
Abstract
Detrimental effects of ionizing radiation (IR) are correlated to the varying efficiency of IR to induce complex DNA damage. A double strand break (DSB) can be considered the simpler form of complex DNA damage. These types of damage can consist of DSBs, single strand breaks (SSBs) and/or non-DSB lesions such as base damages and apurinic/apyrimidinic (AP; abasic) sites in different combinations. Enthralling theoretical (Monte Carlo simulations) and experimental evidence suggests an increase in the complexity of DNA damage and therefore repair resistance with linear energy transfer (LET). In this study, we have measured the induction and processing of DSB and non-DSB oxidative clusters using adaptations of immunofluorescence. Specifically, we applied foci colocalization approaches as the most current methodologies for the in situ detection of clustered DNA lesions in a variety of human normal (FEP18-11-T1) and cancerous cell lines of varying repair efficiency (MCF7, HepG2, A549, MO59K/J) and radiation qualities of increasing LET, that is γ-, X-rays 0.3–1 keV/μm, α-particles 116 keV/μm and 36Ar ions 270 keV/μm. Using γ-H2AX or 53BP1 foci staining as DSB probes, we calculated a DSB apparent rate of 5–16 DSBs/cell/Gy decreasing with LET. A similar trend was measured for non-DSB oxidized base lesions detected using antibodies against the human repair enzymes 8-oxoguanine-DNA glycosylase (OGG1) or AP endonuclease (APE1), that is damage foci as probes for oxidized purines or abasic sites, respectively. In addition, using colocalization parameters previously introduced by our groups, we detected an increasing clustering of damage for DSBs and non-DSBs. We also make correlations of damage complexity with the repair efficiency of each cell line and we discuss the biological importance of these new findings with regard to the severity of IR due to the complex nature of its DNA damage.
Introduction
Ionizing radiation (IR) is the type of radiation that transfers sufficient energy to free electrons from atoms or molecules, thereby ionizing them. The essence of the detrimental effects of IR lies on the clustering of these ionizations at the nm scale and the consequent complexity of cellular damage induced [Citation1]. This differentiates IR from other types of stresses such as oxidative or replication stress that also induce cellular damage [Citation2]. Although the biological effects of IR are centered on DNA, one cannot disregard the deleterious effects on proteins and/or lipid membranes that can modulate radioresistance [Citation3]. Exposure to high-linear energy transfer (LET) IR qualities, such as α-particles and accelerated ions, introduces clustered damaged sites within the DNA. The induced DNA damage is believed to be more compound than that produced by low-LET radiation and leads to more critical biological effects [Citation4]. In addition to double strand breaks (DSBs), IR also induces a diversity of non-DSB lesions, including single strand breaks (SSBs), change or lose of bases (AP; abasic sites) and deoxyriboses, cross-links formed between two complementary DNA strands, as well as the occurrence of alkali- and heat-labile sites [Citation5]. Such types of lesions can occur separately or closely to each other (a few bp) resulting in complex or clustered DNA damage. Despite the fact that DSBs are considered to be the most critical lesions relating to cell survival or other late cellular effects, non-DSB clusters, that is the combination of two or more DNA lesions that do not form a DSB [Citation6] adds to damage complexity of DSBs, significantly affecting their repair [Citation7,Citation8]. One major hypothesis is that this temporal simultaneous activation of different repair pathways like non-homologous end joining (NHEJ) for DSBs, base excision repair (BER) for non-DSB lesions, signifies a “stress” genome region [Citation9] or a DNA repair center [Citation10,Citation11]. Furthermore, additional DSBs may also be generated during processing of the primary damage that cannot be easily discerned from the prompt DSBs formed directly by IR exposure [Citation12]. Radiation-induced non-DSB lesions such as SSBs and base modifications occur more frequently than DSBs although tend to decrease in total with LET [Citation13]. The same stands for non-DSB bistranded clusters for which one can expect a ratio of non-DSB clusters to DSBs ∼3–5 or even higher verified experimentally as reviewed in [Citation14,Citation15]. Similar numbers are also predicted by Monte Carlo simulations for the complexity of damage to increase with LET [Citation16]. Although this last “dogmatic” hypothesis, that is the increase in clustering of damage with increasing LET, has been consistently suggested by older and recent Monte Carlo studies [Citation17,Citation18], experimentally it has not been always verified [Citation2]. This applies to DSB and non-DSB clusters and results obtained by the traditional modified DNA electrophoresis with repair enzymes as damage probes and more recent approaches using immunofluorescence and colocalization approaches. This disparity between theory and experiment can be safely attributed to experimental limitations and specifically to the inability of all these methodologies to discern lesions located closely to each other (1–10 bp apart) [Citation19].
Therefore and based on the above, there are various technical challenges associated with clustered damage detection. In this study, we apply recent advances of immunofluorescence foci colocalization methodologies [Citation19] in a variety of human cells after exposure to different types of radiation with varying dose and LET in order to calculate complex DNA damage (DSBs and non-DSB clusters). We observed increasing complexity of DNA damage with high- (α-particles, 36Ar ions) compared to low-LET radiation qualities (X- and γ-rays). Based on these results, we propose “DNA damage complexity” parameters that can be used to safely document the clustering of IR-induced DNA lesions in situ and at the cellular level.
Materials and methods
Cell lines and culture conditions
We have chosen a variety of cell lines and irradiation conditions in order to test the applicability of our DNA damage detection protocols and optimize parameters for different experimental conditions. All listed cell lines were cultured at 37 °C in a humidified atmosphere (5% CO2, 95% air) and allowed to reach 70–80% confluency before irradiation. MCF7 (ATCC; HTB22) human mammary tumor cells were grown in Dulbecco?s modified Eagle?s medium (DMEM) (Sigma-Aldrich Corp., St. Louis, MO) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% l-glutamine and 1% penicillin–streptomycin [penicillin (100 U/ml)–streptomycin (100 μg/ml)]. Microscope slides were prepared by setting physical boundaries to cell colonies using hydrophobic ink (Liquid Blocker super pap pen, Sigma-Aldrich, Steinheim, Germany). Slides were then immersed in 100% ethanol for at least 24 h followed by drying in sterile conditions. Cells were seeded on the drawn area in approximately 200 μl of culture medium and slides were incubated in Petri dishes until cells attached. Extra culture medium (10 ml) was added after the cells were firmly attached on the slides. For the DNA-PKcs inhibitor experiment, NU7026 (Sigma-Aldrich Corp., St. Louis, MO) was added to the medium 24 h before irradiation to the final concentration of 10 μmol/l. After irradiation, the cells were incubated in fresh medium (for more details see Supplementary data).
Primary normal human immortalized keratinocyte FEP18-11-T1 (Gibco, Paisley, UK; Cat. No. 12332-011) cells were cultured in keratinocyte growth medium (Keratinocyte–SFM; Invitrogen, Paisley, UK; Cat. No. 17005-042) including medium (Cat. No. 17024-011) supplemented with human epidermal growth factor rEGF (2.5 μg; Cat. No. 10450-013) and bovine pituitary extract (25 mg; Cat. No. 13026-014) in total volume 500 ml. Cells were seeded in chamber slides 2 days before irradiation.
Human hepatocellular carcinoma HepG2 cells (HepG2, ATCC; HB-8065) were cultured in α-medium (modified MEM, Pan-Biotech, Aidenbach, Germany) with 10% FBS. For weekly sub-cultivation, cells were washed with PBS before detaching with trypsin/EDTA solution (PAN-Biotech). Subsequently, cells were seeded at a density 3 × 104 cells/cm2 in new flasks. Medium was changed every 4 days. HepG2 cells were seeded 2 days before irradiation, the cells were seeded into 8.6 cm2 slide flasks (Nunc, Roskilde, Denmark) with 3 ml cell culture medium (3 × 104 cells/cm2). To avoid detachment during irradiation, the flasks were coated with poly-l-lysine (0.6 μg/cm2) for 15 min at 37 °C and washed three times with sterile distilled water. After seeding, the cells adhered to the bottom of the flasks and reached ∼70% confluence at the time of irradiation. The slide flasks were completely filled with cell culture medium immediately before irradiation, closed and irradiated as described below.
Human lung carcinoma epithelial A549 cells (ATCC, CCL-185) were cultured on 18 mm poly-l-lysine coated coverslips (0.15 × 106 per 35 mm dish) in McCoy’s 5A medium supplemented with 10% fetal calf serum (FCS).
Human glioblastoma MO59K-MO59J cells (ATCC, CRL-2365/2366) were grown in Dulbecco’s modified Eagle’s medium-F-12 supplemented with 10% iron-supplemented calf serum (HyClone, Logan, UT). Cells were seeded on 18 mm poly-l-lysine coated coverslips (0.3 × 106 and 0.2 × 106 per 35 mm dish). M059K/J are considered a useful model system to study the role of DNA protein kinase (DNA-PK) in cellular and molecular processes involving DNA damage recognition and repair. MO59J cells completely lack expression of the catalytic subunit of DNA-PK (DNA-PKcs) and therefore are significantly less efficient in DSB repair, in contrast to their isogenic MO59K cells, because of a frameshift mutation [Citation20].
IR exposures
Irradiations were performed at different research centers and radiation facilities as described below for each type of radiation. Attention was given that culturing was performed under optimum conditions and similar procedures wherever possible. Controls were treated as the irradiated samples except that they were not exposed to radiation and stored in the irradiation room in upright position for the same time as the irradiated samples.
γ-Rays
Exponentially growing MCF7 cells cultures were grown on petri dishes containing a sterile slide. Confluent cultures on the slides were irradiated at room temperature with a 60Co GammaCell 220 irradiator (Atomic Energy of Canada Ltd., Ottawa, Canada) with 1 Gy (LET = 0.3 keV/μm, dose rate = 0.3 Gy/min). FEP18-11-T1 cells were irradiated with 0.1, 1 and 2 Gy using a 137Cs source (GammaCell40 Irradiator, Nordion International, Ottawa, ON, Canada), at a dose rate of 0.531 Gy/min. Cells were fixed with 4% PFA at 1, 6 and 24 h post-IR and washed with PBS before immunostaining.
X-rays
HepG2 cells were exposed to low LET (0.3–3.0 keV/μm) X-rays using the Gulmay X-ray source RS225 (X-Strahl, Surrey, UK) at DLR Cologne, Germany. The X-ray tube was adjusted to 200 kV and 15 mA. To eliminate soft X-rays, a copper (Cu) filter with a thickness of 0.5 mm was used. Dose and dose rate were determined using the dosimeter UNIDOSwebline with the ionization chamber TM30013 (PTW, Freiburg, Germany). The distance of the sample from the X-ray source was set to 450 mm to provide a constant dose rate of 1.0 Gy/min. After exposure, the samples were transferred to an incubator. As the X-ray source was located above the samples, cells in flasks were exposed in horizontal position below the exit window.
A549, MO59K, MO59J cells were exposed to X-rays on ice using an X-ray machine (Seifert-Pantak, East Haven, CT) operated at 320 kV, 10 mA with a 1.65-mm Al filter (effective photon energy, ∼80 keV), at a dose rate of 3 Gy/min and a distance of 50 cm. Dosimetry was carried out using a calibrated ionization chamber and a chemical dosimeter. The mean LET of this type of radiation is, approximately, 2 keV/μm. A549 cells were exposed for 5 min to X-rays as described above, but also they were covered with a metal mask (2μ GaAs; Micro Léman X-ray collimator, Geneva, Switzerland).
α-Particles
A549 cells were exposed to α-particle emitted by an 241Am source (Amersham) at Institute of Medical Radiation Biology, Essen, Germany. It consists of two cylindrical chambers, one containing the cell targets, the other one containing the source and collimator. The chambers are separated by a window of 50 mm in diameter, sealed with a polyester foil (of 1.5 μm thickness) which allowed flushing the chambers with different gases. The 241Am α-particle source (activity: 1.2 108 Bq) was plated as Am2O3 compound into a silver foil and was covered with a gold layer of 2 μm thickness. The chamber containing the source is flushed with He gas, while the target chamber contains normal air. Helium gas was added to reduce the loss of particle energy between the source and the target. Calibration measurements gave the dose rate of 79 Gy/h =1.32 Gy/min and an average LET ∼116 keV/μm.
Argon ions
HepG2 exposure to 36Ar (energy 95 MeV/n, energy on target 84.7 MeV/n, LET ∼270 keV/μm) ions was performed at the Grand Accélérateur National d'Ions Lourds (GANIL, Caen, France). The slide flasks were exposed to the beam in upright position through the bottom of the vessel. The sample holders were inserted into the biological sample racks (6 samples per rack). Four sample racks were inserted in the biological sample transporter and moved in front of the beam. As temperature control was not available at GANIL, samples were irradiated at room temperature. The irradiation time per sample was up to 10 min, depending on dose, and the total time in the irradiation room was 20–30 min. Dosimetry was performed by the staff at the accelerator facility and dose rates were adjusted to ∼1 Gy/min. The fluence (F) of heavy ions (particle/cm2, P/cm2) was converted to the absorbed dose in Gy by the following formula under consideration of the LET for each heavy ion:
(1)
The number of hit cells and the average hits per cell nucleus after exposure to four different argon ion fluencies are given in .
Table 1. Parameters of the argon ion exposure (95 MeV/n, LET 270.3 keV/μm)Table Footnotea for HepG2 cells.
Detection of DNA damage by immunofluorescence
For γ-H2AX foci measurements, MCF7 cells were seeded on slides, irradiated and after the repair times of 1, 6, 24, 48 and 72 h were fixed with 4% formaldehyde for 20 min at room temperature and washed three times over a 5-min period with PBS. To permeabilize the cells, the slides were washed in a Coplin jar three times over a 5-min period with 0.25% Triton X in PBS. The slides were washed in PBS for 5 min. Then, the slides were incubated in blocking solution (10% bovine serum albumin; 6% FBS and 0.02% Triton X in PBS) for 1 h at 37 °C. Primary γ-H2AX antibody incubation was carried out in blocking solution (1:1000 monoclonal rabbit anti-γ-H2AX; NB100-79967; Novus Biologicals, Abingdon, UK), for 1 h at 37 °C. After one wash with Triton X 0.1% in PBS for 5 min and three washing steps for 3 min in PBS, the incubation in blocking solution was repeated. Cells were then incubated in the secondary antibody for 20 min at 37 °C (dilution 1/4000 in blocking solution, Rhodamin Red-X Goat, Novus Biologicals, Abingdon, UK). The slides were washed once with Triton X 0.1% in PBS, three times in PBS solution and once for 10 min in 100% ice-cold ethanol. The nuclei were stained with DAPI (final working concentration of 1 μg/ml in Vectashield). The slides were covered with coverslips and stored in the dark prior to analysis under a fluorescent microscope (Axioplan 2, Carl Zeiss Microscopy GmbH, Hamburg, Germany), using the Isis imaging software (Metasystems, Altlussheim, Germany). The number of foci in 200 nuclei was analyzed for each experimental point.
For DSB and non DSB detection, MCF7 and HepG2 cells, attached on slides or slide flasks, were fixed with 4% formaldehyde for 15 min at room temperature and washed three times over a 5-min period with PBS. To permeabilize the cells, the slides were washed in a Coplin jar three times over a 5-min period with 0.25% Triton X. The slides were washed in PBS for 5 min. Cells were incubated with primary antibodies rabbit anti-γ-H2AX (1:1000; NB100-79967) and mouse anti-AP endonuclease (APE1) (1:800; 13B8E5C2; NB100-116), diluted in 5% non-fat dry milk (Regilait) for 2 h at 37 °C. After one wash with Triton X 0.1% for 5 min and three washing steps with 3 min in PBS, the incubation with secondary antibodies, goat anti-rabbit (1:4000 Rhodamin Red-X Goat, Novus Biologicals, Abingdon, UK) and goat anti-mouse (1:1000 IgG-FITC: sc-2010, Santa Cruz Biotechnology Inc., Santa Cruz, CA), in 5% non-fat dry milk for 0.5 h at 37 °C was carried out. After three washes with Triton X 0.1% and 2 × 5 min with 1% BSA in PBS and incubation for 15 min in ice-cold ethanol 100%, the nuclei were stained with DAPI. DAPI (final working concentration of 1 μg/ml in Vectashield), the slides covered with coverslips and stored in the dark prior to analysis under a fluorescent microscope (Axioplan 2, Zeiss; Isis, Metasystems).
A549, MO59K-MO59J and FEP cells were fixed with 2% PFA, 2% sucrose in PBS for 15 min room temperature. After one wash with PBS, permeabilization was performed with 100 mmol/l Tris–HCl, 50 mmol/EDTA, 0.5% Triton in PBS for 5 min. After one wash with PBS, the cells were incubated in PBG blocking buffer (0.5% BSA fraction V, 0.2% gelatin) for 1 h at room temperature. Cells were incubated for 1.5 h at room temperature with primary antibodies in PBG. Depending on the experiment, the antibodies were γ-H2AX [1:400 mouse monoclonal (ab22551), Abcam, Cambridge, UK], 53BP1 [1:400 (H-300): sc-22760, Santa Cruz)], 8-oxoguanine-DNA glycosylase (OGG1) (1:500 rabbit polyclonal; NB100-106, Novus Biologicals, Abingdon, UK) and APE1 (1:500; mouse anti-APE1; 13B8E5C2; NB100-116, Novus Biologicals, Abingdon, UK). After 3 × 5 min PBS, cells were incubated for 1 h at room temperature with the secondary antibodies: anti-rabbit (1:400, Alexa Fluor® 568 Goat Anti-Rabbit; Molecular Probes®-Thermo Fisher Scientific, Waltham, MA) and anti-mouse (1:400 Alexa Fluor® 488 Goat Anti-Mouse; Molecular Probes®) diluted in PBG. Nuclei were stained with 50 ng/ml DAPI in PromoFluor Antifade (PromoKine, Heidelberg, Germany). Images were acquired with a laser scanning confocal microscope DMI6000B TCS SP5 (Leica Microsystems, Mannheim, Germany), using the microscope imaging platform LAS AF (Leica Microsystems, Mannheim, Germany).
Data analysis
DNA damage parameters
For estimation of damage yields and level of damage clustering, we used the percentages and levels of colocalization between DSB and non-DSB repair proteins, using our recently introduced Pclc parameter [Citation19]. For estimation of colocalization between γ-Η2ΑΧ and 53BP1, a “classic” colocalization approach has been adopted that it is expressed as the percentage (denoted as “% clc”) of colocalized γ-H2AX foci to total γ-Η2ΑΧ foci number in the nucleus.
The Pclc value, described analytically in [Citation19], provides a reliable estimate of the relative localization of a (non-DSB repair) protein on the (DSB) foci area, compared to its localization in the rest of the cell nucleus (endogenous levels of non-DSB protein). Increase in the Pclc can be a good indication of DNA repair translocating to a DSB repair center.
(2)
Equation (2) is an expression for one cell, where the index 'i' runs through each DSB focus of the given cell. The rest of the cell nucleus is defined as the nucleus volume, excluding the volume occupied by DSB foci. Although and as described above, different culture, irradiation and fixing protocols were followed in various cases, the Pclc parameter depends only on the relative intensities and therefore has minimum dependence on different experimental approaches [Citation19].
Software
For images captured by an epifluorescent microscope the utilized software was JQuantPro developed by Dr. Pavel Lobachevsky, which is based on an earlier JCount Pro version [Citation21].
For images obtained by confocal microscope, Imaris V8.0.2 (Bitplane, Zürich, Switzerland) software was utilized. In order to quantify colocalization, we introduced an additional focus type, to express colocalization events. Depending on the protein of interest, for example A, we defined the colocalization (coloc) focus as the focus having in priority the focus A “segmentation” parameters, and additionally the intensity threshold for the second channel same as the threshold of focus B. For this analysis, the % clc= 100* # coloc foci/# foci A.
The “coloc” focus first has to “obligate” to the focus A segmentation parameters under the condition of focus B intensity. To calculate Pclc values, meta-analysis of the Imaris results was performed and a parallel analysis using JQuantPro.
Monte Carlo DNA damage simulations
Theoretical modeling of DNA damage by low-energy electrons was carried by our in-house Monte Carlo track-structure computer code [Citation22]. The code simulates interaction-by-interaction the transport of the primary electron and all its secondaries (generated in ionization events) in water medium until their energy drops below 10 eV (about the minimum ionization potential of water molecules in the liquid phase). The main output of the code is the spatial co-ordinates of all ionization and excitation collisions and the resulting energy deposition. A detailed description of the physics models used to calculate the total, differential and partial cross sections that are necessary to simulate the elastic and inelastic collisions of electrons in a liquid water medium is provided elsewhere [Citation22,Citation23]. DNA damage calculations presented in this work follow the “jump-the-details” approach first illustrated by Goodhead and Nikjoo [Citation24]. The approach is based on Monte Carlo simulated microdosimetric spectra in DNA-sized volumes coupled with empirical (or heuristic) values of damage activation energies. In the present work, track-structure simulations were carried out for several initial electron energies from 100 eV to 10 keV corresponding to the low to medium LET range of 1–20 keV/μm. These electrons cover most of the energy range of the so-called track-ends (of any type of IR), which are associated with increased biological effectiveness. The concept of the associate volume of the track was used to compute the (single-event) probability distribution of energy deposition, f(E), in 2 nm diameter (d) spherical volumes irradiated uniformly and isotropically by monoenergetic electrons. The linear dimensions chosen (2 nm) correspond to the diameter of B-DNA, which is a characteristic length in theoretical radiobiology. From the scored distribution, f(E), the (average) dose in the target volume can be determined through the relation D = E/m, where E = ∫ Ef(E)dE and m = ρ V is the mass of the spherical target with ρ the density (here ρ = 1 g/cm3) and the volume. The present microdosimetric simulations have been benchmarked against the KURBUC code by comparing the frequency- and dose-mean values of the f(E) distributions. The present values differ by up to 40% from those reported by Nikjoo et al. [Citation25]. Specifically, the present simulations include inter-molecular effects, such as screening and polarization in inelastic electron–electron scattering, through the use of a model dielectric response function [Citation17,Citation21]. The dielectric response function, which is specific to the medium, results in larger inelastic mean free paths and a harder energy-loss spectrum [Citation21,Citation22]. The neglect of the above effects (commonly denoted as the gas-phase approximation) has a sizeable impact on microdosimetric distributions [Citation24,Citation25].
The Monte Carlo generated f (>E) distribution is the main input for the damage calculations in the present approach. Specifically, the yield of the ith type of damage per gray per cell is given by the expression Yi = f (>Ei) × (N/D), where f (>Ei) is the cumulative probability distribution for energy deposition in the target volume greater than the activation energy Ei, and N is the number of target volumes per genome [Citation26]. For a 2- nm target and assuming 6 Gbp/cell and 0.34 nm/bp, we obtain (approximately) N∼109 targets/genome. The distribution f (>Ei) depends upon the track structure (type and energy of primary particle), the size of the target volume and the value of the activation energy Ei. Three types of DNA damage are considered, namely, a base damage (BD), a single-strand-break (SSB) and a double-strand-break (DSB) with activation energies EBD ≥ 10 eV, ESSB ≥ 17.5 eV and EDSB ≥ 67.5 eV, respectively. The activation energies for BD and SSB are taken from Nikjoo et al. [Citation1], whereas the activation energy of a DSB is deduced after normalization of our 10 keV results to the low-LET value suggested also by Semenenko and Stewart [Citation27]. The results presented below are based on Monte Carlo simulations of a large number (≥100,000) of electron histories for each primary energy, which result in a statistical uncertainty of less than 1%.
Results
Simulating DNA damage by low-LET radiation track-ends
In an attempt to approach some of our results on a theoretical basis, we performed Monte Carlo simulations for the case of low-LET radiation of low-energy electrons, which represent the so-called track-ends of any IR. Covering an LET range from ∼1 to ∼20 keV/μm, these particles can be used to explain many of the characteristics associated with the transition from low- to high-LET radiation. In , we present estimates of the SSB (panel a) and DSB (panel b) yields per Gy per cell, as well as the SSB-to-DSB ratio (panel c) as a function of electron energy. Despite the simplicity of the present methodology, the results compare reasonably well against the more elaborate simulations by other researchers [Citation1,Citation28,Citation29]. Interestingly, whereas the variation of the SSB yield with electron energy is less than 10% (with a mean around YSSB = 620/Gy/cell), the DSB yields vary by almost 100% (YDSB varies from 35 to 65 per Gy/cell). As a result, over the examined electron energy range, the SSB-to-DSB ratio varies significantly (from 9 to 19) with a minimum at electron energies between 200 and 300 eV, which roughly coincides with the electron LET maximum [Citation30]. The change in the trend of the curves below 300 eV (increase of the SSB yield with a concomitant decrease of the DSB yield) is due to the very small penetration capacity of these electrons (up to a few nanometers), which renders them particularly effective for SSB production but somewhat ineffective for DSB. Although in the present simulations only the “direct” effect has been considered (energy transfer to the DNA by mainly electron-impact ionization and excitation), one can predict also the indirect effect based on the expected contributions. Using the values reported in Nikjoo et al. [Citation1], we obtain from , a tentative maximum for the total (direct + indirect) DSB yield of ∼90 DSBs/Gy/cell at 300 eV and for the SSB yield of ∼1000 SSB/Gy/cell.
Figure 1. Complex DNA damage simulation. Simulations of single and complex DNA damage (per Gy/cell) using Monte Carlo methodologies for the direct effect of low-energy electrons (track-ends) as described in the text. The double strand breaks (DSB), single strand breaks (SSB), base damages (BD) and total strand breaks (TSB) are shown for electron energies 100–10,000 eV. We assume here a diploid cell of DNA size ∼6 Gbp.
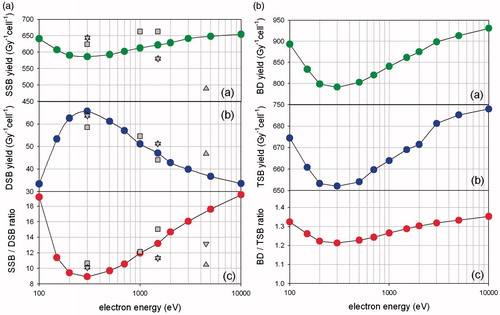
In , we present similar estimates of the BD yield per gray per cell and compare it with the total-strand-break (TSB) yield. The latter is simply the sum of the SSB and DSB yields (YTSB = YSSB + YDSB). The trend of the TSB curve is similar to that for the SSB () because YSSB ≫ YDSB, so YTSB ≈ YSSB. Interestingly, the BD and SSB yields share the same dependence from electron energy with a minimum around 200–300 eV. The absolute yield of BD though is much higher than the yield of SSB due to the lower activation energy. Finally, the BD-to-TSB ratio remains rather constant with electron energy at ∼1.3. This is smaller than the value of ∼2 reported in the literature when both the direct and indirect effects are considered [Citation28]. The above difference can be explained by the fact that the indirect effect is expected to have a greater impact on the BD yield than the TSB yield, thus, raising the BD-to-TSB ratio.
As a general observation, complex damage (e.g. DSBs), which is generally associated with higher activation energy (e.g. compared to SSBs or BD), increases significantly as the electron energy decreases because of a sizeable increase of LET; from less than ∼1 keV/μm for high-energy electrons (≥10 keV) to a maximum of ∼20 keV/μm at ∼200 eV. Advances in computer simulation and theoretical modeling of DNA damage complexity along the above line have been recently reviewed by Nikjoo and co-workers [Citation17]. In relation to our experimental results, such a phenomenon is expected to increase the colocalization (Pclc) slightly for low-LET, where all these different breaks and BDs are evenly spread throughout the whole genome ∼6 Gbp.
Induction and repair of DSBs and non-DSB lesions for different LET radiations
X-rays and γ-rays as low-LET radiations generate ionization clusters mostly at the ends of the produced low-energy electron tracks but still they deposit 50–70% of their energy in well-defined ionization clusters that generate a relatively homogenous ionization pattern within the cell [Citation24,Citation31]. It also accepted that ∼30% of DSBs contain additional lesions following exposure to low-energy electrons but this fraction can increase up to 70% at the same dose of α-particles [Citation32], therefore increasing the damage complexity and DSB clustering [Citation33]. Our results presented in for DSB induction and the response of a variety of mammalian cells to different IRs and doses show an expected almost linear dependence of DSB with dose for low-LET and high-LET. As far as it concerns DSBs (γ-H2AX foci), a saturation at higher doses 2–4 Gy is detected as previously shown by other researchers and reviewed in [Citation34]. Based on the linear fittings presented in , we detected much higher yields for low-LET (X- and γ-rays) compared to high-LET α-particles and Ar ions. In general, we got a range of 5–16 DSBs/Gy/cell depending on the cell line and radiation quality. The yields for X- and γ-rays are similar and are ∼16 foci/Gy/cell on average. In all cases, a variation can be found in literature due to the different cell lines, time points and other technical factors. Comparison of DSB yields for radiations of different LETs shows an unexpected lower yield for DSBs for higher LET, which is probably explained by the DSB clustering and the underestimation of the actual true DSB number under one focus [Citation35]. In order to improve the statistics of foci measurements due to the inherent heterogeneity of cell sub-populations (G0–G1, S-G2), we applied a specific statistical analysis methodology in the case of HepG2 cells as described in the Supplementary data section. Results, although not statistically different from the ones presented in , show less variation and most importantly are presented in relationship with G0–G1 or G2 cell subpopulations (Figure S4).
Figure 2. DSB foci induction and repair for varying doses and radiations. We show the dose dependence of induction and repair for DSBs in human DNA repair proficient cells and at different post-irradiation time points. Results of immunofluorescence experiments using two different DSB probes, γ-H2AX and 53BP1. DSB foci numbers have been counted using Imaris software for confocal images (A549, MO59K, MO59J, FEP cell lines) and with JQuantPro (developed by Dr. Pavel N. Lobachevsky based on a previous JCount version [Citation21]) for epifluorescence images (MCF7, HepG2 cell lines). “#” denotes number. Antibodies against γ-H2AX and 53BP1 proteins have been used as DSB surrogate markers. (a) A549 cells irradiated with X-rays or α-particles. (i) γ-Η2ΑΧ foci already for the time point of 2 h have been reduced in numbers. (ii) 53BP1 foci have similar levels with γ-H2AX up to the dose of 2 Gy, but for 4 Gy their number is smaller probably due to 53BP1 protein cellular levels “consumption”. (iii) A549 cells irradiated with α-particles: α-particles induce bigger but fewer γ-H2AX foci. For the 2-h time point, foci are still forming, because of DSB clustering. At the 1-h time point foci are present but not big enough in size to be detected. (b) HepG2 cells irradiated with X-rays (i) or Argon ions (ii). (c) Dose dependence of γ-Η2ΑΧ foci number per cell, regarding the first time point (≤ 1 h), for several cell lines. Linear fitting is presented for γ-rays, X-rays, α-particles and 36Ar ions. The trendlines and linear fitting coefficients are also included in each case. Values have been obtained out of 2–3 independent experiments. “*” Denotes statistical difference at the p < .05 level between irradiated and non-irradiated (0 Gy) samples. “**” Denotes statistical difference at the p < .05 level between various repair time points and initial “0” h time point before actual repair samples.
![Figure 2. DSB foci induction and repair for varying doses and radiations. We show the dose dependence of induction and repair for DSBs in human DNA repair proficient cells and at different post-irradiation time points. Results of immunofluorescence experiments using two different DSB probes, γ-H2AX and 53BP1. DSB foci numbers have been counted using Imaris software for confocal images (A549, MO59K, MO59J, FEP cell lines) and with JQuantPro (developed by Dr. Pavel N. Lobachevsky based on a previous JCount version [Citation21]) for epifluorescence images (MCF7, HepG2 cell lines). “#” denotes number. Antibodies against γ-H2AX and 53BP1 proteins have been used as DSB surrogate markers. (a) A549 cells irradiated with X-rays or α-particles. (i) γ-Η2ΑΧ foci already for the time point of 2 h have been reduced in numbers. (ii) 53BP1 foci have similar levels with γ-H2AX up to the dose of 2 Gy, but for 4 Gy their number is smaller probably due to 53BP1 protein cellular levels “consumption”. (iii) A549 cells irradiated with α-particles: α-particles induce bigger but fewer γ-H2AX foci. For the 2-h time point, foci are still forming, because of DSB clustering. At the 1-h time point foci are present but not big enough in size to be detected. (b) HepG2 cells irradiated with X-rays (i) or Argon ions (ii). (c) Dose dependence of γ-Η2ΑΧ foci number per cell, regarding the first time point (≤ 1 h), for several cell lines. Linear fitting is presented for γ-rays, X-rays, α-particles and 36Ar ions. The trendlines and linear fitting coefficients are also included in each case. Values have been obtained out of 2–3 independent experiments. “*” Denotes statistical difference at the p < .05 level between irradiated and non-irradiated (0 Gy) samples. “**” Denotes statistical difference at the p < .05 level between various repair time points and initial “0” h time point before actual repair samples.](/cms/asset/6c3606c5-4c5e-4673-89c8-81c964ff351a/ifra_a_1232484_f0002_c.jpg)
In , the effect of repair with time is shown. We have included results for different cell lines and doses up to 4 Gy. In most cases, the DSB levels drop significantly within 24 h as also depicted in where the repair efficiency (% RE) has been calculated. As expected in the case of a DSB repair deficiency (MO59J, or use of DNA-PKcs inhibitors), the RE is significantly reduced even after 24 or 72 h (see for example MCF7 with NU7026 inhibitor). Parallel experiments in our group have shown that MCF7 cells, when treated with NU7026 inhibitor, show a decrease in clonogenic survival and an increase in genomic instability detected using micronuclei assay and chromosomal breaks (Figure S1). The reduction of RE is also verified with some quantitative differences using either γ-H2AX or 53BP1 foci (). In some cases, we observe an increase compared to initial yield like in 8 h for MO59K or 24 h for MO59J. This increase explains also the negative RE values in some cases in where the final values at 24 h are higher than the initial DSBs. Comparison of low-LET X-rays to high-LET 36Ar ions shows real differences only for higher doses (6 h), but unfortunately no data is available for 24 h for 36Ar ions in order to compare. A lower RE is found for α-particles compared to X-rays (1 h).
Figure 3. DSB induction and processing under a status of repair deficiency. Results are shown for immunofluorescence experiments at different post-irradiation time points. DSB foci numbers have been counted with Imaris software for confocal images (A549, MO59K, MO59J, FEP cell lines) and with JQuantPro (developed by Dr. Pavel N. Lobachevsky based on a previous JCount version [Citation21]) for epifluorescence images (MCF7, HepG2 cell lines). “#” denotes number. Antibodies against γ-H2AX and 53BP1 proteins have been used as DSB surrogate markers. (a) A549 cells irradiated with X-rays −/+ DNA-PKcs inhibitor NU7441. (i) and (ii) 53BP1 foci number per cell. (iii) and (iv) γ-H2AX foci numbers. (b) MO59K and MO59J cells irradiated with X-rays. (i) and (ii) 53BP1 foci number per cell. (iii) and (iv) γ-H2AX foci number per cell. (c) MCF7 cells irradiated with γ-rays −/+ DNA-PKcs inhibitor NU7026. Values have been obtained out of 2–3 independent experiments. “*” Denotes statistical difference at the p < .05 level between irradiated and non-irradiated (0 Gy) samples. “**” Denotes statistical difference at the p < .05 level between various repair time points and initial “0” h time point before actual repair samples.
![Figure 3. DSB induction and processing under a status of repair deficiency. Results are shown for immunofluorescence experiments at different post-irradiation time points. DSB foci numbers have been counted with Imaris software for confocal images (A549, MO59K, MO59J, FEP cell lines) and with JQuantPro (developed by Dr. Pavel N. Lobachevsky based on a previous JCount version [Citation21]) for epifluorescence images (MCF7, HepG2 cell lines). “#” denotes number. Antibodies against γ-H2AX and 53BP1 proteins have been used as DSB surrogate markers. (a) A549 cells irradiated with X-rays −/+ DNA-PKcs inhibitor NU7441. (i) and (ii) 53BP1 foci number per cell. (iii) and (iv) γ-H2AX foci numbers. (b) MO59K and MO59J cells irradiated with X-rays. (i) and (ii) 53BP1 foci number per cell. (iii) and (iv) γ-H2AX foci number per cell. (c) MCF7 cells irradiated with γ-rays −/+ DNA-PKcs inhibitor NU7026. Values have been obtained out of 2–3 independent experiments. “*” Denotes statistical difference at the p < .05 level between irradiated and non-irradiated (0 Gy) samples. “**” Denotes statistical difference at the p < .05 level between various repair time points and initial “0” h time point before actual repair samples.](/cms/asset/391b6fe2-7272-493f-b9a6-b2d9d4060765/ifra_a_1232484_f0003_c.jpg)
This apparent increase of DSBs has been also observed in the past in varying repair times and can be attributed to de novo DSBs forming at these time points due to processing of non-DSB lesions or apoptotic events [Citation36]. The concluding remark is that residual number of DSB foci is generally reduced with time, suggesting active repair and processing of these complex DNA lesions with some exceptions as discussed above. Complementary measurements of foci size for X-rays and 36Ar shows a doubling of DSB focus size when one compares the two types of radiation (Figure S6). In general, there is an increase with dose and repair time for both X-rays and 36Ar ions (steeper increase). These findings reflect the previously introduced idea of radiation-induced foci (RIF) and enlarged repair centers for high-LET radiations following a bimodal response with time, that is increase for 0–24 h and then gradually decrease [Citation10,Citation37].
Damage clustering
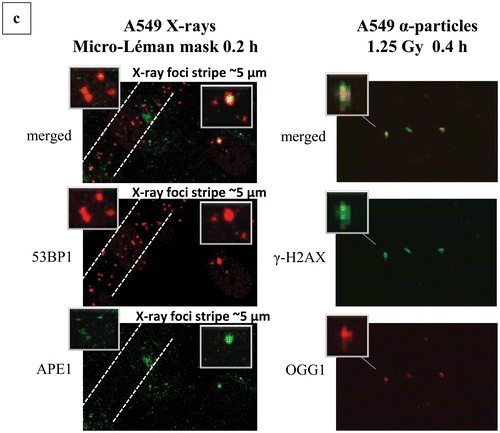
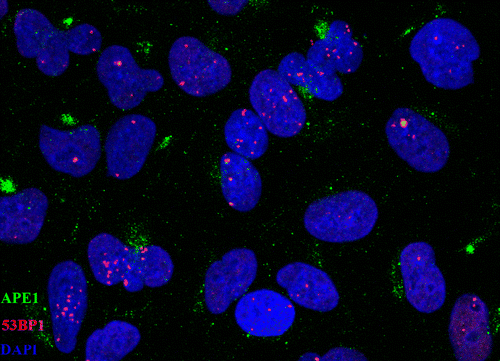
In , we have included the calculations regarding the clustering of damage and particularly the DSB-non-DSB clusters detected by using the parameter Pclc as previously described (please see “Materials and Methods” section). In , representative images and magnification inserts have been included showing the damage foci colocalization by the topological co-existence of two different colors (damage probes antibodies). As a first step, we have calculated were possible the apparent “total” yield of non-DSB lesions (OGG1 or APE1 foci) for low- and high-LET radiations. The basic finding is that the number of non-DSB foci is decreasing with LET from X-rays to α-particles and 36Ar-ions (). Comparison between the ratios of non-DSB lesions/DSB for different radiations, it reveals values ranging from 5–10 (X-rays) to 3–5 for high-LET, which is within the expected and experimental previous findings [Citation6,Citation16]. Although the yield of damage is reducing for high-LET, as already discussed above, the damage clustering is expected to increase. We tested this hypothesis by using our recently introduced methodologies on foci colocalization and Pclc parameter among other “coloc parameters” [Citation19]. Indeed as depicted in , the detected clustering or Pclc increases with dose and LET. This is consistent with the above hypothesis. The Pclc practically depicts the complexity of DSBs, that is DSB foci accompanied in their area by non-DSB foci. For low-LET (X- and γ-rays) values vary from 1 to 2, for higher LET, values increases up to 20 for α-particles, but for 36Ar ions they do not show a significant increase compared to low-LET. This is possibly explained by the very high-LET of these ions and the extreme clustering, which is possibly within the experimental limits of our methodology. A possible alternative explanation can be the fact that as generally accepted above 200 keV/μm, the RBE for biological effects for ions, drops significantly reaching the ranges observed for low-LET radiation due to the pattern of very high ionization clustering ameliorating the overall effects [Citation38]. Based on the dosimetry for 36Ar ions, one would expect for doses >1 Gy most of the cells to be hit (see “Materials and Methods” section). Therefore with lower doses, one can expect a certain level of uncertainty in the calculations of damage and clustering unless directly on the track. A relatively high Pclc value was obtained when A549 cells were exposed to X-rays Micro Léman mask (). In this case, Pclc (APE1/53BP1) values reach the surprising level of ∼8 much higher than everything else found for X-rays in our study. As also shown in and insets, one can see the stripes of damage foci ∼5 μm in width, while the irradiation exposure stripe has only a width of ∼2 μm. In addition the foci size for Ar ions and Micro Léman X-rays is increased as clearly shown in .
Figure 4. Clustering of damage revealed by means of colocalization parameters. Estimation of colocalization between DSB or DSB and non-DSB DNA repair proteins using adaptations of immunofluorescence methodologies. Colocalization levels have been estimated with Imaris software for confocal images (A549, MO59K, MO59J, FEP cell lines) and with JQuantPro (developed by Dr. Pavel N. Lobachevsky based on a JCount version [Citation21]) for epifluorescence images (MCF7, HepG2 cell lines). Antibodies against γ-H2AX and 53BP1 proteins have been used as DSB surrogate markers. For visualization of non-DSB lesions antibodies against OGG1 and APE1 proteins have been used as probes. For estimation of colocalization between γ-Η2ΑΧ and 53BP1, a classic colocalization approach has been adopted. It is expressed as the percentage (denoted as “% clc”) of colocalized γ-H2AX foci to total γ-Η2ΑΧ foci number in the nucleus. For colocalization between DSB and non-DSB proteins, Pclc recently introduced by our group-parameter has been utilized. Pclc expresses the relative concentration of non-DSB protein in the DSB foci area compared to the rest of cell nucleus area. Scale of Pclc: inverse colocalization 0 < Pclc < 1, random colocalization Pclc∼1 and true colocalization Pclc > 1 [Citation19]. (a) Calculation of colocalization between DSB and non-DSB repair proteins using Pclc. (i), (ii) HepG2 cells irradiated with X-rays or Argon ions. (iii)–(v) A549 cells irradiated with X-rays, α-particles or Micro Léman collimated X-rays. (vi) MCF7 cells exposed to γ-rays and (vii) FEP cells irradiated with γ-rays. (b) Non-DSB foci number per cell for various radiations. (c) Calculation of colocalization levels between 53BP1 and γ-Η2ΑΧ DSB repair proteins. (i) and (ii) MO59K and MO59J cells irradiated with X-rays at various doses. Values have been obtained out of 2–3 independent experiments. “*” Denotes statistical difference at the p < .05 level between irradiated and non-irradiated (0 Gy) samples. “**” Denotes statistical difference at the p < .05 level between various repair time points and initial “0” h time point before actual repair samples.
![Figure 4. Clustering of damage revealed by means of colocalization parameters. Estimation of colocalization between DSB or DSB and non-DSB DNA repair proteins using adaptations of immunofluorescence methodologies. Colocalization levels have been estimated with Imaris software for confocal images (A549, MO59K, MO59J, FEP cell lines) and with JQuantPro (developed by Dr. Pavel N. Lobachevsky based on a JCount version [Citation21]) for epifluorescence images (MCF7, HepG2 cell lines). Antibodies against γ-H2AX and 53BP1 proteins have been used as DSB surrogate markers. For visualization of non-DSB lesions antibodies against OGG1 and APE1 proteins have been used as probes. For estimation of colocalization between γ-Η2ΑΧ and 53BP1, a classic colocalization approach has been adopted. It is expressed as the percentage (denoted as “% clc”) of colocalized γ-H2AX foci to total γ-Η2ΑΧ foci number in the nucleus. For colocalization between DSB and non-DSB proteins, Pclc recently introduced by our group-parameter has been utilized. Pclc expresses the relative concentration of non-DSB protein in the DSB foci area compared to the rest of cell nucleus area. Scale of Pclc: inverse colocalization 0 < Pclc < 1, random colocalization Pclc∼1 and true colocalization Pclc > 1 [Citation19]. (a) Calculation of colocalization between DSB and non-DSB repair proteins using Pclc. (i), (ii) HepG2 cells irradiated with X-rays or Argon ions. (iii)–(v) A549 cells irradiated with X-rays, α-particles or Micro Léman collimated X-rays. (vi) MCF7 cells exposed to γ-rays and (vii) FEP cells irradiated with γ-rays. (b) Non-DSB foci number per cell for various radiations. (c) Calculation of colocalization levels between 53BP1 and γ-Η2ΑΧ DSB repair proteins. (i) and (ii) MO59K and MO59J cells irradiated with X-rays at various doses. Values have been obtained out of 2–3 independent experiments. “*” Denotes statistical difference at the p < .05 level between irradiated and non-irradiated (0 Gy) samples. “**” Denotes statistical difference at the p < .05 level between various repair time points and initial “0” h time point before actual repair samples.](/cms/asset/d1198e9d-5f9d-4eb2-b56e-217f58c04645/ifra_a_1232484_f0004_c.jpg)
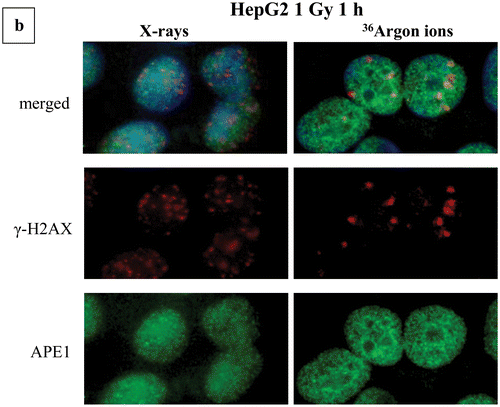
Figure 5. Complex DNA damage foci localization patterns and characteristics. Examples of images are shown used in the calculation of number of foci and localization pattern as described in the “Materials and Methods” section. Original uncut, unedited or merged fluorescence microscopy images and magnification insets have been included showing the damage foci colocalization by the topological co-existence of two different colors (damage probes antibodies). (a) Repair “disappearance” of DSB foci coloc γ-Η2ΑΧ/53BP1 in repair proficient MO59K and DNA-PK deficient MO59J cells X-irradiated with 1 Gy at various post-irradiation time points. Green color, γ-Η2ΑΧ; red color, 53BP1. One can easily see the high intensity and coloc foci even after 24 h for the MO59J cells. Foci size is reduced at 24 h compared to 1 h. (b) γ-Η2ΑΧ (DSB)/APE1 (AP sites) foci colocalization for HepG2 cells X-ray and 36Ar ions irradiated. The increased size of Ar ions foci can be easily detected compared to low-LET radiation. (c) Characteristic 53BP1 (DSB)/APE1 (AP sites) foci colocalization in X-irradiated A549 cells using Micro Léman mask stripes (2 μm width). The wider foci damage stripe has been marked to aid visualization (0.2 h post-irradiation). Characteristic γ-Η2ΑΧ (DSB)/OGG1 (oxidized purine base sites) foci colocalization in α-particle irradiated A549 cells at 1.25 Gy (0.4 h post-irradiation). The increased size DSB foci can be seen with the smaller non-DSB foci lying mainly in the core of the focus (insets). (d) Animation picture of colocalization of APE1 (green) and 53ΒP1 (red) foci on A549 cells exposed to Micro Léman collimated X-rays, 10 min after irradiation as a graphics interchange format (GIF). Different instances depict in order of appearance the staining with each antibody and finally both. This gif file was created using the http://gifmaker.me/.
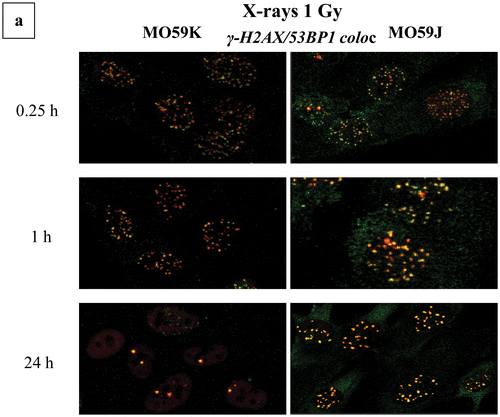
Concerning 53BP1 and γ-H2AX colocalization (), we have detected an increase of colocalization with dose and time in most cases when comparing MO59K and MO59J (DNA-PK deficient). In the case of repair deficient cells MO59J compared to their repair proficient counterpart MO59K cells, we detect interesting enough a high increase of colocalization between these two DSB proteins especially after 8 h post-irradiation with maximum at 24 h, suggesting possibly active repair resistant sites [Citation7].
Discussion
The ability of IR to induce cell lethality relates undoubtedly to the nature of its interaction with cells and the induction of multiply damaged sites [Citation39,Citation40]. Extensive Monte Carlo simulations as well as experimental evidence suggests that this clustering of DNA damage like DSB and/or non-DSB clusters located in a small chromosome region, like a few Mbp (10–20 Mbp) or practically nm in actual length [Citation19], create a DNA damage and repair center. For example for 1 Gy (), the expected DSB and TSB genome density will be ∼0.02 and 0.2 per Mbp, respectively, and for BD ∼0.3 per Mbp. Therefore in the case of a γ-H2AX focus which can spread to a chromosome region up to 10–20 Mbp [Citation19], one can expect roughly ∼0.2–0.4 DSB and 2–4 TSB per focus (low-LET and 1 Gy). DSB clusters are expected for higher doses or high-LET particles where there is a non-homogenous density of breaks along the particle track and one expects even 8 DSBs per focus for α-particles based also on PARTRAC simulations [Citation35]. The electron energies studied cover the low-medium LET range (1–20 keV/μm), so the results can be used to explain some of the characteristics associated with the transition from low- to high-LET radiation. For example, the significant increase of the DSB yield (by a factor of 2) as the LET increases from 1 keV/μm (10 keV electrons) to 20 keV/μm (200 eV electrons) clearly illustrates the increase of clustering with LET, as far as the direct action is concerned. Track-structure effects are essentially related to the secondary electron spectrum. Specifically, the track-structure of X- and γ-rays is determined by sparely distributed high-energy (and low-LET) secondary electrons (>10 keV). On the other hand, the track-structure of an α-particle (or a heavy ion) is determined by densely distributed low-energy (and medium-LET) electrons (<100 eV). Therefore, theoretical simulations of electron tracks represent an important step toward understanding LET effects at the DNA level.
Comparison of DSB clustering using PFGE and nitrogen ions of varying LETs (up to 225 keV/μm), one would expect 1 DSB per cluster for γ-rays and 2–3 DSBs per cluster for ions [Citation41]. The existence of more than one DSB under a focus can be an issue for underestimation and the correct assignment of focus size can improve this situation but up to a point. The yields we found for low-LET () are compatible with previous findings of 10-20 DSB foci/Gy/cell [Citation37,Citation42,Citation43] and the same stands also for α-particles [Citation35,Citation44,Citation45] and Ar ions [Citation46], although in the specific study 40Ar ions of a much lower LET (125 keV/μm) were used. Previous studies using cold lysis PFGE protocols in order to minimize conversion of heat-labile sites to DSBs suggest relatively increased numbers of ∼25 DSBs/Gy/cell for normal human skin fibroblasts (GM5758) for low-LET radiations [Citation47]. We believe that these numbers which can vary even for PFGE-based methodologies are in agreement with our findings since PFGE approaches always tend to give increased numbers compared to γ-H2AX method [Citation34]. In addition and based on , the number of DSB (or rather γ-H2Ax foci) produced per dose decreases with increasing the LET as expected in accordance to the trend observed for cell killing for LETs >200 keV/μm [Citation48]. This can be attributed to the fact that one focus may represent more than one DSB reaching the limitation of the methodology. Another parameter to be considered here is that with increasing LET, due to an increase in recombination of generated radicals, the number of DNA lesions is reduced, as observed for oxidized bases [Citation13].
Our colocalization data presented in and suggest certainly an increase of damage complexity with dose and LET. The increase of “complexity” with increasing doses reflects more correctly the increase of probability to have colocalization of damage since the number of damages increase with dose. In each case, we have analyzed DSB foci colocalizing with either DSB or non-DSB lesions forming repair centers. Our data for DSB and non-DSB oxidized base clusters forming a complex DSB () show a low level of clustering for X-rays compared to α-particles. Based also on our current Monte Carlo calculations for track-end electrons (), one does not expect such a high level of BDs per TSB for low-LET (BD/TSB ∼1.3–2). Previous experimental findings using PFGE suggest usually a similar ratio of 1 DSB to 2–3 non-DSB clusters (including oxidized bases and abasic sites) for low-LET [Citation49]. Our experiment though using X-rays collimated by the Micro Léman mask resulted in an apparently increased localization of APE1 (abasic) sites, suggesting that the very thin (2 μm) openings of the mask induce a highly localized damage effect simulating that of high-LET particle tracks ( animated gif).
We assume that each focus represents an active repair center visualized as a focus of increased size when using immunofluorescence [Citation10,Citation50]. The size of these foci tends to increase with LET and repair time and then decrease, a finding also observed in this study ( and S6). We accept that both proteins are considered to be accurate probes for DSBs. The exact mechanisms for recruitment of 53BP1 to the DSB sites still remain an open question. The most accepted theory is that when the phosphorylated H2AX (γ-H2AX) forms megabase-size foci at a DSB this leads to the recruitment of various downstream DNA damage response (DDR) factors including 53BP1 (as a DSB sensor), BRCA1 and NSB1 enabling correct repair of DNA damage [Citation51]. There are indications that there is a preferential enrolment of 53BP1 in persistent DSBs [Citation52], late repair like for example in heterochromatic regions [Citation51] or DSB clusters [Citation7] and there is an increase of γ-irradiated cells with 53BP1 foci with dose [Citation43]. Our results for MO59J () certainly agree with this hypothesis since we also observe an increase of γ-H2AX/53BP1 colocalization at late repair times, suggesting active repair and recruitment of 53BP1 in these sites. Therefore, one can possibly use the level of γ-H2AX/53BP1 colocalization as a marker of possible repair deficiencies and/or DNA repair resistance. For example, Martin et al. [Citation53] correlated a low colocalization level with Artemis DNA repair deficiency and problems in the DNA repair complexes formed after γ-irradiation. Interestingly, earlier studies suggest ATM as the primary kinase for the phosphorylation of 53BP1 and not DNA-PKcs [Citation54]. In addition, in the specific study, no difference in the time course of 53BP1 foci appearance and disappearance was detected for both cell lines (MO59K/J) after exposure to 1 Gy of γ-radiation.
Our study has identified generally an increased amount of complex DNA damage with dose and LET. These repair centers once formed are considered to be the signature of IR and they are often associated with resistance to repair and proneness to errors and misrepair [Citation55]. Substantial evidence suggests that it is not only the prompt (direct) DSBs formed by IR that have implications on cell survival and genomic instability, but also the conversion of non-DSB clusters into DSBs during processing and attempted repair [Citation56]. This possibility is high especially when the two non-DSB lesions are located in a distance of up to 10 bp [Citation56]. The inaccurate repair of such “hybrid clusters”, consisting of DSBs and non-DSBs, can be attributed to the error-prone DNA repair pathways implicated in this case like NHEJ and its backup subpathways alt-EJ (or B-NHEJ) and BER, respectively [Citation7,Citation8,Citation37]. Previous studies by Peddi et al. [Citation57] have shown, using an adaptation of PFGE and different types and levels of DNA-PK deficiency, that these deficient cells not only exhibit as expected DSB repair problems, but also the non-DSB oxidative clustered DNA lesions processing is significantly impeded. Therefore, these repair resistant DSB sites (foci) will be expected to carry a heavy load of non-DSB damage increasing the possibility for misrepair and chromosomal breaks [Citation50].
Based on the above, one can understand the importance of measuring the induction of complex DNA damage especially in situ. As recently reviewed by Nikitaki et al. [Citation19], the corresponding foci colocalization methodologies are very sensitive compared to earlier PFGE methodologies but cannot provide details on the clustering at the few bp or at least kbp region of the chromosomal DNA. On the other hand, they provide unique in situ probing for the clustering of DNA damage, which is the key of understanding the biological effects of this phenomenon of increased complexity at the cellular level. We believe that based on the different capabilities of each approach (electrophoretic, HPLC, GC/MS, microscopy) [Citation2], one, in order to be able to draw safe conclusions on the levels and types of clustered DNA damage, should apply a combination of these methodologies and obtain a more complete picture on the synthesis and yields of the complex damage sites. Last but not least, based on our currently introduced methodology for analyzing foci frequencies in non-synchronized cell cultures according to G0–G1 or G2 subpopulations, less heterogeneous results and a decrease experimental variation are expected (see also Supplementary data). The use of other DNA repairs foci like RAD51, MDC1 for DSBs and XRCC1, for example for non-DSBs maybe can help toward the optimization of detection according to the type of genotoxic stress [Citation58]. In addition, very recent advances in fluorescence microscopy harnessing previously unused fluorescence or DNA autofluorescence by a 532-nm laser excitation [Citation59] may improve the detection of protein distributions especially located in a complex form [Citation60].
IFRA_1232484_-_SUPP_FILE.pdf
Download PDF (831 KB)Acknowledgements
The authors acknowledge the valuable help of Dr. Pavel N. Lobachevsky (Sir Peter MacCallum Department of Oncology, The University of Melbourne) for providing the “foci” software analysis JQuantPro and his technical support. The authors would like also to sincerely thank the staff at the heavy ions accelerator facility at the Centre Interdisciplinaire de Recherche Ions Lasers (CIRIL) at GANIL, Caen, France, for dosimetry and support of the heavy ion experiments, especially Dr. Isabelle Testard, Dr. Yannick Satigny, Florent Durantel (GANIL), and all the members of the Cellular Biodiagnostics group at DLR for their help during beam times (Prof. Dr. Christa Baumstark-Khan, Dr. Luis F. Spitta, Sebastian Feles, Claudia Schmitz, Sebastian Diegeler, Bikash Konda, Bernd Henschenmacher). We also sincerely thank Dr. Gabriel Pantelias (NCSR “Demokritos”) for helpful discussions and continuous support. C.E.H. acknowledges the support by the European Union program ENSAR for the travel costs to GANIL.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Related Research Data
References
- Nikjoo H, O’Neill P, Terrissol M, Goodhead DT. Quantitative modelling of DNA damage using Monte Carlo track structure method. Radiat Environ Biophys 1999;38:31–38.
- Nikitaki Z, Hellweg C, Georgakilas AG, Ravanat JL. Stress-induced DNA damage biomarkers: applications and limitations. Front Chem 2015;3:35–50.
- Pavlopoulou A, Savva GD, Louka M, Bagos PG, Vorgias CE, Michalopoulos I, Georgakilas AG. Unraveling the mechanisms of extreme radioresistance in prokaryotes: lessons from nature. Mutat Res Rev Mutat Res 2016;767:92–107.
- Goodhead DT. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int J Radiat Biol 1994;65:7–17.
- Ward JF. Biochemistry of DNA lesions. Radiat Res Suppl 1985;8:S103–S111.
- Georgakilas AG, O’Neill P, Stewart RD. Induction and repair of clustered DNA lesions: what do we know so far? Radiat Res 2013;180:100–109.
- Schipler A, Mladenova V, Soni A, Nikolov V, Saha J, Mladenov E, Iliakis G. Chromosome thripsis by DNA double strand break clusters causes enhanced cell lethality, chromosomal translocations and 53BP1-recruitment. Nucleic Acids Res 2016;44:7673–7690.
- Hegde ML, Dutta A, Yang C, Mantha AK, Hegde PM, Pandey A, et al. Scaffold attachment factor A (SAF-A) and Ku temporally regulate repair of radiation-induced clustered genome lesions. Oncotarget 2016;7:54430–54444.
- Pateras IS, Havaki S, Nikitopoulou X, Vougas K, Townsend PA, Panayiotidis MI, et al. The DNA damage response and immune signaling alliance: is it good or bad? Nature decides when and where. Pharmacol Ther 2015;154:36–56.
- Neumaier T, Swenson J, Pham C, Polyzos A, Lo AT, Yang P, et al. Evidence for formation of DNA repair centers and dose–response nonlinearity in human cells. Proc Natl Acad Sci USA 2012;109:443–448.
- Costes SV, Ponomarev A, Chen JL, Nguyen D, Cucinotta FA, Barcellos-Hoff MH. Image-based modeling reveals dynamic redistribution of DNA damage into nuclear sub-domains. PLoS Comput Biol 2007;3:e155.
- Yang N, Galick H, Wallace SS. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair 2004;3:1323–1334.
- Pouget JP, Frelon S, Ravanat JL, Testard I, Odin F, Cadet J. Formation of modified DNA bases in cells exposed either to gamma radiation or to high-LET particles. Radiat Res 2002;157:589–595.
- Hada M, Georgakilas AG. Formation of clustered DNA damage after high-LET irradiation: a review. J Radiat Res 2008;49:203–210.
- Georgakilas AG. Processing of DNA damage clusters in human cells: current status of knowledge. Mol Biosyst 2008;4:30–35.
- Watanabe R, Rahmanian S, Nikjoo H. Spectrum of radiation-induced clustered non-DSB damage – a Monte Carlo track structure modeling and calculations. Radiat Res 2015;183:525–540.
- Nikjoo H, Emfietzoglou D, Liamsuwan T, Taleei R, Liljequist D, Uehara S. Radiation track, DNA damage and response. Rep Prog Phys 2016;79:116601 (55pp).
- Nikjoo H, Uehara S, Wilson WE, Hoshi M, Goodhead DT. Track structure in radiation biology: theory and applications. Int J Radiat Biol 1998;73:355–364.
- Nikitaki Z, Nikolov V, Mavragani IV, Plante I, Emfietzoglou D, Iliakis G, Georgakilas AG. Non-DSB clustered DNA lesions. Does theory colocalize with the experiment? Radiat Phys Chem 2016;128:26–35.
- Anderson CW, Dunn JJ, Freimuth PI, Galloway AM, Allalunis-Turner MJ. Frameshift mutation in PRKDC, the gene for DNA-PKcs, in the DNA repair-defective, human, glioma-derived cell line M059J. Radiat Res 2001;156:2–9.
- Ivashkevich AN, Martin OA, Smith AJ, Redon CE, Bonner WM, Martin RF, Lobachevsky PN. γH2AX foci as a measure of DNA damage: a computational approach to automatic analysis. Mutat Res 2011;711:49–60.
- Emfietzoglou D, Karava K, Papamichael G, Moscovitch M. Monte Carlo simulation of the energy loss of low-energy electrons in liquid water. Phys Med Biol 2003;48:2355–2371.
- Emfietzoglou D, Nikjoo H. The effect of model approximations on single-collision distributions of low-energy electrons in liquid water. Radiat Res 2005;163:98–111.
- Goodhead DT, Nikjoo H. Track structure analysis of ultrasoft X-rays compared to high- and low-LET radiations. Int J Radiat Biol 1989;55:513–529.
- Liamsuwan T, Uehara S, Emfietzoglou D, Nikjoo H. Physical and biophysical properties of proton tracks of energies 1 keV to 300 MeV in water. Int J Radiat Biol 2011;87:141–160.
- Nikjoo H, Uehara S, Emfietzoglou D, Pinsky L. A database of frequency distributions of energy depositions in small-size targets by electrons and ions. Radiat Prot Dosimetry 2011;143:145–151.
- Semenenko VA, Stewart RD. Fast Monte Carlo simulation of DNA damage formed by electrons and light ions. Phys Med Biol 2006;51:1693–1706.
- Nikjoo H, O’Neill P, Wilson EW, Goodhead D. Computational approach for determining the spectrum of DNA damage induced by ionizing radiation. Radiat Res 2001;156:577–583.
- Nikjoo H, O’Neill P, Terrissol M, Goodhead DT. Modelling of radiation-induced DNA damage: the early physical and chemical event. Int J Radiat Biol 1994;66:453–457.
- Emfietzoglou D, Papamichael G, Pathak A, Fotopoulos A, Nikjoo H. Monte-Carlo study of energy deposition by heavy charged particles in sub-cellular volumes. Radiat Prot Dosimetry 2007;126:457–462.
- Nikjoo H, Goodhead DT. Track structure analysis illustrating the prominent role of low-energy electrons in radiobiological effects of low-LET radiations. Phys Med Biol 1991;36:229–238.
- Friedland W, Dingfelder M, Kundrát P, Jacob P. Track structures, DNA targets and radiation effects in the biophysical Monte Carlo simulation code PARTRAC. Mutat Res 2011;711:28–40.
- Mladenova V, Mladenov E, Iliakis G. Novel biological approaches for testing the contributions of single-DSBs and DSB-clusters to the biological effects of high-LET-radiation. Front Oncol 2016;6:163.
- Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res 2008;36:5678–5694.
- Antonelli F, Campa A, Esposito G, Giardullo P, Belli M, Dini V, et al. Induction and repair of DNA DSB as revealed by H2AX phosphorylation foci in human fibroblasts exposed to low- and high-LET radiation: relationship with early and delayed reproductive cell death. Radiat Res 2015;183:417–431.
- Georgakilas AG, Bennett PV, Wilson III DM, Sutherland BM. Processing of bistranded abasic DNA clusters in gamma-irradiated human hematopoietic cells. Nucleic Acids Res 2004;32:5609–5620.
- Niimi A, Yamauchi M, Limsirichaikul S, Sekine R, Oike T, Sato H, et al. Identification of DNA double strand breaks at chromosome boundaries along the track of particle irradiation. Genes Chromosomes Cancer 2016;55:650–660.
- Okayasu R. Repair of DNA damage induced by accelerated heavy ions – a mini review. Int J Cancer 2011;130:991–1000
- Ward JF, Blakely WF, Joner EI. Mammalian cells are not killed by DNA single-strand breaks caused by hydroxyl radicals from hydrogen peroxide. Radiat Res 1985;103:383–392.
- Verkhovtsev A, Surdutovich E, Solov’yov AV. Multiscale approach predictions for biological outcomes in ion-beam cancer therapy. Sci Rep 2016;6:27654.
- Fakir H, Sachs RK, Stenerlow B, Hofmann W. Clusters of DNA double-strand breaks induced by different doses of nitrogen ions for various LETs: experimental measurements and theoretical analyses. Radiat Res 2006;166:917–927.
- Nakajima NI, Brunton H, Watanabe R, Shrikhande A, Hirayama R, Matsufuji N, et al. Visualisation of gammaH2AX foci caused by heavy ion particle traversal; distinction between core track versus non-track damage. PLoS One 2013;8:e70107.
- Asaithamby A, Chen DJ. Cellular responses to DNA double-strand breaks after low-dose gamma-irradiation. Nucleic Acids Res 2009;37:3912–3923.
- Staaf E, Brehwens K, Haghdoost S, Czub J, Wojcik A. Gamma-H2AX foci in cells exposed to a mixed beam of X-rays and alpha particles. Genome Integr 2012;3:8.
- Leatherbarrow EL, Harper JV, Cucinotta FA, O’Neill P. Induction and quantification of gamma-H2AX foci following low and high LET-irradiation. Int J Radiat Biol 2006;82:111–118.
- Burns FJ, Tang MS, Wu F, Schmid E. Linking gamma-H2AX foci and cancer in rat skin exposed to heavy ions and electron radiation. Health Phys 2015;109:157–170.
- Stenerlow B, Karlsson KH, Cooper B, Rydberg B. Measurement of prompt DNA double-strand breaks in mammalian cells without including heat-labile sites: results for cells deficient in nonhomologous end joining. Radiat Res 2003;159:502–510.
- Durante M. New challenges in high-energy particle radiobiology. Br J Radiol 2014;87:20130626.
- Francisco DC, Peddi P, Hair JM, Flood BA, Cecil AM, Kalogerinis PT, et al. Induction and processing of complex DNA damage in human breast cancer cells MCF-7 and non-malignant MCF-10A cells. Free Radic Biol Med 2008;44:558–569.
- Asaithamby A, Hu B, Chen DJ. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc Natl Acad Sci USA 2011;108:8293–8298.
- Kleiner RE, Verma P, Molloy KR, Chait BT, Kapoor TM. Chemical proteomics reveals a γH2AX-53BP1 interaction in the DNA damage response. Nat Chem Biol 2015;11:807–814.
- Minakawa Y, Atsumi Y, Shinohara A, Murakami Y, Yoshioka K-i. Gamma-irradiated quiescent cells repair directly induced double-strand breaks but accumulate persistent double-strand breaks during subsequent DNA replication. Genes Cells 2016;21:789–797.
- Martin OA, Ivashkevich A, Choo S, Woodbine L, Jeggo PA, Martin RF, Lobachevsky P. Statistical analysis of kinetics, distribution and co-localisation of DNA repair foci in irradiated cells: cell cycle effect and implications for prediction of radiosensitivity. DNA Repair (Amst) 2013;12:844–855.
- Rappold I, Iwabuchi K, Date T, Chen J. Tumor suppressor P53 binding protein 1 (53bp1) is involved in DNA damage-signaling pathways. J Cell Biol 2001;153:613–620.
- Schipler A, Iliakis G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res 2013;41:7589–7605.
- Eccles LJ, Lomax ME, O’Neill P. Hierarchy of lesion processing governs the repair, double-strand break formation and mutability of three-lesion clustered DNA damage. Nucleic Acids Res 2010;38:1123–1134.
- Peddi P, Loftin CW, Dickey JS, Hair JM, Burns KJ, Aziz K, et al. DNA-PKcs deficiency leads to persistence of oxidatively induced clustered DNA lesions in human tumor cells. Free Radic Biol Med 2010;48:1435–1443.
- Rothkamm K, Barnard S, Moquet J, Ellender M, Rana Z, Burdak-Rothkamm S. DNA damage foci: Meaning and significance. Environ Mol Mutagen 2015;56:491–504.
- Dong B, Almassalha LM, Stypula-Cyrus Y, Urban BE, Chandler JE, Nguyen T-Q, et al. Superresolution intrinsic fluorescence imaging of chromatin utilizing native, unmodified nucleic acids for contrast. Proc Natl Acad Sci 2016;113:9716–9721.
- Wu Y, Chandris P, Winter PW, Kim EY, Jaumouillé V, Kumar A, et al. Simultaneous multiview capture and fusion improves spatial resolution in wide-field and light-sheet microscopy. Optica 2016;3:897–910.