
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Low density lipoprotein (LDL) might be oxidized by iron in the lysosomes of macrophages in atherosclerotic lesions. We have shown previously that the iron-storage proteinferritin can oxidize LDL at lysosomal pH. We have now investigated the roles of the most important antioxidant contained in LDL, α-tocopherol (the main form of vitamin E) and of ascorbate (vitamin C), a major water-soluble antioxidant, on LDL oxidation by ferritin at lysosomal pH (pH 4.5). We incubated LDL with ferritin at pH 4.5 and 37 °C and measured its oxidation by monitoring the formation of conjugated dienes at 234 n min a spectrophotometer. α-Tocopherol is well known to inhibit LDL oxidation at pH 7.4, but enrichment of LDL with α-tocopherol was unable to inhibit LDL oxidation by ferritin at pH 4.5. Ascorbate had a complex effect on LDL oxidation by ferritin at lysosomal pH and exhibited both antioxidant and pro-oxidant effects. It had no antioxidant effect on partially oxidized LDL, only a pro-oxidant effect. Ascorbate completely inhibited LDL oxidation by copper at pH 7.4 for a long period, but in marked contrast did not inhibit LDL oxidation by copper at lysosomal pH. Dehydroascorbate, the oxidation product of ascorbate, had a pronounced pro-oxidant effect on LDL incubated with ferritin at pH 4.5. The inability of α-tocopherol and ascorbate to effectively inhibit LDL oxidation by ferritin at lysosomal pH might help to explain why the large clinical trials with these vitamins failed to show protection against cardiovascular diseases.
Introduction
There has been a great deal of interest in oxidized low density lipoprotein (LDL), which has many potentially atherogenic effects [Citation1–3]. A beneficial effect of antioxidants in decreasing cardiovascular disease would provide further support for the oxidized LDL hypothesis of atherosclerosis. Some clinical trials have reported efficacy of antioxidants, mainly vitamins E and C, in reducing cardiovascular events [Citation4,Citation5] but the large clinical trials have shown no protection [Citation6–10]. The role of antioxidants and oxidized LDL in atherosclerosis therefore remains controversial.
Vitamin E has eight forms, but α-tocopherol is the main one in the body and lipoproteins [Citation11] and is the most abundant lipid-soluble antioxidant contained in LDL, with each LDL particle containing about five to nine molecules [Citation12,Citation13]. α-Tocopherol has a chain breaking effect on free radical reactions, due to its ability to scavenge alkoxyl and peroxyl radicals formed from lipid peroxidation [Citation14,Citation15]. Increasing the α-tocopherol content of LDL has been shown to decrease its oxidisability by macrophages or copper [Citation16,Citation17], but α-tocopherol can increase the oxidation of LDL if the oxidative stress is low because the α-tocopherol radical can abstract a hydrogen atom from a polyunsaturated lipid and promote peroxidation [Citation18,Citation19]. Supplementation of LDL with α-tocopherol does not protect LDL from oxidation by copper or ferric iron or a low concentration of ferrous iron at lysosomal pH [Citation20]. α-Tocopherol has been shown to reduce diet-induced atherosclerosis in rabbits [Citation21,Citation22] and decreased atherosclerotic lesion formation in apoE-deficient mice fed an atherogenic diet [Citation23] and LDL receptor-deficient mice on a low [Citation24] or high fat diet [Citation25], but it sometimes had no effect on atherosclerosis or even increased it [Citation26].
Ascorbate (vitamin C) is an important water-soluble vitamin and can protect LDL from oxidation by copper [Citation27–29], iron [Citation30,Citation31], a radical generator, neutrophils or cigarette smoke [Citation32]. Ascorbate can increase the oxidation, however, of partially oxidized LDL [Citation29,Citation30]. Some studies have shown an anti-atherogenic effect of ascorbate in cholesterol fed-rabbits [Citation33,Citation34], but others have demonstrated that ascorbate had no protective effect in these animals [Citation35]. Similarly to α-tocopherol, there is no strong evidence that ascorbate decreases cardiovascular disease in clinical trials [Citation8,Citation9,Citation36,Citation37].
Dehydroascorbate (the oxidation product of ascorbate when it acts as an antioxidant) inhibits the oxidation of fresh nonoxidised LDL [Citation38,Citation39], but increases the oxidation of partially oxidized LDL [Citation29,Citation39].
The iron hypothesis of atherosclerosis was proposed by Sullivan in 1981 [Citation40], but this hypothesis is controversial [Citation41] possibly because plasma iron concentrations are not always a good indication of iron levels inside cells. Iron is increased greatly in human [Citation42,Citation43] and rabbit atherosclerotic lesions [Citation44] and macrophage foam cells in human atheromas are high in iron and ferritin [Citation45]. Ferritin light chain levels are increased in human diseased coronary arteries [Citation46] and high plasma ferritin concentrations are associated with an increase in myocardial infarctions [Citation47] and increased progression of human carotid atherosclerosis [Citation48]. Interestingly, the macrophage-specific knockout of ferroportin, which transports iron out of cells, increased atherosclerosis substantially in mice [Citation49].
We have shown that LDL can be oxidized in the lysosomes of macrophages, that this oxidation is catalyzed by iron [Citation50] and that it can be inhibited by the antioxidant cysteamine, which accumulates in lysosomes [Citation51,Citation52]. The lysosomal oxidation of LDL causes the secretion of inflammatory cytokines by macrophages and an increase in the pH of lysosomes, which can be protected against by cysteamine [Citation53]. Inhibiting the lysosomal oxidation of LDL by cysteamine decreases the development of atherosclerosis [Citation54] and causes the regression of preexisting lesions [Citation52] in mice.
LDL can also be oxidized by the main iron-storage protein ferritin at acidic pH, much faster than at pH 7.4 [Citation55]. There was a large increase in hydroperoxides in the LDL, including cholesteryl linoleate hydroperoxide, and an increase in 7-ketocholesterol at pH 4.5 [Citation55]. This raises the possibility that ferritin delivered to lysosomes by autophagy or endocytosis might play a role in oxidizing LDL in these organelles. When cells are deficient in iron the protein NCOA4 binds to ferritin in the cytosol and delivers it to autophagosomes which fuze with lysosomes where the ferritin is degraded and iron made available [Citation56,Citation57]. Plasma ferritin, although not totally saturated with iron [Citation58], binds to transferrin receptor-1 and is endocytosed and delivered to lysosomes [Citation59].
In the present study, we have investigated the effects of α-tocopherol and ascorbate on LDL oxidation by ferritin at lysosomal pH because α-tocopherol is the main antioxidant in LDL, ascorbate would be delivered to lysosomes when cells endocytose interstitial fluid and because these antioxidants have been investigated in a number of very large clinical trials of cardiovascular disease.
Materials and methods
Materials
All reagents used were purchased from Sigma Aldrich Limited. Ferritin was from equine spleen and had a molecular weight of 440 kDa. Ferritin, ascorbate and dehydroascorbate was dissolved freshly before each experiment.
LDL Isolation
LDL was isolated by sequential ultracentrifugation, as previously described [Citation60], from pooled plasma from normal human blood. LDL was stored in the presence of 100 µM EDTA at 4 °C and used within one month of storage. Ethical permission to take human blood for the isolation of LDL was obtained from the University of Reading Research Ethics Committee.
Enrichment of LDL with α-tocopherol
The α-tocopherol content of native LDL was increased as described by Esterbauer et al. [Citation61]. Plasma from blood collected from healthy volunteers was obtained by centrifugation at 1500 g for 30 min at 4 °C. The pooled plasma was then incubated for 3 h at 37 °C with 1% (v/v) dimethylsulfoxide containing 100 mM α-tocopherol (final concentration 1 mM) or 1% (v/v) dimethylsufoxide as a control. LDL was then isolated by sequential ultracentrifugation [Citation60]. (The dimethylsulfoxide would have been removed from the LDL during dialysis.) We quantified the α-tocopherol content of LDL by HPLC, as described previously [Citation62].
Measurement of conjugated dienes
LDL (50 µg protein/ml) was oxidized with 0.1 µM ferritin in 150 mM NaCl/10 mM sodium acetate buffer, pH 4.5 or 150 mM NaCl/10 mM MOPS (3-(N-morpholino) propanesulphonic acid) buffer, pH 7.4 treated with prewashed Chelex-100 (1%, w/v) to remove contaminating transition metal ions. The formation of conjugated dienes was monitored by measuring attenuance (absorbance plus UV scattering) at 234 nm at 37 °C overnight in a dual beam Lambda Bio 40 8-cell position spectrophotometer (PerkinElmer) with UV WinLab software. The test cuvettes were placed against appropriate reference cuvettes that did not contain LDL and the attenuance of the reference cuvettes was automatically subtracted from that of the test cuvettes. The initial attenuance was subtracted from the later time points.
It was important to wash the cuvettes very carefully between use to avoid contamination by transition metal ions. After an experiment, they were rinsed with water several times and cleaned with warm dilute detergent (washing-up liquid) with a cotton bud. They were then rinsed in purified water and soaked in ethanol, rinsed with purified water and then filled with 100 µM diethylenetriaminepentaacetic acid (DTPA) for 1 h, to remove any firmly bound metal ions, rinsed thoroughly with purified water followed by ethanol and left to dry.
Statistics
The mean ± SEM of the specified number of independent experiments are shown. Treatments were compared by a one-way ANOVA with a Tukey’s post-hoc test. A p value of < 0.05 was considered statistically significant.
Results
Effect of enriching LDL with α-tocopherol on oxidation by ferritin at pH 7.4 and 4.5
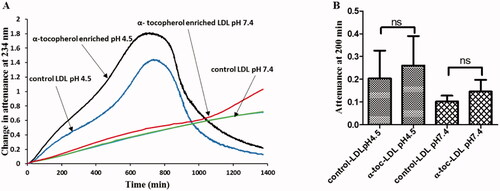
Enriching human LDL with α-tocopherol by taking α-tocopherol orally [Citation16,Citation17] or by adding α-tocopherol to plasma and then isolating LDL [Citation63] inhibits its oxidation by macrophages or copper at pH 7.4, the pH of normal interstitial fluid or plasma. We have investigated the effect of α-tocopherol enrichment of LDL by adding α-tocopherol to plasma and isolating the LDL by ultracentrifugation on LDL oxidation by ferritin at lysosomal pH (pH 4.5). The α-tocopherol content of LDL was increased from 15 ± 0.4 to 26 ± 0.8 nmol/mg protein (mean ± SEM), leading to an increase in the average number of α-tocopherol molecules contained in each LDL particle from about 8 to 13. Control and α-tocopherol-enriched LDL were incubated with ferritin at pH 4.5 and 7.4 and the formation of conjugated dienes was monitored at 234 nm (). The oxidation was faster at acidic pH, as expected [Citation55]. As described previously [Citation55], LDL oxidation at pH 4.5 had a short lag phase, a rapid oxidation phase, a slower oxidation phase, an aggregation phase (in which LDL aggregates and scatters the beam of UV resulting in an increase in attenuance) and a sedimentation phase (in which the aggregates sink beneath the beam of UV in the spectrophotometer and are not detected). The rate of increase of attenuance was not significantly decreased by α-tocopherol enrichment.
Figure 1. Effect of α-tocopherol enrichment on LDL oxidation by ferritin. (A) Control LDL or LDL enriched with α-tocopherol (50 µg protein/ml) was oxidized by ferritin (0.1 µM) in a sodium acetate buffer of pH 4.5 or a MOPS buffer of pH 7.4. The formation of conjugated dienes and at later times aggregation was monitored as attenuance (absorbance plus UV scattering) at 234 nm. This result is representative of four independent experiments. (B) The mean ± SEM of attenuance at 200 min (which was still in the oxidation phase, rather than the aggregation phase) were compared by one-way ANOVA (n = 4) followed by a Tukey’s post-hoc test (p > 0.05). ns indicates not significantly different to the indicated comparison.
Effects of ascorbate on LDL oxidation by ferritin at lysosomal pH
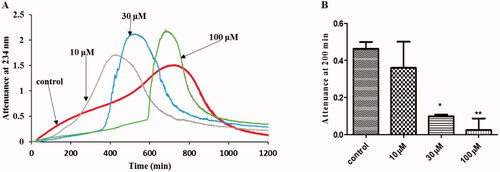
We tested the effect of varying concentrations (10–100 μM) of ascorbate on LDL oxidation by ferritin at lysosomal pH. The recommended plasma ascorbate concentration is 50 μM [Citation64]. Ascorbate exhibited an antioxidant effect on the initial phase of oxidation of LDL, followed by an abrupt pro-oxidant effect such that the extent of oxidation overtook that of the control LDL (). The duration of the inhibition phase was increased as the concentration of ascorbate increased.
Figure 2. Effect of ascorbate on LDL oxidation by ferritin at lysosomal pH. (A) LDL (50 µg protein/ml) was oxidized with ferritin (0.1 µM) in the absence or presence of ascorbate (10 µM–100µM) pH 4.5 and 37 °C. The formation of conjugated dienes and at later times LDL aggregation was monitored by measuring attenuance at 234 nm. This is representative of four independent experiments. (B) The attenuance at 200 min (which was still in the oxidation phase, rather than the aggregation phase) was compared by one-way ANOVA (n = 4) followed by a Tukey’s post-hoc test. *Indicates p < 0.05 and ** indicates p < 0.01 compared to the control.
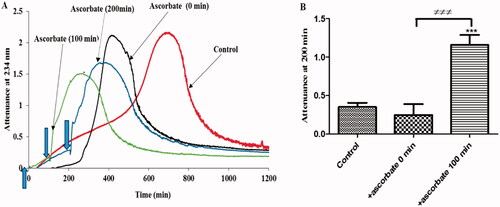
We showed previously that the antioxidant activity of ascorbate on LDL oxidation by copper at pH 7.4 is lost when LDL is partially oxidized [Citation29]. We therefore tested the effects of preexisting oxidized lipids on the antioxidant and pro-oxidant activities of ascorbate when LDL is oxidized by ferritin at pH 4.5. When ascorbate was added at the start of the experiment, there was an antioxidant effect followed by a pro-oxidant effect, as expected (). When ascorbate was added at 100 and 200 min, when the oxidation was already underway, it caused an immediate and rapid oxidation of LDL.
Figure 3. The effect of existing oxidized lipids on the effect of ascorbate on LDL oxidation by ferritin at lysosomal pH. (A) LDL (50 µg protein/ml) was oxidized by ferritin (0.1 µM) at pH 4.5 and 37 °C in the absence or presence of 30 µM ascorbate added at different time points (0–200 min). The formation of conjugated diene and at later times LDL aggregation was monitored by measuring attenuance at 234 nm. This result represents three independent experiments. (B) The attenuance at 200 min (when the LDL in the absence of ascorbate was in its oxidation phase, but not yet in the aggregation phase) was compared by one-way ANOVA (n = 3) followed by a Tukey’s post-hoc test. *** indicates p < 0.001 compared to the control. ≠≠≠ indicates p < 0.001 for the indicated comparison.
Effects of ascorbate on LDL oxidation by copper at pH 4.5 and 7.4
We compared the effects of ascorbate on LDL oxidation by ferritin to the much better studied system of LDL oxidation by copper (). The oxidation profile of LDL oxidation by copper was very different at pH 4.5 and 7.4. At pH 7.4, there was the well-known lag phase, propagation phase and decomposition phase [Citation27]. At pH 4.5, the oxidation was much slower and had a lag phase, rapid oxidation phase, slow oxidation phase, aggregation phase and sedimentation phase, somewhat similar to the oxidation profile given by ferritin () or ferrous and ferric ions [Citation62] at pH 4.5. The attenuance reached a much higher peak due to LDL aggregation and therefore UV scattering.
Figure 4. The effect of ascorbate on LDL oxidation by copper at pH 4.5 and 7.4. (A) LDL (50 µg protein/ml) was oxidized by CuSO4 (5 µM) in the presence or absence of ascorbate (30 µM) at 37 °C in a sodium acetate buffer of pH 4.5 or a MOPS buffer of pH 7.4. The formation of conjugated dienes was measured by attenuance of 234 nm. This result is representative of three independent experiments. (B) The attenuance at 200 min (when the LDL was in its oxidation phase, but not yet in the aggregation phase at pH 4.5) was compared by one-way ANOVA (n = 3) followed by a Tukey’s post-hoc test. ns indicates not significant, *** indicates p < 0.001 for the indicated comparison.
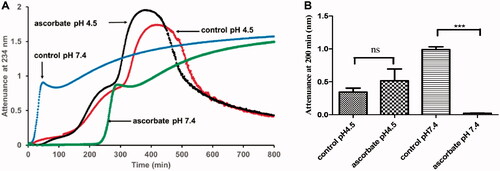
Ascorbate (30 μM) had very different effects at the two pH values. At pH 7.4, it completely prevented the oxidation of LDL for a prolonged period and then the oxidition proceeded at the uninhibited rate. At pH 4.5, however, ascorbate had little effect on LDL oxidation.
Effects of dehydroascorbate on LDL oxidation by ferritin at lysosomal pH
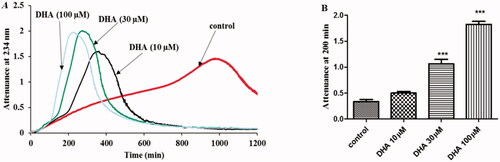
We investigated the effect of the oxidation product of ascorbate, dehydroascorbate, on LDL oxidation by ferritin at lysosomal pH. Dehydroascorbate did not protect LDL from oxidation. It initially had no effect on the oxidation, but later had a pro-oxidant effect, which occurred sooner the higher the concentration of dehydroascorbate ().
Figure 5. Effect of dehydroascorbate on LDL oxidation by ferritin at lysosomal pH. (A) LDL (50 µg protein/ml) was oxidized with ferritin (0.1 µM) in the absence or presence of dehydroascorbate (10–100 µM) pH 4.5 at 37 °C. The formation of conjugated dienes was monitored by measuring attenuance at 234 nm. This result is representative of four independent experiments. (B) The attenuance at 200 min (when the LDL was in its oxidation phase, but not yet in the aggregation phase in the absence of dehydroascorbate) was compared by one-way ANOVA (n = 4) followed by a Tukey’s post hoc-test. *** indicates p < 0.001 compared to the control.
Discussion
Preventing LDL oxidation was considered one of the most impoprtant strategies to reduce cardiovascular diseases, but the large clinical trials of antioxidants, mainly α-tocopherol and ascorbate, showed no protection [Citation6–10]. We previously showed that α-tocopherol-enrichment of LDL did not protect LDL effectively against oxidation by ferric iron, copper ions or a low concentration of ferrous iron at lysosomal pH (pH 4.5), but as expected it protected LDL against oxidation by copper at pH 7.4 [Citation20]. In the present study, enrichment of LDL with α-tocopherol did not protect LDL from oxidation by ferritin at lysosomal pH (). The fold enrichment of α-tocopherol we obtained by adding α-tocopherol to plasma followed by isolating LDL was 1.73, which is comparable to the enrichment obtained in the clinical trials (1.60–2.42) [Citation8,Citation9,Citation65–67].
α-Tocopherol might sometimes have an antioxidant effect on LDL by scavenging radicals, such as lipid peroxyl radicals (LOO•).
(1)
(1)
α-Tocopherol might also have pro-oxidant effects by reducing ferric iron (ferritin contains ferric iron) to ferrous iron itself becoming the α-tocopheroxyl radical, which is not entirely stable and can abstract a hydrogen atom from a bisallylic methylene group in a polyunsaturated fatty acyl moiety promoting lipid peroxidation [Citation68,Citation69].
(2)
(2)
(3)
(3)
(4)
(4)
In addition, ferrous iron would break down lipid hydroperoxide much faster than ferric iron would [Citation70].
(5)
(5)
(6)
(6)
These pro-oxidant effects might counterbalance the antioxidant effect and lead to a net lack of effect of α-tocopherol on LDL oxidation by ferritin at pH 4.5.
We have postulated that LDL is oxidized by iron at lysosomal pH by the hydroperoxyl radical (HO2•), which is more reactive and hydrophobic than the superoxide radical (O2•−) [Citation51]. Ferrous iron might transfer an electron to oxygen forming the superoxide radical, which protonates at acidic pH (pKa 4.8) to form the hydroperoxyl radical. The hydroperoxyl radical might abstract a hydrogen atom from a polyunsaturated fatty acyl moiety to cause its oxidation.
(7)
(7)
(8)
(8)
(9)
(9)
(10)
(10)
We investigated the effects of ascorbate (10–100 μM), whose recommended plasma concentration is 50 μM [Citation64], on LDL oxidation by ferritin at lysosomal pH as it would be delivered to lysosomes when cells endocytose extracellular fluid. Ascorbate had complex antioxidant and pro-oxidant effects on LDL oxidation by ferritin at lysosomal pH. It initially inhibited the oxidation of LDL in a concentration-dependent manner and then had a very abrupt pro-oxidant effect ().
Ascorbate might have inhibited LDL oxidation by ferritin in at least three ways.
Ascorbate might have scavenged superoxide or hydroperoxyl radicals.
(11)
The ascorbyl radical formed by this reaction might be stabilized by dismutation to ascorbate and dehydroascorbate (DHA).
Ascorbate might have acted as a co-antioxidant for α-tocopherol by rejuvenating α-tocopherol from the α-tocopheroxyl radical, restoring the antioxidant ability of α-tocopherol and preventing the pro-oxidant effect of the α-tocopheroxyl radical [Citation71].
It should be noted, however, that the rate of reaction of ascorbate with α-tocopheroxyl radicals in micelles is lower at more acidic pH values [Citation72].
Ascorbate might possibly have bound to iron released from ferritin, decreasing the ability of this iron to oxidize LDL.
Ascorbate’s antioxidant effect on LDL incubated with ferritin very suddenly switched to a pro-oxidant effect and the rate of LDL oxidation then exceeded that of LDL incubated in the absence of ascorbate (). We propose that the pro-oxidant effect of ascorbate might be explained by two mechanims.
Ascorbate might reduce the ferric iron of ferritin to ferrous iron.
The ferrous iron would react with oxygen to form superoxide and hydroperoxyl radicals (Equations 7 and 8), which would oxidize the LDL.
Ferrous iron released from ferritin might react rapidly with the lipid hydroperoxides that have build up in the LDL to form lipid alkoxyl radicals (Equations 5), giving a burst of LDL oxidation.
We do not know why the switch from an antioxidant to a pro-oxidant effect on ferritin was so abrupt with ascorbate, but it appears to be related to the build up of lipid hydroperoxides in LDL because when ascorbate was added to partially oxidized LDL there was an immediate burst of oxidation ().
To investigate the effect of pH on the antioxidant and pro-oxidant activities of ascorbate, we investigated the effects of ascorbate on LDL oxdation by copper, as copper can oxidize LDL well at pH 7.4. Copper, in marked contrast to ferritin () and ferrous iron [Citation50,Citation62], oxidized LDL much faster at pH 7.4 than at pH 4.5 and with very different kinetics (). The kinetics at pH 7.4 were as expected [Citation27]. At pH 4.5, the kinetics were similar to those observed with ferritin () and ferrous or ferric iron at this pH [Citation62]. The mechanism of LDL oxidation by copper might well be different at pH 7.4 and 4.5. At pH 7.4, it might involve copper interacting with certain amino acids, for instance tryptophan residues, in apolipoprotein B-100 of LDL [Citation73], preexisting lipid hydroperoxides [Citation74] or α-tocopherol [Citation68]. At pH 4.5, copper might oxidize LDL by forming the hydroperoxyl radical, as shown below (where the identity of XH is uncertain), followed by EquationEquations 8–10.
(16)
(16)
(17)
(17)
The inhibition by ascorbate of LDL oxidation by CuSO4 at pH 7.4 is similar to what has previously been observed [Citation28,Citation30]. The mechanism of the inhibition might involve scavenging free radicals, decreased binding of copper ions to LDL [Citation75], the reduction of Cu2+ to Cu+ thus preventing α-tocopherol-mediated lipid peroxidation by Cu2+ or a co-antioxidant effect with α-tocopherol [Citation71].
Ascorbate had little effect on LDL oxidation by CuSO4 at pH 4.5 because the above antioxidant mechanisms might not apply at this pH. It is surprising that ascorbate did not inhibit LDL oxidation by scavenging hydroperoxyl/superoxide radicals because its optimal pH for scavenging these radicals is pH 4.5 [Citation76]. It is possible that copper ions generate these radicals in a site-specific manner on the surface of LDL where ascorbate might not have access to them.
We investigated the effects of dehydroascorbate (10–100 μM), the oxidation product of ascorbate, on LDL oxidation by ferritin at lysosomal pH (). Very different plasma concentrations of dehydroascorbate has been reported by a number of studies and range from 2–29 µM (see [Citation39] for references). The local concentrations would be expected to increase when ascorbate acts as an antioxidant. Dehydroascorbate initially had no effect on LDL oxidation by ferritin at pH 4.5, but then had a sudden pro-oxidant effect (). The pro-oxidant effect occurred sooner and the rate of oxidation was greater at the higher dehydroascorbate concentrations. The effects of dehydroascorbate on LDL oxidation by ferritin at pH 4.5 are in marked contrast to the effects with copper at pH 7.4, where dehydroascorbate inhibited very effectively the oxidation of unoxidized LDL (but increased the oxidation of mildly-oxidized LDL) [Citation39]. The pro-oxidative effect of dehydroascorbate might be due to the conversion of Fe3+ in ferritin to Fe2+ by ascorbate or erythroascorbate, both of which can be generated from dehydroascorbate [Citation39], followed by Equations 5 and 7–10.
In conclusion, the lack of effective inhibition by α-tocopherol and ascorbate of LDL oxdation by ferritin at lysosomal pH might help to explain why vitamins E and C did not reduce cardiovascular disease in the large clinical trials [Citation6–10].
Author contributions
O.O.O. carried out the experiments and O.O.O. and D.S.L. wrote the paper.
Acknowledgements
We thank our colleagues who donated blood for LDL isolation, Dr Kim G. Jackson and Ms Rada G. Mihaylova for skilfully taking blood and Dr Hadeel K.M. Alboaklah for measuring α-tocopherol levels in LDL.O.O.O. was supported by the Tertiary Education Trust Fund (TETfund), Nigeria.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Steinberg D. The LDL modification hypothesis of atherogenesis: an update. J Lipid Res. 2009;50 Suppl:S376–S381.
- Itabe H. Oxidative modification of LDL: its pathological role in atherosclerosis. Clin Rev Allergy Immunol. 2009;37(1):4–11.
- Alique M, Luna C, Carracedo J, et al. LDL biochemical modifications: a link between atherosclerosis and aging. Food Nutr Res. 2015;59:29240.
- Stephens NG, Parsons A, Schofield PM, et al. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet. 1996;347(9004):781–786.
- Boaz M, Smetana S, Weinstein T, et al. Others. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): randomised placebo-controlled trial. Lancet. 2000;356(9237):1213–1218.
- Yusuf S, Phil D, Dagenais G, et al. Vitamin E supplementation and cardiovascular events in high-risk patients. New Eng J Med . 2000;342(3):154–160.
- de Gaetano G. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet. 2001;357(9250):89–95.
- Collins R, Armitage J, Parish S, et al. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360(9326):23–33.
- Cook NR, Albert CM, Gaziano JM, et al. A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women: results from the Women’s Antioxidant Cardiovascular Study. Arch Intern Med. 2007;167(15):1610–1618.
- Sesso HD, Buring JE, Christen WG, et al. Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2008;300(18):2123–2133.
- Stocker R. The ambivalence of vitamin E in atherogenesis. Trends Biochem Sci. 1999;24(6):219–223.
- Esterbauer H, Dieber-Rotheneder M, Waeg G, et al. Endogenous antioxidants and lipoprotein oxidation. Biochem Soc Trans. 1990;18(6):1059–1061.
- Meydani M. Vitamin E and atherosclerosis: beyond prevention of LDL oxidation. J Nutr. 2001;131(2):366S–368s.
- Sies H, Stahl W, Sundquist AR. Antioxidant functions of vitamins. Vitamins E and C, beta-carotene, and other carotenoids. Ann N Y Acad Sci. 1992;669:7–20.
- Liebler DC. The role of metabolism in the antioxidant function of vitamin E. Crit Rev Toxicol. 1993;23(2):147–169.
- Jessup W, Rankin SM, de Whalley CV, et al. Alpha-tocopherol consumption during low-density-lipoprotein oxidation. Biochem J. 1990;265(2):399–405.
- Dieber-Rotheneder M, Puhl H, Waeg G, et al. Effect of oral supplementation with D-α-tocopherol on the vitamin E content of human low density lipoproteins and resistance to oxidation. J Lipid Res. 1991;32(8):1325–1332.
- Bowry VW, Stocker R. Tocopherol-mediated peroxidation. The prooxidant effect of vitamin E on the radical-initiated oxidation of human low-density lipoprotein. J Am Chem Soc. 1993;115(14):6029–6044.
- Kontush A, Finckh B, Karten B, et al. Antioxidant and prooxidant activity of alpha-tocopherol in human plasma and low-density-lipoprotein. J Lipid Res. 1996;37(7):1436–1448.
- Alboaklah HKM, Leake DS. Effect of vitamin E on low density lipoprotein oxidation at lysosomal pH. Free Radic Res. 2020;54(8–9):574–584.
- Schwenke DC, Rudel LL, Sorci-Thomas MG, et al. α-Tocopherol protects against diet induced atherosclerosis in New Zealand white rabbits. J Lipid Res . 2002;43(11):1927–1938.
- Ozer NK, Sirikci O, Taha S, et al. Effect of vitamin E and probucol on dietary cholesterol-induced atherosclerosis in rabbits. Free Radic Biol Med. 1998;24(2):226–233.
- Peluzio MCG, Homem APP, Cesar GC, et al. Influences of alpha-tocopherol on cholesterol metabolism and fatty streak development in apolipoprotein E-deficient mice fed an atherogenic diet . Braz J Med Biol Res. 2001;34(12):1539–1545.
- Meydani M, Kwan P, Band M, et al. Long-term vitamin E supplementation reduces atherosclerosis and mortality in ldlr-/- mice, but not when fed Western style diet. Atherosclerosis. 2014;233(1):196–205.
- Cyrus T, Yao YM, Rokach J, et al. Vitamin E reduces progression of atherosclerosis in low-density lipoprotein receptor-deficient mice with established vascular lesions. Circulation. 2003;107(4):521–523.
- Upston JM, Kritharides L, Stocker R. The role of vitamin E in atherosclerosis. Prog Lipid Res. 2003;42(5):405–422.
- Esterbauer H, Striegl G, Puhl H, et al. Continuous monitoring of in vitro oxidation of human low density lipoprotein. Free Radic Res Commun. 1989;6(1):67–75.
- Jialal I, Vega GL, Grundy SM. Physiologic levels of ascorbate inhibit the oxidative modification of low density lipoprotein. Atherosclerosis. 1990;82(3):185–191.
- Stait SE, Leake DS. The effects of ascorbate and dehydroascorbate on the oxidation of low-density lipoprotein. Biochem J. 1996;320(Pt2):373–381.
- Stait SE, Leake DS. Ascorbic acid can either increase or decrease low density lipoprotein modification. FEBS Lett. 1994;341(2-3):263–267.
- Alul RH, Wood M, Longo J, et al. Vitamin C protects low-density lipoprotein from homocysteine-mediated oxidation. Free Radical Biol Med . 2003;34(7):881–891.
- Frei B. Ascorbic acid protects lipids in human plasma and low-density-lipoprotein against oxidative damage. Am J Clin Nutr. 1991;54(6):S1113–S1118.
- Verlangieri AJ, Hollis TM, Mumma RO. Effects of ascorbic acid and its 2-sulfate on rabbit aortic intimal thickening. Blood Vessels. 1977;14(3):157–174.
- Beetens JR, Coene MC, Verheyen A, et al. Influence of vitamin C on the metabolism of arachidonic acid and the development of aortic lesions during experimental atherosclerosis in rabbits. Biomed Biochim Acta . 1984;43(8-9):S273–S276.
- Morel DW, de la Llera-Moya M, Friday KE. Treatment of cholesterol-fed rabbits with dietary vitamins E and C inhibits lipoprotein oxidation but not development of atherosclerosis. J Nutr. 1994;124(11):2123–2130.
- Lee DH, Folsom AR, Harnack L, et al. Does supplemental vitamin C increase cardiovascular disease risk in women with diabetes? Am J Clin Nutr. 2004;80(5):1194–1200.
- Al-Khudairy L, Flowers N, Wheelhouse R, et al. Vitamin C supplementation for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2017;3(3):CD011114.
- Retsky KL, Freeman MW, Frei B. Ascorbic acid oxidation product(s) protect human low density lipoprotein against atherogenic modification – anti- rather than prooxidant activity of vitamin C in the presence of transition metal ions. J Biol Chem. 1993;268(2):1304–1309.
- Horsley ETM, Burkitt MJ, Jones CM, et al. Mechanism of the antioxidant to pro-oxidant switch in the behavior of dehydroascorbate during LDL oxidation by copper(II) ions. Arch Biochem Biophys. 2007;465(2):303–314.
- Sullivan JL. Iron and the sex difference in heart-disease risk. Lancet. 1981;1(8233):1293–1294.
- Sempos CT. Do body iron stores increase the risk of developing coronary heart disease? Am J Clin Nutr. 2002;76(3):501–503.
- Stadler N, Lindner RA, Davies MJ. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: evidence for the presence of elevated levels of iron and copper. ATVB. 2004;24(5):949–954.
- Lee FY, Lee TS, Pan CC, et al. Colocalization of iron and ceroid in human atherosclerotic lesions. Atherosclerosis. 1998;138(2):281–288.
- Watt F, Rajendran R, Ren MQ, et al. A nuclear microscopy study of trace elements Ca, Fe, Zn and Cu in atherosclerosis. Nucl Instrum Methods Phys Res B-Beam Interact Mater Atoms. 2006;249(1–2):646–652.
- Yuan XM. Apoptotic macrophage-derived foam cells of human atheromas are rich in iron and ferritin, suggesting iron-catalysed reactions to be involved in apoptosis. Free Radic Res. 1999;30(3):221–231.
- You SA, Archacki SR, Angheloiu G, et al. Proteomic approach to coronary atherosclerosis shows ferritin light chain as a significant marker: evidence consistent with iron hypothesis in atherosclerosis. Physiol Genomics. 2003;13(1):25–30.
- Salonen JT, Nyyssonen K, Korpela H, et al. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men . Circulation. 1992;86(3):803–811.
- Kiechl S, Willeit J, Egger G, et al. Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation. 1997;96(10):3300–3307.
- Cai J, Zhang M, Liu YT, et al. Iron accumulation in macrophages promotes the formation of foam cells and development of atherosclerosis. Cell Biosci. 2020;10(1):137.
- Wen Y, Leake DS. Low density lipoprotein undergoes oxidation within lysosomes in cells. Circ Res. 2007;100(9):1337–1343.
- Ahmad F, Leake DS. Antioxidants inhibit low density lipoprotein oxidation less at lysosomal pH: a possible explanation as to why the clinical trials of antioxidants might have failed. Chem Phys Lipids. 2018;213:13–24.
- Ahmad F, Mitchell RD, Houben T, et al. Cysteamine decreases low density lipoprotein oxidation, causes regression of atherosclerosis and improves liver and muscle function in LDL receptor deficient mice JAHA. J Am Heart Assoc. 2021 (in press).
- Ahmad F, Leake DS. Lysosomal oxidation of LDL alters lysosomal pH, induces senescence, and increases secretion of pro-inflammatory cytokines in human macrophages. J Lipid Res. 2019;60(1):98–110.
- Wen Y, Ahmad F, Mohri Z, et al. Cysteamine inhibits lysosomal oxidation of low density lipoprotein in human macrophages and reduces atherosclerosis in mice. Atherosclerosis. 2019;291:9–18.
- Ojo OO, Leake DS. Low density lipoprotein oxidation by ferritin at lysosomal pH. Chem Phys Lipids. 2018;217:51–57.
- Mancias JD, Wang XX, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–109.
- del Rey MQ, Mancias JD. NCOA4-mediated ferritinophagy: a potential link to neurodegeneration. Front Neurosci. 2019;13:238.
- tenKate J, Wolthuis A, Westerhuis B, et al. The iron content of serum ferritin: Physiological importance and diagnostic value. Eur J Clin Chem Clin Biochem . 1997;35(1):53–56.
- Li L, Fang CJ, Ryan JC, others, et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc Natl Acad Sci USA. 2010;107(8):3505–3510.
- Wilkins GM, Leake DS. The effect of inhibitors of free radical generating-enzymes on low-density lipoprotein oxidation by macrophages. Biochim Biophys Acta. 1994;1211(1):69–78.
- Esterbauer H, Dieber-Rotheneder M, Striegl G, et al. Role of vitamin E in preventing the oxidation of low-density lipoprotein. Am J Clin Nutr. 1991;53(1 Suppl):314S–321s.
- Satchell L, Leake DS. Oxidation of low-density lipoprotein by iron at lysosomal pH: implications for atherosclerosis. Biochemistry. 2012;51(18):3767–3775.
- Esterbauer H, Dieber-Rotheneder M, Striegl G, et al. Role of vitamin E in preventing the oxidation of low-density lipoprotein. Am J Clin Nutr. 1991;53(1):S):314S–321S.
- Anonymous. New reference values for vitamin C intake. Ann Nutr Metabol. 2015;67:13–20.
- Hodis HN, Mack WJ, LaBree L, et al. Alpha-Tocopherol supplementation in healthy individuals reduces low-density lipoprotein oxidation but not atherosclerosis – the vitamin E atherosclerosis prevention study (VEAPS). Circulation. 2002;106(12):1453–1459.
- Keith ME, Jeejeebhoy KN, Langer A, et al. A controlled clinical trial of vitamin E supplementation in patients with congestive heart failure. Am J Clin Nutr. 2001;73(2):219–224.
- Lonn E, Bosch J, Yusuf S, et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer – a randomized controlled trial. JAMA. 2005;293(11):1338–1347.
- Bowry VW, Ingold KU, Stocker R. Vitamin E in human low-density lipoprotein. When and how this antioxidant becomes a pro-oxidant. Biochem J. 1992;288(Pt2):341–344.
- Thomas SR, Stocker R. Molecular action of vitamin E in lipoprotein oxidation: implications for atherosclerosis. Free Radic Biol Med. 2000;28(12):1795–1805.
- Esterbauer H, Gebicki J, Puhl H, et al. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med. 1992;13(4):341–390.
- Thomas SR, Neuzil J, Mohr D, et al. Coantioxidants make alpha-tocopherol an efficient antioxidant for low-density lipoprotein. Am J Clin Nutr. 1995;62(6 Suppl):1357S–1364S.
- Mukai K, Nishimura M, Kikuchi S. Stopped-flow investigation of the reaction of vitamin C with tocopheroxyl radical in aqueous triton X-100 micellar solutions – the structure-activity relationship of the regeneration reaction of tocopherol by vitamin C. J Biol Chem. 1991;266(1):274–278.
- Gieβauf A, Steiner E, Esterbauer H. Early destruction of tryptophan residues of apolipoprotein B is a vitamin E-independent process during copper-mediated oxidation of LDL. Biochim Biophys Acta-Lipids Lipid Metabol. 1995;1256(2):221–232.
- Frei B, Gaziano JM. Content of antioxidants, preformed lipid hydroperoxides, and cholesterol as predictors of the susceptibility of human LDL to metal ion-dependent and ion-independent oxidation. J Lipid Res. 1993;34(12):2135–2145.
- Retsky KL, Chen K, Zeind J, et al. Inhibition of copper-induced LDL oxidation by vitamin C is associated with decreased copper-binding to LDL and 2-oxo-histidine formation. Free Radic Biol Med. 1999;26(1–2):90–98.
- Cabelli DE, Bielski BHJ. Kinetics and mechanism for the oxidation of ascorbic acid/ascorbate by HO2/O2− radicals. A pulse radiolysis and stopped-flow photolysis study. J Phys Chem. 1983;87(10):1809–1812.