
Abstract
Bardoxolone-methyl (BAR) is reported to have anti-inflammatory, anti-proliferative and anti-fibrotic effects. BAR activates Nrf2 and may ameliorate oxidative stress through induction of antioxidant genes. However, off-target effects, probably concentration and NFkB-dependent, have limited the clinical use of BAR. Nrf2 regulates expression of antioxidant and mitochondrial genes and has been proposed as a target for both obesity and breast cancer. Therefore, we explored whether BAR can alter migration and proliferation in the MCF7 cell line and whether metabolic function is affected by BAR. Incubation with BAR caused a time-dependent migratory inhibition and an associated decrease in mitochondrial respiration. Both migratory and mitochondrial inhibition by BAR were further enhanced in the presence of fatty acids. In addition to the activation of Nrf2, BAR altered the expression of target mRNA GCLC and UCP1. After 24 h, BAR inhibited both glycolytic capacity, reserve (p < 0.05) and oxidative phosphorylation (p < 0.001) with an associated increase in mitochondrial ROS and loss of intracellular glutathione in MCF7 cells; however, impairment of mitochondrial activity was prevented by N-acetyl cysteine. The fatty acid, palmitate, increased mitochondrial ROS, impaired migration and oxidative phosphorylation but palmitate toxicity towards MCF7 could not be inhibited by N-acetyl cysteine suggesting that they exert effects through different pathways. BAR-activated AKT, induced DNA damage and inhibited cell proliferation. When the proteasome was inhibited, there was loss of BAR-mediated changes in p65 phosphorylation and SOD2 expression suggesting non-canonical NFkB signaling effects. These data suggest that BAR-induced ROS are important in inhibiting MCF7 migration and metabolism by negatively affecting glycolytic capacity and mitochondrial function.
Introduction
Despite the decline in the mortality of patients with breast cancer (BC) in developed countries, it remains the second leading cause of death among all cancers [Citation1]. BC, particularly the estrogen receptor (ER)-positive BC, is strongly linked to obesity and with the worldwide increase in prevalence of obesity, and the incidence of BC is expected to rise [Citation2]. Alterations to cell metabolism are common between obesity and BC; on the one hand, obesity associates with elevated circulating concentrations of saturated fatty acids that can drive cells towards inflammation and glycolysis [Citation2]. On the other hand, transformed cells predominantly display high rates of aerobic glycolysis [Citation3–5]. Aerobic glycolysis is essential for cancer cell growth and proliferation, where the metabolic switch from oxidative phosphorylation (OXPHOS) to glycolysis is commonly known as the Warburg effect [Citation6]. Malignant cells typically meet their energy needs by producing ATP through aerobic glycolysis, however, surrounding normal cells produce energy through “aerobic” OXPHOS in the mitochondrion using both fatty acids and glucose as substrates [Citation7]. Migration from primary tumor sites to distant sites is a pre-requisite for metastatic potential and is absolutely dependent on energy availability. As the major cause of morbidity in cancer is due to metastases within distant tissues and organs [Citation8], it is pertinent to study the effects of anti-cancer compounds on energy and migration.
Complex 1 is an important site for generation of ROS when the respiratory chain is blocked [Citation9]. A recent study has shown that loss of an accessory subunit of Complex 1 promoted BC cell proliferation, migration, and invasion [Citation10]. This was associated with an increase in production of mitochondrial ROS, loss of mitochondrial DNA, and modulation of AKT signaling and autophagy. At the same time, cells that were depleted of the accessory subunit of Complex 1 were able to meet their metabolic energy requirements by increased uptake of glucose and reduced lactate secretion indicative of aerobic glycolysis. In addition to providing energy for cell metabolism, mitochondria are increasingly recognized as an important target in BC because they are a source of ROS, and regulate apoptosis, autophagy, and migration.
Triterpenoids have been demonstrated in many studies as promising anticancer agents against BC, particularly the ER-positive MCF7 cells [Citation11]. Bardoxolone-methyl (BAR), or RTA402, is a synthetic triterpenoid that has been under clinical investigation for the treatment of chronic kidney disease associated with type 2 diabetes and pulmonary hypertension. However, off-target cardiovascular effects led to the termination of Phase III clinical trials [Citation12]. More recently, the off-target effect was explained as being due to inhibition of NFkB by BAR which mediates the loss of endothelin and its receptor ET1A leading to hypertension and adverse outcomes [Citation13]. Improved understanding the off-target effects of BAR may facilitate the design of more specific molecules.
Previous studies have shown that BAR specifically targets BC epithelial cells rather than non-malignant cells [Citation14]. A number of existing BC drugs target PI3K/AKT [Citation15]. Although the mechanism for the anticancer activities of BAR are not fully understood, many signaling pathways, including mitochondrial regulation via the KEAP1/Nrf2 pathway and glycolytic regulation via the PI3K/mTOR pathway, have been implicated in the anticancer effects of BAR [Citation16]. In a Phase I clinical trial of solid tumors and lymphomas, BAR achieved nanomolar concentrations in plasma, activated the Nrf2 pathway as determined by an increase in NQO1 in circulating leukocytes; however, a paradoxical increase in DNA fragmentation was also observed [Citation17]. Whether BAR induces an effective antioxidant response and whether the increase in DNA fragmentation induced by BAR is attributable to oxidative damage was not investigated.
In this study, we have investigated the hypothesis that BAR impairs migration, glycolytic metabolism and mitochondrial function in MCF7 cells by increasing oxidative stress.
Materials and methods
Cell culture
MCF7 (HPA Cultures Collections, Salisbury, UK), human BC epithelial cell line, was cultured in RPMI 1640 with stable glutamine (PAA, Hampshire, UK) supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin (50 U/ml) and incubated at 37 °C with 5% CO2. Cells were seeded at a density of 2 × 105 cells per ml in T75 and allowed to reach 80–90% confluence prior to sub-culture.
Mitochondrial metabolism: MTT assay
Mitochondrial metabolic activity was determined using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] colorimetric assay. Cells were seeded at a density of 5 × 104 cells/ml in tissue culture plates and allowed to adhere overnight. Cells were exposed to treatment for up to 48 h prior to addition of MTT reagent [0.5 mg/ml in phosphate-buffered saline (PBS)] and incubation for 4 h at 37 °C with 5% CO2. Cells were lysed and formazan solubilised over 24 h with lysis buffer [0.7 M sodium dodecyl sulfate (SDS), 50% N,N-dimethylformamide (DMF), pH 4.7] and absorbance at 570 nm determined using a spectrophotometer [Citation18].
Western blot
Protein expression was measured by Western blot. Cells were exposed to treatments for up to 24 h and then were collected in lysis buffer [150 mM NaCl, 1% Triton X-100, 0.5% SDS, 50 mM Tris–base, 0.1% protease inhibitor cocktail (AEBSF 104 mM, aprotinin 80 μM, bestatin 4 mM, E-64 1.2 mM, leupeptin 2 mM, pepstatin A 1.5 mM) pH 8.0] and DNA was sheared by passing 10 times through a 25-gauge needle. Protein concentration was determined using BCA reagent and 25 μg total protein used for an immunoblot. Samples were prepared with Laemmli buffer (1:1 v/v) and heated at 95 °C for 5 min before loading into SDS–PAGE gels (16%). SDS–PAGE was carried out in running buffer (25 mM Tris–base, 190 mM glycine, 0.1% SDS) for 120 min at 115 V. Separated proteins were transferred to Hybond™-P membrane (GE Healthcare, Amersham, UK) in transfer buffer (25 mM Tris–Base, 190 mM glycine, 20% methanol) for 70 min at 240 mA. Membranes were blocked with 3% bovine serum albumin (BSA) in Tris-buffered saline (TBS)–Tween (50 mM Tris–base, 200 mM NaCl, 0.05% Tween-20, pH 7.5) overnight at 4 °C. Membranes were probed with primary antibody for 2 h in TBS–Tween (0.2% BSA) followed by probing with secondary detection antibody. Phosphospecific antibodies against ERK (Thr-202, Tyr-204), p38 (Thr-180, Tyr-182), AKT (Ser-473), p65 (Ser-536), SAPK/JNK (Thr183/Tyr185), H2AX (Ser139) in addition to PARP-1 (9542) and Mcl-1 (5453) were purchased from Cell Signaling Technology (Danvers, MA). Antibodies against AKT1/2 (N-19) and β-actin (C-11) were obtained from Santa Cruz Biotechnologies (Dallas, TX). Secondary antibodies were HRP-conjugated anti-mouse IgG (Sigma, Dorset, England, UK) and anti-rabbit IgG (Abcam, Cambridge, UK). Visualization of bound secondary antibody was by enhanced chemiluminescence [Citation18].
Mitochondrial superoxide generation
Mitochondrial superoxide was determined in MCF7 by incubating with MitoSOX Red (1.25 μM; Invitrogen, Paisley, UK) for 30 min exactly, trypsinised, pelleted and was analysed immediately by flow cytometry according to the manufacturer’s instructions using a Beckman Coulter FC500 flow cytometer (High Wycombe, UK) [Citation19].
Cell migration scratch assay
Prior to the assay, cells were seeded (3 × 105 cells/well) in a 12-well plate and incubated overnight. The next day, cells were scratched and synchronized using serum-free media for 2 h. BAR and fatty acids were added in a low-serum media (2% FCS) and placed in the Cell-IQ system. Cell migration was measured by a Cell-IQ cell culturing platform (Chip-Man Technologies, Tampere, Finland). Each analysis included three replicate image sites.
Fatty acid preparation, cell treatments, times, and concentrations
Fatty acid stock solutions, 200 mM were prepared by dissolving each fatty acid in a 0.1 M NaOH in 70% ethanol solution, and left on a hot plate stirrer (50–60 °C) until completely dissolved. Working solutions were freshly prepared by mixing the stock solutions in 5% BSA solution (final concentration 5 mM). The mixture was then equilibrated by being. The pH was adjusted to 7.4 and sterile filtered.
Analysis of metabolic phenotype
MCF7 cells in 24-well plates were incubated in bicarbonate-free PBS supplemented with 5 mM glucose, 2 mM l-glutamine, and 1 mM sodium pyruvate 1 h prior to analysis. Extracellular acidification rates (ECAR) and oxygen consumption rates (OCR) were measured under basal conditions and the MitoStress test using XF-24 Extracellular Flux Analyzer (Seahorse Bioscience, Agilent Technologies, Santa Clara, CA).
RNA extraction and quantitative polymerase chain reaction
Nrf2-regulated oxidoreductive genes, UCP1 and GCLC and the NFkB target gene, SOD2, were measured in MCF7 cells with quantitative polymerase chain reaction (qPCR). Total RNA was extracted from the cells (1 × 106) by using TriZol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. cDNA was synthesised according with the manufacturer’s instructions (Quanta Biosciences Inc., Gaithersburg, MD). Reverse-transcription qPCR was performed in an Agilent Mx3000PTM QPCR System (Agilent Technologies LDA UK Ltd, Stockport, UK) of selected genes with commercially available primers from Primer Design (Chalfont's Foot, UK) for UCP1, GCLC, SOD2, β-actin and YWHAZ. YWHAZ expression was used for normalization. Relative fold change for each gene was calculated using the delta delta Ct method.
Statistics
Statistical analyses were carried out using one-way analysis of variance (ANOVA) or two-way ANOVA with Dunnett’s or Bonferroni’s multiple comparison post-test. Statistics were calculated using Prism 7.0.
Results
We have investigated the effects of BAR on MCF7 migration as a surrogate for metastatic potential. We show for the first time that BAR inhibits MCF7 migration in a time-dependent manner (); the wound size reduced to only 85 ± 10.5% of original size after 24 h in the presence of BAR compared to 55 ± 8.3% of wound size due to cell migration in untreated controls (p < 0.05). As the microenvironment in BC is associated with increased free fatty acids that affect metabolism, we examined the effects of BAR on migration in the presence of the major saturated fatty acid (palmitate) and monounsaturated fatty acid (oleate). Both palmitate and oleate added to the effects of BAR and significantly inhibited cell migration over 24 h ().
Figure 1. BAR and fatty acids impair MCF7 migration. MCF7 cells were seeded (3 × 105 cells/well) in a 12-well plate and incubated overnight. The next day, cells were scratched and synchronized using serum-free media. After 2 h, a low-serum medium was added with and without BAR (50 and 100 nM) and with and without fatty acid (25 μM). Plates were incubated and migration was monitored in the Cell-IQ system for up to 72 h. (A) Increasing BAR for up to 24 h; (B) BAR in the presence and absence of oleate; and (C) BAR in the presence and absence of palmitate. Results are expressed as the mean ± standard error of mean (SEM) of at least three independent experiments where asterisk represents p < 0.05 compared to untreated control and hash represents p < 0.05 by two-way ANOVA for time and treatment.
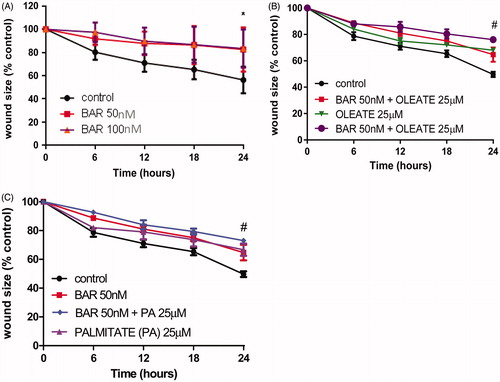
To explore whether mitochondrial metabolism may be a target for the effects of BAR, we used the MTT assay to examine the effect of time and BAR concentration on metabolic activity. BAR (100 nM) inhibited MTT reduction significantly after 24 h and after incubation for 48 h a significant inhibitory effect of BAR was observed at 50 and 100 nM (). Palmitate added to the effect of BAR and caused further inhibition of MTT reduction ().
Figure 2. BAR impairs MCF7 mitochondrial activity. MCF7 cells were seeded at a density of 5 × 104 cells/ml, allowed to adhere overnight, then exposed to treatment for up to 48 h. MTT reagent (0.5 mg/ml in PBS) was added and incubated with cells for the final 4 h. (A) Increasing BAR concentration over 24 h. (B) Increasing BAR concentration over 24 h. (C) Co-incubation with increasing BAR concentration in the presence of palmitate (25 μM) for 24 h; and (D) 48 h. Results are expressed as the mean ± standard error of mean (SEM) of at least three independent experiments where, **p < 0.01, ***p < 0.005 and ****p < 0.0001. Hash represents p < 0.05 for palmitate treatment compared to no palmitate treatment.
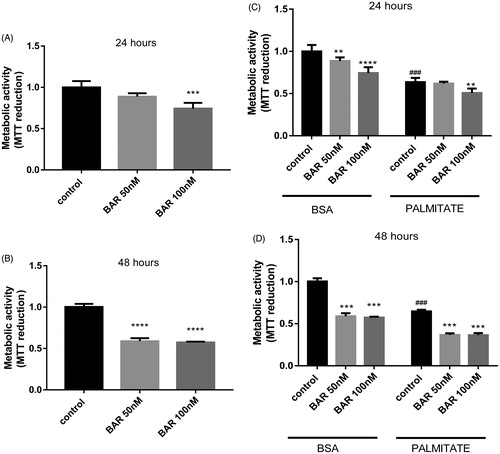
BAR has been reported to affect mitochondrial regulation via the KEAP1/Nrf2 pathway. Therefore, we validated Nrf2 activation in MCF7 cells by examining the expression of two Nrf2 target genes, UCP1 and GCLC. As predicted, we observed a concentration dependent increase in GLCL and a decrease in expression of UCP1 after 24 h (). Palmitate caused significant down-regulation of UCP1 but had no effect on GCLC expression (Supplementary Figure S1(a,b)). To explore whether the effect of palmitate was due to PPARγ inhibition, we co-incubated with rosiglitazone, a PPARγ agonist. Rosiglitazone did not prevent the effect of palmitate. Despite the increase in GCLC expression, we observed a significant increase in Mitosox oxidation and loss of glutathione (GSH) in the presence of BAR at 24 h (). Interestingly, BAR in combination with palmitate caused an additional increase of mitochondrial superoxide production. In parallel, the mitochondria-expressed SOD2 gene was also reduced significantly by BAR ().
Figure 3. BAR affects MCF7 antioxidant gene expression relative to YWHAZ and increases oxidative stress. MCF7 cells were seeded (3 × 105 cells/well) overnight prior to treatment with BAR (0–100 nM), palmitate (25 μM) and rosiglitazone for 24 h. Cells were removed by scraping and RNA was extracted for qPCR analysis of (A) GCLC; (B) UCP1; and (E) SOD2 mRNA. (C) For measuring oxidative stress, superoxide was determined in MCF7 by incubating with MitoSOX Red (1.25 μM) for 30 min prior to scraping and analysis by flow cytometry and (D) GSH was determined using the recycling assay. Results are expressed as the mean ± standard error of mean of at least three independent experiments where asterisk represents p < 0.05, **p < 0.01, ***p < 0.005 and ****p < 0.0001. Hash represents p < 0.05 for palmitate treatment compared to no palmitate treatment.
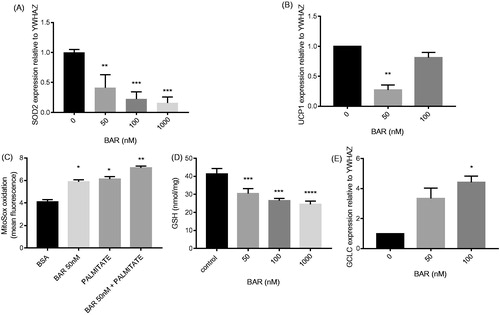
Our data showed that BAR elicited changes in mitochondrial gene expression and loss of mitochondrial reductive activity. Therefore, we used an extracellular flux analyzer to measure in vitro OCR and extracellular acidification rate (ECAR) to measure OXPHOS and glycolysis, respectively. BAR-inhibited OXPHOS in a concentration-dependent manner by 24 h which was measured as a significantly slower rate of oxygen consumption (). After 24 h BAR (50 nM) caused significant loss in glycolytic activity but not glycolysis in MCF7 cells (p < 0.05), measured as glycolytic capacity and glycolytic reserve. However, at 100 nM, only the glycolytic reserve was significantly reduced (). We have previously shown in MCF7 cells that an increase in ROS production can impair mitochondrial activity and induce DNA damage. Here, we have also observed a decrease in GSH in association with loss of OCR and MTT reduction during BAR treatment. N-acetyl cysteine (NAC) offered protection against BAR and significantly restored metabolic activity for BAR exposure in MCF7 cells (). In contrast, NAC did not protect against the inhibitory effect of palmitate (Supplementary Figure S1(c)).
Figure 4. BAR impairs metabolism in MCF7 cells and this effect is prevented by incubation with NAC. ECAR and OCRs were measured in MCF7 cells under basal conditions and using the MitoStress test using XF-24 Extracellular Flux Analyzer (A) OCR with increasing concentration of BAR for 24 h; (B) ECAR with increasing concentration of BAR for 24 h; (C) metabolic activity with increasing concentration of BAR in the presence of 3 mM NAC for 24 h; and (D) for 48 h. Results are expressed as the mean ± standard error of mean of at least three independent experiments where asterisk represents p < 0.05, **p < 0.01 and ***p < 0.005.
In order to identify the concentrations of BAR that would impair proliferation over 24 h, we used a crystal violet assay to titre the effects of BAR. While lower concentrations of BAR (≤100 nM) affected MCF7 metabolism they did not inhibit proliferation; however, at 500 nM and above, BAR-inhibited metabolism and cell proliferation (). The AKT/mTOR pathway regulates autophagy and metabolism. We therefore studied early signaling events of BAR. The effect of 1 μM BAR for short- and long-exposure time on the morphology, expression levels of phosphorylated AKT, as well as apoptosis were investigated. showed the activation of AKT by 1 μM BAR after 1 h which associated with pJNK expression and loss of PARP suggesting DNA damage. By 24 h after BAR treatment, AKT activity returned to baseline. In contrast, P38 and the ERK pathways were not activated after 1 h 1 μM BAR but increased phosphorylation of P38 and ERK was evident at 24 h. Maximal activation of AKT by 1 μM BAR occurred after 6 h exposure; however, low concentrations of BAR (50 or 100 nM) did not induce AKT phosphorylation. p38 and ERK expression continued to increase until 9 h (). In contrast, BAR (1 μM) inhibited p65 expression at 6 h (). Based on those results showing the AKT phosphorylation together with the absence of signs for cell death and/or apoptosis at 6 h, we believe that AKT activation is a cause rather than a consequence for cell death. We hypothesized that targeting the AKT survival signalling together with BAR would halt the cells proliferation.
Figure 5. BAR activates AKT within 6 h and later activates the DNA damage response and NFkB signalling. MCF7 cells were seeded (3 × 105 cells/well) overnight prior to treatment with BAR in the presence and absence of MG132 (proteasome inhibitor) and LY294002 (AKT inhibitor). MCF7 cells were exposed to BAR (1 μM) for up to 24 h and then were collected in lysis buffer for light microscopy (A) or Western blotting to interrogate key signalling pathways. (B) Signalling events were studied by Western blotting for up to 9 h BAR treatment (1 μM). (C) Effect of MG132 and LY294002 co-incubation with BAR for 6 h on cell morphology viewed by light microscopy. (D) Crystal violet proliferation assay for BAR (10–1000 nM) incubated with increasing concentrations of MG132 and results are expressed as the mean ± standard error of mean of at least three independent experiments where asterisk represents p < 0.05, and (E) the synergistic effect of NFkB inhibition on the DNA damage response.
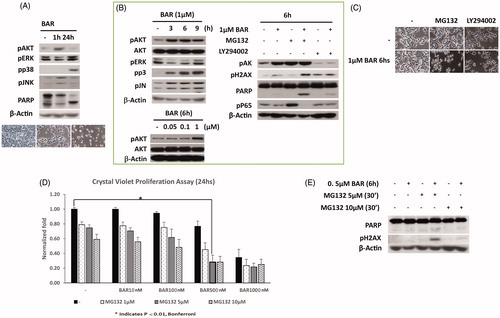
To explore whether the effect of BAR on AKT promotes survival, we investigated the effects of co-incubation of BAR for 6 h with LY294002 which inhibits AKT activity. LY294002 alone elicited morphological changes consistent with autophagy within 6 h as shown in terms of reduced number of cells, whereas the proteasome inhibitor MG132 alone had little effect (). In the presence of BAR, MG132 but not LY294002 exerted morphological changes in MCF7 cells. We examined the potential downstream signalling pathways and observed that AKT inhibition was not responsible for the changes in DNA damage signalling induced by BAR; however, in the presence of MG132 there was an increase in PARP and DNA damage foci (measured as γH2AX) accumulation. MG132 alone inhibited the canonical NFkB pathway as phosphorylated p65 levels were increased; the increase in phospho-p65 level by MG132 was eliminated in the presence of BAR suggesting that the non-canonical p65 signalling pathway is inhibited by BAR and this may explain the decrease in SOD2 expression with BAR.
Finally, we confirmed a synergistic effect for the combined BAR (500 nM) and MG132 (5 μM) exposure () with evidence of increased PARP degradation and phosphorylated gamma H2AX compared to single treatments only after 6 h ().
Discussion
For the first time, we show that sub-lethal concentrations of BAR inhibit MCF7 migration, probably by impairing energy supply from glycolytic and mitochondrial metabolism. BAR exposure increased Mitosox oxidation and depleted GSH, indicative of an oxidative stress environment. In a self-perpetuating loop, the associated increase in mitochondrial superoxide following BAR exposure may contribute to metabolic inhibition and probably impaired migration as previously described in ovarian carcinoma [Citation20]. In combination, the loss of glycolytic capacity that was seen after BAR treatment in our study, suggests that in conditions of increased metabolic demand, such as migration, the cell is compromised. The importance of oxidative stress for the metabolic inhibition of induced by BAR in MCF7 cells was demonstrated by the protective effect of NAC.
Cell migration determines the energetic requirements of cancer cells [Citation21] and in turn, migration is modulated by energy availability [Citation20]. Mitochondria are important players for the maintenance of biosynthetic and energetic capacities of cells. They also act as one of the primary sites of apoptosis and autophagy regulation. For the present study of BAR on metabolism, we selected concentrations of BAR and exposed MCF7 for increasing times that did not induce cell death to avoid complicating the data interpretation by death-dependent changes in mitochondria.
The KEAP1-Nrf2 pathway is a master regulator of cellular homeostasis and BAR is an agonist of Nrf2; it binds to KEAP1 and prevents interaction with CUl1 thereby releasing active Nrf2 [Citation22]. Once activated by an electrophile, Nrf2 mediates expression of antioxidant genes including GCLC and heme oxygenase-1 (HO-1) that stimulates mitochondrial biogenesis to increase the efficiency of OXPHOS and reduce ROS [Citation23]. Due to its antioxidant properties, Nrf2 activation is considered to be protective against cancer development by preventing DNA damage. Indeed, low or undetectable expression of Nrf2 has been observed in breast tumors and this was associated with greater cell migratory potential [Citation24]. Effectors of the antioxidant response include uncoupling protein-1 (UCP1), an Nrf2 target gene that is used to dissipate energy by generating heat [Citation25], and GCLC, which is the rate limiting enzyme in the formation of GSH. However, lower expression of UCP1 as seen here in response to BAR may reduce lipid metabolism, a condition favored by BC cells.
In our study, we have identified that BAR increased GCLC expression which supports Nrf2 involvement in the mechanism of BAR effect. However, contrary to the switch away from mitochondrial metabolism and the increase in GSH that was expected, we observed an increase in Mitosox oxidation and depletion of GSH in the presence of BAR. This suggests that Nrf2 activation might not be the only pathway that is activated by BAR as an overall antioxidant effect was not induced. Others have examined the multiple target effects of BAR and suggest that it may also modulate AKT, PPARγ, FOXO3a and NFkB signalling according to its concentration [Citation26]. The pattern of UCP1 suppression at 50 nM and then its recovery at 100 nM matches the pattern of change in the ECAR () for both glycolysis and glycolytic capacity. A potential explanation for this lies in the concentration-dependent activation of signalling pathways. Liby et al. (2012) illustrates that low concentrations of BAR target Nrf2 and would be predicted to suppress UCP1 expression, whereas intermediate BAR concentrations target PPARγ [Citation27]. PPARγ with PGC1α is an activator of UCP1 [Citation28]. Higher concentrations of BAR are therefore predicted to increase UCP1 expression and this was supported by our own experiments (data not shown). BAR may therefore be exerting important off-target effects in MCF7 cells to inhibit migration that relate to changes in mitochondrial metabolism.
To investigate putative BAR targets, we used Western blotting to examine ERK, MAPK, JNK, AKT, the DNA damage response and NFkB signalling. Consistent with an increase in oxidative stress, we observed that BAR induces DNA damage in MCF7 cells (PARP, H2AX and JNK signalling); this may be a consequence of mitochondrial and nuclear DNA damage.
Synthetic oleanane triterpenoids such as BAR are also potent inhibitors of the NF-κB inflammatory pathway through direct inhibition of IKKβ kinase activity and indirectly through inducing an antioxidant response [Citation26]. Previous studies have described that SOD2 expression is upregulated in aggressive BC via constitutive NFkB activation and it prevents the induction of a DNA damage response [Citation29]. Here, we observed that BAR is anti-inflammatory and decreases the phosphorylation of p65 and expression of SOD2. We would therefore predict that mitochondrial rather than nuclear DNA maybe an important target for oxidation because mitochondrial SOD2 expression is reduced by BAR and there was no detectable increase in GSH following BAR exposure. DNA damage has been shown previously to associate with impaired mitochondrial function and cell migration [Citation30] lending support to our observation of an increase in DNA damage, decreased migration and impaired mitochondrial activity in MCF7 cells after BAR treatment.
AKT is reported to be a target for BAR [Citation26]. AKT is an upstream activator of mTOR that increases the glycolytic rate and suppresses autophagy. By 24 h of BAR treatment, AKT and glycolytic reserve were impaired. After 6 h, we observed that the AKT inhibitor LY294002 introduced morphological changes in MCF7 cells that are suggestive of an increase in autophagy. In summary, our data show that after BAR exposure, the AKT pathway is activated but later (after 24 h) AKT signalling was negligible. Moreover, when AKT was inhibited with the pan-inhibitor LY294002, there was no effect on the DNA damage pathway, indicating that AKT was not a key pathway affected by BAR in MCF7 cells.
MCF7, in common with primary cancer cells, are reported to have a high glycolytic rate, supporting the use of this cell line as a good candidate for studying effects on the Warburg effect [Citation31]. We have previously shown in monocytes that saturated fatty acids can induce mitochondrial ROS, GSH depletion and can direct metabolism towards glycolysis [Citation19,Citation32]. Here, we have shown that the fatty acids, palmitate or oleate, inhibit mitochondrial metabolism in MCF7 cells and this inhibitory effect was enhanced by BAR; palmitate also elicited a significant increase in Mitosox oxidation and the effect of co-incubation with BAR was additive. Recent studies have reported that circulating fatty acids can influence the outcome to BC therapy [Citation33]; this may be due to fatty acid effects on metabolic programming. Evidence from large scale, well-executed epidemiological studies with long-term follow-up have highlighted an increased incidence of BC in obese post-menopausal women and in women with high dietary fat intake [Citation34]. In addition, BC cells overexpressing enzymes involved in fatty acid synthesis show poor prognosis [Citation35]. Together these data suggest that fatty acid metabolism merits further investigation in therapeutic targeting for BC.
Others have shown that a triterpenoid from wild gourd can elicit metabolic changes leading to autophagy and apoptosis in MCF7 cells via PPARγ activation [Citation36]. While PPARγ is upstream of many of the effectors that are influenced by BAR, the PPARγ agonist rosiglitazone did not recapitulate the effects of BAR on gene expression or prevent loss of metabolic activity. Others have shown that FOXO3a is a target for triterpenoids in cancer [Citation15,Citation37].
Our data suggests that BAR is anti-anti-inflammatory and anti-proliferative in MCF7 cells and by acting on an unknown target causes inhibition of non-canonical NFkB signalling that causes; (i) the decrease in phosphorylation of p65, leading indirectly to the loss of MnSOD and inhibition of migration; (ii) the inhibition of OXPHOS, ATP production and migration; and (iii) the inhibition of AKT and indirectly, a decrease in glycolysis and an increase in autophagy. This may offer an explanation for the effects of BAR on BC cell migration, glycolysis and mitochondrial metabolism, and open up new avenues for future therapeutic developments.
Acknowledgements
Alaa Refaat gratefully acknowledges the British Council (Researcher Links: Newton Fund, Project No. 18610) and the International Fellowship Program (IFP), Takeda Science Foundation. Chathyan Pararasa gratefully acknowledges support from HEIF.
Additional information
Funding
References
- DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin 2014;64:52–62.
- Gilbert CA, Slingerland JM. Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annu Rev Med 2013;64:45–57.
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 2011;11:325–337.
- Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem 2002;277:23111–23115.
- Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol 2009;19:12–16.
- Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 2006;66:8927–8930.
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008;452:230–233.
- Liberko M, Kolostova K, Bobek V. Essentials of circulating tumor cells for clinical research and practice. Crit Rev Oncol Hematol 2013;88:338–356.
- da Cunha FM, Torelli NQ, Kowaltowski AJ. Mitochondrial retrograde signaling: triggers, pathways, and outcomes. Oxid Med Cell Longev 2015;2015:482582.
- Li L-D, Sun H-F, Liu X-X, Gao S-P, Jiang H-L, Hu X, Jin W. Down-regulation of NDUFB9 promotes breast cancer cell proliferation, metastasis by mediating mitochondrial metabolism. PLoS One 2015;10:e0144441.
- Fang ZZ, Nian Y, Li W, Wu JJ, Ge GB, Dong PP, et al. Cycloartane triterpenoids from Cimicifuga yunnanensis induce apoptosis of breast cancer cells (MCF7) via p53-dependent mitochondrial signaling pathway. Phytother Res 2011;25:17–24.
- de Zeeuw D, Akizawa T, Agarwal R, Audhya P, Bakris GL, Chin M, et al. Rationale and trial design of bardoxolone methyl evaluation in patients with chronic kidney disease and type 2 diabetes: the occurrence of renal events (BEACON). Am J Nephrol 2013;37:212–222.
- Chin MP, Reisman SA, Bakris GL, O’Grady M, Linde PG, McCullough PA, et al. Mechanisms contributing to adverse cardiovascular events in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. Am J Nephrol 2014;39:499–508.
- Kim EH, Deng C, Sporn MB, Royce DB, Risingsong R, Williams CR, Liby KT. CDDO-methyl ester delays breast cancer development in BRCA1-mutated mice. Cancer Prev Res (Phila) 2012;5:89–97.
- Taylor S, Lam M, Pararasa C, Brown JE, Carmichael AR, Griffiths HR. Evaluating the evidence for targeting FOXO3a in breast cancer: a systematic review. Cancer Cell Int 2015;15:1.
- Liby KT, Sporn MB. Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol Rev 2012;64:972–1003.
- Hong DS, Kurzrock R, Supko JG, He X, Naing A, Wheler J, et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin Cancer Res 2012;18:3396–3406.
- Grant MM, Barber VS, Griffiths HR. The presence of ascorbate induces expression of brain derived neurotrophic factor in SH-SY5Y neuroblastoma cells after peroxide insult, which is associated with increased survival. Proteomics 2005;5:534–540.
- Pararasa C, Ikwuobe J, Shigdar S, Boukouvalas A, Nabney IT, Brown JE, et al. Age-associated changes in long-chain fatty acid profile during healthy aging promote pro-inflammatory monocyte polarization via PPARgamma. Aging Cell 2016;15:128–139.
- Hemachandra LP, Shin DH, Dier U, Iuliano JN, Engelberth SA, Uusitalo LM, et al. Mitochondrial superoxide dismutase has a protumorigenic role in ovarian clear cell carcinoma. Cancer Res 2015;75:4973–4984.
- D’Silva H, Gothoskar BP, Jain VK, Advani SH. Cell migration and energy generating metabolic activities. Eur J Cancer 1978;14:1243–1248.
- Cleasby A, Yon J, Day PJ, Richardson C, Tickle IJ, Williams PA, et al. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS One 2014;9:e98896.
- Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med 2015;88:179–188.
- Thangasamy A, Rogge J, Krishnegowda NK, Freeman JW, Ammanamanchi S. Novel function of transcription factor Nrf2 as an inhibitor of RON tyrosine kinase receptor-mediated cancer cell invasion. J Biol Chem 2011;286:32115–32122.
- Krauss S, Zhang CY, Lowell BB. The mitochondrial uncoupling-protein homologues. Nat Rev Mol Cell Biol 2005;6:248–261.
- Liby KT, Sporn MB. Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol Rev 2012;64:972–1003.
- Wang Y, Porter WW, Suh N, Honda T, Gribble GW, Leesnitzer LM, et al. A synthetic triterpenoid, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO), is a ligand for the peroxisome proliferator-activated receptor gamma. Mol Endocrinol 2000;14:1550–1556.
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998;92:829–839.
- Ennen M, Minig V, Grandemange S, Touche N, Merlin J-L, Besancenot V, et al. Regulation of the high basal expression of the manganese superoxide dismutase gene in aggressive breast cancer cells. Free Radic Biol Med 2011;50:1771–1779.
- Zhang Q, Cui C, Chen CQ, Hu XL, Liu YH, Fan YH, et al. Anti-proliferative and pro-apoptotic activities of Alpinia oxyphylla on HepG2 cells through ROS-mediated signaling pathway. J Ethnopharmacol 2015;169:99–108.
- Mazurek S, Michel A, Eigenbrodt E. Effect of extracellular AMP on cell proliferation and metabolism of breast cancer cell lines with high and low glycolytic rates. J Biol Chem 1997;272:4941–4952.
- Gao D, Pararasa C, Dunston CR, Bailey CJ, Griffiths HR. Palmitate promotes monocyte atherogenicity via de novo ceramide synthesis. Free Radic Biol Med 2012;53:796–806.
- Chauvin L, Goupille C, Blanc C, Pinault M, Domingo I, Guimaraes C, et al. Long chain n-3 polyunsaturated fatty acids increase the efficacy of docetaxel in mammary cancer cells by downregulating Akt and PKCɛ/δ-induced ERK pathways. Biochim Biophys Acta 2016;1861:380–390.
- Carmichael AR. Obesity as a risk factor for development and poor prognosis of breast cancer. BJOG 2006;113:1160–1166.
- Alo PL, Visca P, Marci A, Mangoni A, Botti C, Di Tondo U. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer 1996;77:474–482.
- Weng JR, Bai LY, Chiu CF, Hu JL, Chiu SJ, Wu CY. Cucurbitane triterpenoid from momordica charantia induces apoptosis and autophagy in breast cancer cells, in part, through peroxisome proliferator-activated receptor gamma activation. Evid Based Complement Alternat Med 2013;2013:935675.
- Yang LJ, Tang Q, Wu J, Chen Y, Zheng F, Dai Z, Hann SS. Inter-regulation of IGFBP1 and FOXO3a unveils novel mechanism in ursolic acid-inhibited growth of hepatocellular carcinoma cells. J Exp Clin Cancer Res 2016;35:59.