Abstract
Despite the long history of water research, understanding how the drinking water microbiome is shaped at the user end is rather challenging owing to the complexity in community assembly, water matrices, physical structures, and chemical gradients from source to tap. The application of molecular tools that primarily base on the use of rRNA gene sequences has substantially expanded our view of the drinking water microbiome. In this review, we critically evaluate currently available cultivation-independent tools for monitoring the drinking water microbiome and summarize the ecological patterns we have observed so far on the longitudinal and temporal dynamics, geographical distributions, and structural and functional characteristics of the drinking water microbiome (including those in recycled water systems). Studies on four full-scale systems in the United States and Europe further exemplify the application of ecological theory into drinking water microbiome studies. Finally, we discuss how meta-omics are able to provide new perspectives on microbial function and interspecies relationship within the drinking water ecosystem. This review promotes an integral understanding of the drinking water microbiome and the transformation of drinking water microbiology from a descriptive discipline to an ecology-driven science that attempts to elucidate mechanisms for predicting and shaping the microbiome at the user end.
1. Introduction
Sanskrit and Greek writings, from as early as 4000 BC, document drinking water treatment technologies ranging from filtering through charcoal, exposing to sunlight, boiling, and other forms of straining. The Egyptians even documented using chemical alum to facilitate the settling of suspended particles around 1500 BC (Halliday, Citation2004). This water treatment history also documents the waterborne disease outbreaks including cholera and typhoid fever that occurred during medieval Europe when waste and wastewater were discharged in cities and previous treatment technologies were forgotten or ignored (USEPA, Citation2000). It wasn’t until the 1700s that Europe reestablished the regular use of drinking water treatment filtration technologies as an effective means of removing particles from water (USEPA, Citation2000). The effectiveness of these water treatment technologies could not be successfully measured until after the invention of cultivation technique and the germ theory of disease between 1850s and 1890s (Koch & Duncan, Citation1894; Twisselmann, Citation2003). The concept of microbial ecology in drinking water distribution system (DS) was introduced in 1945 by Wilson (Wilson, Citation1945), and suggested that the ecological niches inside a DS could be determined by knowing the type and number of bacteria developed. After more than 70 years, our understanding of the microbial ecology of drinking water systems has been transformed in a remarkable way with cultivation-independent approaches that primarily base on the use of rRNA gene sequences ().
Current drinking water production plants apply physical and chemical means to remove unwanted chemicals and microorganisms, and some use microbiological processes such as sand filters and granular activated carbon filters to biologically remove excessive nutrients and break down soluble organic matters present in source water (). The evolution of these systems has resulted in two distinct practices related to chlorination for biological safety and the exposure to harmful substances from disinfection by-products (DBPs). One (most countries in the world) proposes to maintain a minimal level of residual disinfectants to suppress (APHA, Citation1905; Bartlett and Stirling, Citation2003; Fischer et al., Citation2005; Herzenberg et al., Citation2002; Koch and Duncan, Citation1894; Langer-Safer, Levine, & Ward, Citation1982; Leeuwenhoek, Liu & Stahl, 2007; Manz et al., Citation1993; Maxam and Gilbert, Citation1977; Metzker, Citation2010; Payment et al., Citation2003; PRT, Citation1996; Schardinger, Citation1892; Smith, Citation1895; Stein, Marsh, Wu, Shizuya, & DeLong, Citation1996; Twisselmann, Citation2003; USEPA, Citation1989a, Citation1989b, Citation1991, Citation1998, Citation2002, Citation2006a, Citation2006b, Citation2013; Vanderkooij et al., Citation1982, Woese and Fox, Citation1977) microbial regrowth in DS and the other (the Netherlands, Switzerland, Austria, and Germany) tries to substantially reduce available carbon for regrowth during distribution instead of adding disinfectant residuals ().
Figure 1. History on the development of major microbiological methods (upper half), and the introduction of key regulations and rules in drinking water production, primarily based on the US system (bottom half). Source: Zhang and Liu, Citationforthcoming. Reprinted with permission. © Water Research Foundation.
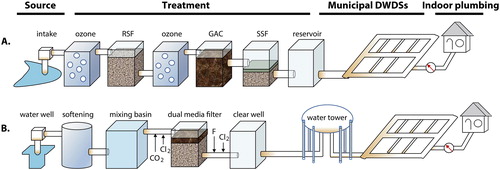
Figure 2. The configuration of typical drinking water systems. A) A complex system in Switzerland, B) a simple system in a mid-size town in the US. Source: Zhang and Liu, Citationforthcoming. Reprinted with permission. © Water Research Foundation.
The assemblage of microbes within drinking water is referred to as the drinking water microbiome (106–108 cells/L) (Henne, Kahlisch, Brettar, & Hofle, Citation2012; Lautenschlager, Boon, Wang, Egli, & Hammes, Citation2010; Vanderwende, Characklis, & Smith, Citation1989). As these microorganisms play critical roles in treatment plant operation and drinking water quality, regulatory agencies have implemented rules and regulations for the microbiological quality of drinking water to protect public health. However, these standards are outdated because they still rely on heterotrophic plate count (HPC) and indicator microorganisms (Escherichia coli and total coliforms) proposed more than 100 years ago to determine the adequacy of water treatment and the integrity of the DS.
To advance our understandings of the drinking water microbiome, this review concentrates on describing: i) what cultivation-independent tools have been developed for the analyses of the drinking water microbiome; ii) what knowledge we have gained on population dynamics and the microbial ecology of the drinking water microbiome; iii) how meta-omics techniques can provide new perspectives on the complexity of the microbial community within drinking water ecosystems; and iv) what are the current challenges that need to be overcome to enable critical research advances. The goal is to guide the operational practices in drinking water utilities so that they can create and maintain a ‘healthy’ or common drinking water microbiome in the DS, and to enlighten future research on the drinking water microbiome. This review focuses on water production and distribution system up to the point where it enters a building. Due to significant differences in the challenges faced by premise plumbing, it is not discussed in this review. Readers can refer to the following reviews published dedicatedly to premise plumbing (Dai et al., Citation2017; Wang et al., Citation2017; Wang, Edwards, Falkinham, & Pruden, Citation2013).
2. Current methods for monitoring the drinking water microbiome
To routinely monitor and characterize the drinking water microbiome, a suite of methods has been developed and used to measure microbial density, microbial composition and structure (including spatial arrangement and the presence/absence and concentrations of specific opportunistic pathogens), and microbial activities (). Detailed description is reported below. It should be noted that no single method can provide all the information. The current strategy is to combine several methods to improve the view of the drinking water microbiome in the studied systems. Also, all the methods have known biases associated with and should be used with caution.
Table 1. Summary of methods used to measure microbial density, microbial composition, and microbial activities in drinking water microbiome studies.
2.1. Methods for measuring microbial density
Cultivation is still the most widely-used technique to quantify microbial density in drinking water sector since late 18th century (). HPC and bacterial indicators (i.e., total coliforms and E. coli) were introduced to determine the adequacy of water treatment and the integrity of the DS (Bartram, Cotruvo, Exner, Fricker, & Glasmacher, Citation2004; Frankland, Citation1894; Koch & Duncan, Citation1894; Payment, Sartory, & Reasoner, Citation2003). As isolating and enumerating disease-causing microorganisms are primary concerns in drinking water studies, selective and differential media techniques have been developed to cultivate almost all known pathogens. For example, the buffered charcoal yeast extract agar is a selective medium developed to isolate the once difficult-to-culture Legionella pneumophila in 1980s that caused the outbreak of Legionnaires’ disease in 1976 (Edelstein, Citation1981; Pasculle et al., Citation1980).
An alternative and most direct way to quantify microbial density is cell counting. Cells in water samples can be directly counted using microscopy or stained with fluorescent dyes and counted under an epifluorescence microscope or flow cytometry (FCM) as total cell count. Fully automated online FCM can be useful in discerning temporal bacterial dynamics at high frequency (Besmer et al., Citation2016; Besmer & Hammes, Citation2016). The usage of FCM in DSs is still limited to systems without residual disinfectants. For drinking water containing residual disinfectants, pretreatment using membrane filtration to concentrate bacteria at an appropriate density is required due to low cell number and the interference of bacteria-like particles (Besmer et al., Citation2016; Lautenschlager et al., Citation2013; Van Nevel et al., Citation2017), which is time-consuming and subjective (Santic, Krstulovic, & Solic, Citation2007).
Since the mid-1980s, various forms of molecular methods, e.g., quantitative PCR, viable quantitative PCR and digital droplet PCR, have been developed to provide qualitative and quantitative information related to total or specific bacterial cells, and to the ratio of live and dead cells (Chen & Chang, Citation2010; Lee, Lee, & Kim, Citation2015; Yanez et al., Citation2011) (). These molecular tools are mostly based on the use of the 16S rRNA gene that is conserved among the domains Bacteria and Archaea and composed of regions that are variable enough to differentiate microorganisms at different levels of specificities (i.e., species, genera … phyla and domains) (Liu & Stahl, Citation2007).
2.2. Methods measuring microbial composition
Characterizing microbial populations is an important first step to elucidate the complexity of microbial ecology in drinking water systems. Two major types of PCR amplification methods can be carried out. The first type of PCR-based methods is generally termed ‘community fingerprint’. It analyzes the amplified 16S rRNA genes and generates a pattern-based profile of community structure, most commonly represented by a banding pattern of nucleic acid fragments resolved by gel electrophoresis. In general, these community fingerprinting methods allow one to rapidly examine the microbial diversity within a microbial ecosystem, or compare the differences and similarities on the microbial community structure among different ecosystems (Liu & Stahl, Citation2007). Many microbial fingerprinting methods were developed in 1990s, including DGGE (denaturing gradient gel electrophoresis), T-RFLP (terminal restriction fragment length polymorphism), PCR-ALH (amplicon length heterogeneity), and SSCP (single-strand-conformation polymorphism) (Liu & Stahl, Citation2007).
The second type of molecular method is to obtain a dataset of 16S rRNA gene sequences from the extracted community genomic DNA. Initially, this was achieved through clone library construction of 16S rRNA gene sequences. In the recent years, the composition of 16S rRNA gene sequences in a microbial sample can be obtained using the next-generation sequencing (NGS) technology. Both approaches describe the microbial composition based on the number of unique 16S rRNA sequences and the abundance of individual 16S rRNA sequences. The 16S rRNA sequences can be further compared with all 16S rRNA sequences stored in a public database. This allows one to infer the phylogeny affiliation of individual 16S rRNA sequences, and determine whether the sequences are novel or related to known organisms based on the similarity of sequence homology (e.g., <97% similarity for defining a new species). Furthermore, based on the sequence information, one can design oligonucleotide probes specific for a target organism or a group of organisms, and then apply them in whole-cell hybridization or membrane hybridization for confirming the presence of the targeted organisms or for quantitative measurement of those targeted organisms in the environment. Likewise, the active members within the microbial community can be determined using the corresponding RNA-based analysis instead of DNA-based methods.
DNA microarray technology has emerged as a high-throughput platform for nucleic acid analysis in environmental microbiology studies (Bodrossy & Sessitsch, Citation2004; Li & Liu, Citation2003; Zhou, Citation2003). For microbial identification and community analysis, this platform generally uses rRNA genes as the phylogenetic marker (Guschin et al., Citation1997; Liu, Mirzabekov, & Stahl, Citation2001; Loy et al., Citation2002; Peplies, Glockner, & Amann, Citation2003; Small, Call, Brockman, Straub, & Chandler, Citation2001; Wilson et al., Citation2002). Initially, hundreds up to thousands of rRNA-based oligonucleotide probes with a length between 15 and 25 nt are designed to target the rRNA gene sequences of interested microorganisms at different levels of specificities, and then spotted or in-situ synthesized onto a microarray substrate (Loy et al., Citation2002; Wilson et al., Citation2002). Followed by hybridization with fluorescently labeled rRNA/rDNA targets and washing at optimal conditions, signals are measured and statistically analyzed to infer microbial community structures in complex environmental samples (El Fantroussi et al., Citation2003; Peplies, Lau, Pernthaler, Amann, & Glockner, Citation2004; Small et al., Citation2001).
Finally, the microbial composition can also be determined by examining the spatial distribution of microorganisms in-situ. Fluorescence in-situ hybridization (FISH) and scanning electron microscopy (SEM) are the most commonly used methods to show the spatial structure and arrangement of microbial communities. The development of SEM in the 1950s (Fischer, Hansen, Nair, Hoyt, & Dorward, Citation2005) has enabled the visualization of 3-dimensional topography of biofilm in water meters (Hong et al., Citation2010). Using 16S rRNA gene as a biomarker, FISH technique was introduced in the late 1980s to identify and quantify microbial populations at different phylogenetic levels and in combination with other techniques to determine microbial functions in their natural positions (Amann, Krumholz, & Stahl, Citation1990; Delong, Wickham, & Pace, Citation1989).
2.3. Methods measuring microbial activities
To measure microbial activity, currently available methods include ATP assay, enzymatic activity tests, and assimilable organic carbon (AOC) tests (Henne et al., Citation2012; Lautenschlager et al., Citation2014; Manz et al., Citation1993; Stutz, Leki, & Lopez Pila, Citation1986; Vanderkooij, Visser, & Hijnen, Citation1982). ATP assay determines all biologically active bacteria based on the total amount of ATP measured through bioluminescence assay (Stutz et al., Citation1986). Enzymatic activity tests quantify specific enzymes by monitoring the increase of fluorescence intensities or absorbance owing to the degradation of substrates by specific enzymatic activities such as polysaccharide-degrading enzymes (α- and β-glucosidase, cellobiohydrolase, xylosidase, chitinase) as a function of time (Lautenschlager et al., Citation2014; Roskoski, Citation2007). AOC concentration represents the fraction of dissolved organic carbon that may readily support microbial growth and is determined by measuring the maximum level of growth of two bacterial isolates (Pseudomonas fluorescens P-17 and Spirillum sp. strain NOX) in a water sample, which usually takes 5–7 days (Vanderkooij et al., Citation1982). However, activity measurements have not been widely incorporated in drinking water microbiome studies because they are time-consuming and labor-intensive.
2.4. Limitations of current methods
Current methods for the monitoring of the drinking water microbiome do have limitations. Cultivation-based methods are known to be time-consuming, low in sensitivity, and ineffective in recovering the majority of microorganisms in the community (Berney et al., Citation2008; Hammes et al., Citation2008; Staley & Konopka, Citation1985). Furthermore, culture-dependent enumeration methods, though widely used in HPC and E. coli testing, cannot effectively detect disinfectant-injured or genetically modified bacterial indicators with inserted antibiotic resistance genes (ARGs) (Li et al., Citation2017). These microorganisms are commonly found in drinking water environment but largely overlooked, which can regain activity under the right test conditions and consequently pose potential health risks to end users. Thus, current studies mostly rely on molecular tools to gain insights into the drinking water microbiome. systematically summarizes biases that can occur or be associated with a few key steps from sampling to data interpretation in those molecular based methods. Firstly, experimental design in technical and biological replicates is critical to statistically determine variations between samples, but is often not considered due to time, manpower, and cost (Brooks et al., Citation2015). For example, sample-to-sample heterogeneity often occur during biofilm samplings. Sampling biofilm in replicates in full-scale systems can be difficult because of limited access, high cost, and high chances of contamination (Gomez-Smith, LaPara, & Hozalski, Citation2015; Ling, Hwang, LeChevallier, Andersen, & Liu, Citation2016). The sample-to-sample heterogeneity can become more significant when integrated analyses of molecules at different metabolic levels (i.e., DNA, RNA, proteins, and metabolites) are involved (Muller, Glaab, May, Vlassis, & Wilmes, Citation2013). Sample volume and concentration methods also play a crucial role in determining whether enough biomass can be successfully obtained for downstream molecular analyses. At present, no standard practice has been established for the minimal sampling volume required and the concentration method used. Sample volume ranging from 100 mL to 2000 L is necessary and often dependent on downstream analyses used and the objectives asked. The extraction efficiency of DNA, RNA, and protein from collected samples can vary from 35 to 85%, and can sometime influence the taxonomic outcomes of microbiota assessments (Brooks et al., Citation2015; Guo & Zhang, Citation2013; Henderson et al., Citation2013; Hwang, Ling, Andersen, LeChevallier, & Liu, Citation2012b; Moran et al., Citation2013; Sinha et al., Citation2017; Stark, Giersch, & Wunschiers, Citation2014; Tsementzi, Poretsky, Rodriguez, Luo, & Konstantinidis, Citation2014). Biases can also occur during PCR amplification (30–50%) (Brooks et al., Citation2015), due to differences in GC content of microorganisms (Duhaime, Deng, Poulos, & Sullivan, Citation2012), rRNA gene copy number (Klappenbach, Saxman, Cole, & Schmidt, Citation2001), and primer annealing efficiency (Wu, Hong, & Liu, Citation2009). All these factors can lead up to 10% variation in estimating the relative abundance of specific microbial groups (Angly et al., Citation2014). During DNA sequencing, various degrees of error, ranging from <1% up to 14%, can occur depending on the sequencing platform used (Feng, Zhang, Ying, Wang, & Du, Citation2015; Jain, Olsen, Paten, & Akeson, Citation2016; Roberts, Carneiro, & Schatz, Citation2013; Ross et al., Citation2013). Last but not the least, when using the short 16S rRNA sequences to identify microbial populations, caution should be taken in relation to its limitations in differentiating closely-related populations at lower phylogenetic levels (e.g., genus or species) (Schloss, Citation2010). Also, 16S rRNA gene sequence cannot accurately provide the physiological function unlike whole genome-based methods (Jain, Rodriguez-R, Phillippy, Konstantinidis, & Aluru, Citation2017). This can be a significant problem in distinguishing pathogenic strains from commensals (Edberg, Citation2009; Steele & Streit, Citation2005).
Table 2. Limitations of current methods as shown by systematic errors reported in the literature.
3. Current understanding on the drinking water microbiome
3.1. Longitudinal shifts from source to tap in conventional drinking water systems
A typical drinking water system spans from source water, the treatment train in the production process, the distribution network, to indoor premise plumbing. The continuous water flow carries upstream microbial assemblages downstream in the distribution system and premise plumbing due to seeding from source water, breakthrough during treatment, seeding from filters, and growth in the distribution network ().
Table 3. Treatment effects on drinking water microbiomes derived from surface water and groundwater.
3.1.1. Seeding from source water
Surface water and groundwater are the two types of source water most commonly used to produce drinking water and can contain distinct microbial populations owing to differences in physical and chemical gradients. These microbial populations can ‘seed’ downstream microbiota in the treatment train, the DS, and premise plumbing. Groundwater systems are mostly known to be anoxic or anaerobic with relatively high concentrations of compounds (i.e., iron, manganese, ammonia, sulfur compounds, methane, and dissolved organic carbon) supporting the growth of anaerobic communities (Albers et al., Citation2015; Bruno et al., Citation2017; Holinger et al., Citation2014; Ling et al., Citation2016; van der Wielen, Voost, & van der Kooij, Citation2009; Zhang, Oh, & Liu, Citation2017). Due to aeration on abstraction, most of these anaerobes cannot survive under the oxidative stress and are generally not detected downstream (Roeselers et al., Citation2015; Zhang, Oh, et al., Citation2017). In comparison, the seeding effect, or treatment breakthrough, is more significant in systems using surface waters as source water (Gomez-Alvarez, Humrighouse, Revetta, & Domingo, Citation2015; Pinto, Xi, & Raskin, Citation2012).
3.1.2. Breakthrough during treatment
Softening is used to remove calcium and magnesium cations that contribute to hardness, with concurrent benefit to the removal of heavy metals, natural organic matter, turbidity, and pathogens (Peters, Citation2011). During the softening process, lime and soda ash are added to raise pH rapidly to 10.3 for calcium precipitation or 11.0 for magnesium precipitation. A recent study (Zhang, Oh, et al., Citation2017) showed a drastic change in the composition and reduction in diversity of microbial communities before and after the softening process, with syntrophs and methanogens abundant in raw water and Exiguobacterium-related populations predominant in the softening effluent. In contrast, a wide variety of different microorganisms were observed to colonize the calcite pellets in a full-scale pellet softening reactor preceded by ozonation. They proliferated as soon as the pH in the water was neutralized due to calcite crystallization in the presence of highly biodegradable nutrients (Hammes et al., Citation2011).
The processes of coagulation, flocculation, and sedimentation are used to remove suspended solids including small particulars and colloids (0.001–1.0 µm), improve water turbidity color, and reduce the level of microbial pathogens and DBP precursors (Peters, Citation2011). Coagulation and flocculation turn small particles present in source water into larger particles called ‘flocs’, which are then removed during sedimentation and filtration. Mostly, these processes are reported to have no observable effect on microbial community structure (Eichler et al., Citation2006; Lin, Yu, Zhang, & Thompson, Citation2014; Poitelon et al., Citation2010), except that one study observed significant community shift during sedimentation (Zeng et al., Citation2013).
Ozonation is used as a disinfection and oxidation process to enhance microbial removal, control taste and odor, and eliminate micropollutants from water (von Gunten, Citation2003). The strong oxidative stress imposed by ozonation causes a significant reduction of the total cell counts and community diversity (Lautenschlager et al., Citation2014; Vaz-Moreira, Egas, Nunes, & Manaia, Citation2013; Zeng et al., Citation2013). It also oxidizes natural organic matter into low-molecular-weight and possibly biodegradable AOC, which is then removed by biological filters. Ozonation can cause a drastic change in the drinking water microbiome as shown by comparing the microbial community before ozonation and in the effluent of the biofilters following ozonation (Lautenschlager et al., Citation2014). Biomass recovery and community analysis during ozonation is difficult due to strong oxidative stress.
Free chlorination and chloramination are two major types of disinfection treatments used to inactivate pathogens during drinking water production and transportation processes. Disinfection treatments can lead to decreases in microbial diversity for systems maintaining a disinfectant residual (Bautista-de los Santos, Schroeder, Sevillano-Rivera, et al., Citation2016; Chao et al., Citation2013; Gomez-Alvarez, Revetta, & Domingo, Citation2012; Sun, Shi, Bai, & Wang, Citation2014). Due to the difference in the inactivation mechanisms, chlorination and chloramination were reported to select for different bacterial populations in a drinking water system with alternating disinfection treatment between chlorination and chloramination (Hwang, Ling, Andersen, LeChevallier, & Liu, Citation2012a; Wang, Proctor, et al., Citation2014). However, this trend was not observed when more systems were incorporated and compared (Bautista-de los Santos, Schroeder, Sevillano-Rivera, et al., Citation2016).
3.1.3. Seeding from filters
Filtration separates suspended or colloidal impurities from water by passing it through a porous medium (e.g., a bed of sand, coal, activated carbon, or garnet) to reduce turbidity, and remove pathogens and many organic and inorganic contaminants. Depending on the source water quality, one or a series of filters are used at a drinking water system, including rapid sand filters (RSFs), granular activated carbon (GAC) filters, and slow sand filters (SSFs). Filtration is a key step in shaping DS microbiota by removing incoming microbes as a form of particles through mechanical screening and by seeding outflow with microbes as planktonic cells or aggregates detached from filter media (Peters, Citation2011; Pinto et al., Citation2012). Microbial biomass can be enriched on the filters and reach up to a density of 109 copies of 16S rRNA gene per g-filter material or 1015–1016 cells per m3 filter material. Depending on water quality, these microbes have various metabolic functions, including oxidation of ammonia, iron, and manganese, metabolism of sulfur compounds, and degradation of dissolved organic carbon and trace organic micropollutants (Albers et al., Citation2015; Cerrato et al., Citation2010; de Vet, Dinkla, Muyzer, Rietveld, & van Loosdrecht, Citation2009; Feng et al., Citation2013; Gulay et al., Citation2016; Holinger et al., Citation2014; Kasuga, Nakagaki, Kurisu, & Furumai, Citation2010a; Kasuga, Nakagaki, Kurisu, & Furumai, Citation2010b; Lautenschlager et al., Citation2014; Liao et al., Citation2013; Magic-Knezev & van der Kooij, Citation2004; Marcus et al., Citation2017; van der Wielen et al., Citation2009; Velten, Hammes, Boller, & Egli, Citation2007; White, DeBry, & Lytle, Citation2012; Zearley & Summers, Citation2012). Dense bacterial cells and protozoa are frequently observed at the top layer of SSFs (known as Schmutzdecke), and eukaryotic predation has been shown to play a critical role in the dynamics of the bacterial community in filters (Haig et al., Citation2015; Lautenschlager et al., Citation2014).
3.1.4. Growth in the distribution network
DS pipes carry drinking water from a centralized treatment plant or well supplies to consumers’ taps, providing the required water quantity and quality at a suitable pressure. Managing the network is a primary challenge from both an operational and public health standpoint due to the expansive physical infrastructure (Snoeyink et al., Citation2006). Microbial regrowth with spatiotemporal variation is the major concern in distribution as the physicochemical and nutritional conditions provided by pipe walls are very different from those found during treatment. Recent studies were able to identify the microbial community and dominant species associated with many important processes in DSs. These processes included the formation of biofilms and loose-deposits on pipe walls (Kelly, Minalt, Culotti, Pryor, & Packman, Citation2014; Liu et al., Citation2014; Wang, Masters, Edwards, Falkinham, & Pruden, Citation2014), harboring pathogens in biofilm (Ling et al., Citation2016; Wang et al., Citation2013), nitrification (Regan, Harrington, Baribeau, De Leon, & Noguera, Citation2003; van der Wielen et al., Citation2009; Wang, Proctor, et al., Citation2014; Zhang, Griffin, & Edwards, Citation2008), oxidation of manganese (Marcus et al., Citation2017) and methane (Kelly et al., Citation2014; Ling et al., Citation2016), and inducing pipe corrosion (Beech & Sunner, Citation2004; Chen, Jia, & Li, Citation2013; Jin, Wu, & Guan, Citation2015; Li et al., Citation2010; Li, Wang, et al., Citation2015; Zhang et al., Citation2008). Many factors have been determined to play a role in the microbial regrowth in DSs, including temperature, especially warm water conditions, the amount of usable carbon, flow regime (hydrodynamics), water residence time, pipe materials, and the presence of corrosion products (Bautista-de los Santos, Schroeder, Sevillano-Rivera, et al., Citation2016; Berry, Xi, & Raskin, Citation2006; Camper, Burr, Ellis, Butterfield, & Abernathy, Citation1998; Dai et al., Citation2017; LeChevallier, Welch, & Smith, Citation1996; Liu, Verberk, & Van Dijk, Citation2013; Proctor, Edwards, & Pruden, Citation2015). For premise plumbing, pipe diameter was reported as a key critical factor (Ji, Parks, Edwards, & Pruden, Citation2015; Lautenschlager et al., Citation2010).
3.2. Temporal dynamics
Temporal dynamics are at the core of understanding the assembly mechanism and succession trajectory of the drinking water microbiome and crucial for developing models to predict changes in water quality and potential microbial contamination events. The drinking water microbiome within a single water distribution system undergoes temporal changes from season to season due to the changing environments provided by pipe walls and from different time points of the day due to hydraulic changes with water demand in the DS. Seasonal variation was found to be the main factor leading to changes in microbial community structure, especially during the summer with elevated temperature (Ling et al., Citation2016; Prest, Weissbrodt, Hammes, van Loosdrecht, & Vrouwenvelder, Citation2016). It has been observed not only in the bulk water phase, but in the biofilm phase and the cold and hot waterlines in DSs (Henne, Kahlisch, Hofle, & Brettar, Citation2013; Ling et al., Citation2016; Pinto, Schroeder, Lunn, Sloan, & Raskin, Citation2014; Prest et al., Citation2016). As a rule of thumb, every 10 °C increase in water temperature leads to a two-fold increase in microbial activity (Baribeau, Citation2006). These seasonal variations have substantial influence on plant operation, including the type and dose of disinfectants and the frequency of positive detection of bacterial indicators. Capturing these changes are critical for establishing mathematical models to predict changes in microbiological water quality. In comparison, short-term fluctuations, such as the diurnal pattern within a day, are difficult to capture due to low frequency in sampling. This diurnal pattern corresponds with shifts in the bulk water community owing to the presence/absence of low-abundance populations and the abundance change of dominant populations and thus has less impact on the drinking water microbiome (Bautista-de los Santos, Schroeder, Blakemore, et al., Citation2016; Besmer et al., Citation2016).
3.3. Biogeography
Biogeography refers to the geographical distributions of organisms over the Earth in both space and time (Beijerinck, Citation1913; Horner-Devine, Carney, & Bohannan, Citation2004). Geographical differences in microbiomes have been observed for waste-treating ecosystems like anaerobic digester sludge (Mei et al., Citation2017). However, few studies have attempted to verify the existence of geographic difference with the drinking water microbiome (Roeselers et al., Citation2015) or understand which environmental factors exert the strongest influences (Bautista-de los Santos, Schroeder, Sevillano-Rivera, et al., Citation2016; Horner-Devine et al., Citation2004). A survey of drinking water microbiomes along the Mississippi River found that the drinking water microbiota of New Orleans, LA differed from other communities with high relative abundances of phylotypes indicative of fresh and saltwater infiltration (e.g., Planctomycetes and Bacteroidetes) and potential opportunistic pathogens (e.g., Legionella and Mycobacterium spp.) (Holinger et al., Citation2014; Hull et al., Citation2017). This survey further observed that the abundant taxa were generally shared among all systems and system-specific taxa were not particularly abundant (Holinger et al., Citation2014). Similar findings were reported among systems across a restricted area. Roeselers et al., (Roeselers et al., Citation2015) surveyed 32 drinking water distribution networks in the Netherlands, all using groundwater from (un)confined sandy aquifers as the source water and no disinfectant residual in the networks, and observed network-specific taxa, which were of low abundances. However, these studies investigated diversity only through 16S rRNA gene amplicon analyses. Future studies revealing diversity at different scales of resolution are needed for accurate description of the biogeography of the drinking water microbiome.
3.4. The core microbiome
Identifying a core microbiome (i.e., shared microbial taxa) is an important step for gaining insights into the microbial function associated with an ecosystem, and is often used to provide guidance on how to manipulate the drinking water microbiome at the user end. A core microbiome is typically defined as the suite of members shared among microbial consortia from similar habitats (Shade & Handelsman, Citation2012). As the water-phase microflora within a given system are relatively stable irrespective of the sampling locations over short-time scales (Lautenschlager et al., Citation2013; Pinto et al., Citation2014; Roeselers et al., Citation2015), studies have attempted to define a core microbiome in the bulk water phase after the production process across different drinking water systems. At high taxonomical levels, such as phyla and classes, the core microbiome is made up primarily of Alpha- and Beta-proteobacteria, and to a lesser extent of Gamma-proteobacteria, Nitrospirae, Planctomycetes, Acidobacteria, Bacteroidetes and Chloroflexi (Bautista-de los Santos, Schroeder, Sevillano-Rivera, et al., Citation2016; Eichler et al., Citation2006; Lautenschlager et al., Citation2014; Lin et al., Citation2014; Pinto et al., Citation2012; Vaz-Moreira et al., Citation2013; Zeng et al., Citation2013). Families of Burkholderiaceae, Methylophilaceae, Comamonadaceae, and Rhodocyclaceae were abundant among Beta-proteobacteria, whereas Sphingomonadaceae, Caulobacteraceae, and Methylobacteriaceae were dominant in Alpha-proteobacteria (Douterelo et al., Citation2014; Eichler et al., Citation2006; Pinto et al., Citation2012; Vaz-Moreira et al., Citation2013; Zeng et al., Citation2013). It is however difficult to define a core microbiome down to genus or species levels across systems using different disinfectants (chlorine, chloramine, and without disinfectants), likely due to the substantial differences in selection pressures from these disinfectants (Bautista-de los Santos, Schroeder, Sevillano-Rivera, et al., Citation2016). Few studies have attempted to define the active core microbiome. Furthermore, it might be impossible to define a core microbiome in the biofilm phase of DSs owing to the numerous but spatially heterogeneous ecological niches and continual ecological succession (Ling et al., Citation2016).
3.5. Microbial exchange between multi-phases
Microbes in a drinking water system are present in bulk water, suspended solids (i.e., particulate matter transported throughout the network), pipe wall biofilm, and loose deposits (i.e., particulate matter accumulated on the pipe bottom) and the exchange of microorganisms between these four phases is one crucial mechanism influencing the water quality received by the end users (Liu et al., Citation2014). Loose deposits and biofilm were observed to host the majority of microorganisms (98%) compared with bulk water and suspended solids combined (2%) (Liu et al., Citation2014). The number of cells attached to each particle in suspended solids and loose deposits almost doubled after passing through the distribution network (Liu, Ling, et al., Citation2013; Liu et al., Citation2016). Unlike water-phase and particle-associated populations, which have a relatively short transit time within a drinking water system, biofilm-phase microbes can be viewed as indigenous populations in a drinking water system. They are organized in highly-structured habitats and exhibit considerable structural, chemical and biological heterogeneity (Allen, Taylor, & Geldreich, Citation1980; Ridgway & Olson, Citation1981; Stewart & Franklin, Citation2008; Wimpenny, Manz, & Szewzyk, Citation2000). In previous studies, biofilm was taken as a composite sample along the pipe wall for microbial community analysis. However, biofilm likely exhibited heterogeneity along the radial direction in the pipe wall. Contrary to intuition, the findings by Ridgway and Olson (Liu, Ren, et al., Citation2017) revealed that biofilm located in the middle part of pipe walls possessed the highest diversity and harbored the highest abundance of possible pathogens. Studies have shown that biofilm assemblages could influence the bulk water communities in the DS, and the effect was dependent on the level of biofilm sloughing from the pipe surface (Douterelo, Jackson, Solomon, & Boxall, Citation2017; Douterelo, Sharpe, & Boxall, Citation2013; Henne et al., Citation2012; Ling et al., Citation2016; Roeselers et al., Citation2015). A recent study estimated that the sloughing of 20% biofilm from PVC pipes or 10% biofilm from HDPE pipes would significantly alter the bulk water community (Liu, Tao, et al., Citation2017).
3.6. Prokaryote-eukaryote interactions
Why do opportunistic pathogens never stop reoccurring in the distribution network even after chlorine treatment? The underlying reason is probably due to pathogens living within eukaryotes embedded in surface-attached biofilms (Brown & Barker, Citation1999). Eukaryotes are an important component of the drinking water microbiome, many of which are resistant to disinfection processes and can be reservoirs for pathogens, such as Mycobacterium, Legionella, and Chlamydia (Buse et al., Citation2017; Cervero-Arago, Rodriguez-Martinez, Puertas-Bennasar, & Araujo, Citation2015; Delafont, Bouchon, Hechard, & Moulin, Citation2016; Delafont et al., Citation2014; Fonseca & Swanson, Citation2014; Kilvington & Price, Citation1990; Sauer, Bachman, & Swanson, Citation2005). Furthermore, eukaryotes can be the ‘hub’ facilitating the genetic exchange between viruses, opportunistic pathogens and their closely-related species, and the eukaryotic hosts themselves (Zhang, Oh, et al., Citation2017). In fact, the co-evolution between amebae and L. pneumophila has ‘trained’ bacteria both for environmental survival and virulence towards human (Gomez-Valero et al., Citation2011; Smith, Citation2005). Reported eukaryotic groups in drinking water systems included amebae, nematodes, fungi, flagellates, segmented worms, arthropods, and flat worms (Delafont et al., Citation2016; Oh, Hammes, & Liu, Citation2018; Zhang, Oh, et al., Citation2017).
3.7. Water recycling and its impact on the microbiome
Water reuse either intended or de facto is gaining momentum in dealing with water scarcity. Indirect or direct potable reuse has become an inevitable choice for certain regions in addition to the traditional non-potable reuse for irrigation and rain water harvest. Depending on the purpose of reuse, a multi-barrier treatment framework is required to achieve a targeted level of water quality, in particular, concerning microbiological safety and public health as people are under direct exposure to reused water through consumption or aerosol. Consequently, several critical questions on the microbiome in recycled water need to be addressed to provide scientific support for technological and regulatory guidance: how effective is the current treatment train in reducing the loading of microorganisms, including antibiotic-resistant bacteria (ARB), ARGs, and viruses? How does each reuse step alter the microbiome? Can we find biomarkers that can be used for day-day or online monitoring of water treatment efficiency and water quality of reused water?
summarizes the current research status on the impact of treatment technologies on the microbiome in recycled water. For centralized potable water reuse (either directly or indirectly) and water desalination, treatment processes commonly involve ultrafiltration (UF)/microfiltration (MF)/cartridge filters (CFs) followed by reverse osmosis (RO). These two membrane processes can significantly alter the downstream microbial community by retaining microorganisms adapted to biofilm mode of growth on membrane surfaces that are continuously in contact with a disinfectant residual (Al Ashhab, Herzberg, & Gillor, Citation2014; Leddy et al., Citation2017; Levi, Bar-Zeev, Elifantz, Berman, & Berman-Frank, Citation2016; Miller, Nelson, & Rodriguez, Citation2017). Processes upstream of RO serve as inocula for the RO biofouling layer and those downstream, such as GAC, play a primary role in seeding the DS (Levi et al., Citation2016; Miller et al., Citation2017). In most cases, recycled water needs to be combined with treated water from conventional drinking water treatment plant before entering the DS. The transition to combined water potentially causes a change of water chemistry in the DS, which in turn can profoundly impact the nature and composition of biofilms and trigger the release of pathogens into drinking water (Liu, Zhang, et al., Citation2017). For decentralized rainwater harvest, opportunistic pathogens is the primary research interest in the recycled water after certain period of sedimentation in the tank and their presence is shown to be correlated with water chemistry (Ahmed, Brandes, Gyawali, Sidhu, & Toze, Citation2014; Campisano et al., Citation2017; Dobrowsky, Khan, Cloete, & Khan, Citation2017). For reclaimed water used for irrigation after tertiary wastewater treatment, ARB and ARGs are major concerns as wastewater treatment plants are hotspots for the dissemination of ARGs and ARB in the downstream DS and soil environments (Garner, Chen, et al., Citation2018; Garner, McLain, et al., Citation2018; Hong et al., Citation2018).
Table 4. Treatment technologies for recycled water and their impact on the microbiome.
4. Ecological patterns of the drinking water microbiome exemplified by four full-scale systems in Western countries
Numerous physical, hydraulic, chemical, and biological processes occur in such a complex system, all of which might impact the drinking water microbiome either locally or system-wise. Meanwhile, utility managers, scientists, and regulators view this complex ecosystem from different perspectives. For managers in water utilities, they are interested in forecasting changes in drinking water microbiome under different conditions and improving the efficiency of existing monitoring practices. Researchers are attracted by mechanistic understanding of phenomena occurring in drinking water ecosystems and establishing predictive models for these phenomena. Regulators consider microbiological water safety as a priority, in particular, how to minimize microbial contamination events through engineering practices. Therefore, we summarize four in-depth studied systems with different levels of residual chlorine to illustrate the current understanding developed on drinking water microbiomes from different perspectives to enlighten future studies (the Braunschweig and Zurich systems without residual disinfectants; the Ann Arbor system with chloramine; and the Champaign system mainly with chlorine).
The city of Braunschweig, Germany represents systems using surface water as source water with low chlorine dose during treatment and no residual chlorine in the distribution network. Highlights of the study series include comparing the impact of treatment processes on both the total community (by DNA analysis) and the active community (by RNA analysis), relating the microbial community in the biofilm phage with those from the water phase, and tracking Legionella diversity and dynamics from source water to cold and hot waterlines (Eichler et al., Citation2006; Henne et al., Citation2012; Henne et al., Citation2013; Lesnik, Brettar, & Hofle, Citation2016). The city uses two surface water reservoirs to produce drinking water through two separate systems with coagulation-flocculation, sand barriers, and chlorination (0.2–0.7 mg/L), and final mixing at a constant ratio (Lesnik et al., Citation2016). The findings suggested that microflora in the DS were influenced by both water sources and chlorination with the latter having a more profound impact on the active community than the overall microbial community (Eichler et al., Citation2006). The bulk water community was observed to have a large number of the low-abundance bacterial populations and the biofilm community had a reduced diversity and did not share any core microbial population with bulk water. It was hypothesized that low-abundance bacterial populations in the bulk water could function as an inoculum to seed the biofilm community (Henne et al., Citation2012). Seasonal dynamics observed in the drinking water microbiome were highly influenced by source water (Henne et al., Citation2013). Regarding Legionella species, a high diversity with over 40 phylotypes was observed in source waters, which was remarkably reduced during the treatment process but completely recovered in the mixing and the followed distribution network. It was further observed that different phylotypes of Legionella species dominated in cold waterlines and hot waterlines with substantially more L. pneumophila, the pathogenic species, in hot waterlines (accounting for as high as 40% of Legionella spp.) (Lesnik et al., Citation2016). The abundance of phylotypes belonging to L. longbeachae and L. pneumophila increased significantly with temperature, indicating the thermophilic nature of these pathogenic species and their enrichment by water heaters.
The city of Zurich, Switzerland, is a typical European system using sequential ozonation and filtration steps (RSFs, GAC filters, and SSFs) without residual disinfectants in the distribution network. Studies on this system underscore a multi-parametric approach to capture as complete a picture as possible of the drinking water microbiome by integrating conventional microbial detection methods with modern molecular techniques, including microbial growth potential (TOC and AOC), community abundance and composition (HPC, FCM, and 16S rRNA gene-based community analysis), and activity and function (ATP and extracellular enzyme activities) (Lautenschlager et al., Citation2010; Lautenschlager et al., Citation2014; Lautenschlager et al., Citation2013). The system uses surface water (Lake Zürich) to produce 50% of daily drinking water demand through the treatment process, mixed with untreated groundwater (49%) and spring water (1%). Microbial community shifts were observed at each stage of the treatment step based on phylogenetic, enzymatic, and metabolic analyses. Filter microbial communities in RSFs, GAC filters, and SSFs differed among each other, and from those observed in the effluent of individual filters. The microbial community in SSF effluent, the subsequent reservoir, and most locations in the DS remained remarkably stable during a two-year consecutive sampling (Lautenschlager et al., Citation2014). Exceptions were locations with the longest water retention times, as shown by a significantly higher total cell concentration by FCM and a shift in the community profile, which could not be observed with traditional detection methods (Lautenschlager et al., Citation2013). In premise plumbing, cell concentration increased in the first liter of tap water after overnight stagnation, followed by step-wise decrease with the water volume flushed (the first 2 liters). This increase in cell concentration could only be partially explained by the growth due to available AOC (Lautenschlager et al., Citation2010). Future studies can further explore the contribution of biofilm growth and detachment to the total regrowth during stagnation.
The city of Ann Arbor, Michigan, USA represents surface water systems with residual disinfectants in the DS, exhibiting strong seasonal cycling. The study series identified the primary role of sand filters in shaping the microbial community in the downstream DS, which provided a possibility to control the drinking water microbiome in the DS and at tap through manipulating the filter microbial community by proper plant operation (Pinto et al., Citation2012). A highlight of the studies was the use of modeling tools to describe and generalize the trend observed with the drinking water microbiome, translating research results to information that could potentially be used by water utilities (Pinto et al., Citation2014). Furthermore, cultivation was carried out to investigate a dominant group of bacteria in the system indicated by 454 pyrosequencing (i.e., Mn(II)‐oxidizing bacteria). The integration of cultivation dependent and independent approaches to decipher the ecology and function of dominant bacterial groups greatly promoted mechanistic understanding of drinking water microbiomes. This drinking water system uses surface water (Huron River) and local wells (groundwater) as raw water at approximately 2:1 in the winter and 8:1 in the summer. The water is treated through a process including lime softening, coagulation, flocculation, sedimentation, ozonation, dual media filtration, and chloramination. The finished water contains approximately 3 mg Cl2/L chloramine as the residual disinfectant with a pH between 9.1 and 9.3. The findings revealed that bacterial taxa that colonized the filter and sloughed off in the filter effluent persisted in the DS, indicating the importance of the filter. The microbiome in the distribution network exhibited a strong temporal trend of seasonal cycling correlating with temperature and source water usage patterns, and weaker spatial dynamics. The relative abundance of a taxon in the studied DS was positively correlated with the frequency of its detection.
The cities of Champaign and Urbana, Illinois, USA is a typical groundwater system with dissolved methane in the reservoir to produce drinking water subjected to chlorine conversion (i.e., shifts between chloramine and chlorine as residual disinfectants) due to the implementation of Stage 2 Disinfectants and Disinfection Byproducts Rules or dealing with the problems of nitrification and microbial regrowth in the DS. A unique feature of this system is the presence of dissolved methane throughout the entire system, leading to the prevalence and a high abundance of methane-oxidizing bacteria and the accompanying species feeding on the intermediate metabolites of methane oxidation in finished water and the DS (in both the water phase and the biofilm phase) (Ling et al., Citation2016). Furthermore, eukaryotes could be a critical factor driving the diversification of the bacterial community as a variety of eukaryotic groups were detected throughout the system (Zhang, Oh et al., Citation2017). Because it is difficult to sample biofilms from pipe surface in the DS, this study series introduces biofilms obtained from water meters as a good alternative to those from pipe surface, enabling extensive investigation of both the water phase and the biofilm phase (Hong et al., Citation2010; Ling et al., Citation2016). In building water supply system, long-term stagnation led to the regrowth of microorganisms, in which stochastic process explained the community composition at the proximal end but deterministic process played a more important role at the distal end (Ling, Whitaker, LeChevallier, & Liu, Citation2018). The source water containing methane went through two-stage lime softening, recarbonation, chlorination and filtration (Flynn et al., Citation2013; Gunsalus, Zeikus, & Wolfe, Citation1972; Hwang et al., Citation2012a). Chloramine was used as residual disinfectant for many years but was switched to free chlorine in 2012. Major microbial community shifts occurred during abstraction and softening processes, whereas the shifts in the DS were shown to be correlated with disinfectant types and sampling time for the water-phase samples but not for the biofilm-phase samples from water meters.
5. Current accomplishments of safe drinking water in third-world countries
Safe and consistent water supply is a significant concern in third-world countries, especially in many rural and peri-urban areas. The problems associated with low-quality drinking water are not only technological, such as poor operation and management of drinking water infrastructure and insufficient monitoring, but also financial (Mackintosh & Colvin, Citation2003; Momba, Tyafa, Makala, Brouckaert, & Obi, Citation2006). For instance, a survey of 55 plants in the rural communities of the Eastern Cape identified that inefficient chemical (coagulant and chlorine) dosing, which led to high turbidity and low chlorine residuals, was the major cause of unacceptable microbiological quality (Momba et al., Citation2006). These facts suggest strong needs for continual funds for operation and maintenance, and technical support and professional training for local operators.
Alternatively, self-sustaining water supply systems, which require minimum human and financial input, is a potential solution for people in rural and peri-urban communities in third-world countries. Existing and recently-developed technologies can be incorporated into these self-sustaining systems, such as biological sand filtration (Wang, Li, Brockman, & Nguyen, Citation2016), gravity-driven membrane technology (Peter-Varbanets, Gujer, & Pronk, Citation2012), handpumps (Marks, Citation2012), and solar water pumps (Ramos & Ramos, Citation2009).
6. Potential of using meta-omics techniques to study the drinking water microbiome
Current studies in the drinking water microbiome primarily investigate ‘who are there and under what conditions?’ by integrating 16S rRNA gene amplicon analysis and environmental metadata. However, it is hard to use the knowledge to guide operational practice as i) classification using 16S rRNA gene amplicon analysis is accurate only to the genus or family level, and ii) 16S rRNA gene-based phylogeny and metabolic functioning in situ do not always agree with each other (Janda & Abbott, Citation2007; Zhang, Oh, et al., Citation2017). This lack of direct linkage between microbes and their functions in drinking water systems can be systematically addressed by next-generation sequencing (NGS) and meta-omics technologies (). We can ask and provide answers to fundamental ecological questions that include but are not limited to ‘what are they doing?’, ‘why are they there?’ and more critically ‘who is doing what?’, and ‘what are the interrelationships among them, and between them and their environment?’ (Rittmann et al., Citation2006).
Figure 3. Types of meta-omics and the key questions that can be addressed by the meta-omics tools in the study of drinking water microbiome. Source: Zhang and Liu, Citationforthcoming. Reprinted with permission. © Water Research Foundation.
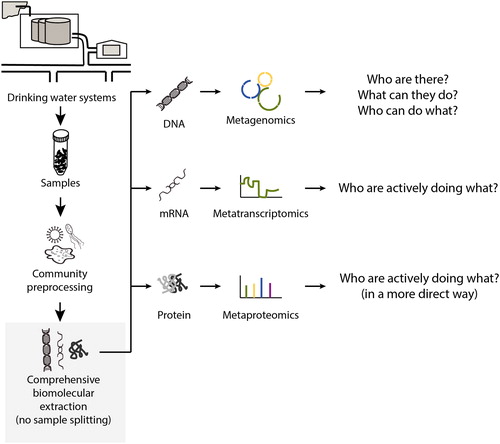
6.1. NGS and Meta-omics technologies
The advance in NGS technology in the last decade serves as the pivotal force to the development of omics tools. The Sanger method is considered as a ‘first-generation’ technology, and newer methods using new sequencing chemistry are referred to as NGS, which includes ‘454’, ‘Illumina’, PacBio SMRT and the Oxford Nanopore MinION. ‘454’ and Illumina platforms can produce a large number of sequences that are low in cost, high in throughput and accuracy, and short in run times (Metzker, Citation2010). They produce short reads (Ross et al., Citation2013), which makes downstream bioinformatics analyses difficult. For this reason, technologies such as PacBio SMRT and the Oxford Nanopore MinION were developed to produce long reads (>5 kb) but at a relatively higher error rate (12–14% for PacBio and 8% for Nanopore) than Illumina platforms (Feng et al., Citation2015; Jain et al., Citation2016; Roberts et al., Citation2013). To take advantage of the strength of both short- and long-read technologies, hybrid de novo assembly at the chromosome-level is performed using long-read technologies with sequencing errors corrected by Illumina data (Goodwin et al., Citation2015; Risse et al., Citation2015). These NGS technologies has enabled the development of metagenomics and metatranscriptomics to study microbial functions and activities in various microbial ecosystems (Béjà et al., Citation2000; Giannoukos et al., Citation2012; Tyson et al., Citation2004; Urich et al., Citation2008; Venter et al., Citation2004; Xiong et al., Citation2012). NGS platforms generate millions or billions of reads in parallel, which require multiple bioinformatic steps, such as assembly, binning, mapping and quantification to convert into useful information for researchers. While dealing with bioinformatic software is challenging, especially with integrated data from metagenomics, metatranscriptomics, and proteomics, several web-based pipelines are accessible to scientists, including MG-RAST (Meyer et al., Citation2008), KBase (Arkin et al., Citation2016), CyVerse/iPlant Discovery Environment (Goff et al., Citation2011), IMG-ER (Markowitz et al., Citation2012), and PATRIC (Wattam et al., Citation2017).
Metagenomics or environmental genomics is the genomic analysis of microorganisms in a microbial community that can provide insights into community physiology (Handelsman, Citation2004; Sharpton, Citation2014), and enables the discovery of new microbial taxa and genes without cultivation. The procedure involves extracting DNA from all cells in a community, shearing DNA into fragments, sequencing fragmented DNA (Handelsman, Citation2004; Tyson et al., Citation2004; Venter et al., Citation2004), assembling all sequences into an ecosystem genome comprised of many genomes of the innate microbial populations (‘metagenome’) (Handelsman, Citation2004), and phylogenetically classifying the genomic fragments to specific microorganisms (‘binning’) (McHardy, Martin, Tsirigos, Hugenholtz, & Rigoutsos, Citation2007). This approach has been greatly improved by using novel assemblers (e.g., metaSPAdes and MEGAHIT) (Li, Liu, Luo, Sadakane, & Lam, Citation2015; Namiki, Hachiya, Tanaka, & Sakakibara, Citation2012; Nurk, Meleshko, Korobeynikov, & Pevzner, Citation2017; Peng, Leung, Yiu, & Chin, Citation2012) and binning methods (McHardy et al., Citation2007; Pati, Heath, Kyrpides, & Ivanova, Citation2011; Patil, Roune, & McHardy, Citation2012; Wu, Tang, Tringe, Simmons, & Singer, Citation2014) together with software that integrate information from essential single-copy genes (e.g., MaxBin) and multiple metagenomes of related samples (e.g., MetaBAT and GroopM) (Albertsen et al., Citation2013; Imelfort et al., Citation2014; Kang, Froula, Egan, & Wang, Citation2015; Wu, Simmons, & Singer, Citation2016). Researchers can now determine individual bins’ phylogeny (‘phylogenomics’) using software such as PhyloPhlAn (Segata, Bornigen, Morgan, & Huttenhower, Citation2013) and genome completeness/contamination with marker genes using software such as CheckM (Parks, Imelfort, Skennerton, Hugenholtz, & Tyson, Citation2015). These advancements enable accurate metagenomic assembly, binning, and recovery of genomes for phylogenetically novel organisms without cultivating them (Kang et al., Citation2015; Wrighton et al., Citation2012; Wu et al., Citation2014).
Metatranscriptomics is based on sequencing the total message RNA (mRNA) in a microbial community to identify genes or pathways that are actively expressed. This process involves extracting total RNA from microbial communities, removing ribosome RNA (rRNA) to obtain high levels of mRNA transcripts, reverse transcribing mRNA into cDNAs, ligating to adapters, and then sequencing using NGS (He, Wurtzel, et al., Citation2010; Sorek & Cossart, Citation2010). Metagenomics is often used together with metatranscriptomics to provide assembled genomes as templates on which transcripts are mapped. Fast and sensitive alignment software such as TopHat (Kim et al., Citation2013) and HISAT (Kim, Langmead, & Salzberg, Citation2015) are developed for this purpose, which are followed by transcript assemble and abundance estimate by Cufflinks (Trapnell et al., Citation2010) and HTSeq (Anders, Pyl, & Huber, Citation2015). Metatranscriptomics has been widely used in a variety of environments, including soil (Urich et al., Citation2008), sediment (Dumont, Pommerenke, & Casper, Citation2013), gut microbiomes (Giannoukos et al., Citation2012; Xiong et al., Citation2012), and activated sludge (He, Kunin, et al., Citation2010; Yu & Zhang, Citation2012). It is a powerful tool to provide insights into community function and activity and identify novel pathways in uncultured microorganisms (Haroon et al., Citation2013).
Metaproteomics is developed to evaluate microbial activity within an ecosystem at a specific time based on protein expression (Wilmes & Bond, Citation2004; Zampieri, Chiapello, Daghino, Bonfante, & Mello, Citation2016). Unlike metagenomics and metatranscriptomics that use NGS technologies, metaproteomics uses liquid chromatography tandem mass spectrometry (LC-MS/MS). The process starts with extracting protein, followed by LC-MS/MS to generate MS spectra, and then comparing spectra with peptides from thousands of proteins of diverse taxonomic groups. These comparisons can be achieved in two ways: through searching against existing protein/peptide databases or by matching to theoretical peptide spectra generated in silico from metagenomes of the same sample or of similar environments (Timmins-Schiffman et al., Citation2017; Zampieri et al., Citation2016). Metaproteomics is a powerful tool to unravel the active metabolic processes in different environments in a more direct way than metagenomics or metatranscriptomics. This approach has been applied to complicated environments, including soils (Benndorf, Balcke, Harms, & von Bergen, Citation2007; Wang et al., Citation2011; Williams, Taylor, & Mula, Citation2010), sediments (Benndorf et al., Citation2009; Bruneel et al., Citation2011), marine habitats (Morris et al., Citation2010; Sowell et al., Citation2011), freshwater systems (Habicht et al., Citation2011; Lauro et al., Citation2011; Ng et al., Citation2010), and activated sludge (Wilmes, Wexler, & Bond, Citation2008).
6.2. Applications of Meta-omics in drinking water microbiome studies
While omics tools have been applied to various microbial ecosystems by many studies, application to the drinking water microbiome is limited to a few studies. Some of these studies are based on the use of cosmid library construction that is low in sequence throughput [212] or early NGS technologies that cannot derive long assembled contigs to provide correct linkage between microbes and functionalities (Chistoserdova, Citation2014; Schmeisser et al., Citation2003). Using early NGS techniques, two studies (Chao et al., Citation2013; Gomez-Alvarez et al., Citation2012) investigated the impact of water treatment on the drinking water microbiome. Their results revealed that chlorine and chloramine treatments caused differences in community structures, disinfectant mechanisms, and virulence genes (Gomez-Alvarez et al., Citation2012). Changes in protective functions (i.e., glutathione synthesis) were observed in treated water compared with raw water (Chao et al., Citation2013). In contrast, the use of the latest NGS technology has provided in-depth information into the metabolic and geochemical potential of groundwater-fed RSFs (Palomo et al., Citation2016). Dominance of Nitrospira was observed to co-occur with high abundance of genes in nitrification and carbon fixation pathways. Genomic analysis revealed that the Nitrospira genome harbored complete ammonia monooxygenase (amoCAB), particularly, the atypical amoA gene similar to the complete ammonia oxidation bacteria Nitrospira (i.e., comammox). This novel finding suggested that Nitrospira in RSF had the potential for complete ammonium oxidation. Moreover, other recovered draft genomes had the capability to oxidize ammonium, nitrite, hydrogen sulfide, methane, and potentially iron and manganese, as well as to assimilate organic compounds. Similarly, Pinto et al. (Citation2016) reported metagenomic evidence for the presence of a comammox Nitrospira genome in biologically active filters. The recovered bin from metagenomes contained the full suite of ammonia oxidation genes on a single scaffold. Genome-resolved metagenomics could also be used to differentiate pathogens and closely related species and identify new biomarkers, such as spacers located in the clustered regularly interspaced short palindromic repeats (CRISPR) regions for the monitoring of ‘true’ pathogenic strains across different drinking water systems (Zhang, Kitajima, Whittle, & Liu, Citation2017).
6.3. Limitations of Meta-omics technologies
Applying meta-omics in drinking water microbiome studies can face several challenges. The first one can be related to sample preparation, as a large quantity of genomic DNA, RNA and protein is required for downstream sequencing and LC-MS/MS analyses. For the water-phase drinking water microbiome, sampling a large volume of water (i.e., over 1000 L) is often required for systems containing residual disinfectants. As conventional concentration devices are not suitable for this purpose (Chao et al., Citation2013; Zhang, Oh, et al., Citation2017), studies have used point-of-use water purifiers (Chao et al., Citation2013; Zhang, Oh, et al., Citation2017), which involve more than one mechanism to concentrate cells and the biases remain unknown. Thus, there is a need to standardize a device for concentrating large volumes of drinking water. Likewise, studying biofilm-phase drinking water microbiome in full-scale drinking water systems can be challenging (Gomez-Smith et al., Citation2015; Ling et al., Citation2016). Currently, two approaches are used: one is to cut pipes and the other is to insert coupons into pipes and retrieve them after biofilms develop (Douterelo et al., Citation2014). The former is labor-intensive, expensive, and prone to contamination from surrounding environments, and the latter can distort hydraulic conditions in pipes and cause deviations from real pipes. An alternative solution is to sample biofilms from the inner surface of water meters (Hong et al., Citation2010). For metatranscriptomics and metaproteomics studies, sampling preparation needs to be carefully evaluated because mRNA is liable to degradation by RNases that are ubiquitously present in the environment (Hansen, Stensballe, Nielsen, & Herbst, Citation2014). Proper stabilization and storage procedures are critical to obtain sufficient quantities of high-quality mRNA. Finally, meta-omics studies can generate huge datasets that require vigorous bioinformatics analyses and computing capacity (Thomas, Gilbert, & Meyer, Citation2012). However, errors associated with bioinformatics can be problematic and substantially influence the final interpretation (Kunin, Copeland, Lapidus, Mavromatis, & Hugenholtz, Citation2008; Timmins-Schiffman et al., Citation2017). Most studies have to establish curated databases of their interests, partially because of the scattered data submitted and stored in various databases. Future studies will require advanced tools to simultaneously interact with multiple databases for microbial genomes, metagenomes, protein, antibiotic resistance genes, and viral genomes. A systematic data management and cross-platform reference framework for the drinking water microbiome is lacking, which requires leveraging current and future plans to expand our understanding of microbiomes on earth.
7. Future research needs
We are at a critical stage in advancing our understanding of the drinking water microbiome that requires critical actions and fundamental questions to be investigated. To address these needs, the following three topics have been identified as research priorities.
7.1. Topic 1: Current drinking water microbiome monitoring techniques
Challenge: Current practices in drinking water treatment, distribution, and monitoring in the USA are regulated by standards and guidelines set by the United States Environmental Protection Agency. However, such regulations are generally based on fecal coliform and HPC, which may have limited direct relevance to the public health. While the understanding of the microbiome composition of drinking water systems is still not complete, recent surveys utilizing 16S rRNA gene amplicon analysis have revealed an astounding level of microbial diversity. In particular, fine differences in water chemistry associated with different geographic regions and water treatments, as well as seasonal variation, appear to result in vast differences in the water microbiome composition. At the same time, studies have strongly suggested that variation in microbiomes from different samples could be caused by biases associated with procedures in sampling, DNA extraction, sequencing, and data analysis. Action: This highlights an urgent need to call for collaborative actions to standardize analytical procedures for future data comparison among studies on drinking water microbiomes. Questions: How can the standardization of analytical procedures be achieved? What are the limitations and gaps that should be overcome?
7.2. Topic 2: A multi-perspective understanding of the drinking water microbiome using the state-of-art meta-omics techniques
Challenge: Most of the current efforts focus on known functions of individual populations detected within the drinking water microbiome using 16S rRNA gene amplicon analysis. However, we have limited knowledge on system-level properties of the microbial assemblage, the function of microorganisms belonging to novel phyla as well as the undiscovered function of existing microorganisms. Examples of system-level characteristics mechanisms of community assembly (e.g., stochastic and deterministic processes), microorganisms or functions stabilizing the community (e.g., keystone species), and interactions occurring among different microorganisms and different groups, which are different from the properties of individual populations. Meta-omics techniques become vital as they can provide information at the community level using different information-carrying biopolymers (i.e., DNA, mRNA, protein and metabolites). Action: Increasing efforts are needed to elucidate many crucial processes that the drinking water microbiome mediate by investigating both the individual components of microbial assemblages and the emergent properties of systems using the state-of-art meta-omics tools. Questions: How do drinking water microbiota assemble spatiotemporally? Are the assemblages primarily influenced by the environmental conditions in water systems, i.e., deterministic processes? Among the suite of meta-omics techniques, which methods are effective and suitable for establishing the linkage between community composition and function for drinking water microbiomes? In addition to 16S rRNA gene as the most commonly used biomarker, are there other biomarkers that can be used for the monitoring of water quality at the user end?
7.3. Topic 3: Novel concepts guiding the control of opportunistic pathogens
Challenge: The persistence of opportunistic pathogens in DSs and building plumbing systems despite various cleaning practices indicates the limitation of bacterial indicators and the ineffectiveness of current practices in controlling pathogen proliferation. The growth of opportunistic pathogens in drinking water environments is attributed to two mechanisms: i) activating certain pathways when growing in the attached form (e.g., biofilms and sediments); and ii) interacting with the rest microbiota through predatory, antagonistic, competitive, symbiotic, and mutualistic relationships. However, few engineering practices based on these mechanisms have been developed and experimentally validated. Action: It is urgent to introduce novel concepts that are effective in engineering a desirable drinking water microbiome. Questions: Is it possible that a ‘healthy’ or common drinking water microbiome is determined or defined, and used to differentiate the ‘unhealthy’ ones? Can these findings be used to develop a signature that indicates the potential for disease outbreaks in the distribution system and premise plumbing, particularly concerning opportunistic pathogens (Legionella, mycobacteria, Pseudomonas)? As the assemblage of the drinking water microbiome can be substantially influenced by the environmental conditions in water systems, what additional metadata should be obtained to link meta-omics data to the drinking water microbiome? Eventually, can engineering intervention and treatment process changes be used to intentionally shift those ‘unhealthy’ microbiomes to ‘healthy’ ones?
| Abbreviations | ||
| AOC | = | Assimilable organic carbon |
| ARB | = | Antibiotic resistant bacteria |
| ARGs | = | Antibiotic resistance genes |
| CFs | = | Cartridge filters |
| CRISPR | = | Clustered regularly interspaced short palindromic repeats |
| DBPs | = | Disinfection by-products |
| DGGE | = | Denaturing gradient gel electrophoresis |
| DSs | = | Distribution systems |
| FCM | = | Flow cytometry |
| FISH | = | Fluorescence in-situ hybridization |
| GAC | = | Granular activated carbon |
| HPC | = | Heterotrophic plate count |
| LC-MS/MS | = | Liquid chromatography tandem mass spectrometry |
| MF | = | Microfiltration |
| NGS | = | Next-generation sequencing |
| PCR-ALH | = | PCR-amplicon length heterogeneity |
| RO | = | Reverse osmosis |
| RSFs | = | Rapid sand filters |
| SEM | = | Scanning electron microscopy |
| SSCP | = | Single strand conformation polymorphism |
| SSFs | = | Slow sand filters |
| T-RFLP | = | Terminal restriction fragment length polymorphism |
| UF | = | Ultrafiltration |
| USA | = | The United States of America |
Acknowledgements
This study is funded in part by Water Research Foundation (WRF) Project 4700 and in kind contribution from University of Illinois at Urbana-Champaign.
References
- Ahmed, W., Brandes, H., Gyawali, P., Sidhu, J. P. S., & Toze, S. (2014). Opportunistic pathogens in roof-captured rainwater samples, determined using quantitative PCR. Water Research, 53, 361–369. doi:10.1016/j.watres.2013.12.021
- Al Ashhab, A., Herzberg, M., & Gillor, O. (2014). Biofouling of reverse-osmosis membranes during tertiary wastewater desalination: Microbial community composition. Water Research, 50, 341–349. doi:10.1016/j.watres.2013.10.044
- Albers, C. N., Ellegaard-Jensen, L., Harder, C. B., Rosendahl, S., Knudsen, B. E., Ekelund, F., & Aamand, J. (2015). Groundwater chemistry determines the prokaryotic community structure of waterworks sand filters. Environmental Science & Technology, 49, 839–846. doi:10.1021/es5046452
- Albertsen, M., Hugenholtz, P., Skarshewski, A., Nielsen, K. L., Tyson, G. W., & Nielsen, P. H. (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nature Biotechnology, 31, 533–538. doi:10.1038/nbt.2579
- Allen, M. J., Taylor, R. H., & Geldreich, E. E. (1980). The occurrence of microorganisms in water main encrustations. Journal - American Water Works Association, 72, 614–625. doi:10.1002/j.1551-8833.1980.tb04598.x
- Amann, R. I., Krumholz, L., & Stahl, D. A. (1990). Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental-studies in microbiology. Journal of Bacteriology, 172, 762–770. doi:10.1128/jb.172.2.762-770.1990
- Anders, S., Pyl, P. T., & Huber, W. (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England), 31, 166–169.
- Angly, F. E., Dennis, P. G., Skarshewski, A., Vanwonterghem, I., Hugenholtz, P., & Tyson, G. W. (2014). CopyRighter: A rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction. Microbiome, 2, 11.
- APHA. (1905). Standard Methods of Water Analysis, 1st edition, 84–85.
- APHA, AWWA, and WEF. (1998). Standard Methods for the Examination of Water and Wastewater. Washington, DC: American Public Health Association.
- Arkin, A. P., Stevens, R. L., Cottingham, R. W., Maslov, S., Henry, C. S., Dehal, P., …., Yoo, S. (2016). The DOE systems biology knowledgebase (KBase). bioRxiv.
- Baribeau, H. (2006). Growth and inactivation of nitrifying bacteria. In Fundamentals and control of nitrification in chloraminated drinking water distribution systems - Manual of water supply practices M56. Denver, CO: American Water Works Association.
- Bartlett, J. M., & Stirling, D. (2003). A short history of the polymerase chain reaction. Methods Mol Biol, 226, 3–6.
- Bartram, J., Cotruvo, J., Exner, M., Fricker, C., & Glasmacher, A. (2004). Heterotrophic plate count measurement in drinking water safety management – Report of an Expert Meeting Geneva, 24–25 April 2002. International Journal of Food Microbiology, 92, 241–247. doi:10.1016/j.ijfoodmicro.2003.08.005
- Bautista-de los Santos, Q. M., Schroeder, J. L., Blakemore, O., Moses, J., Haffey, M., Sloan, W., & Pinto, A. J. (2016). The impact of sampling, PCR, and sequencing replication on discerning changes in drinking water bacterial community over diurnal time-scales. Water Research, 90, 216–224. doi:10.1016/j.watres.2015.12.010
- Bautista-de los Santos, Q. M., Schroeder, J. L., Sevillano-Rivera, M. C., Sungthong, R., Ijaz, U. Z., Sloan, W. T., & Pinto, A. J. (2016). Emerging investigators series: Microbial communities in full-scale drinking water distribution systems – A meta-analysis. Environmental Science: Water Research & Technology, 2, 631–644. doi:10.1039/C6EW00030D
- Beech, W. B., & Sunner, J. (2004). Biocorrosion: Towards understanding interactions between biofilms and metals. Current Opinion in Biotechnology, 15, 181–186. doi:10.1016/j.copbio.2004.05.001
- Beijerinck, M. (1913). De Infusies en de Ontdekking der Backterien, Jaarboek van de Koninklijke Akademie v. Wetenschappen. Muller, Amsterdam, the Netherlands.
- Béjà, O., Aravind, L., Koonin, E. V., Suzuki, M. T., Hadd, A., Nguyen, L. P., … DeLong, E. F. (2000). Bacterial rhodopsin: Evidence for a new type of phototrophy in the sea. Science (New York, N.Y.), 289, 1902–1906.
- Benndorf, D., Balcke, G. U., Harms, H., & von Bergen, M. (2007). Functional metaproteome analysis of protein extracts from contaminated soil and groundwater. The ISME Journal, 1, 224–234.
- Benndorf, D., Vogt, C., Jehmlich, N., Schmidt, Y., Thomas, H., Woffendin, G., … von Bergen, M. (2009). Improving protein extraction and separation methods for investigating the metaproteome of anaerobic benzene communities within sediments. Biodegradation, 20, 737–750. doi:10.1007/s10532-009-9261-3
- Berney, M., Vital, M., Hulshoff, I., Weilenmann, H. U., Egli, T., & Hammes, F. (2008). Rapid, cultivation-independent assessment of microbial viability in drinking water. Water Research, 42, 4010–4018. doi:10.1016/j.watres.2008.07.017
- Berry, D., Xi, C., & Raskin, L. (2006). Microbial ecology of drinking water distribution systems. Current Opinion in Biotechnology, 17, 297–302.
- Besmer, M. D., Epting, J., Page, R. M., Sigrist, J. A., Huggenberger, P., & Hammes, F. (2016). Online flow cytometry reveals microbial dynamics influenced by concurrent natural and operational events in groundwater used for drinking water treatment. Scientific Reports, 6
- Besmer, M. D., & Hammes, F. (2016). Short-term microbial dynamics in a drinking water plant treating groundwater with occasional high microbial loads. Water Research, 107, 11–18. doi:10.1016/j.watres.2016.10.041
- Bodrossy, L., & Sessitsch, A. (2004). Oligonucleotide microarrays in microbial diagnostics. Current Opinion in Microbiology, 7, 245–254.
- Brooks, J. P., Edwards, D. J., Harwich, M. D., Rivera, M. C., Fettweis, J. M., Serrano, M. G., … Consortium, V. M. (2015). The truth about metagenomics: Quantifying and counteracting bias in 16S rRNA studies. BMC Microbiology, 15, 66.
- Brown, M. R., & Barker, J. (1999). Unexplored reservoirs of pathogenic bacteria: Protozoa and biofilms. Trends in Microbiology, 7(1), 46–50. doi:10.1016/S0966-842X(98)01425-5
- Bruneel, O., Volant, A., Gallien, S., Chaumande, B., Casiot, C., Carapito, C., … Arsène-Ploetze, F. (2011). Characterization of the active bacterial community involved in natural attenuation processes in arsenic-rich creek sediments. Microbial Ecology, 61, 793–810. doi:10.1007/s00248-011-9808-9
- Bruno, A., Sandionigi, A., Rizzi, E., Bernasconi, M., Vicario, S., Galimberti, A., … Casiraghi, M. (2017). Exploring the under-investigated “microbial dark matter” of drinking water treatment plants. Scientific Reports, 7, 44350.
- Buse, H. Y., Ji, P., Gomez-Alvarez, V., Pruden, A., Edwards, M. A., & Ashbolt, N. J. (2017). Effect of temperature and colonization of Legionella pneumophila and Vermamoeba vermiformis on bacterial community composition of copper drinking water biofilms. Microbial Biotechnology, 10, 773–788. doi:10.1111/1751-7915.12457
- Camper, A., Burr, M., Ellis, B., Butterfield, P., & Abernathy, C. (1998). Development and structure of drinking water biofilms and techniques for their study. Journal of Applied Microbiology, 85(S1), 1s–12s. doi:10.1111/j.1365-2672.1998.tb05277.x
- Campisano, A., Butler, D., Ward, S., Burns, M. J., Friedler, E., DeBusk, K., … Han, M. (2017). Urban rainwater harvesting systems: Research, implementation and future perspectives. Water Research, 115, 195–209. doi:10.1016/j.watres.2017.02.056
- Cerrato, J. M., Falkinham, J. O., Dietrich, A. M., Knocke, W. R., McKinney, C. W., & Pruden, A. (2010). Manganese-oxidizing and -reducing microorganisms isolated from biofilms in chlorinated drinking water systems. Water Research, 44, 3935–3945. doi:10.1016/j.watres.2010.04.037
- Cervero-Arago, S., Rodriguez-Martinez, S., Puertas-Bennasar, A., & Araujo, R. M. (2015). Effect of common drinking water disinfectants, chlorine and heat, on free Legionella and Amoebae-associated Legionella. PLoS One, 10, e0134726.
- Chao, Y., Ma, L., Yang, Y., Ju, F., Zhang, X. X., Wu, W. M., & Zhang, T. (2013). Metagenomic analysis reveals significant changes of microbial compositions and protective functions during drinking water treatment. Scientific Reports, 3, 3550.
- Chen, L., Jia, R. B., & Li, L. (2013). Bacterial community of iron tubercles from a drinking water distribution system and its occurrence in stagnant tap water. Environmental Science: Processes & Impacts, 15, 1332–1340. doi:10.1039/c3em00171g
- Chen, N. T., & Chang, C. W. (2010). Rapid quantification of viable legionellae in water and biofilm using ethidium monoazide coupled with real-time quantitative PCR. Journal of Applied Microbiology, 109, 623–634.
- Chiao, T. H., Clancy, T. M., Pinto, A., Xi, C. W., & Raskin, L. (2014). Differential resistance of drinking water bacterial populations to monochloramine disinfection. Environmental Science & Technology, 48, 4038–4047. doi:10.1021/es4055725
- Chistoserdova, L. (2014). Is metagenomics resolving identification of functions in microbial communities? Microbial Biotechnology, 7(1), 1–4. doi:10.1111/1751-7915.12077
- Dai, D., Prussin, A. J., 2nd, Marr, L. C., Vikesland, P. J., Edwards, M. A., & Pruden, A. (2017). Factors shaping the human exposome in the built environment: Opportunities for engineering control. Environmental Science & Technology, 51, 7759–7774. doi:10.1021/acs.est.7b01097
- de Vet, W. W. J. M., Dinkla, I. J. T., Muyzer, G., Rietveld, L. C., & van Loosdrecht, M. C. M. (2009). Molecular characterization of microbial populations in groundwater sources and sand filters for drinking water production. Water Research, 43(1), 182–194. doi:10.1016/j.watres.2008.09.038
- Delafont, V., Bouchon, D., Hechard, Y., & Moulin, L. (2016). Environmental factors shaping cultured free-living amoebae and their associated bacterial community within drinking water network. Water Research, 100, 382–392. doi:10.1016/j.watres.2016.05.044
- Delafont, V., Mougari, F., Cambau, E., Joyeux, M., Bouchon, D., Hechard, Y., & Moulin, L. (2014). First evidence of amoebae-mycobacteria association in drinking water network. Environmental Science & Technology, 48, 11872–11882. doi:10.1021/es5036255
- Delong, E. F., Wickham, G. S., & Pace, N. R. (1989). Phylogenetic stains – Ribosomal RNA-based probes for the identification of single cells. Science, 243, 1360–1363. doi:10.1126/science.2466341
- Dobrowsky, P. H., Khan, S., Cloete, T. E., & Khan, W. (2017). Microbial and physico-chemical characteristics associated with the incidence of Legionella spp. and Acanthamoeba spp. in rainwater harvested from different roofing materials. Water, Air, & Soil Pollution, 228
- Douterelo, I., Boxall, J. B., Deines, P., Sekar, R., Fish, K. E., & Biggs, C. A. (2014). Methodological approaches for studying the microbial ecology of drinking water distribution systems. Water Research, 65, 134–156. doi:10.1016/j.watres.2014.07.008
- Douterelo, I., Jackson, M., Solomon, C., & Boxall, J. (2017). Spatial and temporal analogies in microbial communities in natural drinking water biofilms. Science of the Total Environment, 581, 277–288. doi:10.1016/j.scitotenv.2016.12.118
- Douterelo, I., Sharpe, R. L., & Boxall, J. B. (2013). Influence of hydraulic regimes on bacterial community structure and composition in an experimental drinking water distribution system. Water Research, 47, 503–516. doi:10.1016/j.watres.2012.09.053
- Duhaime, M. B., Deng, L., Poulos, B. T., & Sullivan, M. B. (2012). Towards quantitative metagenomics of wild viruses and other ultra-low concentration DNA samples: A rigorous assessment and optimization of the linker amplification method. Environmental Microbiology, 14, 2526–2537. doi:10.1111/j.1462-2920.2012.02791.x
- Dumont, M. G., Pommerenke, B., & Casper, P. (2013). Using stable isotope probing to obtain a targeted metatranscriptome of aerobic methanotrophs in lake sediment. Environmental Microbiology Reports, 5, 757–764.
- Edberg, S. C. (2009). Does the possession of virulence factor genes mean that those genes will be active? Journal of Water and Health, 7(S1), S19–S28. doi:10.2166/wh.2009.066
- Edelstein, P. H. (1981). Improved semiselective medium for isolation of Legionella pneumophila from contaminated clinical and environmental specimens. Journal of Clinical Microbiology, 14, 298–303.
- Eichler, S., Christen, R., Holtje, C., Westphal, P., Botel, J., Brettar, I., … Hofle, M. G. (2006). Composition and dynamics of bacterial communities of a drinking water supply system as assessed by RNA- and DNA-based 16S rRNA gene fingerprinting. Applied and Environmental Microbiology, 72, 1858–1872. doi:10.1128/AEM.72.3.1858-1872.2006
- El Fantroussi, S., Urakawa, H., Bernhard, A. E., Kelly, J. J., Noble, P. A., Smidt, H., … Stahl, D. A. (2003). Direct profiling of environmental microbial populations by thermal dissociation analysis of native rRNAs hybridized to oligonucleotide microarrays. Applied and Environmental Microbiology, 69, 2377–2382. doi:10.1128/AEM.69.4.2377-2382.2003
- Feng, S., Chen, C., Wang, Q. F., Zhang, X. J., Yang, Z. Y., & Xie, S. G. (2013). Characterization of microbial communities in a granular activated carbon-sand dual media filter for drinking water treatment. International Journal of Environmental Science and Technology, 10, 917–922. doi:10.1007/s13762-013-0188-1
- Feng, Y. X., Zhang, Y. C., Ying, C. F., Wang, D. Q., & Du, C. L. (2015). Nanopore-based fourth-generation DNA Sequencing technology. Genomics Proteomics Bioinformatics, 13, 200–201.
- Fischer, E. R., Hansen, B. T., Nair, V., Hoyt, F. H., & Dorward, D. W. (2005). Scanning electron microscopy. In Current protocols in microbiology. Hoboken, NJ: John Wiley & Sons, Inc.
- Flynn, T. M., Sanford, R. A., Ryu, H., Bethke, C. M., Levine, A. D., Ashbolt, N. J., & Domingo, J. W. S. (2013). Functional microbial diversity explains groundwater chemistry in a pristine aquifer. BMC Microbiology, 13, 146.
- Fonseca, M. V., & Swanson, M. S. (2014). Nutrient salvaging and metabolism by the intracellular pathogen Legionella pneumophila. Frontiers in Cellular and Infection Microbiology, 4
- Frankland, P. (1894). Micro-organisms in water: Their significance, identification and removal, together with an account of the bacteriological methods employed in their investigation, specially designed for the use of those connected with the sanitary aspects of water-supply. Harlow, UK: Longmans, Green & Co.
- Garner, E., Chen, C., Xia, K., Bowers, J., Engelthaler, D. M., McLain, J., … Pruden, A. (2018). Metagenomic characterization of antibiotic resistance genes in full-scale reclaimed water distribution systems and corresponding potable systems. Environmental Science & Technology, 52, 6113–6125. doi:10.1021/acs.est.7b05419
- Garner, E., McLain, J., Bowers, J., Engelthaler, D. M., Edwards, M. A., & Pruden, A. (2018). Microbial ecology and water chemistry impact regrowth of opportunistic pathogens in full-scale reclaimed water distribution systems. Environmental Science & Technology, 52, 9056–9068. doi:10.1021/acs.est.8b02818
- Giannoukos, G., Ciulla, D. M., Huang, K., Haas, B. J., Izard, J., Levin, J. Z., … Gnirke, A. (2012). Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biology, 13
- Goff, S. A., Vaughn, M., McKay, S., Lyons, E., Stapleton, A. E., Gessler, D., … Stanzione, D. (2011). The iPlant collaborative: Cyberinfrastructure for plant biology. Frontiers in Plant Science, 2
- Gomez-Alvarez, V., Humrighouse, B. W., Revetta, R. P., & Domingo, J. W. S. (2015). Bacterial composition in a metropolitan drinking water distribution system utilizing different source waters. Journal of Water and Health, 13(1), 140–151. doi:10.2166/wh.2014.057
- Gomez-Alvarez, V., Revetta, R. P., & Domingo, J. W. S. (2012). Metagenomic analyses of drinking water receiving different disinfection treatments. Applied and Environmental Microbiology, 78, 6095–6102. doi:10.1128/AEM.01018-12
- Gomez-Smith, C. K., LaPara, T. M., & Hozalski, R. M. (2015). Sulfate reducing bacteria and mycobacteria dominate the biofilm communities in a chloraminated drinking water distribution system. Environmental Science & Technology, 49, 8432–8440. doi:10.1021/acs.est.5b00555
- Gomez-Valero, L., Rusniok, C., Jarraud, S., Vacherie, B., Rouy, Z., Barbe, V., … Buchrieser, C. (2011). Extensive recombination events and horizontal gene transfer shaped the Legionella pneumophila genomes. BMC Genomics, 12, 536.
- Goodwin, S., Gurtowski, J., Ethe-Sayers, S., Deshpande, P., Schatz, M. C., & McCombie, W. R. (2015). Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Research, 25, 1750–1756. doi:10.1101/gr.191395.115
- Gulay, A., Musovic, S., Albrechtsen, H. J., Abu Al-Soud, W., Sorensen, S. J., & Smets, B. F. (2016). Ecological patterns, diversity and core taxa of microbial communities in groundwater-fed rapid gravity filters. The ISME Journal, 10, 2209–2222. doi:10.1038/ismej.2016.16
- Gunsalus, R. P., Zeikus, J. G., & Wolfe, R. S. (1972). Microbial modification of ground water.
- Guo, F., & Zhang, T. (2013). Biases during DNA extraction of activated sludge samples revealed by high throughput sequencing. Applied Microbiology and Biotechnology, 97, 4607–4616. doi:10.1007/s00253-012-4244-4
- Guschin, D. Y., Mobarry, B. K., Proudnikov, D., Stahl, D. A., Rittmann, B. E., & Mirzabekov, A. D. (1997). Oligonucleotide microchips as genosensors for determinative and environmental studies in microbiology. Applied and Environmental Microbiology, 63, 2397–2402.
- Habicht, K. S., Miller, M., Cox, R. P., Frigaard, N.-U., Tonolla, M., Peduzzi, S., … Andersen, J. S. (2011). Comparative proteomics and activity of a green sulfur bacterium through the water column of Lake Cadagno, Switzerland. Environmental Microbiology, 13(1), 203–215. doi:10.1111/j.1462-2920.2010.02321.x
- Haig, S. J., Schirmer, M., D'Amore, R., Gibbs, J., Davies, R. L., Collins, G., & Quince, C. (2015). Stable-isotope probing and metagenomics reveal predation by protozoa drives E-coli removal in slow sand filters. The ISME Journal, 9, 797–808. doi:10.1038/ismej.2014.175
- Halliday, S. (2004). Water: A turbulent history. London, UK: Sutton.
- Hammes, F., Berney, M., Wang, Y. Y., Vital, M., Koster, O., & Egli, T. (2008). Flow-cytometric total bacterial cell counts as a descriptive microbiological parameter for drinking water treatment processes. Water Research, 42(1–2), 269–277. doi:10.1016/j.watres.2007.07.009
- Hammes, F., Boon, N., Vital, M., Ross, P., Magic-Knezev, A., & Dignum, M. (2011). Bacterial colonization of pellet softening reactors used during drinking water treatment. Applied and Environmental Microbiology, 77, 1041–1048. doi:10.1128/AEM.02068-10
- Handelsman, J. (2004). Metagenomics: Application of genomics to uncultured microorganisms. Microbiology and Molecular Biology Reviews: MMBR, 68, 669–685.
- Hansen, S. H., Stensballe, A., Nielsen, P. H., & Herbst, F. A. (2014). Metaproteomics: Evaluation of protein extraction from activated sludge. PROTEOMICS, 14, 2535–2539. doi:10.1002/pmic.201400167
- Haroon, M. F., Hu, S., Shi, Y., Imelfort, M., Keller, J., Hugenholtz, P., … Tyson, G. W. (2013). Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature, 500, 567. doi:10.1038/nature12375
- He, S., Kunin, V., Haynes, M., Martin, H. G., Ivanova, N., Rohwer, F., … McMahon, K. D. (2010). Metatranscriptomic array analysis of ‘Candidatus Accumulibacter phosphatis’-enriched enhanced biological phosphorus removal sludge. Environmental Microbiology, 12, 1205–1217. doi:10.1111/j.1462-2920.2010.02163.x
- He, S., Wurtzel, O., Singh, K., Froula, J. L., Yilmaz, S., Tringe, S. G., … Hugenholtz, P. (2010). Validation of two ribosomal RNA removal methods for microbial metatranscriptomics. Nature Methods, 7, 807–U858. doi:10.1038/nmeth.1507
- Henderson, G., Cox, F., Kittelmann, S., Miri, V. H., Zethof, M., Noel, S. J., … Janssen, P. H. (2013). Effect of DNA extraction methods and sampling techniques on the apparent structure of cow and sheep rumen microbial communities. PLoS One, 8, e74787.
- Henne, K., Kahlisch, L., Brettar, I., & Hofle, M. G. (2012). Analysis of structure and composition of bacterial core communities in mature drinking water biofilms and bulk water of a citywide network in Germany. Applied and Environmental Microbiology, 78, 3530–3538. doi:10.1128/AEM.06373-11
- Henne, K., Kahlisch, L., Hofle, M. G., & Brettar, I. (2013). Seasonal dynamics of bacterial community structure and composition in cold and hot drinking water derived from surface water reservoirs. Water Research, 47, 5614–5630. doi:10.1016/j.watres.2013.06.034
- Herzenberg, L. A., Parks, D., Sahaf, B., Perez, O., Roederer, M. & Herzenberg, L. A. (2002). The history and future of the fluorescence activated cell sorter and flow cytometry: A view from Stanford. Clin Chem, 48, 1819–1827.
- Holinger, E. P., Ross, K. A., Robertson, C. E., Stevens, M. J., Harris, J. K., & Pace, N. R. (2014). Molecular analysis of point-of-use municipal drinking water microbiology. Water Research, 49, 225–235. doi:10.1016/j.watres.2013.11.027
- Hong, P.-Y., Julian, T., Pype, M.-L., Jiang, S., Nelson, K., Graham, D., … Manaia, C. (2018). Reusing treated wastewater: Consideration of the safety aspects associated with antibiotic-resistant bacteria and antibiotic resistance genes. Water, 10, 244. doi:10.3390/w10030244
- Hong, P. Y., Hwang, C. C., Ling, F. Q., Andersen, G. L., LeChevallier, M. W., & Liu, W. T. (2010). Pyrosequencing analysis of bacterial biofilm communities in water meters of a drinking water distribution system. Applied and Environmental Microbiology, 76, 5631–5635. doi:10.1128/AEM.00281-10
- Horner-Devine, M. C., Carney, K. M., & Bohannan, B. J. M. (2004). An ecological perspective on bacterial biodiversity. Proceedings. Biological Sciences, 271, 113–122.
- Hull, N. M., Holinger, E. P., Ross, K. A., Robertson, C. E., Harris, J. K., Stevens, M. J., & Pace, N. R. (2017). Longitudinal and source-to-tap New Orleans, LA, USA drinking water microbiology. Environmental Science & Technology, 51, 4220–4229.
- Hwang, C., Ling, F. Q., Andersen, G. L., LeChevallier, M. W., & Liu, W. T. (2012a). Microbial community dynamics of an urban drinking water distribution system subjected to phases of chloramination and chlorination treatments. Applied and Environmental Microbiology, 78, 7856–7865. doi:10.1128/AEM.01892-12
- Hwang, C., Ling, F. Q., Andersen, G. L., LeChevallier, M. W., & Liu, W. T. (2012b). Evaluation of methods for the extraction of DNA from drinking water distribution system biofilms. Microbes and Environments, 27(1), 9–18. doi:10.1264/jsme2.ME11132
- Imelfort, M., Parks, D., Woodcroft, B. J., Dennis, P., Hugenholtz, P., & Tyson, G. W. (2014). GroopM: An automated tool for the recovery of population genomes from related metagenomes. PeerJ, 2, e603. doi:10.7717/peerj.603
- Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., & Aluru, S. (2017). High-throughput ANI Analysis of 90K prokaryotic genomes reveals clear species boundaries. bioRxiv,
- Jain, M., Olsen, H. E., Paten, B., & Akeson, M. (2016). The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biology, 17, 239.
- Janda, J. M., & Abbott, S. L. (2007). 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. Journal of Clinical Microbiology, 45, 2761–2764. doi:10.1128/JCM.01228-07
- Ji, P., Parks, J., Edwards, M. A., & Pruden, A. (2015). Impact of Water Chemistry, Pipe Material and Stagnation on the Building Plumbing Microbiome. PLoS One, 10, e0141087. doi:10.1371/journal.pone.0141087
- Jin, J., Wu, G., & Guan, Y. (2015). Effect of bacterial communities on the formation of cast iron corrosion tubercles in reclaimed water. Water Research, 71, 207–218.
- Kang, D. D., Froula, J., Egan, R., & Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ, 3, e1165. doi:10.7717/peerj.1165
- Kasuga, I., Nakagaki, H., Kurisu, F., & Furumai, H. (2010a). Abundance and diversity of ammonia-oxidizing archaea and bacteria on biological activated carbon in a pilot-scale drinking water treatment plant with different treatment processes. Water Science and Technology, 61, 3070–3077. doi:10.2166/wst.2010.204
- Kasuga, I., Nakagaki, H., Kurisu, F., & Furumai, H. (2010b). Predominance of ammonia-oxidizing archaea on granular activated carbon used in a full-scale advanced drinking water treatment plant. Water Research, 44, 5039–5049. doi:10.1016/j.watres.2010.07.015
- Keiblinger, K. M., Wilhartitz, I. C., Schneider, T., Roschitzki, B., Schmid, E., Eberl, L., … Zechmeister-Boltenstern, S. (2012). Soil metaproteomics – Comparative evaluation of protein extraction protocols. Soil Biology and Biochemistry, 54, 14–24. doi:10.1016/j.soilbio.2012.05.014
- Kelly, J. J., Minalt, N., Culotti, A., Pryor, M., & Packman, A. (2014). Temporal variations in the abundance and composition of biofilm communities colonizing drinking water distribution pipes. PLoS One, 9, e98542. doi:10.1371/journal.pone.0098542
- Kilvington, S., & Price, J. (1990). Survival of legionella-pneumophila within cysts of acanthamoeba-polyphaga following chlorine exposure. Journal of Applied Bacteriology, 68, 519–525. doi:10.1111/j.1365-2672.1990.tb02904.x
- Kim, D., Langmead, B., & Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nature Methods, 12, 357–360.
- Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., & Salzberg, S. L. (2013). TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14, R36.
- Klappenbach, J. A., Saxman, P. R., Cole, J. R., & Schmidt, T. M. (2001). rrndb: The ribosomal RNA operon copy number database. Nucleic Acids Research, 29(1), 181–184.
- Koch, R., & Duncan, G. (1894). Professor Koch on the bacteriological diagnosis of cholera, water-filtration and cholera, and the cholera in Germany during the Winter of 1892–93. Translated by G. Duncan, et al., [Reprinted from the “Scotsman”]. Edinburgh.
- Kunin, V., Copeland, A., Lapidus, A., Mavromatis, K., & Hugenholtz, P. (2008). A bioinformatician’s guide to metagenomics. Microbiology and Molecular Biology Reviews: MMBR, 72, 557–578.
- Langer-Safer, P. R., Levine, M., & Ward, D. C. (1982). Immunological method for mapping genes on Drosophila polytene chromosomes. Proc Natl Acad Sci USA, 79, 4381–4385.
- Lauro, F. M., DeMaere, M. Z., Yau, S., Brown, M. V., Ng, C., Wilkins, D., … Cavicchioli, R. (2011). An integrative study of a meromictic lake ecosystem in Antarctica. The ISME Journal, 5, 879–895.
- Lautenschlager, K., Boon, N., Wang, Y., Egli, T., & Hammes, F. (2010). Overnight stagnation of drinking water in household taps induces microbial growth and changes in community composition. Water Research, 44, 4868–4877. doi:10.1016/j.watres.2010.07.032
- Lautenschlager, K., Hwang, C., Ling, F., Liu, W.-T., Boon, N., Köster, O., … Hammes, F. (2014). Abundance and composition of indigenous bacterial communities in a multi-step biofiltration-based drinking water treatment plant. Water Research, 62, 40–52. doi:10.1016/j.watres.2014.05.035
- Lautenschlager, K., Hwang, C., Liu, W.-T., Boon, N., Köster, O., Vrouwenvelder, H., … Hammes, F. (2013). A microbiology-based multi-parametric approach towards assessing biological stability in drinking water distribution networks. Water Research, 47, 3015–3025. doi:10.1016/j.watres.2013.03.002
- LeChevallier, M. W., Welch, N. J., & Smith, D. B. (1996). Full-scale studies of factors related to coliform regrowth in drinking water. Applied and Environmental Microbiology, 62, 2201–2211.
- Leddy, M. B., Hasan, N. A., Subramanian, P., Heberling, C., Cotruvo, J., & Colwell, R. R. (2017). Characterization of microbial signatures from advanced treated wastewater biofilms. Journal - American Water Works Association, 109, E503–E512. doi:10.5942/jawwa.2017.109.0116
- Lee, C. K., Herbold, C. W., Polson, S. W., Wommack, K. E., Williamson, S. J., McDonald, I. R., & Cary, S. C. (2012). Groundtruthing next-gen sequencing for microbial ecology-biases and errors in community structure estimates from PCR amplicon pyrosequencing. PLoS One, 7
- Lee, E. S., Lee, M. H., & Kim, B. S. (2015). Evaluation of propidium monoazide-quantitative PCR to detect viable Mycobacterium fortuitum after chlorine, ozone, and ultraviolet disinfection. International Journal of Food Microbiology, 210, 143–148. doi:10.1016/j.ijfoodmicro.2015.06.019
- Lesnik, R., Brettar, I., & Hofle, M. G. (2016). Legionella species diversity and dynamics from surface reservoir to tap water: From cold adaptation to thermophily. The ISME Journal, 10, 1064–1080. doi:10.1038/ismej.2015.199
- Levi, A., Bar-Zeev, E., Elifantz, H., Berman, T., & Berman-Frank, I. (2016). Characterization of microbial communities in water and biofilms along a large scale SWRO desalination facility: Site-specific prerequisite for biofouling treatments. Desalination, 378, 44–52. doi:10.1016/j.desal.2015.09.023
- Li, D., Li, Z., Yu, J., Cao, N., Liu, R., & Yang, M. (2010). Characterization of bacterial community structure in a drinking water distribution system during an occurrence of red water. Applied and Environmental Microbiology, 76, 7171–7180. doi:10.1128/AEM.00832-10
- Li, D., Liu, C.-M., Luo, R., Sadakane, K., & Lam, T.-W. (2015). MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics, btv033.
- Li, E. S. Y., & Liu, W. T. (2003). DNA microarray technology in microbial ecology studies- principle, application and current limitations. Microbes and Environments, 18, 175–187. doi:10.1264/jsme2.18.175
- Li, J., Liu, L., Yang, D., Liu, W.-L., Shen, Z.-Q., Qu, H.-M., … Jin, M. (2017). Culture-dependent enumeration methods failed to simultaneously detect disinfectant-injured and genetically modified Escherichia coli in drinking water. Environmental Science: Processes & Impacts, 19, 720–726. doi:10.1039/C6EM00625F
- Li, X. X., Wang, H. B., Hu, C., Yang, M., Hu, H. Y., & Niu, J. F. (2015). Characteristics of biofilms and iron corrosion scales with ground and surface waters in drinking water distribution systems. Corrosion Science, 90, 331–339. doi:10.1016/j.corsci.2014.10.028
- Liao, X. B., Chen, C., Wang, Z., Wan, R., Chang, C. H., Zhang, X. J., & Xie, S. G. (2013). Pyrosequencing analysis of bacterial communities in drinking water biofilters receiving influents of different types. Process Biochemistry, 48, 703–707. doi:10.1016/j.procbio.2013.02.033
- Lin, W. F., Yu, Z. S., Zhang, H. X., & Thompson, I. P. (2014). Diversity and dynamics of microbial communities at each step of treatment plant for potable water generation. Water Research, 52, 218–230. doi:10.1016/j.watres.2013.10.071
- Ling, F., Hwang, C., LeChevallier, M. W., Andersen, G. L., & Liu, W. T. (2016). Core-satellite populations and seasonality of water meter biofilms in a metropolitan drinking water distribution system. The ISME Journal, 10, 582–595. doi:10.1038/ismej.2015.136
- Ling, F., Whitaker, R., LeChevallier, M. W., & Liu, W.-T. (2018). Drinking water microbiome assembly induced by water stagnation. The ISME Journal, 12, 1520–1531. doi:10.1038/s41396-018-0101-5
- Liu, G., Bakker, G. L., Li, S., Vreeburg, J. H. G., Verberk, J. Q. J. C., Medema, G. J., … Van Dijk, J. C. (2014). Pyrosequencing reveals bacterial communities in unchlorinated drinking water distribution system: An integral study of bulk water, suspended solids, loose deposits, and pipe wall biofilm. Environmental Science & Technology, 48, 5467–5476. doi:10.1021/es5009467
- Liu, G., Ling, F. Q., Magic-Knezev, A., Liu, W. T., Verberk, J. Q. J. C., & Van Dijk, J. C. (2013). Quantification and identification of particle-associated bacteria in unchlorinated drinking water from three treatment plants by cultivation-independent methods. Water Research, 47, 3523–3533.
- Liu, G., Ling, F. Q., van der Mark, E. J., Zhang, X. D., Knezev, A., Verberk, J. Q. J. C., … van Dijk, J. C. (2016). Comparison of particle-associated bacteria from a drinking water treatment plant and distribution reservoirs with different water sources. Scientific Reports, 6
- Liu, G., Tao, Y., Zhang, Y., Lut, M., Knibbe, W.-J., van der Wielen, P., … van der Meer, W. (2017). Hotspots for selected metal elements and microbes accumulation and the corresponding water quality deterioration potential in an unchlorinated drinking water distribution system. Water Research, 124, 435–445.
- Liu, G., Verberk, J. Q., & Van Dijk, J. C. (2013). Bacteriology of drinking water distribution systems: An integral and multidimensional review. Applied Microbiology and Biotechnology, 97, 9265–9276. doi:10.1007/s00253-013-5217-y
- Liu, G., Zhang, Y., Knibbe, W. J., Feng, C. J., Liu, W. S., Medema, G., & van der Meer, W. (2017). Potential impacts of changing supply-water quality on drinking water distribution: A review. Water Research, 116, 135–148.
- Liu, J., Ren, H., Ye, X., Wang, W., Liu, Y., Lou, L., … Hu, B. (2017). Bacterial community radial-spatial distribution in biofilms along pipe wall in chlorinated drinking water distribution system of East China. Applied Microbiology and Biotechnology, 101, 749–759.
- Liu, W. T., Mirzabekov, A. D., & Stahl, D. A. (2001). Optimization of an oligonucleotide microchip for microbial identification studies: A non-equilibrium dissociation approach. Environmental Microbiology, 3, 619–629. doi:10.1046/j.1462-2920.2001.00233.x
- Liu, W. T., & Stahl, D. A. (2007). Molecular approaches for the measurement of density, diversity and phylogeny: Manual of environmental microbiology (3rd ed., pp. 139–156). Washington, DC: American Society for Microbiology.
- Loy, A., Lehner, A., Lee, N., Adamczyk, J., Meier, H., Ernst, J., … Wagner, M. (2002). Oligonucleotide microarray for 16S rRNA gene-based detection of all recognized lineages of sulfate-reducing prokaryotes in the environment. Applied and Environmental Microbiology, 68, 5064–5081. doi:10.1128/AEM.68.10.5064-5081.2002
- Mackintosh, G., & Colvin, C. (2003). Failure of rural schemes in South Africa to provide potable water. Environmental Geology, 44(1), 101–105. doi:10.1007/s00254-002-0704-y
- Magic-Knezev, A., & van der Kooij, D. (2004). Optimisation and significance of ATP analysis for measuring active biomass in granular activated carbon filters used in water treatment. Water Research, 38, 3971–3979. doi:10.1016/j.watres.2004.06.017
- Manz, W., Szewzyk, U., Ericsson, P., Amann, R., Schleifer, K. H., & Stenstrom, T. A. (1993). In situ identification of bacteria in drinking water and adjoining biofilms by hybridization with 16S and 23S rRNA-directed fluorescent oligonucleotide probes. Applied and Environmental Microbiology, 59, 2293–2298.
- Marcus, D. N., Pinto, A., Anantharaman, K., Ruberg, S. A., Kramer, E. L., Raskin, L., & Dick, G. J. (2017). Diverse manganese(II)-oxidizing bacteria are prevalent in drinking water systems. Environmental Microbiology Reports, 9, 120–128. doi:10.1111/1758-2229.12508
- Markowitz, V. M., Chen, I.-M. A., Palaniappan, K., Chu, K., Szeto, E., Grechkin, Y., … Kyrpides, N. C. (2012). IMG: The Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Research, 40(D1), D115–D122. doi:10.1093/nar/gkr1044
- Marks, S. J. (2012). Water System Sustainability in Rural Sub-Saharan Africa: Participation, Sense of Ownership, and Performance: Stanford University.
- Maxam, A. M., & Gilbert, W. (1977). A new method for sequencing DNA. Proceedings of the National Academy of Sciences of the United States of America, 74, 560–564.
- McHardy, A. C., Martin, H. G., Tsirigos, A., Hugenholtz, P., & Rigoutsos, I. (2007). Accurate phylogenetic classification of variable-length DNA fragments. Nature Methods, 4(1), 63–72. doi:10.1038/nmeth976
- Mei, R., Nobu, M. K., Narihiro, T., Kuroda, K., Muñoz Sierra, J., Wu, Z., … Liu, W.-T. (2017). Operation-driven heterogeneity and overlooked feed-associated populations in global anaerobic digester microbiome. Water Research, 124, 77–84. doi:10.1016/j.watres.2017.07.050
- Metzker, M. L. (2010). Sequencing technologies – the next generation. Nat Rev Genet, 11, 31–46.
- Meyer, F., Paarmann, D., D'Souza, M., Olson, R., Glass, E. M., Kubal, M., … Edwards, R. A. (2008). The metagenomics RAST server – A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics, 9, 386.
- Miller, S., Nelson, K., & Rodriguez, R. (2017). Microbiological stability in direct potable reuse systems: Insights from pilot-scale research using flow cytometry and high-throughput sequencing. Proceedings of the Water Environment Federation, 2017, 1016–1023. doi:10.2175/193864717822153779
- Momba, M., Tyafa, Z., Makala, N., Brouckaert, B., & Obi, C. (2006). Safe drinking water still a dream in rural areas of South Africa. Case Study: The Eastern Cape Province. Water SA, 32, 47864.
- Moran, M. A., Satinsky, B., Gifford, S. M., Luo, H., Rivers, A., Chan, L.-K., … Hopkinson, B. M. (2013). Sizing up metatranscriptomics. The ISME Journal, 7, 237–243.
- Morris, R. M., Nunn, B. L., Frazar, C., Goodlett, D. R., Ting, Y. S., & Rocap, G. (2010). Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction. The ISME Journal, 4, 673–685. doi:10.1038/ismej.2010.4
- Muller, E. E. L., Glaab, E., May, P., Vlassis, N., & Wilmes, P. (2013). Condensing the omics fog of microbial communities. Trends in Microbiology, 21, 325–333.
- Namiki, T., Hachiya, T., Tanaka, H., & Sakakibara, Y. (2012). MetaVelvet: An extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Research, 40, e155. doi:10.1093/nar/gks678
- Ng, C., DeMaere, M. Z., Williams, T. J., Lauro, F. M., Raftery, M., Gibson, J. A. E., … Cavicchioli, R. (2010). Metaproteogenomic analysis of a dominant green sulfur bacterium from Ace Lake, Antarctica. The ISME Journal, 4, 1002–1019.
- Nurk, S., Meleshko, D., Korobeynikov, A., & Pevzner, P. (2017). metaSPAdes: A new versatile metagenomic assembler. Genome Research, 27, 824–834.
- Oh, S., Hammes, F., & Liu, W. T. (2018). Metagenomic characterization of biofilter microbial communities in a full-scale drinking water treatment plant. Water Research, 128, 278–285. doi:10.1016/j.watres.2017.10.054
- Palomo, A., Jane Fowler, S., Gulay, A., Rasmussen, S., Sicheritz-Ponten, T., & Smets, B. F. (2016). Metagenomic analysis of rapid gravity sand filter microbial communities suggests novel physiology of Nitrospira spp. The ISME Journal, 10, 2569–2581. doi:10.1038/ismej.2016.63
- Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., & Tyson, G. W. (2015). CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research, 25, 1043–1055.
- Pasculle, A. W., Feeley, J. C., Gibson, R. J., Cordes, L. G., Myerowitz, R. L., Patton, C. M., … Dowling, J. N. (1980). Pittsburgh pneumonia agent: Direct isolation from human lung tissue. The Journal of Infectious Diseases, 141, 727–732.
- Pati, A., Heath, L. S., Kyrpides, N. C., & Ivanova, N. (2011). ClaMS: A Classifier for Metagenomic Sequences. Standards in Genomic Sciences, 5, 248–253. doi:10.4056/sigs.2075298
- Patil, K. R., Roune, L., & McHardy, A. C. (2012). The PhyloPythiaS web server for taxonomic assignment of metagenome sequences. PLoS One, 7, e38581.
- Payment, P., Sartory, D., & Reasoner, D. (2003). The history and use of HPC in drinking-water quality management In J. Bartram (Eds.), Heterotrophic plate counts and drinking-water safety, 20–48). Alliance House, UK: IWA Publishing.
- Peng, Y., Leung, H. C., Yiu, S. M., & Chin, F. Y. (2012). IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics (Oxford, England), 28, 1420–1428.
- Peplies, J., Glockner, F. O., & Amann, R. (2003). Optimization strategies for DNA microarray-based detection of bacteria with 16S rRNA-targeting oligonucleotide probes. Applied and Environmental Microbiology, 69, 1397–1407. doi:10.1128/AEM.69.3.1397-1407.2003
- Peplies, J., Lau, S. C., Pernthaler, J., Amann, R., & Glockner, F. O. (2004). Application and validation of DNA microarrays for the 16S rRNA-based analysis of marine bacterioplankton. Environmental Microbiology, 6, 638–645. doi:10.1111/j.1462-2920.2004.00588.x
- Peter-Varbanets, M., Gujer, W., & Pronk, W. (2012). Intermittent operation of ultra-low pressure ultrafiltration for decentralized drinking water treatment. Water Research, 46, 3272–3282. doi:10.1016/j.watres.2012.03.020
- Peters, R. W. (2011). Water and wastewater engineering: Design principles and practice. Hoboken, NJ: Wiley Online Library.
- Pinto, A. J., Marcus, D. N., Ijaz, U. Z., Santos, Q. M. B. D., Dick, G. J., & Raskin, L. (2016). Metagenomic evidence for the presence of comammox Nitrospira-Like Bacteria in a drinking water system. mSphere, 1, e00054-15.
- Pinto, A. J., Schroeder, J., Lunn, M., Sloan, W., & Raskin, L. (2014). Spatial-temporal survey and occupancy-abundance modeling to predict bacterial community dynamics in the drinking water microbiome. MBio, 5, e01135–e01114.
- Pinto, A. J., Xi, C. W., & Raskin, L. (2012). Bacterial community structure in the drinking water microbiome is governed by filtration processes. Environmental Science & Technology, 46, 8851–8859. doi:10.1021/es302042t
- Poitelon, J. B., Joyeux, M., Welte, B., Duguet, J. P., Prestel, E., & DuBow, M. S. (2010). Variations of bacterial 16S rDNA phylotypes prior to and after chlorination for drinking water production from two surface water treatment plants. Journal of Industrial Microbiology & Biotechnology, 37, 117–128. doi:10.1007/s10295-009-0653-5
- Prest, E. I., Weissbrodt, D. G., Hammes, F., van Loosdrecht, M. C. M., & Vrouwenvelder, J. S. (2016). Long-term bacterial dynamics in a full-scale drinking water distribution system. PLoS One, 11, e0164445.
- Proctor, C. R., Edwards, M. A., & Pruden, A. (2015). Microbial composition of purified waters and implications for regrowth control in municipal water systems. Environmental Science: Water Research & Technology, 1, 882–892. doi:10.1039/C5EW00134J
- Prosser, J. I. (2010). Replicate or lie. Environmental Microbiology, 12, 1806–1810.
- PRT, S. (1996). The safe drinking water act. Public law, 104, 182.
- Ramos, J., & Ramos, H. M. (2009). Solar powered pumps to supply water for rural or isolated zones: A case study. Energy for Sustainable Development, 13, 151–158. doi:10.1016/j.esd.2009.06.006
- Regan, J. M., Harrington, G. W., Baribeau, H., De Leon, R., & Noguera, D. R. (2003). Diversity of nitrifying bacteria in full-scale chloraminated distribution systems. Water Research, 37(1), 197–205. doi:10.1016/S0043-1354(02)00237-3
- Revetta, R. P., Gomez-Alvarez, V., Gerke, T. L., Domingo, J. W., & Ashbolt, N. J. (2016). Changes in bacterial composition of biofilm in a metropolitan drinking water distribution system. Journal of Applied Microbiology, 121(1), 294–305. doi:10.1111/jam.13150
- Ridgway, H. F., & Olson, B. H. (1981). Scanning electron microscope evidence for bacterial colonization of a drinking water distribution system. Applied and Environmental Microbiology, 41, 274–287.
- Risse, J., Thomson, M., Patrick, S., Blakely, G., Koutsovoulos, G., Blaxter, M., & Watson, M. (2015). A single chromosome assembly of Bacteroides fragilis strain BE1 from Illumina and MinION nanopore sequencing data. Gigascience, 4, 60.
- Rittmann, B. E., Hausner, M., Loffler, F., Love, N. G., Muyzer, G., Okabe, S., … Wagner, M. (2006). A vista for microbial ecology and environmental biotechnology. Environmental Science & Technology, 40, 1096–1103. doi:10.1021/es062631k
- Roberts, R. J., Carneiro, M. O., & Schatz, M. C. (2013). The advantages of SMRT sequencing. Genome Biology, 14, 405.
- Roeselers, G., Coolen, J., van der Wielen, P. W. J. J., Jaspers, M. C., Atsma, A., de Graaf, B., & Schuren, F. (2015). Microbial biogeography of drinking water: Patterns in phylogenetic diversity across space and time. Environmental Microbiology, 17, 2505–2514. doi:10.1111/1462-2920.12739
- Roskoski, R. (2007). Enzyme assays xPharm: The comprehensive pharmacology reference (pp. 1–7). New York, NY: Elsevier.
- Ross, M. G., Russ, C., Costello, M., Hollinger, A., Lennon, N. J., Hegarty, R., … Jaffe, D. B. (2013). Characterizing and measuring bias in sequence data. Genome Biology, 14, R51.
- Santic, D., Krstulovic, N., & Solic, M. (2007). Comparison of flow cytometric and epifluorescent counting methods for marine heterotrophic bacteria. Acta Adriatica, 48, 107–114.
- Sauer, J. D., Bachman, M. A., & Swanson, M. S. (2005). The phagosomal transporter A couples threonine acquisition to differentiation and replication of Legionella pneumophila in macrophages. Proceedings of the National Academy of Sciences of the United States of America, 102, 9924–9929. doi:10.1073/pnas.0502767102
- Schloss, P. D. (2010). The effects of alignment quality, distance calculation method, sequence filtering, and region on the analysis of 16S rRNA gene-based studies. PLOS Computational Biology, 6, e1000844.
- Schardinger, F. (1892). Uber das Vorkommen Gahrung erregender Spaltpilze im Trinkwasser und ihre Bedeutung fur die hygienische Beurtheilung desselben. Wiener klinische Wochenschrift, 5, 403–405.
- Schmeisser, C., Stockigt, C., Raasch, C., Wingender, J., Timmis, K. N., Wenderoth, D. F., … Streit, W. R. (2003). Metagenome survey of biofilms in drinking-water networks. Applied and Environmental Microbiology, 69, 7298–7309. doi:10.1128/AEM.69.12.7298-7309.2003
- Segata, N., Bornigen, D., Morgan, X. C., & Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nature Communications, 4, 2304.
- Shade, A., & Handelsman, J. (2012). Beyond the Venn diagram: The hunt for a core microbiome. Environmental Microbiology, 14(1), 4–12.
- Sharpton, T. J. (2014). An introduction to the analysis of shotgun metagenomic data. Frontiers in Plant Science, 5, 209.
- Sinha, R., Abu-Ali, G., Vogtmann, E., Fodor, A. A., Ren, B., Amir, A., … Huttenhower, C. (2017). Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nature Biotechnology, 35, 1077–1086.
- Small, J., Call, D. R., Brockman, F. J., Straub, T. M., & Chandler, D. P. (2001). Direct detection of 16S rRNA in soil extracts by using oligonucleotide microarrays. Applied and Environmental Microbiology, 67, 4708–4716. doi:10.1128/AEM.67.10.4708-4716.2001
- Smith, A. (2005). Protozoa and pathogenic bacteria: Lessons learned from Legionella pneumophila. The Journal of Eukaryotic Microbiology, 52, 27S–28S. doi:10.1111/j.1550-7408.2005.05202003_2_4.x
- Smith, T. (1895). Notes on Bacillus coli commune and related forms, together with some suggestions concerning the bacteriological examination of drinking water. American Journal of Medical Science, 110, 283–302.
- Snoeyink, V., Hass, C., Boulos, P., Burlinghame, G., Camper, A., Clark, R., … Moe, C. (2006). Drinking water distribution systems: Assessing and reducing risks. Washington, DC: National Academics Press.
- Sorek, R., & Cossart, P. (2010). Prokaryotic transcriptomics: A new view on regulation, physiology and pathogenicity. Nature Reviews Genetics, 11(1), 9–16.
- Sowell, S. M., Abraham, P. E., Shah, M., Verberkmoes, N. C., Smith, D. P., Barofsky, D. F., & Giovannoni, S. J. (2011). Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. The ISME Journal, 5, 856–865. doi:10.1038/ismej.2010.168
- Staley, J. T., & Konopka, A. (1985). Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annual Review of Microbiology, 39, 321–346.
- Stark, L., Giersch, T., & Wunschiers, R. (2014). Efficiency of RNA extraction from selected bacteria in the context of biogas production and metatranscriptomics. Anaerobe, 29, 85–90. doi:10.1016/j.anaerobe.2013.09.007
- Steele, H. L., & Streit, W. R. (2005). Metagenomics: Advances in ecology and biotechnology. FEMS Microbiology Letters, 247, 105–111.
- Stein, J. L., Marsh, T. L., Wu, K. Y., Shizuya, H., & DeLong, E. F. (1996). Characterization of uncultivated prokaryotes: isolation and analysis of a 40-kilobase-pair genome fragment from a planktonic marine archaeon. Journal of Bacteriology, 178, 591–599.
- Stewart, P. S., & Franklin, M. J. (2008). Physiological heterogeneity in biofilms. Nature Reviews. Microbiology, 6, 199–210.
- Stutz, W., Leki, G., & Lopez Pila, J. M. (1986). The ATP concentration of drinking water compared to the colony count. Zentralbl Bakteriol Mikrobiol Hyg B, 182, 421–429.
- Sun, H., Shi, B., Bai, Y., & Wang, D. (2014). Bacterial community of biofilms developed under different water supply conditions in a distribution system. Science of the Total Environment, 472, 99–107. doi:10.1016/j.scitotenv.2013.11.017
- Thomas, T., Gilbert, J., & Meyer, F. (2012). Metagenomics – A guide from sampling to data analysis. Microbial Informatics and Experimentation, 2(1), 3.
- Timmins-Schiffman, E., May, D. H., Mikan, M., Riffle, M., Frazar, C., Harvey, H. R., … Nunn, B. L. (2017). Critical decisions in metaproteomics: Achieving high confidence protein annotations in a sea of unknowns. The ISME Journal, 11, 309–314. doi:10.1038/ismej.2016.132
- Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., … Pachter, L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology, 28, 511–515. doi:10.1038/nbt.1621
- Tsementzi, D., Poretsky, R., Rodriguez, L. M., Luo, C. W., & Konstantinidis, K. T. (2014). Evaluation of metatranscriptomic protocols and application to the study of freshwater microbial communities. Environmental Microbiology Reports, 6, 640–655. doi:10.1111/1758-2229.12180
- Twisselmann, B. (2003). The discovery of the germ. London, UK: BMJ Publishing Group Ltd.
- Tyson, G. W., Chapman, J., Hugenholtz, P., Allen, E. E., Ram, R. J., Richardson, P. M., … Banfield, J. F. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature, 428, 37–43. doi:10.1038/nature02340
- Urich, T., Lanzen, A., Qi, J., Huson, D. H., Schleper, C., & Schuster, S. C. (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS One, 3.
- USEPA. (1989a). National Primary Drinking Water Regulations; Filtration, Disinfection; Turbidity, Giardia, Viruses, Legionella, and Heterotrophic Bacteria; Final Rule. Federal Register, 54, 27486.
- USEPA. (1989b). National Primary Drinking Water Regulations; Total Coliforms (Including Fecal Coliforms and E. Coli); Final Rule. Federal Register, 54, 27544.
- USEPA. (1991). Maximum Contaminant Level Goals and National Primary Drinking Water Regulations for Lead and Copper; Final Rule. Federal Register, 56, 26460.
- USEPA. (1998). National Primary Drinking Water Regulations: Disinfectants and Disinfection Byproducts. Federal Register, 63, 69390.
- USEPA. (2000). The history of drinking water treatment. Washington, DC: Office of Water.
- USEPA. (2002). National Primary Drinking Water Regulations: Long Term 1 Enhanced Surface Water Treatment Rule; Final Rule. Federal Register, 67, 1812.
- USEPA. (2006a). National Primary Drinking Water Regulations: Long Term 2 Enhanced Surface Water Treatment Rule; Final Rule. Federal Register, 71, 654.
- USEPA. (2006b). National Primary Drinking Water Regulations: Stage 2 Disinfectants and Disinfection Byproducts Rule. Federal Register, 71, 388.
- USEPA. (2013). National Primary Drinking Water Regulations: Revisions to the Total Coliform Rule; Final Rule. Federal Register, 78, 10269.
- van der Wielen, P. W. J. J., Voost, S., & van der Kooij, D. (2009). Ammonia-oxidizing bacteria and Archaea in groundwater treatment and drinking water distribution systems. Applied and Environmental Microbiology, 75, 4687–4695. doi:10.1128/AEM.00387-09
- van Leeuwenhoek, A. (1800). The Select Works of Antony Van Leeuwenhoek, Containing His Microscopical Discoveries in Many of the Works of Nature. Translated from the Dutch and Latin Editions Published by the Author, by Samuel Hoole: London: Printed by Henry Fry, for the translator, and sold by George Nicol.
- Van Nevel, S., Koetzsch, S., Proctor, C. R., Besmer, M. D., Prest, E. I., Vrouwenvelder, J. S., … Hammes, F. (2017). Flow cytometric bacterial cell counts challenge conventional heterotrophic plate counts for routine microbiological drinking water monitoring. Water Research, 113, 191–206. doi:10.1016/j.watres.2017.01.065
- Vanderkooij, D., Visser, A., & Hijnen, W. A. M. (1982). Determining the concentration of easily assimilable organic carbon in drinking water. Journal - American Water Works Association, 74, 540–545. doi:10.1002/j.1551-8833.1982.tb05000.x
- Vanderwende, E., Characklis, W. G., & Smith, D. B. (1989). Biofilms and Bacterial Drinking-Water Quality. Water Research, 23, 1313–1322. doi:10.1016/0043-1354(89)90193-0
- Vaz-Moreira, I., Egas, C., Nunes, O. C., & Manaia, C. M. (2013). Bacterial diversity from the source to the tap: A comparative study based on 16S rRNA gene-DGGE and culture-dependent methods. FEMS Microbiology Ecology, 83, 361–374. doi:10.1111/1574-6941.12002
- Velten, S., Hammes, F., Boller, M., & Egli, T. (2007). Rapid and direct estimation of active biomass on granular activated carbon through adenosine tri-phosphate (ATP) determination. Water Research, 41, 1973–1983. doi:10.1016/j.watres.2007.01.021
- Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., … Smith, H. O. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science, 304, 66–74. doi:10.1126/science.1093857
- Vetrovsky, T., & Baldrian, P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One, 8, e57923.
- von Gunten, U. (2003). Ozonation of drinking water: Part I. Oxidation kinetics and product formation. Water Research, 37, 1443–1467.
- Wang, H., Bedard, E., Prevost, M., Camper, A. K., Hill, V. R., & Pruden, A. (2017). Methodological approaches for monitoring opportunistic pathogens in premise plumbing: A review. Water Research, 117, 68–86. doi:10.1016/j.watres.2017.03.046
- Wang, H., Edwards, M. A., Falkinham, J. O., & Pruden, A. (2013). Probiotic approach to pathogen control in premise plumbing systems? A review. Environmental Science & Technology, 47, 10117–10128. doi:10.1021/es402455r
- Wang, H., Masters, S., Edwards, M. A., Falkinham, J. O., & Pruden, A. (2014). Effect of disinfectant, water age, and pipe materials on bacterial and eukaryotic community structure in drinking water biofilm. Environmental Science & Technology, 48, 1426–1435. doi:10.1021/es402636u
- Wang, H., Proctor, C. R., Edwards, M. A., Pryor, M., Santo Domingo, J. W., Ryu, H., … Pruden, A. (2014). Microbial community response to chlorine conversion in a chloraminated drinking water distribution system. Environmental Science & Technology, 48, 10624–10633. doi:10.1021/es502646d
- Wang, H.-B., Zhang, Z.-X., Li, H., He, H.-B., Fang, C.-X., Zhang, A.-J., … Lin, W.-X. (2011). Characterization of metaproteomics in crop rhizospheric soil. Journal of Proteome Research, 10, 932–940.
- Wang, H. T., Li, M. M., Brockman, K., & Nguyen, T. H. (2016). Reduction of MS2 bacteriophage and rotavirus in biosand filters. Environmental Science: Water Research & Technology, 2, 483–491. doi:10.1039/C5EW00297D
- Wattam, A. R., Davis, J. J., Assaf, R., Boisvert, S., Brettin, T., Bun, C., … Stevens, R. L. (2017). Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Research, 45(D1), D535–D542. doi:10.1093/nar/gkw1017
- White, C. P., DeBry, R. W., & Lytle, D. A. (2012). Microbial survey of a full-scale, biologically active filter for treatment of drinking water. Applied and Environmental Microbiology, 78, 6390–6394. doi:10.1128/AEM.00308-12
- Williams, M. A., Taylor, E. B., & Mula, H. P. (2010). Metaproteomic characterization of a soil microbial community following carbon amendment. Soil Biology and Biochemistry, 42, 1148–1156. doi:10.1016/j.soilbio.2010.03.021
- Wilmes, P., & Bond, P. L. (2004). The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environmental Microbiology, 6, 911–920. doi:10.1111/j.1462-2920.2004.00687.x
- Wilmes, P., Wexler, M., & Bond, P. L. (2008). Metaproteomics provides functional insight into activated sludge wastewater treatment. PLoS One, 3, e1778.
- Wilson, C. (1945). Bacteriology of water pipes. Journal - American Water Works Association, 37(1), 52–58. doi:10.1002/j.1551-8833.1945.tb17462.x
- Wilson, K. H., Wilson, W. J., Radosevich, J. L., DeSantis, T. Z., Viswanathan, V. S., Kuczmarski, T. A., & Andersen, G. L. (2002). High-density microarray of small-subunit ribosomal DNA probes. Applied and Environmental Microbiology, 68, 2535–2541.
- Wimpenny, J., Manz, W., & Szewzyk, U. (2000). Heterogeneity in biofilms. FEMS Microbiology Reviews, 24, 661–671.
- Woese, C. R., & Fox, G. E. (1977). Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci USA, 74, 5088–5090.
- Wrighton, K. C., Thomas, B. C., Sharon, I., Miller, C. S., Castelle, C. J., VerBerkmoes, N. C., … Banfield, J. F. (2012). Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science, 337, 1661–1665. doi:10.1126/science.1224041
- Wu, J. H., Hong, P. Y., & Liu, W. T. (2009). Quantitative effects of position and type of single mismatch on single base primer extension. Journal of Microbiological Methods, 77, 267–275. doi:10.1016/j.mimet.2009.03.001
- Wu, Y.-W., Simmons, B. A., & Singer, S. W. (2016). MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics, 32, 605–607. doi:10.1093/bioinformatics/btv638
- Wu, Y.-W., Tang, Y.-H., Tringe, S. G., Simmons, B. A., & Singer, S. W. (2014). MaxBin: An automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome, 2, 1–18.
- Xiong, X. J., Frank, D. N., Robertson, C. E., Hung, S. S., Markle, J., Canty, A. J., … Parkinson, J. (2012). Generation and analysis of a mouse intestinal metatranscriptome through Illumina based RNA-sequencing. PLoS One, 7, e36009.
- Yanez, M. A., Nocker, A., Soria-Soria, E., Murtula, R., Martinez, L., & Catalan, V. (2011). Quantification of viable Legionella pneumophila cells using propidium monoazide combined with quantitative PCR. Journal of Microbiological Methods, 85, 124–130. doi:10.1016/j.mimet.2011.02.004
- Yu, K., & Zhang, T. (2012). Metagenomic and metatranscriptomic analysis of microbial community structure and gene expression of activated sludge. PLoS One, 7, e38183.
- Zampieri, E., Chiapello, M., Daghino, S., Bonfante, P., & Mello, A. (2016). Soil metaproteomics reveals an inter-kingdom stress response to the presence of black truffles. Scientific Reports, 6, 25773.
- Zearley, T. L., & Summers, R. S. (2012). Removal of trace organic micropollutants by drinking water biological filters. Environmental Science & Technology, 46, 9412–9419. doi:10.1021/es301428e
- Zeng, D. N., Fan, Z. Y., Chi, L., Wang, X., Qu, W. D., & Quan, Z. X. (2013). Analysis of the bacterial communities associated with different drinking water treatment processes. World Journal of Microbiology and Biotechnology, 29, 1573–1584. doi:10.1007/s11274-013-1321-5
- Zhang, Y., Griffin, A., & Edwards, M. (2008). Nitrification in premise plumbing: Role of phosphate, pH and pipe corrosion. Environmental Science & Technology, 42, 4280–4284. doi:10.1021/es702483d
- Zhang, Y., Kitajima, M., Whittle, A. J., & Liu, W. T. (2017). Benefits of genomic insights and CRISPR-Cas signatures to monitor potential pathogens across drinking water production and distribution systems. Frontiers in Microbiology 8, 2036.
- Zhang, Y., Oh, S., & Liu, W. T. (2017). Impact of drinking water treatment and distribution on the microbiome continuum: An ecological disturbance’s perspective. Environmental Microbiology, 19, 3163–3174. doi:10.1111/1462-2920.13800
- Zhang, Y., & Liu, W. T. (Forthcoming). Application of Molecular Tools to Study Drinking Water Microbiome. Report 4700. Denver, Colo.: The Water Research Foundation.
- Zhou, J. (2003). Microarrays for bacterial detection and microbial community analysis. Current Opinion in Microbiology, 6, 288–294.