
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Advanced osteosarcoma (OS) has low responsiveness to treatment regimens. Methotrexate (MTX), the first-line chemotherapeutic agent for advanced OS, frequently suffers from severe side effects that lead to chemotherapy interruption. To improve the efficacy and reduce the toxic side effects, we constructed a PF@MTX NP nano-delivery system using MTX-ss-γPGA polymer as a drug carrier loaded with PF. The average size of PF@MTX NPs was approximately 171.9 ± 2.13 nm with a zeta potential of approximately -32.91 ± 0.03 mV, uniformly distributed. We found that the nano showed a significant slowdown in MTX and PF release within 48 h and exhibited GSH-responsive pH sensitivity. In addition, the results of cellular uptake assay, toxicity, scratch and AO/EB staining assays showed that the PF@MTX NPs nanoparticles were able to enter the cells and induced 143B cell death and migration. Thus, the PF@MTX NPs nano-delivery system provides an effective strategy for the combined treatment of solid osteosarcoma.
Introduction
Osteosarcoma (OS) is a kind of bone cancer that primarily affects adolescents and young adults. By 2023, it is anticipated that 1000 people in America will be diagnosed with Osteosarcoma [Citation1]. OS is one of the most aggressive primary malignant bone tumours [Citation2]. Recurrent disease has been reported to be observed in 30–50% of the population, and approximately 68% of patients with localised OS survive for 5 years, but unfortunately metastasis or recurrence occurs in 20–30% of patients [Citation2]. Methotrexate (MTX) is a commonly used drug in the clinical treatment of OS that blocks the conversion of dihydrofolate to tetrahydrofolate by inhibiting dihydrofolate reductase, thereby inhibiting the growth of tumour cells [Citation3]. The strong hydrophobicity, nephrotoxicity, low bioavailability and high tumour tissue resistance of high-dose methotrexate (HD-MTX) have limited the clinical use of MTX [Citation4]. A study found that chemotherapy resistance and lung metastasis in OS patients are associated with low expression of a key tumour suppressor gene, phosphatase and tensin homolog gene (PTEN) [Citation5], which antagonises the oncogenic PI3K/AKT/mTOR pathway [Citation6]. PF-04691502 (PF) is a relatively new ATP-competitive PI3K/mTOR dual inhibitor that has inhibitory effects on various cancers [Citation7]. PF can induce apoptosis in osteosarcoma cells by regulating the PI3K/AKT/mTOR/autophagy pathway [Citation8] or by inducing upregulation of PTEN gene [Citation9], PF inhibits the PI3K pathway, leading to the downregulation of its downstream AKT protein. PF can also directly act on the downstream mTORC1 protein, resulting in the phosphorylation of its downstream targets, including 4EBP1, p70S6K, and S6RP [Citation10]. This also demonstrates that PF may become a potential chemotherapy drug for late-stage OS patients. Currently, single-drug therapy has been gradually replaced by combination therapy to reduce the damage of chemotherapy drugs to normal cells and to decrease the resistance of tumour cells to chemotherapy drugs. The most active clinical drug combinations against OS are mainly doxorubicin, cisplatin, and high-dose methotrexate [Citation11–13] in combination with formyltetrahydrofolate and ifosfamide [Citation14], but the ideal combination remains to be determined. Inhibition of the PI3K/mTOR pathway by PF can counteract partial resistance of some OS cells to MTX; thus, the combination therapy of MTX and PF will exert greater potential in anti-OS tumours.
Although the combination can reduce MTX and PF doses and improve the efficacy of MTX and PF alone for OS, the doses required to achieve effectiveness remain high due to the lack of drug targeting. How to efficiently transport both drugs to the tumour tissue is the key to further enhancing their in vivo efficacy by combining them. Nanoparticle drug delivery is more effective compared to conventional drug delivery because it has better biocompatibility, stability and fewer toxic side effects [Citation15]. Nanodrug delivery systems provide new ideas for targeted drug delivery by passively targeting drugs to tumours through enhanced permeability and retention (EPR) effects [Citation16], and the high concentration of reduced GSH in solid tumour tissues also enables targeted drug release from nanoconnected disulphide bonds [Citation17]. Polymers are safe, non-toxic, water soluble and highly biocompatible. Currently, the main preparation methods for nanoparticles as drug delivery platforms are the following: thin-film hydration [Citation18], thin-film dispersion [Citation19], emulsion and volatilisation [Citation20], wet chemistry [Citation21], high-pressure homogenisation [Citation22], and ultrasonic dispersion. Ultrasound-assisted technology is considered to be a fast, environmentally friendly and green synthesis method. Nanomaterials are subjected to localised high temperature, high energy and high-pressure bubble generation in the medium, and the bubbles generated by cavitation cause the nanoparticles to converge to a non-aggregated state, with intermittent pulses more conducive to nanoparticle self-assembly [Citation23]. Therefore, ultrasound-assisted methods are considered as a viable option for nanoparticle synthesis. We grafted the hydrophobic drug MTX onto the disulphide bond-containing cystamine dihydrochloride via a polymer carrier, grafted hydrophilic polyglutamic acid at the other end to form MTX-SS-PGA polymers, and finally encapsulated PF via ultrasound-assisted technology to form PF@MTX NPs, which can improve the water solubility of single-drug MTX and PF as well as avoid metabolic inactivation of the drugs before they can reach the target site. In recent studies in OS, various targets, such as folate receptors [Citation24], vascular endothelial growth factor receptors [Citation25] and integrins [Citation26], have been applied to target OS. The structure of MTX is similar to that of folic acid, which gives MTX a strong affinity for the folate receptor. However, because of the poor water solubility of free MTX, it is mostly metabolised before reaching the tumour tissue. As in , PF@MTX NPs deliver MTX to tumour tissue and release MTX in response to interstitial GSH in tumour tissue, resulting in enrichment of MTX in tumour tissue and an increased anti-OS effect by MTX binding to folate receptors, at which point MTX can be both a targeted tumour biomarker and a therapeutic agent [Citation27]. γ-PGA may also bind to γ-glutamyltransferase [Citation28,Citation29] in tumour cells, increasing the uptake of the drug by tumour cells, thus delivering one or several biochemical drugs to the tumour site in a targeted manner, which may ultimately improve the anticancer efficacy in OS therapy.
Figure 1. Design of the PF@MTX NP nanodelivery system and its process of exerting an anti-OS effect.
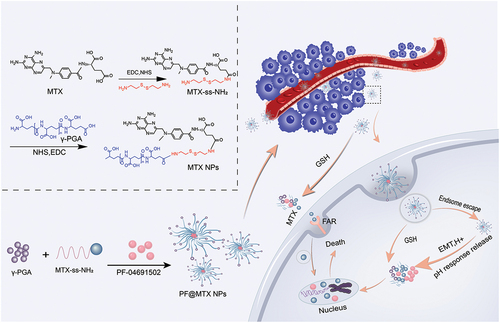
In this study, as in we used a methotrexate-disulphide-polyglutamate polymer designed in preresearch as a drug carrier while forming polymeric nanoparticles using ultrasound-assisted self-assembly of loaded PF inhibitors. We measured the synthesis, characterisation, nanoparticle morphology, drug loading and drug release of the nanocarriers. In addition, we investigated the cellular uptake, tumour cell viability, metastasis inhibition and apoptosis of the nano by OS cells.
Materials and methods
Materials
Methotrexate hydrate (MTX, Purity greater than 98%) carbodiimide hydrochloride (EDCI) and N-hydroxysuccinimide (NHS) from Shanghai Maclean Biochemical Technology Co; Cystamine dihydrochloride (Purity more than 97%) from Triciai Shanghai Chemical Industry Development Co; Dimethyl sulphoxide (DMSO), hydrochloric acid (HCl) from the national drug group; Polyglutamic acid (γ-PGA, Mw~10000), reduced glutathione (GSH), PF-04691502 inhibitor (PF), from Aladdin (Shanghai, China); Cell Counting Kit (CCK-8) from Biosys; High sugar medium (DMEM), trypsin, double antibody, serum, PBS from gibco (USA); DAPI staining solution, CY5 staining solution from Beyotime Biotechnology Co; 4% paraformaldehyde and crystalline violet staining solution from Biosharp; AO/EB staining solution from Maokang Bio; The serum-free cell lyophilisation solution is from New Saimei Biotechnology Co; The 143B cell line were purchased from Procell (Wuhan, China); The HSF cell were presented by Mr. Wang Guowei (Central South University, China). Ultrasound from Ningbo Scientz Biotechnology Co, China (Power 100W); DLS from Malvern Dynamic Light Scattering Instrument; TEM from FEI F20 model, USA.
Cell culture
Cells were cultured in DMEM with high sugar medium with 10% foetal bovine serum and 1% penicillin‒streptomycin solution. Cells were cultured in a CO2 incubator at 37°C containing 5% CO2 and passaged with trypsin containing 0.25% EDTA.
Cell cytotoxicity
143B cells were evenly spread inside a 96-well plate (3000 cells per well) and dispersed in complete medium with different concentrations of MTX and PF by gradient dilution, and the cells were treated with fresh complete medium containing 10% CCK-8 reagent after 48 h of drug administration. The absorbance was detected at 450 nm with an enzyme marker after 1 h to calculate the IC50 value of a single drug. Four concentrations of each of the dual drugs were set near the IC50, and mixed incubations were performed to de-evaluate the synergistic effect of the combined drugs. A similar method was used to evaluate the toxic effects of the nano group, each free group and the combined group on 143B cells at the same drug concentration. The cell survival rates were calculated as follows:
Ten percent CCK-8 medium was added to the wells without cells, called the blank group, while the same CCK-8 medium was added to the wells without drug treatment, called the control group. To explore the synergistic efficacy of MTX and PF in 143B cell therapy, the synergy score of the two drugs was calculated using synergyfinder 3.0 with reference to our previous study.
Synthesis of nanomaterials
Briefly, for the synthesis of disulphide-bonded grafted MTX and γ-PGA with reference to previous studies, 0.2 g MTX was dissolved with 0.112 g NHS and 0.184 g EDC in 15 mL DMSO and activated for 2 h at 40°C with closed light. Cystamine dihydrochloride (0.218 g) was added and reacted for 24 h under a nitrogen atmosphere and protected from light. Then, the above solution was dialysed in deionised water (3000 Da) for 24 h and further freeze-dried after dialysis to obtain MTX-ss-NH2.
Dissolve 25.2 mg of γ-PGA in 15 mL of ultrapure water, adjust the pH to 4.5 with HCl, add 18 mg of NHS, 27 mg of EDC, activate for 2 h at 40°C, then add 41.1 mg of MTX-ss-NH2 under the protection of nitrogen, react for 24 h under protection from light, dialyse, and lyophilise to obtain MTX-ss-PGA polymers (MTX NPs).
Detection of nanomaterials
Small amounts of MTX, s-s, γ-PGA, MTX-ss-NH2, and MTX-ss-PGA were mixed well with potassium bromide solids, dried with water, and ground, and then the well-mixed powder was pressed and scanned on an FTIR instrument at 400–4000 cm−1 to obtain FTIR spectra. Ten milligrams of γ-PGA was dissolved in 1 mL of deuterated D2O, and 10 mg of MTX, s-s, MTX-ss-NH2, and MTX-ss-PGA were dissolved in 1 mL of deuterated DMSO, Sample concentrations were all 10 mg/mL, loaded into NMR sample tubes and scanned at 0–15 ppm absorption in1H NMR spectra.
Preparation and characterization of nanoparticles
The nanoparticles were prepared by an ultrasonic self-assembly method. Ten milligrams of MTX-ss-PGA was dissolved in 10 mL of DMSO, concentration of 1 mg/mL, treated with ultrasonic waves for 40 min, and dialysed (8000–14000 Da) for 12 h, and the water was changed every 2 h to obtain MTX-ss-PGA nanoparticles (MTX NPs). PF@MTX-ss-PGA nanoparticles (PF@MTX NPs) were prepared using a concentration ratio (MTX:PF concentration ratio of 1:5) with a strong synergistic effect obtained from cytotoxicity experiments. The method was similar to the preparation of MTX NPs, where PF was dissolved and slowly added dropwise to MTX NP solution dissolved in DMSO, and the nanoparticles PF@MTX NPs were synthesised by an ultrasound-assisted self-assembly method. The hydrated particle size and zeta potential of the two nanoparticles were measured by dynamic light scattering. After dropping the two nanoparticles on a copper grid, they were left to stand and air-dried, and the morphology of the nanoparticles was dephotographed by transmission electron microscopy (TEM).
Drug loading and drug release in vitro
MTX and PF were dissolved in DMSO, and the UV spectra were scanned at 200–600 cm−1. MTX had a maximum absorption wavelength at 303 nm, PF had a maximum absorption wavelength at 346 nm, and MTX did not affect the absorption of PF at 346 nm. After dissolving MTX and PF in DMSO, standard curves were prepared by diluting MTX with water at 0, 10, 20, 30, 40 and 50 µg/mL and PF at 0, 40, 80, 90, 100 and 110 µg/mL and measuring the absorbance at 303 nm and 346 nm. To determine the drug loading and encapsulation rate of PF@MTX NPs, the prepared nanoparticles, diluted to a certain concentration so that the absorbance was between 0.3 and 0.7, were used to calculate the drug loading and encapsulation rate. The calculation equations are as follows:
To measure the release rate of PF@MTX NPs, 5 mL of the above dialysate was packed into a 3000 Da dialysis bag, dialysed in 20 mL of PBS (pH 5.5 and containing 10 mM GSH solution and pH 7.4) buffer to simulate the reducing environment within the tumour and the normal physiological environment(The HCl was used to obtain a pH of 5.5), placed on a shaker (100 rpm, 37 °C), and taken at different time points from the release solution 3 mL and supplemented with 3 mL of fresh dialysis solution. The UV absorption at 303 nm and 346 nm was measured, and the amount released was calculated.
where and
represent the intensity at t = 0 and at a specific sampling time, respectively, and
represents the maximum intensity at all sampling points.
Cytotoxicity and colony formation
143B cells were added to 96-well plates at 3000 per well, and after 12 h of cell wall growth, MTX and PF were added at drug concentrations of 0.1 and 0.5 µg/mL, respectively, and various designated equivalent MTX and PF concentrations in the two-drug combination group, MTX NPs and PF@MTXNPs groups, and the cells were treated for 48 h, followed by the addition of fresh CCK-8 reagent containing 10% CCK-8 medium, treated for 1 h, and the absorbance was detected at 450 nm on an enzyme marker.
143B cells were added to 6-well plates at a density of 300 cells per well, and then the cells were treated with MTX NPs and PF@MTXNPs for 48 h, followed by incubation with fresh medium until Day 10. Cells were fixed with 4% paraformaldehyde and then stained with 0.1% crystal violet.
In vitro cell uptake
143B cells were inoculated in 6-well plates (1 × 105 cells), and when the cells grew to 60% confluence, PF@MTX NPs loaded with the fluorescent agent CY5 (concentration of IC50 value) were added at different times (2, 4 and 6 h). After treatment, the cells were fixed with 4% paraformaldehyde for 5–15 min, washed with PBS, stained with DAPI (1 µg/mL) for 10 min, photographed with an inverted fluorescence microscope (at 200× magnification) and then processed with ImageJ software for fluorescence intensity to calculate cell uptake.
Cell migration
The 143B cells were spread in 6-well plates at 1 × 105 cells per well, and when the cells were cultured to 80% cell density, after adding different concentrations (IC50 values of concentration) of MTX, PF, MTX+PF, MTX NPs and PF@MTX NPs, the cells were photographed at 0, 12 and 24 hours (at 40× magnification), and the area was analysed using ImageJ software to calculate the migration rate.
Apoptosis experiments
The 143B cells were inoculated in 6-well plates at 1 × 105 cells per well, and after 12 h of cell wall growth, the cells were treated with equivalent concentrations (concentration of IC50 values) of MTX, PF, MTX+PF, MTX NPs and PF@MTX NPs for 24 h. The cells were fixed with 4% paraformaldehyde for 15–20 min, washed with PBS and stained with A0/EB kit, A0/EB/Dilution buffer = 1:1:8 for 5–15 min, rinsed with PBS and photographed with inverted fluorescence microscopy (at 100× magnification) for apoptosis analysis.
Statistical analysis
All groups included three parallel datasets, expressed as MEAN ± SD, and differences between groups were tested by t test using GRAPHPAD 7.0. Data were plotted by ORIGIN 2022 and GRAPHPAD 7.0. Differences between the two groups were considered significant at *p < 0.05 and **p < 0.01and highly significant at ***p < 0.001 and ****p < 0.0001.
Results
Cytotoxicity and synergistic efficacy of free MTX and PF-04691502 (PF)
First, we investigated the in vitro antitumor effects of the two free drugs. As shown in , the CCK-8 Kit was used to detect cell viability under different concentrations of treatment, and the results showed that 143B cell viability gradually decreased with increasing drug concentration. The results are shown in , the IC50 of MTX (0.5227 ± 0.1141 μg/mL) was calculated to be smaller than that of PF (1.835 ± 0.4186 μg/mL), and the synergistic effect of free MTX with PF was evaluated by treating different concentration combinations of the two drugs. The synergy score was calculated using SynergyFinder 3.0. The cell survival rate decreased to 48.21% for MTX and PF concentrations of 0.1 μg/mL and 2 μg/mL, respectively, while the cell survival rate was 35.96% for 0.4 μg/mL MTX and 2 μg/mL PF treatment. As shown in , MTX and PF had a certain synergistic effect with a synergistic score of 2.993. Meanwhile, MTX and PF at a 1:5 concentration ratio had a more significant synergistic effect, which provided an effective ratio for the subsequent experiments.
Figure 2. Toxicity of MTX (a), PF (b) and both drugs (c) in 143B cells. (d) Combination synergy score analysis using SynergyFinder 3.0. n = 3 independent experiments; *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 represent significant differences.
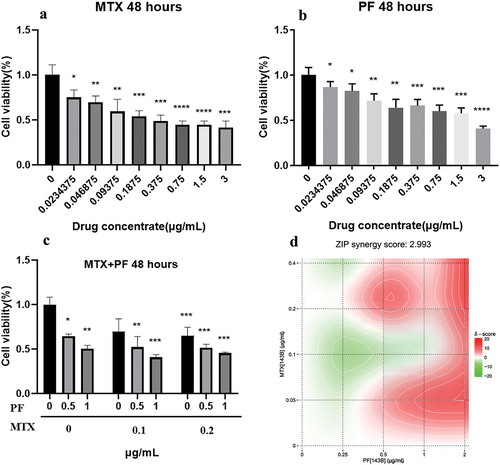
Table 1. IC50 of MTX and PF.
Material synthesis and characterization
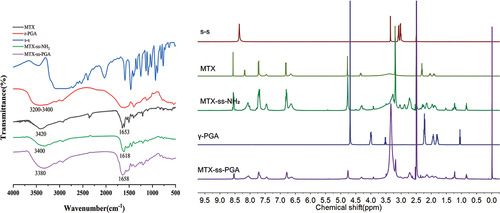
To measure the synthesised nanomaterials, nuclear magnetic resonance (1H NMR) spectroscopy and Fourier transform infrared (FTIR) spectroscopy were used to detect the products in each step. As shown in , FTIR results of MTX-linked cystamine dihydrochloride (MTX-ss-NH2) showed that the carbonyl group of the amide of MTX was at 1653 cm−1, and due to the resonance effect, MTX-ss-NH2 moved towards the lower wavenumber and had an absorption peak at 1618 cm−1. However, MTX-ss-PGA shifted towards a high wavenumber due to the great spatial potential resistance amide with an absorption peak at 1658 cm−1, indicating the successful modification of the γ-PGA moiety. The -OH absorption peak of MTX was at 3420 cm−1, and the hydroxyl absorption peak of γ-PGA had a large conjoined peak at 3200 cm−1 to 3400 cm−1, while the -OH absorption peak of MTX-ss-PGA was at 3380 cm−1, which was less obvious on FTIR, which might be due to the use of γ-PGA with molecular weights of 8000–14000. Meanwhile, as shown in , in the1H NMR spectrum of MTX-ss-NH2, an 8.09 ppm amide shift of MTX with cystamine dihydrochloride appeared, which further proved the successful synthesis of MTX with cystamine dihydrochloride. The chemical shift of the amide bond in MTX is at 8.20 ppm, and in MTX-ss-NH2, the chemical shift of the amide bond is at 8.09 ppm because of the existence of the electron absorption effect of the disulphide bond, which makes the chemical shift move to the low field. In the1H NMR of MTX-ss-PGA, the hydrogen shift of the amide bond was 8.06 ppm due to the presence of hydrogen bonding in the polymer γ-PGA, which caused the chemical shift to move to the low field, and the above data indicated that the nanomaterials were synthesised successfully.
Figure 3. Synthesis of materials, FTIR spectra of MTX, γ-PGA, s-s, MTX-ss-NH2, MTX-ss-PGA on the left and 1 H NMR of MTX, γ-PGA, s-s, MTX-ss-NH2, MTX-ss-PGA on the right.
Characterisation of nanoparticles
Referring to the synergistic effect of the two free drugs in Section 2.1, we chose a ratio of MTX and PF concentrations of 1:5 to prepare nanoparticles and measured the hydrodynamic size and morphology of these nanoparticles by DLS and TEM. Sample concentration of 1 mg/mL for both DLS and TEM measurements. As shown in and , the particle size of MTX NPs was 144.95 ± 1.51 nm, and the PDI was 0.14 ± 0.03, while the particle size of PF-loaded nanoparticles (PF@MTX NPs) was 171.9 ± 2.13 nm, and the PDI was 0.13 ± 0.03. The increased size of drug-loaded nanoparticles compared to blank nanoparticles was also confirmed by TEM. The particle morphology of MTX NPs and PF@MTX NPs observed by TEM has a regular spherical shape with uniform dispersion, and the particle size is consistent with the DLS results. Meanwhile, the zeta potentials of both nanoparticles were as expected; the zeta potential of MTX NPs was -37.05 ± 0.04 mV, while the zeta potential of PF@MTX NPs was -32.91 ± 0.03 mV, which may be due to the amino group on the surface of PF, which carries a positive charge, and the above data indicated that the nanoparticles were prepared successfully.
Figure 4. The particle sizes of MTX NPs (a) and PF@MTX NPs (c) and zeta potential MTX NPs (b) and PF@MTX NPs (d). TEM of MTX NPs (e) and PF@MTX NPs (f). n = 3 independent experiments.
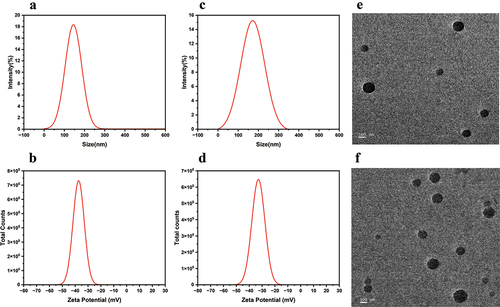
Table 2. Size and zeta of MTX NPs and PF@MTX NPs.
In vitro drug release
We calculated the drug loading efficiency of PF@MTX NPs by a UV measurement standard curve, and the results showed that the drug loadings of MTX and PF were 1.01 ± 0.0002% and 28.15 ± 0.0032%, respectively. We further performed in vitro release assays to detect drug release by using 10 mM GSH at pH 5.5 to simulate the tumour microenvironment (TME) and pH 7.4 to simulate the physiological environment in vivo. As shown in , both free MTX and PF showed high release rates at 8 h. The release rates of MTX were 80.22 ± 3.74% and 77.18 ± 1.89%, while the release rates of PF were 87.08 ± 1.64% and 84.98 ± 5.19%. In both cases, we observed rapid drug release, and drug release under tumour microenvironment conditions compared to the physiological environment during release effects were similar. For PF@MTX NPs, the total release rate of MTX was 74 ± 4.02% and 55.20 ± 0.85% over 48 h in the simulated tumour microenvironment and normal physiological environment, respectively, while the total release rate of PF was 71.17 ± 0.57% and 51.32 ± 4.46% over 48 h. We observed that the drug release rate in the simulated tumour environment was much higher than that in the normal environment, while the release rate of nanoparticles in the normal pH 7.4 state was lower than that in the free state, indicating that under physiological conditions, nanoparticles can effectively delay drug release, while in the tumour microenvironment, the release rate of nanoparticles increased significantly.
Figure 5. MTX (left) and PF (right) release of PF@MTX NPs in physiological conditions (pH 7.4) or simulated tumour endosomal microenvironment (pH 5.5, 10 mM GSH) during 48 hours. n = 3 independent experiments.
Cell cytotoxicity and colony formation
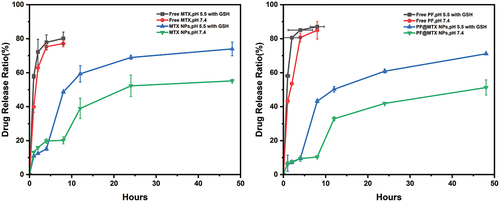
The CCK-8 kit was used to detect the cell viability of the equivalent nanodrug and free drug on 143B cells in vitro for 48 h. As shown in , even at very high MTX (3 µg/mL) concentrations, the cytostatic rate of pure MTX drug was only approximately 58.26 ± 7.04%, and the combination of PF and MTX exhibited significant cytotoxicity with a cytostatic rate of 64.86 ± 8.62% compared to the double free drug. in the inhibition of 143B cells by PF@MTX NPs at equivalent MTX and PF concentrations was 78.25 ± 2.04%. This observation suggests that nanoparticles PF@MTX NPs loaded with MTX and PF dual drugs were more effective in inhibiting the growth of 143B cells compared to MTX NPs. To detect the cytotoxicity of nanoparticles on 143B cells, we also verified it by colony formation assay, and the results are shown in are consistent with the results of the cytotoxicity assay. In the control group with a good colony formation rate of 97.2 ± 1.54% cell status, the colony formation rate in the two-drug combination group was 37.14 ± 1.83%, which was less than the 66.97 ± 3.3% and 52.75 ± 1.05% in the free MTX and PF groups. In contrast, the colony formation rate in the PF@MTX NP group was only 15.62 ± 2.57% at the equivalent administration concentration, which was significantly stronger than the free drug in inhibiting colony formation. The results were consistent with the results of the 143B cytotoxicity assay. The cytotoxicity of free MTX and free PF on fibroblast HSFs is high, as also evidenced in . This is the reason why chemotherapeutic drugs are so injurious to normal tissues. in , at the equivalent administration concentration, the toxicity of MTX NPs and PF@MTX NPs to normal fibroblasts was significantly reduced, indicating that the nanoparticles synthesised in this experiment can reduce the damage of chemotherapeutic drugs to normal tissues while enhancing the toxicity of chemotherapeutic drugs to tumours; therefore, this delivery system can be used as a safe delivery system for chemotherapeutic drugs.
Figure 6. (a) and (d) shows the toxicity of various drugs on 143B cells; (b) shows the colony formation of 143B cells; (c) shows the colony formation rate of each group; (e)MTX NPs and PF@MTX NPs, (f) free MTX and free PF show the cytotoxicity of each group of drugs on HSF fibroblasts. n = 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 indicate significant differences.
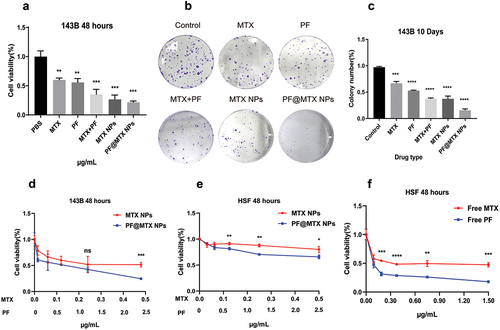
In vitro cellular uptake
To illustrate the relationship between cellular uptake and cytotoxicity, we took fluorescence photographs of the cellular uptake of PF@MTX NPs. We prepared PF@MTX NPs containing CY5 and observed the uptake of CY5/PF@MTX NPs by 143B cells at different time periods using inverted fluorescence microscopy. As shown in , CY5-labelled PF@MTX NPs show red fluorescence, and DAPI staining shows blue fluorescence, with the overlap of the two representing cellular uptake. Analysis of the results shown by inverted fluorescence microscopy showed that the tumour cells had red fluorescence after drug treatment, which implies that PF@MTX NPs have the ability to enter the cells. In addition, the fluorescence intensity of the tumour cells became stronger with increasing time (2–6 h), and cellular uptake was significantly enhanced. The above data indicate that grafted γ-PGA-ss significantly increases the cellular uptake of MTX and PF and maintains the toxic concentrations of MTX and PF in tumour cells, suggesting the potential application of this nanoparticle in tumour therapy.
Figure 7. Cellular uptake of the nanodrug PF@MTX NPs at different times with a CY5/PF@MTX NP concentration of 10 µg/mL and DAPI concentration of 1 µg/mL. n = 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 represent significant differences.
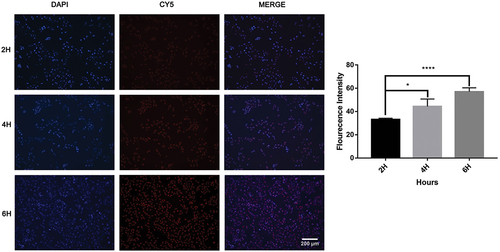
Cell migration
To characterise the inhibitory effect of synergistic drugs and nanoparticles on osteosarcoma, we performed cell migration assays. The migration ability of 143B osteosarcoma cells treated with free MTX, free PF, MTX and PF double combination, MTX NPs and PF@MTX NPs was evaluated at the same equivalent corresponding dosing concentrations. The experimental results are showed in that the control group migrated significantly at 12 h and 24 h in the presence of obvious scratches, with cell migration rates of approximately 35 ± 0.38% and 70.9 ± 7.5%, respectively. The cell migration rate of free MTX was 20.8 ± 1.3% and 45.6 ± 8.3% at 12 h and 24 h, respectively, and the effect of inhibition of migration was weaker than that of 8.8 ± 2.2% and 24.9 ± 3.9% at 12 h and 24 h in the combined drug group. The total migration rate in the combination group at 24 h was 24.9 ± 3.9% less than that in the PF@MTX NPs group, indicating that the inhibitory effect in the combination group was less than that in the PF@MTX NPs group. Meanwhile, there was almost no metastasis in the nano and single-drug groups compared with the control group at 24 h (the cell migration rate was 8.7 ± 1.6% at 24 h in the PF@MTX NPs group), and the MTX and PF dual drug combination group at the same dose was more effective in inhibiting migration than the free MTX and free PF groups. In conclusion, we provide an actively targeted nanoparticle for the treatment of osteosarcoma by inhibiting the metastasis of 143B cells.
Figure 8. 143B cells scratching experiment after free MTX, free PF, dual drug combination group and MTX NPs, PF@MTX NPs treatment. n = 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Significantly different.
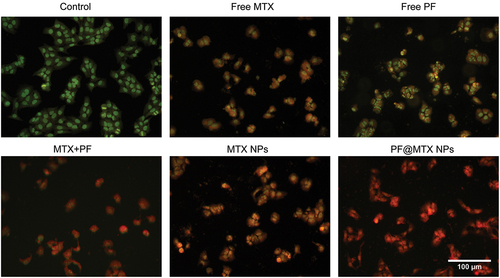
Cell apoptosis
In this experiment, AO/EB double fluorescence staining was used to observe cell colour and nuclear morphology to distinguish normal cells, early apoptotic cells, late apoptotic cells and necrotic cells. In this experiment, drug concentrations corresponding to IC50 values were chosen for all dosing groups. The AO/EB results of the control group showed in that the cells showed uniform green fluorescence, and their nuclear morphology was normal; in the free MTX group, the cells showed yellow fluorescence and normal nuclear morphology, and most of them showed early apoptosis. In the free PF inhibitor group, the nucleus showed green fluorescence and the surrounding cytoplasm showed light yellow fluorescence, with normal nuclear morphology, mainly in the early apoptotic state; the cells in the double drug combination group showed orange‒red fluorescence, with a higher degree of apoptosis compared with the free group, belonging to late apoptotic state, with minor changes in nuclear morphology; MTX NPs group belonging to the process of transition from early to late apoptotic cells; The deformation of cell nuclear volume visible to the naked eye and orange‒red fluorescence in the PF@MTX NPs groups, but the higher apoptotic effect in the PF@MTX NPs group, all indicate that the PF@MTX NPs synthesised in this experiment have a superior potential to induce apoptosis in 143B cells than the free drug, which are attributed to the combination of the dual drug and the cellular uptake of the nanocarrier. Furthermore, these results could also indicate that PF@MTX NPs can significantly reduce cell viability, partly due to the induction of 143B cell death.
Figure 9. AO/EB fluorescence staining results. n = 3 independent experiments.
Discussion
OS is a particularly chemoresistant tumour that responds only to high-dose chemotherapy but rapidly acquires drug resistance, which is commonly caused by changes in drug metabolism or drug targets [Citation30]. The PI3K/mTOR pathway is relatively upstream in cells and regulates many cellular functions, including protein synthesis, apoptosis, angiogenesis and drug resistance, but notably, it is one of the most variable pathways in tumours [Citation31]. There is evidence that downregulation of clathrin heavy-chain gene expression inhibits OS cell proliferation by activating the TGF-β and AKT/mTOR signalling pathways with the tumour-associated protein TFG [Citation32]. The activity of 131 major kinase inhibitors was also evaluated by screening a chemical library of kinase inhibitors, and dual PI3K/mTOR inhibitors were found to have strong anti-OS activity. Patients with primary human OS also showed sensitivity to dual PI3K/mTOR inhibitors [Citation33], all of which suggest that inhibition of the PI3K/mTOR pathway may offer new hope for patients with OS. Combination chemotherapy can enhance chemotherapy exposure by circumventing the dose-limiting toxicity of a single drug, which can avoid the problem of resistance to a single chemotherapeutic agent and thus improve antitumor efficacy; thus, it is clear that monotherapy will be replaced by multidrug synergistic therapy [Citation34]. Multidrug synergistic therapy is necessary to improve targeting to reduce side effects because a lack of effective tumour targeting can lead to serious side effects, which can be achieved with nanodrug delivery systems [Citation35]. The simple preparation of carrier-free nanodrugs and relatively high reticuloendothelial system clearance rates limit their practical application. Lipid nanomedicines have dominated the field of drug delivery, but the widespread application of lipid nanodrugs is still limited due to the self-assembly principle on which they are synthesised [Citation36]. Polymeric carriers are safe, non-toxic, water soluble and biocompatible. The nanodelivery system designed in this experiment helps to increase the tumour targeting ability and is a highly efficient and low toxicity nanotherapeutic approach. In this study, the dual drug-carrying nano-delivery system passively targets the tumour tissue through the EPR effect, which is oxidized by the high concentration of reduced GSH at the tumour tissue site to break the disulphide bond of MTX-ss-PGA, locating the lesion to release the drug, and realising a more sensitive tumour microenvironmental drug-enrichment function. It can improve the solubility of free MTX, reduce the dosage of chemotherapeutic drug MTX, reduce the toxic side effects on normal tissues, and achieve the synergistic delivery of dual-drugs, so as to better exert the anti-tumour efficacy.
In this experiment, grafting was performed by reacting the amino group of cystamine dihydrochloride with the carboxyl group of MTX and the carboxyl group of γ-PGA to form amide bonds, which are usually stable for several days at low temperatures in aqueous solution, and we observed that the lyophilised nanomaterials were stable for several months. In the reaction system, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDCI), N-hydroxy succinimide (NHS), and urea (EDCI-catalysed side reaction) were soluble in water, and these impurities were removed by dialysis after the synthesis. In the synthesis of MTX-ss-PGA nanoparticles, we chose different feeding ratios, and there are many feeding ratios with very large hydrodynamic sizes. Finally, we chose the size under the optimal particle size for the subsequent experiments of encapsulated PF. We demonstrated the successful synthesis of MTX-ss-PGA by Fourier transform infrared spectroscopy (FTIR) and nuclear magnetic resonance hydrogen spectroscopy (1H NMR), and the regular spherical shape of nanoparticles was observed by transmission electron microscopy. The drug loading of MTX was calculated by UV absorption, both of which showed that MTX-ss-PGA polymers were successfully synthesised.
To maximise the efficacy of the MTX and PF combination, we first performed in vitro cellular experiments with free dual-drug synergy. The results showed that the best anti-OS effect was achieved at a ratio of 1:5 MTX:PF, so we chose this ratio of PF:MTX-ss-PGA for the preparation of PF@MTX NPs. The prepared nanoparticles PF@MTX NPs (171.9 ± 2.13) were larger than MTX NPs (144.95 ± 1.51) due to the encapsulation of drug PF. Additionally, because γ-PGA has many carboxyl groups for anionic polymers, the surface of MTX NPs is negatively charged, while PF is positively charged because of its amino group in the structure, so PF@MTX NPs (-32.91 ± 0.03 mV) is greater than MTX NPs (-37.05 ± 0.04 mV). In addition, we observed a significant increase in the total release of PF at pH 5.5 and GSH 10 mM, reaching 71.17 ± 0.57% in nanoparticles at pH 5.5 compared to the cumulative release rate at pH 7.4 (51.32 ± 4.46%). A large number of carbonyl and carboxyl groups on the surface of γ-PGA, with a single molecule having an amino group, are more easily ionised and become more hydrophilic in a PBS environment (pH 7.4). For the -NH2 on the MTX structure, under the condition of PBS 5.5, MTX is more likely to accept protons, and the hydrophilicity increases. The outermost layer of the structure of MTX NPs is hydrophilic γ-PGA in the outer layer of nanoparticles, so at pH 7.4, the ionization of the group on γ-PGA is more hydrophilic, and the cumulative release will be higher than that at pH 5.5. In contrast, the structure of MTX NP nanoparticles is MTX-ss-PGA, and in the presence of reduced GSH, the nanoparticle structure suffers damage from the binding of glutathione to compounds containing disulphide bonds due to the presence of oxidative stress. When the release of γ-PGA reaches a certain amount, the disulphide bond is exposed, and GSH10 mM binds to the disulphide bond, after which the release of MTX will be accelerated, while the release of MTX from PF@MTX NPs, a nanoparticle encapsulated with PF, will be more easily ionised at pH 7.4 due to the hydroxyl group on PF, so the release of MTX from PF@MTX NPs was similar at pH 7.4 and pH 5.5 in the early stage. The cumulative release rates in the drug release profiles of MTX and PF were as expected.
Next, we tested the antitumor effect of our nanoparticles in vitro. The results showed that γ-PGA-ss grafting did not significantly reduce the efficacy of MTX, and the efficacy of PF@MTX NP treatment was superior to that of the free dual drug. In the cell colony formation assay, the colony formation rate in the PF@MTX NP group was significantly smaller than that in the control group. Further scratch experiments demonstrated that the two-drug synergistic treatment with our synthetic nanoparticles almost completely inhibited the metastasis of OS cells, and the free two-drug combination also inhibited the metastasis of OS cells to some extent, but continuous treatment might increase the drug resistance of OS cells and lead to the metastasis or recurrence of OS cells. To further investigate the uptake of nanoparticles in OS cells, we performed cellular uptake experiments, and the results showed that more PF@MTX NPs entered the cells with increasing time, indicating that our prepared nanoparticles achieved effective 143B cell targeting. Finally, AO/EB staining experiments also illustrated that the PF@MTX NPs synthesised in this experiment had a superior potential to induce 143B cell death than the free drug, which was attributed to the combination of the dual drug and the cellular uptake of the nanocarrier. Furthermore, these results could also indicate that PF@MTX NPs can significantly reduce cell viability, partly due to the induction of 143B cell death. Thus, the present work may provide a new direction of choice for the clinical treatment of OS patients.
Conclusions
In the present study, we prepared novel drug-loaded nanoparticles PF@MTX NPs by the optimal dosing concentration ratio MTX:PF = 1:5 derived from cellular synergistic toxicity experiments, which improved the aqueous solubility, stability and anti-OS effect of MTX and PF and demonstrated the credibility of novel PF@MTX NPs based on dual-drug combination and glutathione response for the treatment of OS. PF@MTX NPs have a small size and negatively charged surface, and the release of the drug exhibits GSH-responsive pH sensitivity. PF@MTX NPs are very potent in inhibiting proliferation and metastasis and inducing death in 143B cells in vitro, and thus, this nanodelivery system may represent an emerging therapeutic strategy for osteosarcoma treatment.
Author contributions
Performed experiments, acquired data, writing and revised the paper, M.L.W.; collected experimental samples, L.W.L. and W.Z.; performed statistical analysis, M.L.W. and Z.D.L., drafted the manuscript Z.H.F., S.Y.F., Y.J.H. and X.J.T., funding acquisition, Y.J.H, C.l.H All authors had approved the final manuscript.
List of abbreviation
| MTX | = | Methotrexate |
| PF-14691502 | = | PF |
| OS | = | Osteosarcoma |
| s-s | = | Cystamine dihydrochloride |
| γ-PGA | = | Polyglutamic acid |
| MTX-ss-NH2 | = | Methotrexate grafted cystamine dihydrochloride |
| MTX-ss-PGA | = | Methotrexate grafted cystamine dihydrochloride- polyglutamic acid |
| MTX NPs | = | MTX-ss-PGA nanomicelle |
| PF@MTX NPs | = | PF loaded MTX-ss-PGA nanomicelle |
| FAR | = | Folate receptor |
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors declare that all data supporting the findings of this study are available within the paper.
Additional information
Funding
References
- Shabib Akhtar M, Chandrasekaran K, Saminathan S, et al. Nanoengineered chitosan functionalized titanium dioxide biohybrids for bacterial infections and cancer therapy. Sci Rep. 2024;14(1):3705. doi: 10.1038/s41598-024-52847-1
- Chen C, Xie L, Ren T, et al. Immunotherapy for osteosarcoma: fundamental mechanism, rationale, and recent breakthroughs. Cancer Lett. 2021;500:1–14. doi: 10.1016/j.canlet.2020.12.024
- Kozminski P, Halik PK, Chesori R, et al. Overview of dual-acting drug methotrexate in different neurological diseases, autoimmune pathologies and cancers. Int J Mol Sci. 2020;21(10):3483. doi: 10.3390/ijms21103483
- Zhang W, Zhang Q, Zheng TT, et al. Delayed high-dose methotrexate excretion and influencing factors in osteosarcoma patients. Chin Med J (Engl). 2016;129(21):2530–2534. doi: 10.4103/0366-6999.192781
- Tian Z, Guo B, Yu M, et al. Upregulation of micro-ribonucleic acid-128 cooperating with downregulation of PTEN confers metastatic potential and unfavorable prognosis in patients with primary osteosarcoma. Onco Targets Ther. 2014;7:1601–1608. doi: 10.2147/OTT.S67217
- Zheng C, Tang F, Min L, et al. PTEN in osteosarcoma: recent advances and the therapeutic potential. Biochim Biophys Acta Rev Cancer. 2020;1874(2):188405. doi: 10.1016/j.bbcan.2020.188405
- Tonlaar N, Galoforo S, Thibodeau BJ, et al. Antitumor activity of the dual PI3K/MTOR inhibitor, PF-04691502, in combination with radiation in head and neck cancer. Radiother Oncol. 2017;124(3):504–512. doi: 10.1016/j.radonc.2017.08.001
- Wang J, Yue H, Huang X, et al. Novel nano-platinum induces autophagy through dual pathways in the treatment of osteosarcoma in cell lines with different P53 expression patterns. Chem Eng J. 2024;488:151071. doi: 10.1016/j.cej.2024.151071
- Shang X, Na X, Wang L, et al. Evaluation of a dual PI3K/mTOR inhibitor PF-04691502 against bladder cancer cells. Evid Based Complement Alternat Med. 2022;2022:1–10. doi: 10.1155/2022/8110796
- Yuan J, Mehta PP, Yin MJ, et al. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther. 2011;10(11):2189–2199. doi: 10.1158/1535-7163.MCT-11-0185
- Cersosimo F, Lonardi S, Bernardini G, et al. Tumor-associated macrophages in osteosarcoma: from mechanisms to therapy. Int J Mol Sci. 2020;21(15):5207. doi: 10.3390/ijms21155207
- Marina NM, Smeland S, Bielack SS, et al. Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): an open-label, international, randomised controlled trial. Lancet Oncol. 2016;17(10):1396–1408. doi: 10.1016/S1470-2045(16)30214-5
- Smrke A, Anderson PM, Gulia A, et al. Future directions in the treatment of osteosarcoma. Cells. 2021;10(1):172. doi: 10.3390/cells10010172
- Ashique S, Faiyazuddin M, Afzal O, et al. Advanced nanoparticles, the hallmark of targeted drug delivery for osteosarcoma-an updated review. J Drug Delivery Sci Technol. 2023;87:104753. doi: 10.1016/j.jddst.2023.104753
- Haritha V, Gowri S, Janarthanan B, et al. Biogenic synthesis of nickel oxide nanoparticles using averrhoa bilimbi and investigation of its antibacterial, antidiabetic and cytotoxic properties. Inorg Chem Commun. 2022;144:109930. doi: 10.1016/j.inoche.2022.109930
- Chen D, Liu X, Lu X, et al. Nanoparticle drug delivery systems for synergistic delivery of tumor therapy. Front Pharmacol. 2023;14:1111991. doi: 10.3389/fphar.2023.1111991
- Shi Y, van der Meel R, Chen X, et al. The EPR effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics. 2020;10(17):7921–7924. doi: 10.7150/thno.49577
- Zhu G, Wang B, Feng G, et al. A nano-preparation approach to enable the delivery of daphnoretin to potentiate the therapeutical efficacy in hepatocellular cancer. Front Pharmacol. 2022;13:965131. doi: 10.3389/fphar.2022.965131
- Sun Q, Liang J, Lin Y, et al. Preparation of nano-sized multi-vesicular vesicles (MVVs) and its application in co-delivery of doxorubicin and curcumin. Colloids Surf B Biointerfaces. 2023;229:113471. doi: 10.1016/j.colsurfb.2023.113471
- Chen W, Liu X, Li Y, et al. Preparation and in vitro evaluation of a nano ultrasound contrast agent targeting pancreatic cancer. J Nanosci Nanotechnol. 2021;21(3):1413–1418. doi: 10.1166/jnn.2021.18883
- Li Y, Liu Y, Yao B, et al. Rapid preparation and antimicrobial activity of polyurea coatings with RE-Doped nano-ZnO. Microbiol Biotechnol. 2022;15(2):548–560. doi: 10.1111/1751-7915.13891
- Wang A, Cui J, Wang Y, et al. Preparation and characterization of a novel controlled-release nano-delivery system loaded with pyraclostrobin via high-pressure homogenization. Pest Manag Sci. 2020;76(8):2829–2837. doi: 10.1002/ps.5833
- Barbhuiya RI, Singha P, Asaithambi N, et al. Ultrasound-assisted rapid biological synthesis and characterization of silver nanoparticles using pomelo peel waste. Food Chem. 2022;385:132602. doi: 10.1016/j.foodchem.2022.132602
- Amiryaghoubi N, Abdolahinia ED, Nakhlband A, et al. Smart chitosan-folate hybrid magnetic nanoparticles for targeted delivery of doxorubicin to osteosarcoma cells. Colloids Surf B Biointerfaces. 2022;220:112911. doi: 10.1016/j.colsurfb.2022.112911
- Wang L, Huang X, You X, et al. Nanoparticle enhanced combination therapy for stem-like progenitors defined by single-cell transcriptomics in chemotherapy-resistant osteosarcoma. Signal Transduct Target Ther. 2020;5(1):196. doi: 10.1038/s41392-020-00248-x
- Liu H, Zhang R, Zhang D, et al. Cyclic RGD-decorated liposomal gossypol AT-101 targeting for enhanced antitumor effect. Int J Nanomedicine. 2022;17:227–244. doi: 10.2147/IJN.S341824
- Wong PT, Choi SK. Mechanisms and implications of dual-acting methotrexate in folate-targeted nanotherapeutic delivery. Int J Mol Sci. 2015;16(1):1772–1790. doi: 10.3390/ijms16011772
- Ko S, Park J, Lee Y, et al. Biochemical reprogramming of tumors for active modulation of receptor-mediated nanomaterial delivery. Biomaterials. 2020;262:120343. doi: 10.1016/j.biomaterials.2020.120343
- Wang C, Wang B, Zou S, et al. Cyclo-gamma-polyglutamic acid-coated dual-responsive nanomicelles loaded with doxorubicin for synergistic chemo-photodynamic therapy. Biomater Sci. 2021;9(17):5977–5987. doi: 10.1039/D1BM00713K
- Lilienthal I, Herold N. Targeting molecular mechanisms underlying treatment efficacy and resistance in osteosarcoma: a review of current and future strategies. Int J Mol Sci. 2020;21(18):6885. doi: 10.3390/ijms21186885
- Bertacchini J, Heidari N, Mediani L, et al. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol Life Sci. 2015;72(12):2337–2347. doi: 10.1007/s00018-015-1867-5
- Shijie L, Zhen P, Kang Q, et al. Deregulation of CLTC interacts with TFG, facilitating osteosarcoma via the TGF-beta and AKT/mTOR signaling pathways. Clin Transl Med. 2021;11(6):e377. doi: 10.1002/ctm2.377
- Gupte A, Baker EK, Wan SS, et al. Systematic screening identifies dual PI3K and mTOR inhibition as a conserved therapeutic vulnerability in osteosarcoma. Clin Cancer Res. 2015;21(14):3216–3229. doi: 10.1158/1078-0432.CCR-14-3026
- Plana D, Palmer AC, Sorger PK. Independent drug action in combination therapy: implications for precision oncology. Cancer Discov. 2022;12(3):606–624. doi: 10.1158/2159-8290.CD-21-0212
- Patra JK, Das G, Fraceto LF, et al. Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnol. 2018;16(1):71. doi: 10.1186/s12951-018-0392-8
- Machtakova M, Therien-Aubin H, Landfester K. Polymer nano-systems for the encapsulation and delivery of active biomacromolecular therapeutic agents. Chem Soc Rev. 2022;51(1):128–152. doi: 10.1039/D1CS00686J