency of a compound to accept electrons is. Correlations were also found with electron affinity EA (r2 = 0.834) and one-electron reduction potential E1 (r2 = 0.810). EA represents the energy difference associated with the gain of an electron, which should correlate with the ease or difficulty in the reduction of a compound (Colón et al., Citation2006). E1 is a promising descriptor for dechlorination as it represents the potential of the rate limiting initial electron-transfer step (Scherer et al., Citation1998).
The kred of 13 halogenated aliphatic hydrocarbons can be correctly estimated using a combination of constitutional (MW), and of several quantum-chemical (BO, C, CCR, EE, EE1c, EE1x, EE2, EHOMO, ELUMO, EN1c, EN1x, EN2, HOF, J, K, NN2, q−, q-cx, QH+, qxc, TE, TE2, α, and μ; Table S1) descriptors (r2 = 0.808; Table S9). As for nitroaromatics and chlorinated aliphatics, the greater the ELUMO values, the lower the dehalogenation rate constants of halogenated aliphatic hydrocarbons. Increasing the values of QH+ led to decreasing values of kred, and the higher the BO, the lower the kred, which implies that the stronger carbon-halogen bond, the slower the dehalogenation rate. Halogenated aliphatic compounds with great BO, EE, ELUMO, QH+, and TE tend to be dehalogenated slowly, whereas halogenated aliphatic compounds with high values of CCR, MW, and α values tend to be dehalogenated fast (Zhao et al., Citation2001).
The EOH of 20 nitroaromatics can be estimated with one of these three quantum-chemical descriptors: EA, ELUMO in aqueous or gas phase, or vertical detachment energy (VDE). ELUMO and VDE are alternative measures used to approximate the energy change accompanying the transfer of one electron. EA value incorporates the effect of changes in geometry between the (gas-phase) anion and neutral species, whereas ELUMO and VDE do not account for geometry changes. All correlations have similarly high predictive capabilities (r2 ranges from 0.913 to 0.940; Table S9) suggesting that any reasonable measure of the energy change accompanying the one-electron reduction process will be highly correlated with the reduction potential (Phillips et al., Citation2010).
As a conclusion, ELUMO was found to be the most fundamental descriptor allowing the prediction of the reduction of organic compounds in the environment. Only constitutional, and quantum-chemical descriptors were used, but the number of equations is low.
Oxidation, along with hydrolysis and reduction, accounts for the vast majority of chemical reactions that result in degradation of organic contaminants, especially in aquatic systems. Oxidation of organics can occur by a wide variety of mechanisms: the main ones are loss of electrons or hydrogen atoms (abstraction by the oxidant), addition of an oxidant (OH, 1O2, manganese (III/IV) oxides), or substitution of a hydrogen atom by an electron-withdrawing atom or functional group (Canonica and Tratnyek, Citation2003; Rorije and Peijnenburg, Citation1996). Oxidation by ozone (ozonation) is also considered in this review as it is an effective method for removing residual pollutants such as pesticides and other hazardous chemicals from water during drinking water treatment (Hu et al., Citation2000; Sudhakaran and Amy, Citation2013). Twenty-four QSAR allowing the prediction of oxidation rates are reported in Table S9. They mainly involve one descriptor (with a maximum of three descriptors) and were developed for several classes of organic compounds (Table S9).
EHOMO was the best descriptor to estimate the oxidation of substituted phenols by singlet oxygen, manganese (III/IV) oxides, chlorine dioxide, peroxydisulfate, and potassium dichromate in the aqueous phase (r2 > 0.750; Table S9), and the oxidation reactivity decreases with increasing EHOMO. Indeed, an electron from the HOMO with a large ionization potential has to overcome a larger energy barrier before it can be removed from its orbital (Rorije and Peijnenburg, Citation1996). EHOMO was also strongly related to the reaction rates kHOCl of eight organophosphorous pesticides in the presence of chlorine (r2 > 0.950; Table S9; Duirk et al., Citation2009), and with reaction rates with ozone kO3 for different pesticides (r2 > 0.840; Table S9; Hu et al., Citation2000), but kO3 increases with EHOMO. However, the number of compounds used for the development of these QSAR was very small (from three to a marginal maximum of 24; Table S9). For eight phenoxyalkylacetic pesticides, the rate constant kO3 was very well predicted by a two-parameter QSAR model that used EHOMO and the absolute electronegativity (EN) as predictors (r2 = 0.970; Table S9), but the number of data for the regression was also low. The dependence of rate constants of pesticides on EHOMO shows that the reaction between pesticides and ozone was controlled by the frontier orbital effect connected with partly covalent bonding in the transition state (Hu et al., Citation2000; Ljubic and Sabljic, Citation2002). For 55 miscellaneous compounds, some correlations between kOH and constitutional descriptors such as nC = C or double bond equivalence (DBE), geometric such as SAS, and quantum-chemical such as α were found. With one descriptor, the best estimates of kOH of 55 miscellaneous organic compounds were found with DBE or the number of ring atoms (NR; r2 = 0.767 and 0.902, respectively; Table S9), but the greatest correlation was obtained with a combination of DBE and the weakly polar component of SAS (WPSA; r2 = 0.918; Table S9). Similarly, for 27 organic compounds, the best estimate of kO3 used DBE, WPSA, and IP (r2 = 0.832; Table S9). DBE focuses on the double bond nature of the organic compounds which enhances ozonation efficiency. WPSA focuses on the surface area occupied by halogens, and IP represents the energy required to remove an electron from a neutral atom. An increase in both WPSA and IP decreases ozonation efficiency (Sudhakaran and Amy, Citation2013). For 60 aromatic compounds, two models were found to give good estimate of kOH (r2 > 0.735; Table S9). The four-descriptors model involves EHOMO, the Geary autocorrelation-2 lag weighted by atomic polarizabilities (GATS2p), the leverage-weighted autocorrelation of lag 7 weighted by atomic polarizabilities (HATS7p) and the number of path of length 8 (P8), whereas the five-descriptors model involves EHOMO, MW, P9, and two 3D MoRSE-signal descriptors (Mor(02)e and Mor(26)p). In both cases, the main contribution to the degradation rate was obtained from EHOMO. Indeed, as stated before, EHOMO determines the nucleophilic ability of compound and hence the possibility of reaction by attack of such a strong electrophile as the OH radical. Thus, compounds with higher value of EHOMO are more reactive with OH radical (Kušić et al., 2009).
Considering these results, EHOMO was the most used and the most useful descriptor to estimate the oxidation of organic compounds in the environment. Only constitutional, geometric, geometric-topological, and quantum-chemical descriptors were used, but the number of equations is low.
3.5.2.2 Abiotic Degradation in the Atmosphere
Organic compounds emitted or formed in the troposphere are removed by physical processes such as wet and dry deposition, and by chemical transformation processes that include reaction with photochemically generated oxidants such as hydroxyl radicals and ozone at day time, and nitrate radicals at night time (Güsten, Citation1999; Meylan and Howard, Citation2003; Pompe and Veber, Citation2001). Reaction with chlorine atoms can also be significant due to their high concentrations in the atmosphere (Long and Niu, Citation2007; Meng et al., Citation2005). Güsten et al. (Citation1995) reviewed the tropospheric degradation of chemicals and found that the great majority of published models were only developed for single chemical class and a small number of chemicals.
Only four equations were found to predict the persistence of several persistent organic pollutants (POP) in the atmosphere, and they are related to four different environmental parameters (Table S9).
Atmospheric half-life (T1/2) is one of the criteria commonly used to study air persistence and long-range transport (LRT) potentials of organic compounds. The mean atmospheric half-lives (Mean T1/2) of 59 POP were satisfactorily correlated (r2 = 0.841; Table S9) to two global WHIM descriptors (Ku and Tu), and to two topological descriptors (mean information content index on the distance degree equality IED,deq and Kier flexibility index ø). The WHIM descriptors were the most relevant ones, highlighting that 3D size Tu, and shape Ku have opposite role in determining the persistence of compounds (Tu has a negative sign in the regression and Ku has a positive sign). The same relationship was obtained for the maximum half-lives (Max T1/2), but the results were slightly less satisfactory than for mean half-lives (mean T1/2; r2 = 0.826; Table S9; Gramatica et al., Citation2001).
A principal component analysis performed on the same data allowed the development of an atmospheric persistence index (API), and a LRT index. The API was correlated to nH (this highlights the importance of the number of hydrogen atoms in the molecule influencing the hydroxyl-radical reaction), and to three WHIM descriptors (Ve, η2e and θ2e; r2 = 0.897; Table S9). The LRT index was well correlated to MW and to two directional WHIM (λ2e and λ1p; r2 = 0.952; Table S9; Gramatica et al., Citation2001).
Only two equations were found to estimate the photodegradation of PAH, PCDD, and PCDF in the atmosphere (Table S9). The predicted photodegradation half-lives T1/2p of 11 PAH on aerosols (r2 = 0.960; Chen et al., Citation2001b), and 75 PCDD and PCDF on fly ashes (r2 = 0.704; Niu et al., Citation2004) were mainly correlated to EHOMO, ELUMO, MW, and α (Table S9). Increasing MW values of the PCDD and PCDF leads to increase half-life values; on the contrary, PAH with great MW values tend to photolyze fast. The PAH, PCDD, and PCDF with small absolute electronegativity (EN) values and large absolute (EHOMO – ELUMO) values tend to have lower T1/2p (Chen et al., Citation2001b; Niu et al., Citation2004).
In the atmosphere, the oxidation of organic compounds is mainly due to reactions with OH, O3, NO3, and Cl atoms, and in particular, the reaction with the OH radical is the major chemical loss process for the majority of organic compounds emitted into the troposphere (Medven et al., Citation1996; Sabljic and Peijnenburg, Citation2001). Twenty-nine equations were found to estimate the oxidation reaction rate constants of organic compounds in the atmosphere, the highest number being for reaction with OH (Table S9). The equations involve from 1 to 10 descriptors, and were developed for miscellaneous organic compounds, but not, for example, for PCB or pesticides.
Among the 15 QSAR related to the reaction rates with OH in the atmosphere, kA,OH, eight relationships were developed using only one descriptor (Table S9). The EHOMO allowed good estimate of kA,OH of various compounds (r2 > 0.749; Bartolotti and Edney, Citation1994; Güsten et al., Citation1995), and the ionization potential IP was well correlated to the kA,OH of 15 hydrocarbons and fluorinated hydrocarbons (r2 = 0.846; Percival et al., Citation1995), and of aromatic and aliphatic compounds (r2 > 0.902; Güsten et al., Citation1984; Table S9). Oberg (Citation2005) found that the main sources of variation of kA,OH were directly linked to four constitutional descriptors: number of aromatic bonds (NAB), NDB, nH, and nX. For Bakken and Jurs (Citation1999), the rate constants kA,OH of 52 unsaturated hydrocarbons were estimated using five topological descriptors: number of sp2 hybridized carbon atoms (2SP2, 3SP2) and molecular distance edge (MDE-13, MDE-23, MDE-34), encoding information concerning attack sites for the radical, branching information, and steric considerations (r2 = 0.868; Table S9). This is consistent with the reaction center for radical reactions which is often an unsaturated carbon. Then, they developed a second relation using computational neural networks (CNN). It showed correlation with miscellaneous descriptors: one constitutional (number of multiple-multiple carbon bonds MCB), one topological (PND), one geometric-electronic (FPSA-3), and two quantum-chemical (ELUMO and EN). PND encodes information on branching of the molecules, and quantum-chemical descriptors encode the energetics of the reactant molecular orbitals that will be involved in the reaction. Then, using a very large dataset of 281 miscellaneous compounds, they found a ten-descriptor linear relationship involving two constitutional (NAB, number of lone pairs (NLP)), six topological (3SP2, MDE-14, PND, sum of weighted paths starting from heteroatoms WTPT-3, path-three κ index 3κ, 3χ), and two quantum-chemical descriptors (ELUMO, EN). The CNN model involved three constitutional (nC, NDB, NSB), three topological (1SP2, MDE-33, WTPT-3), one geometric (GEOM-3), one geometric-electronic (CHAA-3), and two quantum-chemical (EHOMO, Hard) descriptors (r2 = 0.876; Table S9). Using descriptors of the different categories, kA,OH of various organic chemicals was correctly correlated to some constitutional (MW, number of atoms in the molecule (NAT), nC, nHD, nOH, NoRING, unsaturation index (UI)), and geometric (WHIM descriptors) and/or topological (information index) descriptors (r2 > 0.733; Table S9; Gramatica et al., Citation1999a). The rate constants of 14 alkylnaphthalene, as for it, depended on MW, and on quantum-chemical (CCR, EE, EHOMO, ELUMO, QCave, QH+, TE, μ, α) descriptors (r2 = 0.879; Table S9; Long and Niu, Citation2007). Finally, using PLS regression, Medven et al. (1996) showed that five descriptors had more pronounced influence than the others on the kA,OH of 57 unsaturated hydrocarbons: EHOMO, ELUMO, Hard, average number of alkyl substituents per unsaturated bond (nAlk), and number of carbon atoms in unsaturated bonds (nCub). Among these, EHOMO was the most influential, which is in accordance with the assumed mechanism of electrophilic addition to multiple bonds in which the singly occupied molecular orbital (SOMO) of the electrophilic radical predominantly interacts with the HOMO of the unsaturated chemical, which means that chemicals with low values of EHOMO are more reactive. Two other descriptors, Hard and ELUMO, have a strong influence on the reactivity of unsaturated compounds with hydroxyl radical. Again, chemicals with lower values of both descriptors are more reactive. Thus a small HOMO-LUMO energy gap has a positive effect on the reactivity of unsaturated chemicals with hydroxyl radical. This result can be rationalized within the framework of the frontier molecular orbital theory: the SOMO of the electrophilic radical also interacts with the LUMO of unsaturated chemicals, and this interaction has a significant effect on reactivity. As expected, the reactivity of unsaturated chemicals increases with the number of carbon atoms in unsaturated bonds or potential reactive centers. Furthermore, the degree of alkyl substitution on unsaturated bonds (nAlk) also has a positive effect on reactivity.
Klamt (Citation1993) proposed to divide the rate constants of degradation of various organic compounds by hydroxyl radicals considering three subreactions: (a) the OH addition to carbon-carbon double bonds (kaddC), which depended on the charge-limited effective HOMO energy at H atom (ECHH); (b) the addition to aromatic rings (karC), which depended on the energy-weighted effective HOMO energy at atom H (EEHH) and on the energy required to deform the molecule in a way to enable the OH-addition (ΔdefC); and (c) the hydrogen abstraction from aliphatic carbon atoms (kabsH), which depended on ECHH. They obtained very good results (r2 > 0.954; Table S9). For 13 halogenated compounds, an hologram method was used to predict half-life of the hydroxyl radical with substituted aromatic compounds: the generated fragments include atoms, bonds, connections, hydrogen atoms, donor and acceptor atoms, and the chiral center (Vrtacnik and Voda, Citation2003).
The QSAR developed by Atkinson (Citation1987), though widely used to estimate the OH reaction rates, were not considered in this study, as they involve an experimental descriptor: the sum of the electrophilic substituent constants.
Two equations are reported for the reaction rates with ozone, kA,O3, and quantum-chemical descriptors were found to be the best predictors in both cases (Table S9). For 117 miscellaneous compounds, all descriptors of the QSAR were quantum-chemical (average electrophilic reaction index for a C atom AERC, EHOMO, fractional hydrogen bonding donor ability of the molecule FHDCA(1), maximum exchange energy for a C‒C bond MaxC‒C, maximum electron-electron repulsion for a C‒C bond MaxeeC-C, minimum (> 0.1) bond order of a C atom MinC; r2 = 0.870; Table S9). The relationship encodes chemical features which are important in the reaction of organic compounds with ozone, that is, information about formation of transition state, cleavage of the chemical bonds, and different conformational changes. The MaxC-C and MaxeeC-C characterize intramolecular energy distribution and may be related to the conformational changes or atomic reactivity in the molecule (Pompe and Veber, Citation2001). The other quantum-chemical descriptors that were used to predict kA,O3 were EHOMO, AERC which represents the interactions between the frontier orbitals of the reacting compounds, and the MinC that relates to the strength of intramolecular bonding interactions, and therefore to the stability of the molecule or its conformational flexibility (Sannigrahi, Citation1992). The FHDCA(1) can describe polar interactions between molecules as well as their chemical reactivity (Pompe and Veber, Citation2001). Among the quantum-chemical (EHOMO – ELUMO gap), constitutional (MW, number of conjugated double bonds nAB, number of isolated double bonds nDB), topological (Moran autocorrelation lag (7) weighted by atomic Sanderson electronegativities (MATS7e), and geometric-electronic (R autocorrelation of lag 3 weighted by atomic Sanderson electronegativities (R3e)) descriptors that allow the prediction of kA,O3 of 125 miscellaneous compounds, the best descriptor was the EHOMO – ELUMO gap. As stated before, this is an important stability index reflecting molecule reactivity as well as polarization: the more reactive chemicals have a smaller gap. The information regarding attack sites for ozone is provided by the constitutional descriptors highlighting the relevance of the double bonds for the molecular cleavage by ozone. Charge distribution factors, in addition to dimensional aspects, are encoded by the different kinds of autocorrelation descriptors selected, all weighted by the atomic electronegativity of Sanderson (Gramatica et al., Citation2003; Table S9).
Eight equations allowing estimate of the rate constants for the reaction with NO3, kNO3, were inventoried in Table S9. The kNO3 are essentially and correctly predicted by EHOMO and/or ELUMO (r2 > 0.677; Table S9; Güsten et al., Citation1995; Long and Niu, Citation2007; Müller and Klein, Citation1991), though the mechanism of NO3 radical addition is complex and that it's maybe not possible to model its reactivity by one single descriptor (Güsten et al., Citation1995). The relationship developed by Long and Niu (Citation2007) for 14 alkylnaphthalene involved, in addition to ELUMO and EHOMO, constitutional (MW) and quantum-chemical (QCave, QH+, total energy TE, standard heat of formation ΔHf, μ) descriptors (r2 = 0.817; Table S9). For 58 aliphatic compounds, the kNO3 is correlated to constitutional (NAT, nHD, UI), and geometric WHIM (Du, λ1s) descriptors (r2 = 0.840), and for 16 aromatics, kNO3 was correlated to two constitutional (MW, hydrophilic factor (HY)), and one topological (mean information content index on the distance degree equality IED,deq) descriptors (r2 = 0.978; Table S9; Gramatica et al., Citation1999a). Sabljic and Güsten (Citation1990) developed two relationships linking kNO3 to the ionization potential (IP): one for 62 aliphatic compounds and one for seven benzene derivatives. For the aliphatic compounds, the correlation was significant (r2 = 0.927; Table S9) but chloroalkenes compounds needed to be approximate by the parent hydrocarbon molecule because of lack of ionization energy in the study. For benzene derivatives, good correlation (r2 = 0.883; Table S9) was found excluding two outliers (tetraline and methoxybenzene), however the dataset was limited.
Only one equation was found to estimate the reaction rate of organic compounds with chlorine kCl (Table S9). For 14 alkylnaphthalene, kCl was again mainly found to depend on energies of orbital (EHOMO, ELUMO), but also on QCave, QH+, and μ (r2 = 0.944; Table S9; Long and Niu, Citation2007).
As a summary, EHOMO was the most relevant descriptor to estimate the oxidation of organic compounds in the atmosphere. No geometric-topological and no electro-topological descriptors were involved in the equations.
3.5.2.3 Abiotic Degradation on Vegetation.
Few QSAR studies are related to the degradation of organic compounds on the surface of plants, and only two equations were found for PCDD and PCDF (Table S9). The photodegradation half-lives T1/2v of 42 PCDF adsorbed to spruce needle surfaces (Niu et al., Citation2005) was mainly correlated to EHOMO, ELUMO, MW, TE, and α (r2 = 0.740; Table S9). But, the stability of PCDF was also shown to increase with the increase in chlorine atoms in the parent molecules, and PCDF with high (ELUMO – EHOMO) values tend to be more stable and difficult to be degraded. For 10 PCDD and PCDF dissolved in cuticular wax from Prunus laurocerasus leaves exposed to sunlight, the degradation rates kv mainly depended on qCl, but the rates also increased with MW and α. PCDD and PCDF with large values of ELUMO, EHOMO, and ELUMO – EHOMO tend to have low kv, however the dataset was limited (r2 = 0.958; Table S9; Chen et al., Citation2001a).
3.6 Absorption by Plants Processes
Organic compounds present in soil, water and air may be taken up by plants (Paterson et al., Citation1994). The chemical can be transferred to the vegetation from the soil and/or water by uptake through the roots (i.e., symplastic way), from the atmosphere by their aboveground parts after wet or dry deposition, or following rain-splash in which soil particles are dispersed onto the leaf surfaces following the impact of raindrops on the soil surface (Hiatt, Citation1998; McKone and Maddalena, 2007; Paterson et al., Citation1994; Sabljic et al., Citation1990). For atmospheric contaminants, the predominant initial site of interception is the plant cuticle, and the adsorption on the cuticles is the first step of the transfer of volatile and nonvolatile organic compounds to plant (Chaumat et al., Citation1992; Hiatt, Citation1998; Welke et al., Citation1998).
Twenty-six QSAR allowing the estimation of the parameters describing the absorption of organic compounds by plants are reported in Table S10. The absorption can be estimated with the bioconcentration factor BCF (ratio between the contaminant concentration in the plant tissue and the concentration in soil), the bioconcentration ratio BCR (the ratio of a tissue concentration to the concentration in a relevant exposure medium), the cuticle-air partition coefficient Kca, the cuticle-water partition coefficient KCW, the polymer matrix membrane-water partition coefficient KMXw, the permeability of the cuticle P, and the sorbed amount by the plant cuticle Q. In most cases, a single descriptor was used to predict these environmental parameters, the maximum being five descriptors. The equations were developed for a wide diversity of organic compounds but mainly with very limited datasets (Table S10).
The BCF (in zucchini) of several POP were correlated to nCl (except for PCB), which is related to the hydrophobicity of the molecule. However, the best correlations were found using combinations of GETAWAY, VolSurf, and quantum-chemical descriptors (r2 > 0.918; Table S10; Bordás et al., Citation2011): POP taken up preferentially from soil are characterized by high values of ELUMO – EHOMO, VOH2 and HB5O (two VolSurf descriptors), and low values of Z-component, GETAWAY (H4p and H5e), and VolSurf (BV31OH2, D3DRY, D6DRY, H5e, W4O) descriptors.
The MCI were involved in six of the 26 relationships: the BCR of several miscellaneous compounds for aboveground plant-soil bioconcentration, root-soil bioconcentration, and plant-air bioconcentration were well correlated with 1χ and the polar correction factors of Meylan et al. (1992; r2 > 0.780; Table S10; Dowdy and McKone, Citation1997). Similarly, for 14 alcohols, the Kca (of tomato cuticle) was well correlated to 1χ (r2 = 0.868; Table S10; Welke et al., Citation1998). For 47 organic chemicals, the KCW was correctly estimated using 3χv and nOHaliph (r2 = 0.984; Table S10). The KCW are primarily influenced by the size of the molecule, which is described by 3χv: larger molecules show a higher affinity for cuticles than smaller ones. However, it has to be underlined that not all parts of a molecule contribute equally to its affinity for plant cuticles. The major contribution is from chlorine substituents, hydrocarbon chains, and benzene rings. Another factor controlling the magnitude of KCW is the presence of aliphatic hydroxy groups. The negative regression coefficient of nOHaliph shows that the association with cuticles decreases with the presence of aliphatic hydroxy groups. Compared with the main factor, the size of the molecule, nOHaliph can be viewed only as a fine-tuning element for the affinity of organic chemicals for plant cuticles (Sabljic, Citation1991; Sabljic et al., Citation1990).
For 5 phenylurea herbicides, significant relationships between KCW, Q, or P (for tomato and pepper), and the steric descriptors ΣD and ΣS were found (r2 > 0.943; Table S10), but the number of data used in the regression is very low. As observed for the 47 organic chemicals (Sabljic et al., Citation1990), the overall dimension of the phenylurea molecules reflected by the steric descriptors is an important factor for cuticular adsorption. Within the same chemical family, penetration in plant tissues is greater for chemicals well adsorbed to cuticles (Chaumat et al., Citation1992).
Platts and Abraham (2002) used the LSER approach to estimate the KMXw for tomato of 62 volatile organic compounds, and found very good result (r2 = 0.981; Table S10). They concluded that the cuticular matrix interacts more through pi- and n-electron pairs, is rather less polar/polarizable and basic, and much less acidic than bulk water, and that cavities are much more easily formed in cuticle than in water.
Finally, a PLS analysis was performed to determine the main descriptors involved in the estimation of the Rhizopus oryzae cell-wall water partition coefficients of only 3 PAH. MW was the principal contributing descriptor, followed by the total information content index with neighborhood symmetry of one-order TIC1 (Ma et al., Citation2011).
As a conclusion, descriptors related to the size of the organic compounds (3χv, steric descriptors such as ΣS and ΣD, and the number of chlorine atoms) were the most relevant ones to estimate their absorption by plants.
4. SYNTHESIS AND DISCUSSION
For the first time, a comprehensive review of QSAR focused on several processes driving the fate of organic compounds in the environment was done. Six main processes were considered: water dissolution, dissociation, volatilization, retention, degradation, and absorption by higher plants. These main processes were then subdivided in 23 environmental subprocesses as shown in Table S2. We chose to focus our work on QSAR based on structural molecular descriptors because QSAR based on KOW or SW, for example, are prone to experimental errors in the input variables, which can result in some statistical problems (Lohninger, Citation1994; Nguyen et al., Citation2005; Sabljic and Piver, Citation1992). Overall, 790 QSAR equations involving 686 different molecular descriptors allowing the assessment of 90 environmental parameters are presented here (Tables S1–S10). The equations were developed for a wide diversity of organic compounds including pesticides, PAH, PCB, PCDD, pharmaceuticals, and hormones. However it has to be underlined that almost no pesticide was up to now included in the development of QSAR for pKa, vapor pressures, KH, KOA, or adsorption on sediments. Similarly, most of the QSAR related to abiotic degradation were developed for restricted classes of organic compounds (Tables S3–S10).
shows the number of equations found for each of the 23 environmental processes (Table S2). The highest number of equations, 145 equations (i.e., 18.3% of the total number of equations), was developed to predict the dissociation of organic compounds (pKa). A fairly large number of equations were found to predict the KOW (115; i.e., 14.5%), and the adsorption of compounds on the soils (102; i.e., 12.9%) as well. The number of equations to predict biodegradation and solubility in water (SW) were also important, being 75 (i.e., 9.5%) and 65 (i.e., 8.2%), respectively. On the contrary, there were only one equation for the soil desorption of organic compounds, two equations for the degradation of compounds on vegetation, four equations for the nonlinear adsorption, and six equations to predict the potential of transfer to groundwater. To the best of our knowledge, there is neither QSAR to estimate the formation of nonextractable (bound) residues, known as an important dissipation route of pesticides and other organic compounds in soil matrices (Barriuso et al., Citation2008), nor to estimate the potential of transfer of organic compounds to surface water. This might be explained by the fact that the estimation of the transfer of organic compounds to ground and surface water is often addressed by mass balance models, which are mechanistic (Mackay et al., Citation2003).
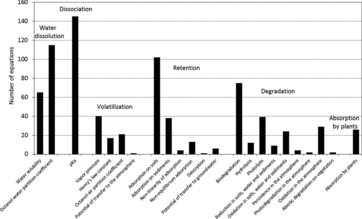
The descriptors that were the most used in the 790 equations are summarized in . Twenty-two descriptors were involved in more than 10 equations: nine were quantum-chemical descriptors related to the energies (CCR, EE, EHOMO, ELUMO, q−, QH+, TE, α, μ), eight were topological and they were all MCI (0χ, 0χv, 1χ, 1χv, 2χ, 2χv, 3χv, 3χvc), three were geometric related to the surface and the volume of the compounds (TSA, VdW, Vm), and two were constitutional (MW and nCl). The most used descriptor is EHOMO, which is involved in 80 (i.e., 10%) equations, followed by α in 68 (i.e., 8.6%) equations, ELUMO in 58 (i.e., 7.3%) equations, MW in 57 (i.e., 7.2%) equations, μ in 46 (i.e., 5.8%) equations, and 1χv in 44 (i.e., 5.6%) equations. The nCl, VdW, Vm, TSA, 0χ, 0χv, 1χ, 2χ, 2χv, 3χv, 3χvc, CCR, EE, q−, QH+, and TE descriptors were included in 11–41 (i.e., 1.4 to 5.4%) equations (). The remaining descriptors of Table S1 were mainly found only in one or two equations.
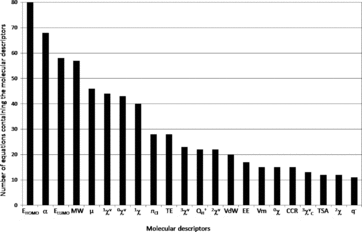
Finally, the descriptors the most frequently used to estimate environmental processes are summarized in (a descriptor involved in several different process will be considered as generic). Nineteen descriptors were found to be involved in six or more processes (): twelve were quantum-chemical (EE, EHOMO, ELUMO, HOF, q−, qC, QH+, TE, α, ΔGs, ΔHf, μ), four were topological (1χ, 1χv, 2χv, 3χv), two were constitutional (MW, nCl) and one was geometric (Vm). Fifteen of these 19 descriptors were among the 22 most used descriptors (Table S1, ); the four remaining descriptors are HOF, qC-, ΔGs, and ΔHf. In particular, the five most generic descriptors were also the five most used descriptors: EHOMO, α, ELUMO, μ, and MW, and among them, EHOMO was the most used descriptor and one of those that allowed the assessment of the highest diversity of environmental processes ( and ).
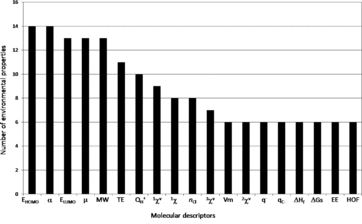
The overall synthesis of the results that are reviewed in this work showed that, in general, an increase in the absolute values of EHOMO led to a decrease in KOW, adsorption on soils, redox reactions in soils, water and sediments, photodegradation in the atmosphere, and degradation on the vegetation. On the contrary an increase in the absolute values of EHOMO led to an increase in biodegradation and BCF (Tables S3–S10). Similar results were found for ELUMO, except that an increase in the absolute values of ELUMO involved an increase in photolysis and atmospheric oxidation of organic compounds (there was no clear relationship between ELUMO and biodegradation or BCF; Tables S3–S10). Orbitals play a major role in a lot of chemical reactions and they are also responsible for the formation of many charge-transfer complexes. The EHOMO is directly related to the ionization potential and characterizes the susceptibility of the molecule toward electrophilic attack (Karelson et al., Citation1996). EHOMO also represents the proton acceptance ability in forming hydrogen bond, while ELUMO represents the proton donation ability in forming hydrogen bond. Therefore, the compounds with large value of EHOMO and ELUMO tend to donate or accept protons easily (Chen et al., Citation2002a; Colón et al., Citation2006; Zhou et al., Citation2005). This could explain why EHOMO and ELUMO were rather good descriptors for estimating several processes such as adsorption, biodegradation, photolysis, redox reactions, and degradation in the atmosphere.
The two quantum-chemical descriptors, α and μ, also allowed the estimate of a wide diversity of environmental processes (; Table S2). They were especially used to assess partition properties such as SW, KOW, PL, PS, KH, KOA, and adsorption on soils and sediments (Tables S3, S4, S6, and S7). An increase in α was related to an increase in KOW, KOA, redox reactions and degradation on vegetation; and to a decrease in SW, vapor pressure and photodegradation in the atmosphere (Tables S3 to S10). The bigger the α is, the more hydrophobic the molecules are (Chen et al., 1996; Katritzky et al., Citation1998; Shi et al., Citation2012; Yang et al., Citation2007). An increase in μ was found to increase KOA, COD, atmospheric photodegradation, and degradation on the vegetation, but to decrease KOW, vapor pressure, adsorption and reduction reactions. As indicated before, molecules with larger μ tend to transfer from octanol phase to water phase, to volatilize and to sorb less because intermolecular dipole-dipole interactions and dipole-induced dipole interactions are in direct proportion to μ2. A larger dipole would imply greater dipole interactions with the polar water molecules (Chen et al., Citation1996a; Dai et al., Citation2000; Shi et al., Citation2012; Zeng et al., Citation2007).
The last of the five most used and generic descriptor is MW ( and ; Tables S3, S4, S6–S9). An increase in MW is correlated with an increase in KOW, KOA, adsorption on soils, reduction reactions, degradation on vegetation, and to a decrease in SW, vapor pressure, biodegradation, photolysis, and persistence in the atmosphere.
Finally, the MCI 1χv was also a useful descriptor (Tables S3, S4, S6 to S8). Solubility in water and vapor pressure decrease when 1χv increases. On the contrary, KOW, KOA, and adsorption on soils increase with 1χv. Several other MCI (0χ, 2χv, 3χv) were involved in the estimate of partition properties and biodegradation (Tables S3, S4, S6–S8, S10).
This review also showed that the combination of descriptors belonging to different categories improves the estimate of the different environmental parameters, probably because it allows to consider simultaneously different representations and properties of the molecule (Basak et al., Citation1997; Boethling and Sabljic, Citation1989; Güsten et al., Citation1991; Huibers and Katritzky, Citation1998; Kim et al., Citation2007; Liang and Gallagher, Citation1998; Lü et al., Citation2007; Makino, Citation1998; Reddy and Locke, Citation1994a; Sabljic et al., Citation1989; Schüürmann, Citation1995; Sudhakaran and Amy, Citation2013; Xie et al., Citation2008; Tables S3–S9). Therefore, one could hypothesize that the combination of the five descriptors that were identified as the most used and most generic ones will be pretty much helpful to develop the next generation of QSAR to predict a range of environmental parameters for a wide diversity of organic compounds.
5. CONCLUSION
A tremendously high number of organic compounds having a wide diversity are still released in the environment. Although it is well known that some of them will cause environmental and health problems in a near future, they cannot be studied on a case by case basis because it is time consuming and cost prohibitive. Therefore, in order to overcome this problem, a lot of QSAR allowing the prediction of the fate of organic compounds in the environment from their molecular properties were developed. This review is the first comprehensive synthesis of QSAR focused on the principal processes governing the behavior of organic compounds in the different compartments of the environment, and using only structural molecular descriptors.
The most important numbers of equations were found for pKa, KOW, adsorption to soils and biodegradation parameters. A lack of QSAR was observed to estimate the desorption, nonequilibrium adsorption, adsorption nonlinearity, potential of transfer to water (especially surface water), or nonextractable residues formation. Five molecular descriptors (energies of orbitals EHOMO and ELUMO, polarizability α, dipole moment μ and molecular weight (MW)) were especially used in the 790 equations, and also involved in the assessment of the highest diversity of environmental processes. Further QSAR development should therefore pay a particular attention to these descriptors. In addition, the combination of descriptors belonging to different categories (e.g., constitutional, topological, quantum-chemical) was generally found to improve the predictions of the environmental parameters as it simultaneously considers different representations and properties of the molecule.
In order to facilitate the broader application of the QSAR in organic compounds risks assessment, it is important to define criteria of acceptability of these predicting models, improving their validation, defining their accuracy, and defining and checking their applicability domain (range of cases for which prediction can be made; Boethling and Costanza, Citation2010; Gramatica, Citation2007; Hermens et al., Citation1995; Jaworska et al., Citation2005). The use of QSAR for regulatory purposes has been increasing steadily (Cronin et al., Citation2003; Mackay et al., Citation2003). This review is delivering relevant QSAR equations to predict the fate of a wide diversity of compounds in the environment. In a near future the development and the implementation of highly powerful QSAR may offer an invaluable insight in various stages of the hazard and risk assessment processes.
ACKNOWLEDGMENTS
The authors acknowledge the anonymous reviewers for their constructive comments.
SUPPLEMENTAL MATERIAL
Supplemental data for this article can be accessed on the publisher*s website.
Mamy_etal_CREST_2015_SI.pdf
Download PDF (1.5 MB)Related Research Data
REFERENCES
- Abraham, M.H. (1993). Hydrogen bonding. XXVII. Solvation parameters for functionally substituted aromatic compounds and heterocyclic compounds, from gas-liquid chromatographic data. Journal of Chromatography 644, 95–139.
- Abraham, M.H., and McGowan, J.C. (1987). The use of characteristic volumes to measure cavity terms in reversed phase liquid chromatography. Chromatographia 23, 243–246.
- Arp, H.P. H., Breedveld, G.D., and Cornelissen, G. (2009). Estimating the in situ sediment-porewater distribution of PAHs and chlorinated aromatic hydrocarbons in anthropogenic impacted sediments. Environmental Science and Technology 43, 5576–5585.
- Atkinson, R. (1987). A structure-activity relationship for the estimation of rate constants for the gas-phase reactions of OH radicals with organic compounds. International Journal of Chemical Kinetics 19, 799–828.
- Bahnick, D.A., and Doucette, W.J. (1988). Use of molecular connectivity indices to estimate soil sorption coefficients for organic chemicals. Chemosphere 17, 1703–1715.
- Baker, J.R., Gamberger, D., Mihelcic, J.R., and Sabljic, A. (2004). Evaluation of artificial intelligence based models for chemical biodegradability prediction. Molecules 9, 989–1004.
- Baker, J.R., Mihelcic, J.R., Luehrs, D.C., and Hickey, J.P. (1997). Evaluation of estimation methods for organic carbon normalized sorption coefficients. Water Environment Research 69, 136–145.
- Baker, J.R., Mihelcic, J.R., and Sabljic, A. (2001). Reliable QSAR for estimating Koc for persistent organic pollutants: correlation with molecular connectivity indices. Chemosphere 45, 213–221.
- Bakken, G.A., and Jurs, P.C. (1999). Prediction of hydroxyl rate constants from molecular structure. Journal of Chemical Information and Computer Science 39, 1064–1075.
- Barriuso, E., Benoit, P., and Dubus, I.G. (2008). Formation of pesticide nonextractable (bound) residues in soil: magnitude, controlling factors and reversibility. Environmental Science and Technology 42, 1845–1854.
- Bartolotti, L.J., and Edney, E.O. (1994). Investigation of the correlation between the energy of the highest occupied molecular orbital (HOMO) and the logarithm of the OH rate constant of hydrofluorocarbons and hydrofluoroethers. International Journal of Chemical Kinetics 26, 913–920.
- Basak, S.C. (1999). Information theoretic indices of neighborhood complexity and their application. In Topological Indices and Related Descriptors in QSAR and QSPR; Devillers, J., and Balaban, A.T., Eds.; Gordon and Breach Science Publishers: The Netherlands 1999; pp 563–593.
- Basak, S.C., Gute, B.D., and Grunwald, G.D. (1996). A comparative study of topological and geometrical parameters in estimating normal boiling point and octanol/water partition coefficient. Journal of Chemical Information and Computer Sciences 36, 1054–1060.
- Basak, S.C., Gute, B.D., and Grunwald, G.D. (1997). Use of topostructural, topochemical, and geometric parameters in the prediction of vapor pressure: a hierarchical QSAR approach. Journal of Chemical Information and Computer Sciences 37, 651–655.
- Beasley, K.K., Gieg, L.M., Suflita, J.M., and Nanny, M.A. (2009). Polarizability and spin density correlate with the relative anaerobic biodegradability of alkylaromatic hydrocarbons. Environmental Science and Technology 43, 1995–2000.
- Beigel, C., Barriuso, E., and Di Pietro, L. (1997). Time dependency of triticonazole fungicide sorption and consequences for diffusion in soil. Journal of Environmental Quality 26, 1503–1510.
- Berger, B.M., and Wolfe, N.L. (1996). Hydrolysis and biodegradation of sulfonylurea herbicides in aqueous buffers and anaerobic water-sediment systems: assessing fate pathways using molecular descriptors. Environmental Toxicology and Chemistry 15, 1500–1507.
- Berger, B.M., Müller, M., and Eing, A. (2001). Quantitative structure-transformation relationships of phenylurea herbicides. Pest Management Science 57, 1043–1054.
- Berger, B.M., Müller, M., and Eing, A. (2002). Quantitative structure-transformation relationships of sulfonylurea herbicides. Pest Management Science 58, 724–735.
- Bhhatarai, B., and Gramatica, P. (2011). Modelling physico-chemical properties of (benzo)triazoles, and screening for environmental partitioning. Water Research 45, 1463–1471.
- Blockeel, H., Dzeroski, S., Kompare, B., Kramer, S., Pfahringer, B., and Van Laer, W. (2004). Experiments in predicting biodegradability. Applied Artificial Intelligence 18, 157–181.
- Bodor, N., and Buchwald, P. (1997). Molecular size based approach to estimate partition properties for organic solutes. Journal of Physical Chemistry B 101, 3404–3412.
- Bodor, N., Gabanyi, Z., and Wong, C.-K. (1989). A new method for the estimation of partition coefficient. Journal of American Chemical Society 111, 3783–3786.
- Boethling, R.S. (1986). Application of molecular topology to quantitative structure-biodegradability relationships. Environmental Toxicology and Chemistry 5, 797–806.
- Boethling, R.S., and Costanza, J. (2010). Domain of EPI suite biotransformation models. SAR and QSAR in Environmental Research 21, 415–443.
- Boethling, R.S., Howard, P.H., Meylan, W., Stiteler, W., Beauman, J., and Tirado, N. (1994). Group-contribution method for predicting probability and rate of aerobic biodegradation. Environmental Science and Technology 28, 459–465.
- Boethling, R.S., and Sabljic, A. (1989). Screening-level model for aerobic biodegradability based on a survey of expert knowledge. Environmental Science and Technology 23, 672–679.
- Bogdanov, B., Nikolić, S., and Trinajstić, N. (1989). On the three-dimensional Wiener number. Journal of Mathematical Chemistry 3, 299–309.
- Bordás, B., Bélai, I., and Kőmives, T. (2011). Theoretical molecular descriptors relevant to the uptake of persistent organic pollutants from soil by Zucchini. A QSAR Study. Journal of Agricultural and Food Chemistry 59, 2863–2869.
- Braekevelt, E., Tittlemier, S.A., and Tomy, G.T. (2003). Direct measurement of octanol–water partition coefficients of some environmentally relevant brominated diphenyl ether congeners. Chemosphere 51, 563–567.
- Brennan, R.A., Nirmalakhandan, N., and Speece, R.E. (1998). Comparison of predictive methods for Henrys law coefficients of organic chemicals. Water Research 32, 1901–1911.
- Briggs, G.G. (1981). Theoretical and experimental relationships between soil adsorption, octanol-water partition coefficients, water solubilities, bioconcentration factors, and the parachor. Journal of Agricultural and Food Chemistry 29, 1050–1059.
- Brown, T.N., and Mora-Diez, N. (2006a). Computational determination of aqueous pKa values of protonated benzimidazoles (Part 1). The Journal of Physical Chemistry B 110, 9270–9279.
- Brown, T.N., and Mora-Diez, N. (2006b). Computational determination of aqueous pKa values of protonated benzimidazoles (Part 2). The Journal of Physical Chemistry B 110, 20546–20554.
- Brunner, S., Hornung, E., Santl, H., Wolff, E., Piringer, O.G., Altschuh, J., and Brüggemann, R. (1990). Henry's law constant for polychlorinated biphenyls: experimental determination and structure-property relationships. Environmental Science and Technology 24, 1751–1754.
- Brusseau, M.L. (1993). Using QSAR to evaluate phenomenological models for sorption of organic compounds by soil. Environment Toxicology and Chemistry 12, 1835–1846.
- Burgos, W.D., and Pisutpaisal, N. (2006). Sorption of naphthoic acids and quinoline compounds to estuarine sediment. Journal of Contaminant Hydrology 84, 107–126.
- Canonica, S., and Tratnyek, P.G. (2003). Quantitative structure-activity relationships for oxidation reactions of organic chemicals in water. Environment Toxicology and Chemistry 22, 1743–1754.
- Cao, Q., Garib, V., Yu, Q., Connell, D.W., and Campitelli, M. (2009). Quantitative structure–property relationships (QSPR) for steroidal compounds of environmental importance. Chemosphere 76, 453–459.
- Chaumat, E., Chamel, A., Taillandier, G., and Tissut, M. (1992). Quantitative relationships between structure and penetration of phenylurea herbicides through isolated plant cuticles. Chemosphere 24, 189–200.
- ChemOffice. (2009). ChemOffice Ultra 12.0 molecular modelling software. Cambridge, MA: Perkin Elmer.
- Chen, J.W., Feng, L., Liao, Y., Han, S., and Wang, L.S. (1996a). Using AM1 Hamiltonian in quantitative structure-properties relationship studies of alkyl(1-phenylsulfonyl)cycloalkane carboxylates. Chemosphere 33, 537–546.
- Chen, J.W., Harner, T., Ding, G., Quan, X., Schramm, K.-W., and Kettrup, A. (2004). Universal predictive models on octanol-air partition coefficients at different temperatures for persistent organic pollutants. Environmental Toxicology and Chemistry 23, 2309–2317.
- Chen, J.W., Harner, T., Schramm, K.-W., Quan, X., Xue, X.Y., and Kettrup, A. (2003b). Quantitative relationships between molecular structures, environmental temperatures and octanol/air partition coefficients of polychlorinated biphenyls. Computational Biology and Chemistry 27, 405–421.
- Chen, J.W., Harner, T., Schramm, K.W., Quan, X., Xue, X.Y., Wu, W.Z., and Kettrup, A. (2002b). Quantitative relationships between molecular structures, environmental temperatures and octanol-air partition coefficients of PCDD/Fs. Science of the Total Environment 300, 155–166.
- Chen, J.W., Harner, T., Yang, P., Quan, X., Chen, S., Schramm, K.-W., and Kettrup, A. (2003c). Quantitative predictive models for octanol-air partition coefficients of polybrominated diphenyl ethers at different temperatures. Chemosphere 51, 577–584.
- Chen, J.W., Kong, L.R., Zhu, C.M., Huang, Q.G., and Wang, L.S. (1996b). Correlation between photolysis rate constants of polycyclic aromatic hydrocarbons and frontier molecular orbital energy. Chemosphere 33, 1143–1150.
- Chen, J.W., Peijnenburg, W.J. G. M., Quan, X., and Yang, F. (2000). Quantitative structure-property relationships for direct photolysis quantum yields of selected polycyclic aromatic hydrocarbons. Science of the Total Environment 246, 11–20.
- Chen, J.W., Peijnenburg, W.J. G. M., Quan, X., Chen, S., Martens, D., Schramm, K.-W., and Kettrup, A. (2001e). Is it possible to develop a QSPR model for direct photolysis half-lives of PAHs under irradiation of sunlight? Environmental Pollution 114, 137–143.
- Chen, J.W., Peijnenburg, W.J. G. M., Quan, X., Zhao, Y., Xue, D., and Yang, F. (1998b). The application of quantum chemical and statistical technique in developing quantitative structure-property relationships for the photohydrolysis quantum yields of substituted aromatic halides. Chemosphere 37, 1169–1186.
- Chen, J.W., Peijnenburg, W.J. G. M., and Wang, L. (1998a). Using PM3 Hamiltonian, factor analysis and regression analysis in developing quantitative structure-property relationships for photohydrolysis quantum yields of substituted aromatic halides. Chemosphere 36, 2833–2853.
- Chen, J.W., Quan, X.F., Schramm, K.-W., Kettrup, A., and Yang, F. (2001c). Quantitative structure-property relationships (QSPR) on direct photolysis of PCDDs. Chemosphere 45, 151–159.
- Chen, J.W., Quan, X., Yan, Y., Yang, F., and Peijnenburg, W.J.G.M. (2001b). Quantitative structure-property relationship studies on direct photolysis of selected aromatic hydrocarbons in atmospheric aerosol. Chemosphere 42, 263–270.
- Chen, J.W., Quan, X., Yang, F., and Peijnenburg, W.J.G.M. (2001a). Quantitative structure-property relationships on photodegradation of PCDD/Fs in cuticular waxes of laurel cherry (Prunus laurocesarus). Science of the Total Environment 269, 163–170.
- Chen, J.W., Quan, X., Zhao, Y.Z., Yang, F.L., Schramm, K.-W., and Kettrup, A. (2001d). Quantitative structure-property relationships for octanol-air partition coefficients of PCDD/Fs. Bulletin of Environmental Contamination and Toxicology 66, 755–761.
- Chen, J.W., Xue, X., Schramm, K.-W., Quan, X., Yang, F., and Kettrup, A. (2003a). Quantitative structure-property relationships for octanol/air partition coefficients of polychlorinated naphthalenes, chlorobenzenes and p,p’-DDT. Computational Biology and Chemistry 27, 165–171.
- Chen, J.W., Xue, X., Schramm, K.W., Quan, X., Yang, F., and Kettrup, A. (2002a). Quantitative structure-property relationships for octanol-air partition coefficients of polychlorinated biphenyls. Chemosphere 48, 535–544.
- Chen, S.-D., Zeng, X.-L., Wang, Z.-Y., and Liu, H.-X. (2007). QSPR modeling of n-octanol/water partition coefficients and water solubility of PCDEs by the method of Cl substitution position. Science of the Total Environment 382, 59–69.
- Cheu, J., Huang, Q., and Wang, L. (1996). Using AM1 Hamiltonian and factor analysis in prediction of partition properties for phenylthio, phenylsulfinyl, and phenylsulfonyl acetates. Chemosphere 33, 2565–2575.
- Citra, M.J. (1999). Estimating the pKa of phenols, carbolxylic acids and alcohols from semi-empirical quantum chemical methods. Chemosphere 38, 191–206.
- Clark, M. (2005). Generalized fragment-substructure based property prediction method. Journal of Chemical Information and Modeling 45, 30–38.
- Colón, D., Weber, E.J., and Anderson, J.L. (2006). QSAR study of the reduction of nitroaromatics by Fe(II) species. Environmental Science and Technology 40, 4976–4982.
- Colón, D., Weber, E.J., and Baughman, G.L. (2002). Sediment-associated reactions of aromatic amines. 2. QSAR development. Environmental Science and Technology 36, 2443–2450.
- Consonni, V., and Todeschini, R. (2010). Molecular descriptors. In T. Puzyn, J. Leszczynski, and M.T.D. Cronin (Eds.), Recent advances in QSAR studies. Methods and applications (pp. 20–102). New York: Springer.
- Consonni, V., Todeschini, R., and Pavan, M. (2002). Structure/response correlations and similarity/diversity analysis by GETAWAY descriptors. 1. Theory of the novel 3D molecular descriptors. Journal of Chemical Information and Computer Science 42, 682–692.
- Cousins, I., and Mackay, D. (2000). Correlating the physical-chemical properties of phthalate esters using the ‘three solubility’ approach. Chemosphere 41, 1389–1399.
- Cramer, C.J., Famini, G.R., and Lowrey, A.H. (1993). Use of calculated quantum chemical properties as surrogates for solvatochromic parameters in structure-activity relationships. Accounts of Chemical Research 26, 599–605.
- Cronin, M.T. D., Walker, J.D., Jaworska, J.S., Comber, M.H. I., Watts, C.D., and Worth, A.P. (2003). Use of QSARs in international decision-making frameworks to predict ecologic effects and environmental fate of chemical substances. Environmental Health Perspectives 111, 1376–1390.
- Cruciani, G., Crivori, P., Carrupt, P.-A., and Testa, B. (2000a). Molecular fields in quantitative structure-permeation relationships: the VolSurf approach. Journal of Molecular Structure (Theochem) 503, 17–30.
- Cruciani, G., Pastor, M., and Guban, W. (2000b). VolSurf: a new tool for the pharmacokinetic optimization of lead compounds. European Journal of Pharmaceutical Sciences 11, S29–S39.
- Cuissart, B., Touffet, F., Cremilleux, B., Bureau, R., and Rault, S. (2002). The maximum common substructure as a molecular depiction in a supervised classification context: experiments in quantitative structure/biodegradability relationships. Journal of Chemical Information and Computer Sciences 42, 1043–1052.
- Dai, J., Jin, L., Wang, L., and Zhang, Z. (1998). Determination and estimation of water solubilities and octanol/water partition coefficients for derivatives of benzanilides. Chemosphere 37, 1419–1427.
- Dai, J., Sun, C., Han, S., and Wang, L. (1999). QSAR for polychlorinated organic compounds (PCOCs). I. Prediction of partition properties for PCOCs using quantum chemical parameters. Bulletin of Environmental Contamination and Toxicology 62, 530–538.
- Dai, J., Xu, M., and Wang, L. (2000). Prediction of octanol/water partitioning coefficient and sediment sorption coefficient for benzaldehydes by various molecular descriptors. Bulletin of Environmental Contamination and Toxicology 65, 190–199.
- Dearden, J.C., and Nicholson, R.M. (1986). The prediction of biodegradability by the use of quantitative structure-activity-relationships: Correlation of biological oxygen-demand with atomic charge difference. Pesticide Science 17, 305–310.
- Dearden, J.C., and Nicholson, R.M. (1987). QSAR study of the biodegradability of environmental pollutants. In D. Hadzi and B. Blazic (Eds.), QSAR in drug design and toxicology (Vol. 45, pp. 307–312), Amsterdam.
- Dearden, J.C., and Schüürmann, G. (2003). Quantitative structure-property relationships for predicting Henrys’law constant from molecular structure. Environmental Toxicology and Chemistry 22, 1755–1770.
- Delisle, R.K., and Dixon, S.L. (2004). Induction of decision trees via evolutionary programming. Journal of Chemical Information and Computer Sciences 44, 862–870.
- Desai, S.M., Govind, R., and Tabak, H.H. (1990). Development of quantitative structure-activity-relationships for predicting biodegradation kinetics. Environmental Toxicology and Chemistry 9, 473–477.
- Dimitriou-Christidis, P., Autenrieth, R.L., and Abraham, M.H. (2008). Quantitative structure-activity relationships for kinetic parameters of polycyclic aromatic hydrocarbon biotransformation. Environmental Toxicology and Chemistry 27, 1496–1504.
- Ding, G., Chen, J., Qiao, X., Huang, L., Lin, J., and Chen, X. (2006). Quantitative relationships between molecular strutures, environmental temperatures and solid vapor pressures of PCDD/Fs. Chemosphere1057–1063.
- Djohan, D., Yu, Q., and Connell, D.W. (2005). Partition isotherms of chlorobenzenes in a sediment-water system. Water, Air and Soil Pollution 161, 157–173.
- Doucette, W.J. (2003). Quantitative structure-activity relationships for predicting soil-sediment sorption coefficients for organic chemicals. Environmental Toxicology and Chemistry 22, 1771–1788.
- Doucette, W.J., and Andren, A.W. (1988). Estimation of octanol/water partition coefficients: evaluation of six methods for highly hydrophobic aromatic hydrocarbons. Chemosphere 17, 345–359.
- Dowdy, D.L., and McKone, T.E. (1997). Predicting plant uptake of organic chemicals from soil or air using octanol/water and octanol/air partition ratios and a molecular connectivity index. Environmental Toxicology and Chemistry 16, 2448–2456.
- 5 (2007). Software for the calculation of molecular descriptors, Talete s.r.l. http://www.talete.mi.it/
- Droge, S.T. J., Yarza-Irusta, L., and Hermens, J.L. M. (2009). Modeling nonlinear sorption of alcohol ethoxylates to sediment: the influence of molecular structure and sediment properties. Environmental Science and Technology 43, 5712–5718.
- Duirk, S.E., Desetto, L.M., and Davis, G.M. (2009). Transformation of organophosphorous pesticides in the presence of aqueous chlorine: kinetics, pathways, and structure-activity relationships. Environmental Science and Technology 43, 2335–2340.
- Dunnivant, F.M., Elzerman, A.W., Jurs, P.C., and Hasan, M.N. (1992). Quantitative structure-property relationships for aqueous solubilities and Henry's law constants of polychlorinated biphenyls. Environmental Science and Technology 26, 1567–1573.
- Edward, J.T. (1998). Calculation of octanol-water partition coefficients of organic solutes from their molecular volumes. Canadian Journal of Chemistry 76, 1294–1303.
- Estrada, E., Delgado, E.J., Alderete, J.B., and Jaña, G.A. (2004). Quantum-connectivity descriptors in modeling solubility of environmentally important organic compounds. Journal of Computational Chemistry 25, 1787–1796.
- Famini, G.R., and Wilson, L.Y. (1997). Using theoretical descriptors in quantitative structure activity relationships: application to partition properties of alkyl (1-phenylsulfonyl)cycloalkane-carboxylates. Chemosphere 35, 2417–2447.
- Feng, L., Han, S., Wang, L., and Wang, Z. (1996). Determination and estimation of partitioning properties for phenylthio-carboxylates. Chemosphere 32, 353–360.
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A. Jr., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, N.J., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, Ö., Foresman, J.B., Ortiz, J.V., Cioslowski, J., and Fox, D.J. (2009). Gaussian 09. Wallingford, CT: Gaussian, Inc.
- Gamberger, D., Horvatic, D., Sekusak, S., and Sabljic, A. (1996). Applications of experts’ judgement to derive structure-biodegradation relationships. Environmental Science and Pollution Research International 3, 224–228.
- Gawlik, B.M., Sotiriou, N., Feicht, E.A., Schulte-Hostede, S., and Kettrup, A. (1997). Alternatives for the determination of the soil adsorption coefficient, Koc, of non-ioninorganic compounds: A review. Chemosphere 34, 2525–2551.
- Geating, J. (1981). Project summary, literature study of the biodegradability of chemicals in water, vol 1 and 2, U.S. Environmental Protection Agency, EPA-600/S2-172/176.
- Gerstl, Z. (1990). Estimation of organic chemical sorption by soils. Journal of Contaminant Hydrology 6, 357–375.
- Gerstl, Z., and Helling, C.S. (1987). Evaluation of molecular connectivity as a predictive method for the adsorption of pesticides by soils. Journal of Environmental Science and Health B 22, 55–69.
- Ghasemi, J., Saaidpour, S., and Brown, S.D. (2007). QSPR study for estimation of acidity constants of some aromatic acids derivatives using multiple linear regression (MLR) analysis. Journal of Molecular Structure: THEOCHEM 805, 27–32.
- Gombar, V.K., and Enslein, K. (1991). A structure-biodegradability relationship model by discrimant analysis. In J. Devillers and W. Karcher (Eds.), Applied multivariate analysis in SAR and environmental studies. Dordrecht, the Netherlands: Kluwer Academic.
- Gombar, V.K., and Enslein, K. (1996). Assessment of n-octanol/water partition coefficient: when is the assessment reliable? Journal of Chemical Information and Computer Science 36, 1127–1134.
- Goss, K.-U. (2006). Prediction of the temperature dependency of Henry's law constant using poly-parameter linear free energy relationships. Chemosphere 64, 1369–1374.
- Goss, K.-U., and Schwarzenbach, R.P. (2001). Linear free energy relationships used to evaluate equilibrium partitioning of organic compounds. Environmental Science and Technology 35, 1–9.
- Goudarzi, N., Goodarzi, M., Ugulino Araujo, M.C., and Harrop Galvao, R.K. (2009). QSPR modeling of soil sorption coefficients (KOC) of pesticides using SPA-ANN and SPA-MLR. Journal of Agricultural and Food Chemistry 57, 7153–7158.
- Gramatica, P. (2007). Principles of QSAR models validation: internal and external. QSAR and Combinatorial Science 26, 694–701.
- Gramatica, P., Consolaro, F., and Pozzi, S. (2001). QSAR approach to POPs screening for atmospheric persistence. Chemosphere 43, 655–664.
- Gramatica, P., Consonni, V., and Todeschini, R. (1999a). QSAR study on the tropospheric degradation of organic compounds. Chemosphere 38, 1371–1378.
- Gramatica, P., Corradi, M., and Consonni, V. (2000). Modelling and prediction of soil sorption coefficients of non-ionic pesticides by molecular descriptors. Chemosphere 41, 763–777.
- Gramatica, P., and Di Guardo, A. (2002). Screening of pesticides for environmental partitioning tendency. Chemosphere 47, 947–956.
- Gramatica, P., Navas, N., and Todeschini, R. (1999b). Classification of organic solvents and modelling of their physico-chemical properties by chemometric methods using different sets of molecular descriptors. Trends in Analytical Chemistry 18, 461–471.
- Gramatica, P., Pilutti, P., and Papa, E. (2003). QSAR prediction of ozone tropospheric degradation. QSAR 22, 364–373.
- Greaves, A.J., Churchley, J.H., Hutchings, M.G., Philipps, D.A. S., and Taylor, J.A. (2001). A chemometric approach to understanding the bioelimination of anionic, water-soluble dyes by a biomass using empirical and semi-empirical molecular descriptors. Water Research 35, 1225–1239.
- Gross, K.C., and Seybold, P.G. (2000). Substituents effects on the physical properties and pKa of aniline. International Journal of Quantum Chemistry 80, 1107–1115.
- Gross, K.C., and Seybold, P.G. (2001). Substituents effects on the physical properties and pKa of phenol. International Journal of Quantum Chemistry 85, 569–579.
- Gross, K.C., Seybold, P.G., Peralta-Inga, Z., Murray, J.S., and Politzer, P. (2001). Comparison of quantum chemical parameters and Hammett constants in correlating pKa values of substituted anilines. Journal of Organic Chemistry 66, 6919–6925.
- Grüber, C., and Buß, V. (1989). Quantum-mechanically calculated properties for the development of quantitative-structure activity relationships (QSAR's). pKa values of phenols and aromatic and aliphatic carboxylic acids. Chemosphere 19, 1595–1609.
- Gupta, K., Roy, D.R., Subramanian, V., and Chattaraj, P.K. (2007). Are strong Brønsted acids necessarily strong Lewis acids? Journal of Molecular Structure 812, 13–24.
- Gupta, S., Singh, M., and Madan, A.K. (1999). Superpendentic index: a novel topological descriptor for predicting biological activity. Journal of Chemical Information and Computer Sciences 39, 272–277.
- Gustafson, D.I. (1989). Groundwater ubiquity score: a simple method for assessing pesticide leachability. Environmental Toxicology and Chemistry 8, 339–357.
- Güsten, H. (1999). Predicting the abiotic degradability of organic pollutants in the troposphere. Chemosphere 38, 1361–1370.
- Güsten, H., Horvatic, D., and Sabljic, A. (1991). Modelling n-octanol/water partition coefficients by molecular topology: polycyclic aromatic hydrocarbons and their alkyl derivatives. Chemosphere 23, 199–213.
- Güsten, H., Klasinc, L., and Maric, D. (1984). Prediction of the abiotic degradability of organic compounds in the troposphere. Journal of Atmospheric Chemistry 2, 83–93.
- Güsten, H., Medven, Z., Sekusak, S., and Sabljic, A. (1995). Predicting tropospheric degradation of chemicals: from estimation to computation. SAR and QSAR in Environmental Research 4, 197–209.
- Habibi-Yangjeh, A., Pourbasheer, E., and Danandeh-Jenagharad, M. (2009). Application of principal component-genetic algorithm-artificial neural network for prediction acidity constant of various nitrogen-containing compounds in water. Monatshefte für Chemie 140, 15–27.
- Hall, L.H., and Story, C.T. (1996). Boiling point and critical temperature of a heterogeneous data set: QSAR with atom type electrotopological state indices using artificial neural networks. Journal of Chemical Information and Computer Sciences 36, 1004–1014.
- Hall, L.H., Mohney, B., and Kier, L.B. (1991). The electrotopological state: an atom index for QSAR. Quantitative Structure Activity Relationships 10, 43–51.
- Han, X.-Y., Wang, Z.-Y., Zhai, Z.-C., and Wang, L.-S. (2006). Estimation of n-octanol/water partition coefficients (KOW) of all PCB congeners by ab initio and a Cl substitution position method. QSAR and Combinatorial Science 25, 333–341.
- Hanai, T. (2003). Quantitative structure–retention relationships of phenolic compounds without Hammett's equations. Journal of Chromatography A 985, 343–349.
- Hance, R.J. (1969). An empirical relationship between chemical structure and the sorption of some herbicides by soils. Journal of Agricultural and Food Chemistry 17, 667–668.
- Hansch, C., and Gao, H. (1997). Comparative QSAR: radical reactions of benzene derivatives in chemistry and biology. Chemical Reviews 97, 2995–3059.
- Hansch, C., and Leo, A. J. (1979). Substituent constants for correlation analysis in chemistry and biology. New York: Wiley.
- Hansen, B.G., Paya-Perez, A.B., Rahman, M., and Larsen, B.R. (1999a). QSARs for KOW and Koc of PCB congeners: a critical examination of data, assumptions and statistical approaches. Chemosphere 39, 2209–2228.
- Hansen, B.G., van Haelst, A.G., van Leeuwen, K., and van der Zandt, P. (1999b). Priority setting for existing chemicals: European Union risk ranking methods. Environmental Toxicology and Chemistry 18, 772–779.
- Hawker, D.W., and Connell, D.W. (1988). Octanol-water partition coefficients of polychlorinated biphenyl congeners. Environmental Science and Technology 22, 382–387.
- Hawthorne, S.B., Grabanski, C.B., Miller, D.J., and Arp, H.P. H. (2011). Improving predictability of sediment-porewater partitioning models using trends observed with PCB-contaminated field sediments. Environmental Science and Technology 45, 7365–7371.
- He, Y., Wang, L., Han, S., Zhao, Y., Zhang, Z., and Zou, G. (1995). Determination and estimation of physicochemical properties for phenylsulfonyl acetates. Chemosphere117–125.
- Hermens, J., Balaz, S., Damborsky, J., Karcher, W., Müller, M., Peijnenburg, W., Sabljic, A., and Sjöström, M. (1995). Assessment of QSARs for predicting fate and effects of chemicals in the environment: an international European project. SAR and QSAR in Environmental Research 3, 223–236.
- Hiatt, M.H. (1998). Bioconcentration factors for volatile organic compounds in vegetation. Analytical Chemistry 70, 850–856.
- Hickey, J.P., and Passino-Reader, D.R. (1991). Linear solvation energy relationships: “rules of thumb” for estimation of variable values. Environmental Science and Technology 25, 1753–1760.
- Hollingsworth, C.A., Seybold, P.G., and Hadad, C.M. (2002). Substituents effects on the electronic structure and pKa of benzoic acid. International Journal of Quantum Chemistry 90, 1396–1403.
- Hou, T.J., Xia, K., Zhang, W., and Xu, X.J. (2004). ADME evaluation in drug discovery. 4. Prediction of aqueous solubility based on atom contribution approach. Journal of Chemical Information and Computer Science 44, 266–275.
- Howard, P.H. (2000). Biodegradation. In R. Boethling and D. Mackay (Eds.), Handbook of property estimation methods for chemicals (pp. 281–310). Boca Raton: Lewis.
- Howard, P.H. (2008). Predicting the persistence of organic compounds. In Handbook of Environmental Chemistry Vol. 2, Springer, Berlin, published online 14 March 2008.
- Howard, P.H., Boethling, R.S., Stiteler, W.M., Meylan, W.M., and Beauman, H.A. (1991). Development of a predictive model for biodegradability based on Biodeg, the evaluated biodegradation database. Science of the Total Environment 109, 635–641.
- Howard, P.H., Boethling, R.S., Stiteler, W.M., Meylan, W.M., Hueber, A.E., Beauman, H.A., and Larosche, M.E. (1992). Predictive model for aerobic biodegradability developed from a file of evaluated biodegradation data. Environmental Toxicology and Chemistry 6, 593–603.
- Howard, P.H., Meylan, W., Aronson, D., Stiteler, W., Tunkel, J., Comber, M., and Parkerton, T. (2005). A new biodegradation prediction model specific to petroleum hydrocarbons. Environmental Toxicology and Chemistry 24, 1847–1860.
- Hsieh, H.-N., and Mukherjee, S. (2003). A QSAR model for desorption of halogenated aliphatics from biosolids. Advances in Environmental Research 7, 511–520.
- Hu, J.-Y., Morita, T., Magara, Y., and Aizawa, T. (2000). Evaluation of reactivity of pesticides with ozone in water using the energies of frontier molecular orbitals. Water Research 34, 2215–2222.
- Hu, Q., Wang, X., and Brusseau, M.L. (1995). Quantitative structure-activity relationships for evaluating the influence of sorbate structure on sorption of organic compounds by soil. Environmental Toxicology and Chemistry 14, 1133–1140.
- Huibers, P.D. T., and Katritzky, A.R. (1998). Correlation of the aqueous solubility of hydrocarbons and halogenated hydrocarbons with molecular structure. Journal of Chemical Information and Computer Sciences 38, 283–292.
- Huuskonen, J.J. (2000). Estimation of aqueous solubility for a diverse set of organic compounds based on molecular topology. Journal of Chemical Information and Computer Science 40, 773–777.
- Huuskonen, J.J. (2001a). Estimation of water solubility from atom-type electrotopological state indices. Environmental Toxicology and Chemistry 20, 491–497.
- Huuskonen, J.J. (2001b). Prediction of biodegradation from the atom-type electrotopological state indices. Environmental Toxicology and Chemistry 20, 2152–2157.
- Huuskonen, J.J. (2003). Prediction of soil sorption coefficient of organic pesticides from the atom-type electrotopological state indices. Environmental Toxicology and Chemistry 22, 816–820.
- Huuskonen, J.J., Salo, M., and Taskinen, J. (1997). Neural network modeling for estimation of the aqueous solubility of structurally related drugs. Journal of Pharmaceutical Sciences 86, 450–454.
- Huuskonen, J.J., Salo, M., and Taskinen, J. (1998). Aqueous solubility prediction of drugs based on molecular topology and neural network modeling. Journal of Chemical Information and Computer Sciences 38, 450–456.
- Huuskonen, J.J., Villa, A.E. P., and Tetko, I.V. (1999). Prediction of partition coefficient based on atom-type electrotopological state indices. Journal of Pharmaceutical Sciences 88, 229–233.
- HyperChem. (2007). Hyperchem 8.0. Gainesville, FL: Hypercube, Inc.
- Jaworska, J., Boethling, R.S., and Howard, P.S. (2003). Recent development in broadly applicable structure-biodegradability relationships. Environmental Toxicology and Chemistry 22, 1710–1723.
- Jaworska, J., Dimitrov, S., Nikolova, N., and Mekenyan, O. (2002). Probabilistic assessment of biodegradability based on metabolic pathways: Catabol system. SAR and QSAR in Environmental Research 13, 307–323.
- Jaworska, J., Nikolova-Jeliazkova, N., and Aldenberg, T. (2005). QSAR applicability domain estimation by projection of the training set in descriptor space: a review. Atla-Alternatives to Laboratory Animals 33, 445–459.
- Jin, L., Dai, J., Wang, L., Wei, Z., and Huang, Q. (1997). Determination and estimation of the sorption of benzaldehydes on soil. Chemosphere 35, 2707–2712.
- Jover, J., Bosque, R., and Sales, J. (2007). Neural network based QSPR study for predicting pKa of phenols in different solvents. QSAR and Combinatorial Science 26, 385–397.
- Jover, J., Bosque, R., and Sales, J. (2008). QSPR prediction of pKa for benzoic acids in different solvents. QSAR and Combinatorial Science 27, 563–581.
- Kallies, B., and Mitzner, R. (1997). pKa values of amines in water from quantum mechanical calculations using a polarized dielectric continuum representation of the solvent. Journal of Physical Chemistry B 101, 2959–2967.
- Kamlet, M.J., Doherty, R.M., Carr, P.W., Mackay, D., Abraham, M.H., and Taft, R.W. (1988). Linear solvation energy relationships. 44. Parameter estimation rules that allow accurate prediction of octanol/water partition coefficients and other solubility and toxicity properties of polychlorinated biphenyls and polycyclic aromatic hydrocarbons. Environmental Science and Technology 22, 503–509.
- Kanazawa, J. (1989). Relationship between the soil sorption constants for pesticides and their physicochemical properties. Environmental Toxicology and Chemistry477–484.
- Karelson, M., Lobanov, V.S., and Katritzky, A.R. (1996). Quantum-chemical descriptors in QSAR/QSPR studies. Chemical Reviews 96, 1027–1043.
- Katayama, A., Bhula, R., Burns, G.R., Carazo, E., Felsot, A., Hamilton, D., Harris, C., Kim, Y.-H., Kleter, G., Koedel, W., Linders, J., Peijnenburg, J.G. M. W., Sabljic, A., Stephenson, R.G., Racke, D.K., Rubin, B., Tanaka, K., Unsworth, J., and Wauchope, D. (2010). Bioavailability of xenobiotics in the soil environment. Reviews of Environmental Contamination and Toxicology 203, 1–86.
- Katritzky, A.R., Lobadov, V.S., and Karelson, M. (2005). CODESSA PRO User's Manual, University of Florida (http://www.codessa-pro.com/).
- Katritzky, A.R., Maran, U., Lobanov, V.S., and Karelson, M. (2000). Structurally diverse quantitative structure-property relationship correlations of technologically relevant physical properties. Journal of Chemical Information and Computer Sciences 40, 1–18.
- Katritzky, A.R., Wang, Y., Sild, S., and Tamm, T. (1998). QSPR studies on vapor pressure, aqueous solubility, and the prediction of water-air partition coefficients. Journal of Chemical Information and Computer Science 38, 720–725.
- Kier, L.B. (1986). Shape indexes of orders one and three from molecular graphs. Quantitative Structure-Activity Relationship, 5, 1–7.
- Kier, L.B., and Hall, L.H. (1999). The electrotopological state: structure modelling for QSAR and database analysis. In Topological Indices and Related Descriptors in QSAR and QSPR; Devillers, J., and Balaban, A.T., Eds.; Gordon and Breach Science Publishers: The Netherlands 1999; pp 491–562.
- Kier, L.B., and Hall, L.H. (2000). Intermolecular accessibility: the meaning of molecular connectivity. Journal of Chemical Information and Computer Sciences 40, 792–795.
- Kier, L.B., and Hall, L.H. (2002). The meaning of molecular connectivity: a bimolecular accessibility model. Croatica Chemica Acta 75, 371–382.
- Kiewiet, A.T., de Beer, K.G. M., Parsons, J.R., and Govers, H.A.J. (1996). Sorption of linear alcohol ethoxylates on suspended sediments. Chemosphere 32, 675–680.
- Kim, J.H., Gramatica, P., Kim, M.G., Kim, D., and Tratnyek, P.G. (2007). QSAR modelling of water quality indices of alkylphenol pollutants. SAR and QSAR in Environmental Research 18, 729–743.
- Klamt, A. (1993). Estimation of gas-phase hydroxyl radical rate constants of organic compounds from molecular orbital calculations. Chemosphere 26, 1273–1289.
- Klamt, A., Eckert, F., and Diedenhofen, M. (2002). Prediction of soil sorption coefficients with a conductor-like screening model for real solvents. Environmental Toxicology and Chemistry 21, 2562–2566.
- Klamt, A., Eckert, F., Diedenhofen, M., and Beck, M.E. (2003). First principles calculations of aqueous pKa values for organic and inorganic acids using COSMO-RS reveal an inconsistency in the slope of the pKa scale. Journal of Physical Chemistry A 107, 9380–9386.
- Klopman, G. (1992). A hierarchical computer automated structure evaluation program.1. Quantitative Structure-Activity Relationships 11, 176–184.
- Klopman, G., Dimayuga, M., and Talafous, J. (1994). Meta.1. a program for the evaluation of metabolic transformation of chemicals. Journal of Chemical Information and Computer Sciences 34, 1320–1325.
- Klopman, G., Zhang, Z.T., Balthasar, D.M., and Rosenkranz, H.S. (1995). Computer-automated predictions of aerobic biodegradation of chemicals. Environmental Toxicology and Chemistry 14, 395–403.
- Kompare, B. (1998). Estimating environmental pollution by xenobiotic chemicals using QSAR (QSBR) models based on artificial intelligence. Water Science and Technology 37, 9–18.
- Kühne, R., Ebert, R.-U., Kleint, F., Schmidt, G., and Schüürmann, G. (1995). Group contribution methods to estimate water solubility of organic chemicals. Chemosphere 30, 2061–2077.
- Kühne, R., Ebert, R.-U., and Schüürmann, G. (1997). Estimation of vapour pressures for hydrocarbons and halogenated hydrocarbons from chemical structure by a neural network. Chemosphere 34, 671–686.
- Kühne, R., Ebert, R.-U., and Schüürmann, G. (2005). Prediction of the temperature dependency of Henry's law constant from chemical structure. Environmental Science and Technology 39, 6705–6711.
- Kušić, H., Rasulev, B., Leszczynska, D., Leszczynski, J., and Koprivanac, N. (2009). Prediction of rate constants for radical degradation of aromatic pollutants in water matrix: A QSAR study. Chemosphere 75, 1128–1134.
- Lara, R., and Ernst, W. (1989). Interaction between polychlorinated biphenyls and marine humic substances. Determination of association coefficients. Chemosphere 19, 1655–1664.
- Lee, A.C., and Crippen, G.M. (2009). Predicting pKa. Journal of Chemical Information and Modeling 49, 2013–2033.
- Leo, AJ.. (1975). In Veith G.D. and Korasevich D.E. (Eds). Proceedings of Symposium on structure-activity correlations in studies of toxicity and bio-concentration with aquatic organisms (p 151). Great Lakes Research Advisory Board: Burlington, Ontario.
- Li, L., Xie, S., Cai, H., Bai, X., and Xue, Z. (2008). Quantitative structure–property relationships for octanol-water partition coefficients of polybrominated diphenyl ethers. Chemosphere 72, 1602–1606.
- Li, Y., and Xi, D.L. (2007). Quantitative structure-activity relationship study on the biodegradation of acid dyestuffs. Journal of Environmental Sciences 19, 800–804.
- Liang, C., and Gallagher, D.A. (1998). QSPR prediction of vapor pressure from solely theoretically-derived descriptors. Journal of Chemical Information and Computer Sciences 38, 321–324.
- Lindner, A.S., Whitfield, C., Chen, N., Semrau, J.D., and Adriaens, P. (2003). Quantitative structure-biodegradation relationships for ortho-substituted biphenyl compounds oxidized by methylosinus trichosporium OB3b. Environmental Toxicology and Chemistry 22, 2251–2257.
- Liu, G., and Yu, J. (2005). QSAR analysis of soil sorption coefficients for polar organic chemicals: substituted anilines and phenols. Water Research 39, 2048–2055.
- Liu, S., Cao, C., and Li, Z. (1998). Approach to estimation and prediction for normal boiling point (NBP) of alkanes based on a novel molecular distance-edge (MDE) vector, λ. Journal of Chemical Information and Computer Sciences 38, 387–394.
- Liu, S., and Pedersen, L.G. (2009). Estimation of molecular acidity via electrostatic potential at the nucleus and valence natural atomic orbitals. Journal of Physical Chemistry A 113, 3648–3655.
- Liu, S., Yin, C., Cai, S., and Li, Z. (2002). Molecular structural vector description and retention index of polycyclic aromatic hydrocarbons. Chemometrics and Intelligent Laboratory Systems 61, 3–15.
- Ljubic, I., and Sabljic, A. (2002). Theoretical study of the mechanism and kinetics of gas-phase ozone additions to ethene, fluoroethene, and chloroethene: A multireference approach. Journal of Physical Chemistry A 106, 4745–4757.
- Lohninger, H. (1994). Estimation of soil partition coefficients of pesticides from their chemical structure. Chemosphere 29, 1611–1626.
- Long, X., and Niu, J. (2007). Estimation of gas-phase reaction rate constants of alkylnaphthalenes with chlorine, hydroxyl and nitrate radicals. Chemosphere 67, 2028–2034.
- Loonen, H., Lindgren, F., Hansen, B., and Karcher, W. (1996). Biodegradability prediction. Kluwer Academic Publishers, Dordrecht, NL 1996, pp105–114.
- Loonen, H., Lindgren, F., Hansen, B., Karcher, W., Niemela, J., Hiromatsu, K., Takatsuki, M., Peijnenburg, W., Rorije, E., and Struijs, J. (1999). Prediction of biodegradability from chemical structure: modeling of ready biodegradation test data. Environmental Toxicology and Chemistry 18, 1763–1768.
- Louchart, X., and Voltz, M. (2007). Aging effects on the availability of herbicides to runoff transfer. Environmental Science and Technology 41, 1137–1144.
- Lu, C. (2009). Prediction of environmental properties in water-soil-air systems for phthalates. Bulletin of Environmental Contamination and Toxicology 83, 168–173.
- Lu, C., Wang, Y., Yin, C., Guo, W., and Hu, X. (2006). QSPR study on soil sorption coefficient for persistent organic pollutants. Chemosphere 63, 1384–1391.
- Lu, G.-N., Dang, Z., Tao, X.-Q., Yang, C., and Yi, X.-Y. (2008). Estimation of water solubility of polycyclic aromatic hydrocarbons using quantum chemical descriptors and partial least squares. QSAR and Combinatorial Science 27, 618–626.
- Lü, W., Chen, Y., Liu, M., Chen, X., and Hu, Z. (2007). QSPR prediction of n-octanol/water partition coefficient for polychlorinated biphenyls. Chemosphere 69, 469–478.
- Ma, B., Chen, H., Xu, M., Hayat, T., He, Y., and Xu, J. (2010). Quantitative structureeactivity relationship (QSAR) models for polycyclic aromatic hydrocarbons (PAHs) dissipation in rhizosphere based on molecular structure and effect size. Environmental Pollution 158, 2773–2777.
- Ma, B., Xu, M., Wang, J., Chen, H., He, Y., Wu, L., Wang, H., and Xu, J. (2011). Adsorption of polycyclic aromatic hydrocarbons (PAHs) on Rhizopus oryzae cell walls: application of cosolvent models for validating the cell wall-water partition coefficient. Bioresource Technology 102, 10542–10547.
- Ma, Y., Gross, K.C., Hollingsworth, C.A., Seybold, P.G., and Murray, J.S. (2004). Relationships between aqueous acidities and computed surface-electrostatic potentials and local ionization energies of substituted phenols and benzoic acids. Journal of Molecular Modeling 10, 235–239.
- MacElroy, N.R., and Jurs, P.C. (2001). Prediction of aqueous solubility of heteroatom-containing organic compounds from molecular structure. Journal of Chemical Information and Computer Science 41, 1237–1247.
- Mackay, D., Hubbarde, J., and Webster, E. (2003). The role of QSARs and fate models in chemical hazard and risk assessment. Environmental Toxicology and Chemistry 22, 106–112.
- Mackay, D., McCarty, L.S., and McLeod, M. (2001). On the validity of classifying chemicals for persistence, bioaccumulation, toxicity, and potential for long-range transport. Environmental Toxicology and Chemistry1491–1498.
- MacKone, T.E., and Maddalena, R.L. (2007). Plant uptake of organic pollutants from soil: bioconcentration estimates based on models and experiments. Environmental Toxicology and Chemistry 26, 2494–2504.
- Makino, M. (1998). Prediction of n-octanol/water partition coefficients of polychlorinated biphenyls by use of computer calculated molecular properties. Chemosphere 37, 13–26.
- Medven, Z., Güsten, H., and Sabljic, A. (1996). Comparative QSAR study on hydroxyl radical reactivity with unsaturated hydrocarbons: PLS versus MLR. Journal of Chemometrics 10, 135–147.
- Meng, J.-X., Wang, X.-B., Ruan, G.-L., Li, G.-Q., and Deng, Z.-X. (2005). Determination of chlorine in atmosphere by kinetic spectrophotometry. Spectrochimica Acta Part A 61, 823–827.
- Meylan, W.M., Boethling, R., Aronson, D., Howard, P., and Tunkel, J. (2007). Chemical structure-based predictive model for methanogenic anaerobic biodegradation potential. Environmental Toxicology and Chemistry 26, 1785–1792.
- Meylan, W.M., and Howard, P.H. (1991). Bond contribution method for estimating Henry's law constants. Environmental Toxicology and Chemistry 10, 1283–1293.
- Meylan, W.M., and Howard, P.H. (1995). Atom/fragment contribution method for estimating octanol-water partition coefficients. Journal of Pharmaceutical Sciences 84, 83–92.
- Meylan, W.M., and Howard, P.H. (2003). A review of quantitative structure-activity relationship methods for the prediction of atmospheric oxidation of organic chemicals. Environmental Toxicology and Chemistry 22, 1724–1732.
- Meylan, W.M., Howard, P.H., and Boethling, R.S. (1992). Molecular topology/fragment contribution method for predicting soil sorption coefficients. Environmental Science and Technology 26, 1560–1567.
- Mill, T. (1989). Structure-activity relationships for photooxidation processes in the environment. Environmental Toxicology and Chemistry 8, 31–43.
- Mill, T. (1999). Predicting photoreaction rates in surface waters. Chemosphere 38, 1379–1390.
- Mitchell, B.E., and Jurs, P.C. (1998). Prediction of aqueous solubility of organic compounds from molecular structure. Journal of Chemical Information and Computer Sciences 38, 489–496.
- Modarresi, H., Modarress, H., and Dearden, J.C. (2007). QSPR model of Henry's law constant for a diverse set of organic chemicals based on genetic algorithm-radial basis function network approach. Chemosphere 66, 2067–2076.
- Mon, J., Flury, M., and Harsh, J.B. (2006). A quantitative structure–activity relationships (QSAR) analysis of triarylmethane dye tracers. Journal of Hydrology 316, 84–97.
- Muir, D.C. G., and Howard, P.H. (2006). Are there other persistent organic pollutants? A challenge for environmental chemists. Environmental Science and Technology 40, 7157–7166.
- Müller, M., and Klein, W. (1991). Estimating atmospheric degradation processes by SARs. Science of the Total Environment261–273.
- Müller, M., and Klein, W. (1992). Comparative evaluation of methods predicting water solubility for organic compounds. Chemosphere 25, 769–782.
- Müller, M., and Kördel, W. (1996). Comparison of screening methods for the estimation of adsorption coefficients on soil. Chemosphere 32, 2493–2504.
- Nandihalli, U.B., Duke, M.V., and Duke, S.O. (1993). Prediction of RP-HPLC log P from semi-empirical molecular properties of diphenyl ether and phenopylate herbicides. Journal of Agricultural and Food Chemistry 41, 582–587.
- Nguyen, T.H., Goss, K.-U., and Ball, W.P. (2005). Polyparameter linear free energy relationships for estimating the equilibrium partition of organic compounds between water and the natural organic matter in soils and sediments. Environmental Science and Technology 39, 913–924.
- Niemi, G.J., Basak, S.C., Veith, G.D., and Grunwald, G. (1992). Prediction of octanol/water partition coefficient (KOW) with algorithmically derived variables. Environmental Toxicology and Chemistry 11, 893–900.
- Nirmalakhandan, N.N., and Speece, R.E. (1988a). Prediction of aqueous solubility of organic chemicals based on molecular structure. Environmental Science and Technology 22, 328–338.
- Nirmalakhandan, N.N., and Speece, R.E. (1988b). QSAR model for predicting Henry's constant. Environmental Science and Technology 22, 1349–1357.
- Nirmalakhandan, N.N., and Speece, R.E. (1989). Prediction of aqueous solubility of organic chemicals based on molecular structure. 2. Application to NPAs, PCBs, PCDDs, etc. Environmental Science and Technology 23, 708–713.
- Nirmalakhandan, N.N., and Speece, R.E. (1990). Response to comment on “Prediction of aqueous solubility of organic chemicals based on molecular structure. 2. Application to NPAs, PCBs, PCDDs, etc.” Environmental Science and Technology 24, 929–930.
- Niu, J., Chen, J., Yu, G., and Schramm, K.-W. (2004). Quantitative structure-property relationships on direct photolysis of PCDD/Fs on surfaces of fly ash. SAR and QSAR in Environmental Research 15, 265–277.
- Niu, J., Huang, L., Chen, J., Yu, G., and Schramm, K.-W. (2005). Quantitative structure-property relationships on photolysis of PCDD/Fs adsorbed to spruce (Picea abis (L.) Karst.) needle surfaces under sunlight irradiation. Chemosphere 58, 917–924.
- Niu, J., Shen, Z., Yang, Z., Long, X., and Yu, G. (2006). Quantitative structure-property relationships on photodegradation of polybrominated diphenyl ethers. Chemosphere 64, 658–665.
- Oberg, T. (2005). A QSAR for the hydroxyl radical reaction rate constant: validation, domain of application, and prediction. Atmospheric Environment 39, 2189–2200.
- Okey, R.W., and Stensel, H.D. (1996). A QSAR-based biodegradability model: A QSBR. Water Research 30, 2206–2214.
- Organization for Economic Cooperation and Development. (1992). OECD guideline for testing of chemicals. Ready biodegradability. Paris, France: OECD.
- Organization for Economic Cooperation and Development. (1993). Application of structure-activity relationships to the estimation of properties important in exposure assessment. Environment monograph No 67. Paris, France: OECD.
- Organization for Economic Cooperation and Development. (2009). OECD guideline for testing of chemicals. Inherent biodegradability: modified MITI test (II), 302 C. Paris, France: OECD.
- Organization for Economic Cooperation and Development. (2013). Introduction to (quantitative) structure activity relationships. Retrieved from http://www.oecd.org/env/ehs/risk-assessment/introductiontoquantitativestructureactivityrelatio-nships.htm
- Papa, E., Kovarich, S., and Gramatica, P. (2009). Development, validation and inspection of the applicability domain of QSPR models for physicochemical properties of polybrominated diphenyl ethers. QSAR and Combinatorial Science 8, 790–796.
- Paris, D.F., and Wolfe, N.L. (1987). Relationships between properties of a series of anilines and their transformation by bacteria. Applied and Environmental Microbiology 53, 911–916.
- Paris, D.F., Wolfe, N.L., Steen, W.C., and Baughman, G.L. (1983). Effect of phenol molecular-structure on bacterial transformation rate constants in pond and river samples. Applied and Environmental Microbiology 45, 1153–1155.
- Parthasarathi, R., Padmanabhan, J., Elango, M., Chitra, K., Subramanian, V., and Chattaraj, P.K. (2006). pKa prediction using group philicity. Journal of Physical Chemistry 110, 6540–6544.
- Paterson, S., Mackay, D., and MacFariane, C. (1994). A model of organic chemical uptake by plants from soil and the atmosphere. Environmental Science and Technology 26, 2259–2266.
- Patil, G.S. (1994). Prediction of aqueous solubility and octanol-water partition coefficient for pesticides based on their molecular structure. Journal of Hazardous Materials 36, 35–43.
- Pavan, M., and Worth, A.P. (2008). Review of estimation models for biodegradation. QSAR and Combinatorial Science 27, 32–40.
- Peijnenburg, W.J. G. M. (1994). Structure-activity relationships for biodegradation: a critical review. Pure and Applied Chemistry 66, 1931–1941.
- Peijnenburg, W.J. G. M., de Beer, K.G. M., de Haan, M.W. A., den Hollander, H.A., Stegeman, M.H. L., and Verboom, H. (1992). Development of a structure-reactivity relationship for the photohydrolysis of substituted aromatic halides. Environmental Science and Technology 26, 2116–2121.
- Percival, C.J., Marston, G., and Wayne, R.P. (1995). Correlations between rate parameters and calculated molecular properties in the reactions of the hydroxyl radical with hydrofluorocarbons. Atmospheric Environment 29, 305–311.
- Philipp, B., Hoff, M., Germa, F., Schink, B., Beimborn, D., and Mersch-Sundermann, V. (2007). Biochemical interpretation of quantitative structure-activity relationships (QSAR) for biodegradation of N-heterocycles: a complementary approach to predict biodegradability. Environmental Science and Technology 41, 1390–1398.
- Phillips, K.L., Sandler, S.I., and Chiu, P.C. (2010). A method to calculate the one-electron reduction potentials for nitroaromatic compounds based on gas-phase quantum mechanics. Journal of Computational Chemistry 32, 226–239.
- Platts, J.A., and Abraham, M.H. (2000). Partition of volatile organic compounds from air and from water into plant cuticular matrix: an LFER analysis. Environmental Science and Technology 34, 318–323.
- Platts, J.A., Abraham, M.H., Butina, D., and Hersey, A. (2000). Estimation of molecular linear free energy relationship descriptors by a group contribution approach. 2. Prediction of partition coefficients. Journal of Chemical Information and Computer Sciences 40, 71–80.
- Platts, J.A., Butina, D., Abraham, M.H., and Hersey, A. (1999). Estimation of molecular linear free energy relation descriptors using a group contribution approach. Journal of Chemical Information and Computer Sciences 39, 835–845.
- Pompe, M., and Randic, M. (2007). Variable connectivity model for determination of pKa values for selected organic acids. Acta Chimica Slovenica 54, 605–610.
- Pompe, M., and Veber, M. (2001). Prediction of rate constants for the reaction of O3 with different organic compounds. Atmospheric Environment 35, 3781–3788.
- Poole, S.K., and Poole, C.F. (1999). Chromatographic models for the sorption of neutral organic compounds by soil from water and air. Journal of Chromatography A 845, 381–400.
- Puzyn, T., Mostrag, A., Falandysz, J., Kholod, Y., and Leszczynski, J. (2009). Predicting water solubility of congeners: Chloronaphthalenes: A case study. Journal of Hazardous Materials 170, 1014–1022.
- Randic, M. (1975). On characterization of molecular branching. Journal of the American Chemical Society 97, 6609–6615.
- Randic, M. (1984). On molecular identification numbers. Journal of Chemical Information and Computer Sciences 24, 164–175.
- Rao, P.S. C., Hornsby, A.G., and Jessup, R.E. (1985). Indices for ranking the potential for pesticide contamination of groundwater. Soil and Crop Science Society of Florida Proceedings 44, 1–8.
- Raymond, J.W., Rogers, T.N., Shonnard, D.R., and Kline, A.A. (2001). A review of structure-based biodegradation estimation methods. Journal of Hazardous Materials B84, 189–215.
- Reddy, K.N., and Locke, M.A. (1994a). Prediction of soil sorption (Koc) of herbicides using semiempirical molecular properties. Weed Science 42, 453–461.
- Reddy, K.N., and Locke, M.A. (1994b). Relationships between molecular properties and logP and soil sorption (Koc) of substituted phenylureas: QSAR models. Chemosphere 28, 1929–1941.
- Reddy, K.N., and Locke, M.A. (1996). Molecular properties as descriptors of octanol-water partition coefficients of herbicides. Water, Air and Soil Pollution 86, 389–405.
- Renaud, F.G., Leeds-Harrison, P.B., Brown, C.D., and Van Beinum, W. (2004). Determination of time-dependent partition coefficients for several pesticides using diffusion theory. Chemosphere 57, 1525–2535.
- Rorije, E., Loonen, H., Muller, M., Klopman, G., and Peijnenburg, W.J.G.M. (1999). Evaluation and application of models for the prediction of ready biodegradability in the MITI-I test. Chemosphere 38, 1409–1417.
- Rorije, E., and Peijnenburg, W.J. G. M. (1996). QSARs for oxidation of phenols in the aqueous environment, suitable for risk assessment. Journal of Chemometrics 10, 79–93.
- Roy, K., Sanyal, I., and Ghosh, G. (2007). QSPR of n-octanol/water partition coefficient of nonionic organic compounds using extended topochemical atom (ETA) indices. QSAR and Combinatorial Science 26, 629–646.
- Rücker, C., and Kümmerer, K. (2012). Modeling and predicting aquatic aerobic biodegradation: a review from a user's perspective. Green Chemistry 14, 875–887.
- Russom, C.L., Breton, R.L., Walker, J.D., and Bradbury, S.P. (2003). An overview of the use of quantitative structure-activity relationships for ranking and prioritizing large chemical inventories for environmental risk assessments. Environmental Toxicology and Chemistry 22, 1810–1821.
- Sabljic, A. (1984). Predictions of the nature and strength of soil sorption of organic pollutants by molecular topology. Journal of Agricultural and Food Chemistry 32, 243–246.
- Sabljic, A. (1987). On the prediction of soil sorption coefficients of organic pollutants from molecular structure: Application of molecular topology model. Environmental Science and Technology 21, 358–366.
- Sabljic, A. (1989). Quantitative modelling of soil sorption for xenobiotic chemicals. Environmental Health Perspectives 83, 179–190.
- Sabljic, A. (1991). Chemical topology and ecotoxicology. Science of the Total Environment197–220.
- Sabljic, A. (2001). QSAR models for estimating properties of persistent organic pollutants required in evaluation of their environmental fate and risk. Chemosphere 43, 363–375.
- Sabljic, A., and Güsten, H. (1989). Predicting Henry's law constant for polychlorinated biphenyls. Chemosphere 19, 1503–1511.
- Sabljic, A., and Güsten, H. (1990). Predicting the night-time NO3 radical reactivity in the troposphere. Atmospheric Environment 24A, 73–78.
- Sabljic, A., Güsten, H., Hermens, J., and Opperhuizen, A. (1993). Modeling octanol/water partition coefficients by molecular topology: chlorinated benzenes and biphenyls. Environmental Science and Technology 27, 1394–1402.
- Sabljic, A., Güsten, H., Schönherr, J., and Riederer, M. (1990). Modeling plant uptake of airborne organic chemicals. 1. Plant cuticle/water partitioning and molecular connectivity. Environmental Science and Technology 24, 1321–1326.
- Sabljic, A., Güsten, H., Verhaar, H., and Hermens, J. (1995). QSAR modelling of soil sorption. Improvements and systematics of log Koc vs. log KOW correlations. Chemosphere 31, 4489–4514.
- Sabljic, A., and Horvatic, D. (1993). GRAPH III: A computer program for calculating molecular connectivity indices on microcomputers. Journal of Chemical Information and Computer Sciences 33, 292–295.
- Sabljic, A., Lara, R., and Ernst, W. (1989). Modelling association of highly chlorinated biphenyls with marine humic substances. Chemosphere 19, 1665–1676.
- Sabljic, A., and Peijnenburg, W. (2001). Modeling lifetime and degradability of organic compounds in air, soil, and water systems. Pure and Applied Chemistry 73, 1331–1348.
- Sabljic, A., and Piver, W.T. (1992). Quantitative modeling of environmental fate and impact of commercial chemicals. Environmental Toxicology and Chemistry 11, 961–972.
- Sabljic, A., and Trinajstic, N. (1981). Quantitative structure-activity relationships: the role of topological indices. Acta Pharmaceutica Jugolsavica 31, 189–214.
- Sannigrahi, A.B. (1992). Ab-initio molecular-orbital calculations of bond index and valency. Advances in Quantitative Chemistry 23, 301–351.
- Scherer, M.M., Balko, B.A., Gallagher, D.A., and Tratnyek, P.G. (1998). Correlation analysis of rate constants for dechlorination by zero-valent iron. Environmental Science and Technology 32, 3026–3033.
- Schüürmann, G. (1995). Quantum chemical approach to estimate physicochemical compound properties: application to substituted benzenes. Environmental Toxicology and Chemistry 14, 2067–2076.
- Schüürmann, G., Ebert, R.-U., and Kühne, R. (2006). Prediction of the sorption of organic compounds into soil organic matter from molecular structure. Environmental Science and Technology 40, 7005–7011.
- Sedykh, A., and Klopman, G. (2007). Data analysis and alternative modelling of MITI-I aerobic biodegradation. SAR and QSAR in Environmental Research 18, 693–709.
- Sekusak, S., and Sabljic, A. (1992). Soil sorption and chemical topology. Journal of Mathematical Chemistry 11, 271–280.
- Seybold, P.G. (2008). Analysis of the pKas of aliphatic amines using quantum chemical descriptors. International Journal of Quantum Chemistry 108, 2849–2855.
- Sharer, M., Park, J.-H., Voice, T.C., and Boyd, S.A. (2003). Aging effects on the sorption-desorption characteristics of anthropogenic organic compounds in soil. Journal of Environmental Quality 32, 1385–1392.
- Sharma, V., Goswami, R., and Madan, A.K. (1997). Eccentric connectivity index: a novel highly discriminating topological descriptor for structure-property and structure-activity studies. Journal of Chemical Information and Computer Sciences 37, 273–282.
- Shi, J., Zhang, X., Qu, R., Xu, Y., and Wang, Z. (2012). Synthesis and QSPR study on environment-related properties of polychlorinated diphenyl sulfides (PCDPSs). Chemosphere 88, 844–854.
- Shiu, W.Y., Doucette, W., Gobas, F.A. P. C., Andren, A., and Mackay, D. (1988). Physical-chemical properties of chlorinated dibenzo-p-dioxins. Environmental Science and Technology 22, 651–658.
- Soriano, E., Cerdán, S., and Ballesteros, P. (2004). Computational determination of pKa values. A comparison of different theoretical approaches and a novel procedure. Journal of Molecular Structure 684, 121–128.
- Soscún Machado, H.J., and Hinchliffe, A. (1995). Relationships between the HOMO energies and pK, values in monocyclic and bicyclic azines. Journal of Molecular Structure. THEOCHEM 339, 255–258.
- Staikova, M., Wania, F., and Donaldson, D.J. (2004). Molecular polarizability as a single-parameter predictor of vapour pressures and octanol-air partitioning coefficients of non-polar compounds: a priori approach and results. Atmospheric Environment 38, 213–225.
- Stanton, D.T., and Jurs, P.C. (1990). Development and use of charged partial surface area structural descriptors in computer-assisted quantitative structure-property relationship studies. Analytical Chemistry 62, 2323–2329.
- Sudhakaran, S., and Amy, G.L. (2013). QSAR models for oxidation of organic micropollutants in water based on ozone and hydroxyl radical rate constants and their chemical classification. Water Research1111–1122.
- Sun, H., Huang, G., and Dai, S. (1996). Adsorption behaviour and QSPR studies of organotin compounds on estuarine sediment. Chemosphere 33, 831–838.
- Sun, L., Zhou, L., Yu, Y., Lan, Y., and Li, Z. (2007). QSPR study of polychlorinated diphenyl ethers by molecular electronegativity distance vector (MEDV-4). Chemosphere 66, 1039–1051.
- Sutter, J.M., and Jurs, P.C. (1996). Prediction of aqueous solubility for a diverse set of heteroatom-containing organic compounds using a quantitative structure-property relationship. Journal of Chemical Information and Computer Sciences 36, 100–107.
- Tabak, H.H., Gao, C., Desai, S., and Govind, R. (1992). Development of predictive structure-biodegradation relationship models with the use of respirometrically generated biokinetic data. Water Science and Technology 26, 763–772.
- Tabak, H.H., and Govind, R. (1993). Prediction of biodegradation kinetics using a nonlinear group contribution method. Environmental Toxicology and Chemistry 12, 251–260.
- Tao, S., and Lu, X. (1999). Estimation of organic carbon normalized sorption coefficient (Koc) for soils by topological indices and polarity factors. Chemosphere 39, 2019–2034.
- Tao, S., Piao, H., Dawson, R., Lu, X., and Hu, H. (1999). Estimation of organic carbon normalized sorption coefficient (Koc) for soils using the fragment constant method. Environmental Science and Technology 33, 2719–2725.
- Tehan, B.G., Lloyd, E.J., Wong, M.G., Pitt, W.R., Gancia, E., and Manallack, D.T. (2002a). Estimation of pKa using semiempirical molecular orbital methods. Part 2: Application to amines, anilines and various nitrogen containing heterocyclic compounds. Quantitative Structure-Activity Relationships 21, 473–485.
- Tehan, B.G., Lloyd, E.J., Wong, M.G., Pitt, W.R., Montana, J.G., Manallack, D.T., and Gancia, E. (2002b). Estimation of pKa using semiempirical molecular orbital methods. Part 1: Application to phenols and carboxylic acids. Quantitative Structure-Activity Relationships 21, 457–472.
- Tetko, I.V., Tanchuk, V.Y., Kasheva, T.N., and Villa, A.E. P. (2001). Estimation of aqueous solubility of chemical compounds using E-state indices. Journal of Chemical Information and Computer Sciences 41, 1488–1493.
- Thomsen, M., Rasmussen, A.G., and Carlsen, L. (1999). SAR/QSAR approaches to solubility, partitioning and sorption of phtalates. Chemosphere 38, 2613–2624.
- Todeschini, R., and Consonni, V. (2000). Handbook of molecular descriptors. Methods and principles in medicinal chemistry. Volume 11. Weinheim, Germany: Wiley.
- Todeschini, R., and Gramatica, P. (1997a). The WHIM theory: new 3D molecular descriptors for QSAR in environmental modeling. SAR and QSAR in Environmental Research 7, 89–115.
- Todeschini, R., and Gramatica, P. (1997b). 3D-modelling and prediction by WHIM descriptors. Part 5. Theory development and chemical meaning of WHIM descriptors. Quantitative Structure Activity Relationships 16, 113–119.
- Todeschini, R., Bettiol, C., Giurin, G., Gramatica, P., Miana, P., and Argese, E. (1996). Modeling and prediction by using WHIM descriptors in QSAR studies: submitochondrial particles (SMP) as toxicity biosensors of chlorophenols. Chemosphere 33, 71–79.
- Topol, I.G., Tawa, G.J., Caldwell, R.A., Eissenstat, A., and Burt, S.K. (2000). Acidity of organic molecules in the gas phase and in aqueous solvent. Journal of Physical Chemistry A 104, 9619–9624.
- TORVS Research Team. (1999). Parameter estimation for the treatment of reactivity applications. Erlangen, Germany: Computer-Chemie-Centrum, University of Erlangen-Nuernberg. Retrieved from http://www2.ccc.uni-erlangen.de/software/petra/
- Tratnyek, P.G., Weber, E.J., and Schwarzenbach, R.P. (2003). Quantitative-structure activity relationships for chemical reductions of organic contaminants. Environmental Toxicology and Chemistry 22, 1733–1742.
- Tunkel, J., Howard, P.H., Boethling, R.S., Stiteler, W., and Loonen, H. (2000). Predicting ready biodegradability in the Japanese Ministry of International Trade and Industry test. Environmental Toxicology and Chemistry 19, 2478–2485.
- Türker Saçan, M., and Balcioğlu, I.A. (1996). Prediction of the soil sorption coefficient of organic pollutants by the characteristic root index model. Chemosphere 32, 1993–2001.
- Türker Saçan, M., and Inel, Y. (1995). Application of the characteristic root index model to the estimation of n-octanol/water partition coefficients. Polychlorinated biphenyls. Chemosphere 30, 39–50.
- Uddameri, V., and Kuchanur, M. (2004). Fuzzy QSAR for predicting logKoc of persistent organic pollutants. Chemosphere 54, 771–776.
- Van Compernolle, R., McAvoy, D.C., Sherren, A., Wind, T., Cano, M.L., Belanger, S.E., Dorn, P.B., and Kerr, K.M. (2006). Predicting the sorption of fatty alcohols and alcohol ethoxylates to effluent and receiving water solids. Ecotoxicology and Environmental Safety 64, 61–74.
- Van Noort, P.C. M., Haftka, J.J. H., and Parsons, J.R. (2010). Updated Abraham solvation parameters for polychlorinated biphenyls. Environmental Science and Technology 44, 7037–7042.
- Von Oepen, B., Kördel, W., Klein, W., and Schüürmann, G. (1991). Predictive QSPR models for estimating soil sorption coefficients: potential and limitations based on dominating processes. Science of the Total Environment 109/110, 343–354.
- Vrtacnik, M., and Voda, K. (2003). HQSAR and CoMFA approaches in predicting reactivity of halogenated compounds with hydroxyl radicals. Chemosphere 52, 1689–1699.
- Walker, A., Rodriguez-Cruz, M.S., and Mitchell, M.J. (2005). Influence of ageing of residues on the availability of herbicides for leaching. Environmental Pollution 133, 43–51.
- Walker, J.D., Carlsen, L., Hulzebos, E., and Simon-Hettich, B. (2002). Global government applications of analogues, SARs, QSARs to predict aquatic toxicity, chemical or physical properties, environmental fate parameters and health effects of organic chemicals. SAR and QSAR in Environmental Research 13, 607–616.
- Walker, J.D., Jaworska, J., Comber, M.H. I., Schultz, T.W., and Dearden, J.C. (2003). Guidelines for developing and using quantitative structure-activity relationships. Environmental Toxicology and Chemistry 22, 1653–1665.
- Wammer, K.H., and Peters, C.A. (2005). Polycyclic aromatic hydrocarbon biodegradation rates: a structure-based study. Environmental Science and Technology 39, 2571–2578.
- Wang, Z.Y., Zeng, X.L., and Zhai, Z.C. (2008). Prediction of supercooled liquid vapor pressures and n-octanol/air partition coefficients for polybrominated diphenyl ethers by means of molecular descriptors from DFT method. Science of the Total Environment 389, 296–305.
- Wania, F., and Dugani, C.B. (2003). Assessing the long-range transport potential of polybrominated diphenyl ethers: a comparison of four multimedia models. Environmental Toxicology and Chemistry 22, 1252–1261.
- Wauchope, R.D., Yeh, S., Linders, J.B. H. J., Kloskowski, R., Tanaka, K., Rubin, B., Katayama, A., Kördel, W., Gerstl, Z., Lane, M., and Unsworth, J.B. (2002). Pesticide soil sorption parameters: theory, measurement, uses, limitations and reliability. Pest Management Science 58, 419–445.
- Welke, B., Ettlinger, K., and Riederer, M. (1998). Sorption of volatile organic chemicals in plant surfaces. Environmental Science and Technology 32, 1099–1104.
- Wilson, L.Y., and Famini, G.R. (1991). Using theoretical descriptors in quantitative structure-activity relationships: some toxicological indices. Journal of Medicinal Chemistry 34, 1668–1674.
- Winget, P., Cramer, C.J., and Truhlar, D.G. (2000). Prediction of soil sorption coefficients using a universal solvation model. Environmental Science and Technology 34, 4733–4740.
- Woodrow, J.E., Seiber, J.N., and Baker, L.W. (1997). Correlation techniques for estimating pesticide volatilization flux and downwind concentrations. Environmental Science and Technology 31, 523–529.
- Worrall, F. (2001). A molecular topology approach to predicting pesticide pollution of groundwater. Environmental Science and Technology 35, 2282–2287.
- Worrall, F., and Thomsen, M. (2004). Quantum vs. topological descriptors in the development of molecular models of groundwater pollution by pesticides. Chemosphere 54, 585–596.
- Xie, Y.J., Liu, H., Liu, H.X., Zhai, Z.C., and Wang, Z.Y. (2008). Determination of solubilities and n-octanol/water partition coefficients and QSPR study for substituted phenols. Bulletin of Environmental Contamination and Toxicology 80, 319–323.
- Xing, L., and Glen, R.C. (2002). Novel methods for the prediction of logP, pKa and logD. Journal of Chemical Information and Computer Sciences 42, 796–805.
- Xu, F., Liang, X., Lin, B., Su, F., Schramm, K.-W., and Kettrup, A. (2002). Linear solvation energy relationships regarding sorption and retention properties of hydrophobic organic compounds in soil leaching column chromatography. Chemosphere 48, 553–562.
- Xu, H.-Y., Zou, J.-W., Hu, G.-X., and Wei, W. (2010). QSPR/QSAR models for prediction of the physico-chemical properties and biological activity of polychlorinated diphenyl ethers (PCDEs). Chemosphere 80, 665–670.
- Xu, H.-Y., Zou, J.-W., Yu, Q.-S., Wang, Y.-H., Zhang, J.-Y., and Jin, H.-X. (2007). QSPR/QSAR models for prediction of the physicochemical properties and biological activity of polybrominated diphenyl ethers. Chemosphere 66, 1998–2010.
- Yan, A., and Gasteiger, J. (2003). Prediction of aqueous solubility of organic compounds based on a 3D structure representation. Journal of Chemical Information and Computer Sciences 43, 429–434.
- Yang, G.-Y., Yu, J., Wang, Z.-Y., Zeng, X.-L., and Ju, X.-H. (2007). QSPR study on the aqueous solubility (-lgSW) and n-octanol/water partition coefficients (lgKOW) of polychlorinated dibenzo-p-dioxins (PCDDs). QSAR and Combinatorial Science 26, 352–357.
- Yang, H., Jiang. Z., and Shi, S. (2004). Anaerobic biodegradability of aliphatic compounds and their quantitative structure biodegradability relationship. Science of the Total Environment 322, 209–219.
- Yang, H., Jiang, Z., and Shi, S. (2006). Biodegradability of nitrogenous compounds under anaerobic conditions and its estimation. Ecotoxicology and Environmental Safety 63, 299–305.
- Yang, P., Chen, J., Chen, S., Yuan, X., Schramm, K.-W., and Kettrup, A. (2003). QSPR models for physicochemical properties of polychlorinated diphenyl ethers. Science of the Total Environment 305, 65–76.
- Yonezawa, Y., and Urushigawa, Y. (1979). Chemico-biological interactions in biological purification systems V. Relation between biodegradation rate constants of aliphatic alcohols by activated sludge and their partition coefficients in a 1-octanol-water system. Chemosphere 8, 139–142.
- Yu, H., Kühne, R., Ebert, R.-U., and Schüürmann, G. (2010). Comparative analysis of QSAR models for predicting pKa of organic oxygen acids and nitrogen bases from molecular structure. Journal of Chemical Information and Modeling 50, 1949–1960.
- Yu, H., Kühne, R., Ebert, R.-U., and Schüürmann, G. (2011). Prediction of the dissociation constant pKa of organic acids from local molecular parameters of their electronic ground state. Journal of Chemical Information and Modeling 51, 2336–2344.
- Zeng, X.-L., Wang, H.-J., and Wang, Y. (2012). QSPR models of n-octanol/water partition coefficients and aqueous solubility of halogenated methyl-phenyl ethers by DFT method. Chemosphere 86, 619–625.
- Zeng, X.-L., Wang, Z., Ge, Z., and Liu, H. (2007). Quantitative structure-property relationships for predicting subcooled liquid vapor pressure (PL) of 209 polychlorinated diphenyl ethers (PCDEs) by DFT and the position of Cl substitution (PCS) methods. Atmospheric Environment 41, 3590–3603.
- Zeng, X.-L., Zhang, X.-L., and Wang, Y. (2013). QSPR modeling of n-octanol/air partition coefficients and liquid vapor pressures of polychlorinated dibenzo-p-dioxins. Chemosphere 91, 229–232.
- Zhang, J., Kleinöder, T., and Gasteiger, J. (2006). Prediction of pKa values for aliphatic carboxylic acids and alcohols with empirical atomic charges descriptors. Journal of Chemical Information and Modeling 46, 2256–2266.
- Zhao, H., Chen, J., Quan, X., Yang, F., and Peijnenburg, W.J.G.M. (2001). Quantitative structure-property relationship study on reductive dehalogenation of selected halogenated aliphatic hydrocarbons in sediment slurries. Chemosphere 44, 1557–1563.
- Zhao, H., Xie, Q., Tan, F., Chen, J., Quan, X., Qu, B., Zhang, X., and Li, X. (2010). Determination and prediction of octanol-air partition coefficients of hydroxylated and methoxylated polybrominated diphenyl ethers. Chemosphere 80, 660–664.
- Zhao, H., Zhang, Q., Chen, J., Xue, X., and Liang, X. (2005). Prediction of octanol-air partition coefficients of semivolatile organic compounds based on molecular connectivity index. Chemosphere 59, 1421–1426.
- Zhao, Y.H., Abraham, M.H., and Zissimos, A.M. (2003). Determination of McGowan volumes for ions and correlation with van der Waals volumes. Journal of Chemical Information and Computer Sciences 43, 1848–1854.
- Zhou, W., Zhai, Z., Wang, Z., and Wang, L. (2005). Estimation of n-octanol/water partition coefficients (KOW) of all PCB congeners by density functional theory. Journal of Molecular Structure: THEOCHEM 755, 137–145.
- Zou, J.-W., Zhao, W.-N., Shang, Z.-C., Huang, M.-L., Guo, M., and Yu, Q.-S. (2002). A quantitative structure-property relationship analysis of logP for disubstituted benzenes. Journal of Physical Chemistry A 106, 11550–11557.