
Abstract
Individuals with sickle cell disease (SCD) are at greater risk of rhabdomyolysis, a potentially life-threatening condition resulting from the breakdown of skeletal muscle fibers. Acute kidney injury (AKI) is one of the most severe complications of rhabdomyolysis. Chronic kidney and cardiovascular disease, which account for SCD mortality, are long-term consequences of AKI. Although SCD elevates the risks of rhabdomyolysis-induced sudden death, the mechanisms that underlie rhabdomyolysis-induced AKI in SCD are unclear. In the present study, we show that, unlike their control non-sickling (AA) counterparts, transgenic homozygous SCD (SS; Townes model) mice exhibited 100% mortality 8–24 h after intramuscular glycerol injection. Five hours after glycerol injection, SS mice showed a more significant increase in myoglobinuria and plasma creatine kinase levels than AA mice. Basal plasma heme and kidney tissue iron levels were significantly higher in SS than in AA mice. In contrast to AA, glycerol-induced rhabdomyolysis aggravated these parameters in SS mice. Rhabdomyolysis also amplified oxidative stress in SS compared to AA mice. Glycerol-treated SS mice exhibited worse renal function, exemplified by a reduction in GFR with a corresponding increase in plasma and urinary biomarkers of early AKI and renal tubular damage. The free radical scavenger and Fenton chemistry inhibitor, TEMPOL, ameliorated rhabdomyolysis-induced AKI in the SS mice. These findings demonstrate that oxidative stress driven by renal iron accumulation amplifies rhabdomyolysis-induced AKI in SCD mice.
Introduction
Sickle cell disease (SCD) generally refers to various inherited disorders caused by different genotypes of the sickle cell gene in which at least half of the hemoglobin (Hb) is made up of Hb S variant [Citation1]. This mutation causes HbS to polymerize upon deoxygenation, thereby changing red blood cell structure into sickled shapes [Citation1]. Fragile, rigid, sickled red blood cells cause cellular injury, hemolytic anemia, and blood flow obstruction [Citation1]. Patients who are homozygous HbSS or those with HbS/β° thalassemia have severe sickle cell anemia complications, including pain crises, acute chest syndrome, and multisystem organ damage [Citation1–3]. SCD is a significant genetic blood disorder in the United States, affecting approximately 1 in every 500 African-American and 1 in every 1000 Hispanic-American births [Citation4]. Despite recent improvements in clinical care, the median life expectancy of patients with severe genotypes of SCD remains less than 50 years [Citation5].
Kidney disease at varying levels of severity is associated with increased SCD mortality [Citation6]. Renal complications in SCD include albuminuria, hematuria, isosthenuria, tubular acidosis, impaired glomerular filtration, and glomerulosclerosis [Citation1,Citation7–9]. Chronic kidney disease (CKD) develops earlier in SCD than in non-SCD patients [Citation1,Citation7–11]. The mechanisms that underlie renal pathology in SCD are not fully resolved. However, the hypoxic, acidotic, and hypertonic renal medulla predisposes the HbS to polymerization in the microvessels [Citation1]. The resulting medullary vasoocclusion may cause renal ischemia and infarction [Citation1]. Glomerulosclerosis, on the other hand, may result from glomerular hyperfiltration [Citation12].
CKD and cardiovascular events, which account for SCD morbidity and mortality, are long-term consequences of acute kidney injury (AKI). AKI indicates a sudden reduction in kidney function, causing a decrease in urine production and a build-up of nitrogenous products in the blood. Although AKI is a clinical syndrome with multiple etiologies, factors predisposing SCD patients to sudden kidney insufficiency include episodes of pain crises, volume depletion, infections, non-steroidal anti-inflammatory drugs, and rhabdomyolysis (rhabdo) [Citation7].
Rhabdo results from skeletal muscle breakdown, releasing muscle cell contents, including creatine kinase and myoglobin, into the bloodstream [Citation13–16]. High concentrations of circulating myoglobin can accumulate in the kidney tubules leading to cast formation, tubular obstruction, oxidative stress, and cellular injury [Citation13–16]. Foremost among the causes of rhabdo in SCD is exertion. Athletes and members of the United States military with sickle cell trait (SCT) have been reported to have an increased risk of exertional rhabdo, which may be complicated by severe dehydration, hyperthermia, and tissue hypoxia [Citation17–22]. Clinical findings indicate that SCD patients hospitalized for AKI frequently have concomitant rhabdo [Citation23]. Although renal pathology is more severe in homozygous SCD (SS), the pathophysiology of rhabdo-induced AKI in SS is poorly understood. Myoglobin-derived iron can bind molecular oxygen, which may produce a hydroxyl radical in the oxidation of ferrous oxide (Fe2+) to ferric oxide (Fe3+) via the Fenton reaction. A combination of iron accessibility and increased production of oxyradicals can contribute to cellular derangement and AKI [Citation24].
In the present study, we employed the model of glycerol-induced muscle damage to investigate the severity of rhabdo in transgenic SS mice. We also tested the hypothesis that the mechanism of myoglobin-induced renal toxicity includes heightened renal iron accumulation and oxidative stress.
Materials and methods
Animals
Male and female 8-week-old Townes transgenic HbSS (SS; sickling) and HbAA (AA; non-sickling) control mice (B6;129-Hbbtm2(HBG1,HBB*)Tow/Hbbtm3(HBG1,HBB)Tow Hbatm1(HBA)Tow/J; Strain #: 013071), were obtained from the Jackson Laboratory (Bar Harbor, ME) [Citation25,Citation26]. The mice were bred, genotyped, and maintained in our animal facilities. The mice were provided ad libitum access to food and water. The Institutional Animal Care and Use Committee at the University of Tennessee Health Science Center approved all experimental protocols. Animal studies followed the ARRIVE guidelines during the design and implementation of the studies.
Glycerol-induced rhabdo protocol
Mice were deprived of water for 18–24h before being anesthetized with isoflurane and intramuscular injection with glycerol. Upon confirmation of anesthesia, glycerol or vehicle (saline) was injected equally into both hindlimbs as follows: (7 mL/kg of glycerol diluted 1:2 in saline or 7 mL/kg of saline for control mice). The TEMPOL-treated group received an intraperitoneal injection of TEMPOL (100 mg/kg) 1 h before glycerol treatment. All mice received Buprenorphine SR (1 mg/kg) for pain management and were allowed to recover in their cages.
Determination of glomerular filtration rate (GFR)
FITC-sinistrin clearance was determined by the transdermal methods to measure the GFR as previously described [Citation27–29]. Briefly, the optical transdermal GFR device (MediBeacon GmbH, Mannheim, Germany) was attached to a shaved flank region of the mice using a dual-sided adhesive patch. FITC-Sinistrin (100 mg/kg) was injected into the tail vein, and the device was left for 1.5 h to record FITC-sinistrin clearance, which was removed at the end of the acquisition. The data was analyzed following the manufacturer’s instructions using the MediBeacon Studio 3 software.
Biochemical assays
Urine myoglobin levels were measured using the mouse myoglobin ELISA kit (80654; Crystal Chem, Elk Grove Village, IL). Plasma creatine kinase levels were detected using the mouse creatine kinase ELISA kit (RK02687; ABclonal, Woburn, MA). Kidney iron content (ab83366), plasma IL-18 (ab216165), urinary NGAL (ab199083), and plasma heme (ab272534) were determined using kits purchased from Abcam (Cambridge, UK). Plasma liver fatty acid binding protein (L-FABP) was detected using the mouse ELISA kit (BG-MUS11491) from Novatein Biosciences (Woburn, MA). Plasma H2O2 levels were determined using the hydrogen peroxide colorimetric detection kit (K034-H1) from Arbor Assays (Ann Arbor, MI). Urine 8-OHdG levels were measured using the DNA damage kit (SKT-120-96S) from StressMarq Biosciences (Victoria, BC). The kits were used following the manufacturer’s instructions. Urinary markers were normalized to urinary creatinine to control for urine flow rate and creatinine clearance variations. Liquid chromatography-tandem mass spectrometry was used to determine creatinine concentrations at the UAB/UCSD O'Brien Core Center for AKI Research at the University of Alabama at Birmingham.
Quantitative RT-PCR (qRT-PCR)
The Direct-zol RNA Miniprep Plus kit (Zymo Research, R2073) was used to isolate total RNA. cDNA was synthesized from the RNA samples using the Applied Biosystems High-Capacity cDNA Reverse Transcription kit (Life Technologies, 4368813). Reactions were performed in an Applied Biosystems QuantStudio 3 system using the Applied Biosystems PowerUp SYBR Green Master Mix kit (Life Technologies, A25742). β-actin was used as the housekeeping gene control. The expression levels of gene transcripts were determined using 2−ΔΔCt, where the Ct values of the gene of interest were first normalized to β-actin and then to the ΔCt of the control. The gene-specific oligonucleotide primers used are listed in .
Table 1. Oligonucleotide primer sequences.
Histology
Kidney samples were fixed in 10% neutral-buffered formalin, processed for paraffin embedding using a HISTOS 3/5 microwave tissue processor, and embedded in paraffin. The samples were then sectioned 6 µm thick, deparaffinized, dehydrated in graded alcohols, stained for Hematoxylin and Eosin, and imaged with a microscope. The sections were examined independently for morphological damage. The slides were scored blindly by assessment of the percentage of damaged tubules: 0–20% (0), 21–40% (1), 41–60% (2), 61–80% (3), and 81–100% (4).
Renal iron stain
Paraffin-embedded kidney sections were deparaffinized and dehydrated, as described above. The sections were stained using an iron kit (Prussian Blue stain; ab150674) purchased from Abcam, per the manufacturer’s protocol. Microscopy images were analyzed with ImageJ software (NIH, Bethesda, MD).
Statistical analysis
Data are presented as means ± SE. GraphPad Prism software (Graph Pad, Sacramento, CA) was used for data analysis. One-way ANOVA with Holm-Šídák’s multiple comparisons test was used. P values < 0.05 were considered significant. For data marked with bars and asterisks (to indicate statistical significance), P values less than 0.05, 0.01, 0.001, and 0.0001 are summarized with one, two, three, and four asterisks, respectively.
Results
Glycerol injection in SCD mice is lethal after 5 h
Unlike the control mice (AA), homozygous SCD mice (SS) exhibited 100% mortality 8-24 h after intramuscular glycerol injection. However, a 100% survival rate was recorded in the SS mice subjected to 5 h after glycerol injection.
Glycerol injection heightens rhabdo in SCD mice
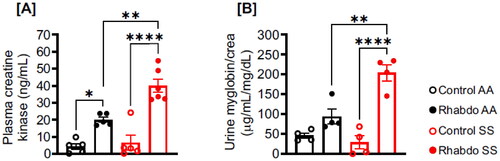
Five hours after glycerol injection, both AA and SS mice showed elevated levels of biomarkers of rhabdo, plasma creatine kinase, and urinary myoglobin (). However, glycerol injection caused a more significant increase in plasma creatine kinase and urinary myoglobin in SS compared to AA mice ().
Figure 1. Glycerol-induced rhabdo is apparent in 5 h and aggravated in SS mice. Five hours after glycerol injection, plasma, and urine samples were harvested to measure rhabdo markers in AA and SS mice. (A) Plasma creatine kinase and (B) urinary myoglobin in control and rhabdo AA and SS mice (one-way ANOVA, with Holm-Šídák’s posthoc test).
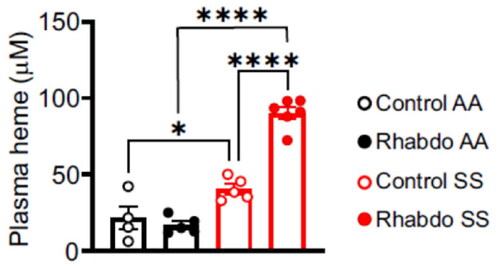
Rhabdo-induced increases in plasma heme are amplified in SCD mice
No significant difference in plasma heme levels was detected between rhabdo AA and control AA mice.
(). The basal plasma heme level in control SS was significantly higher than that of control AA mice (). Upon treatment with glycerol, the plasma heme level was ∼ 2-fold higher in rhabdo SS compared to the control SS mice ().
Figure 2. Rhabdo increases plasma heme levels in SS mice. Plasma heme levels in control and rhabdo AA and SS mice (one-way ANOVA, with Holm-Šídák’s posthoc test).
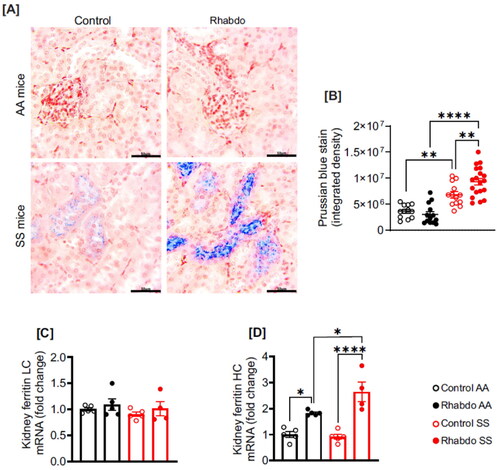
Rhabdo increases iron accumulation in SCD mouse kidneys
Renal iron staining was performed in control and glycerol-treated AA and SS mouse kidney sections (). Control and glycerol-treated AA mice showed no iron accumulation in kidney tissues (). Conversely, control SS mice show moderate but significant basal levels of iron accumulation in kidney tissue, which was exacerbated in the glycerol-treated SS group (). Whereas kidney mRNA expression of ferritin light-chain was unchanged across all groups, ferritin heavy chain was increased in glycerol-treated AA and SS mice (). Glycerol-induced increase in ferritin heavy-chain mRNA was significantly upregulated in SS mice ().
Figure 3. Rhabdo increases kidney iron accumulation and upregulates ferritin heavy chain in SS mice. (A) Kidney sections were stained with Prussian blue to detect iron deposition in AA and SS mice, (B) scatter plots summarizing Prussian blue stain density in the mice, (C-D) relative mRNA expression of ferritin light chain (LC) and heavy chain (HC) in the kidneys of control and rhabdo AA and SS mice (one-way ANOVA, with Holm-Šídák’s posthoc test); scale bar = 50 µm.
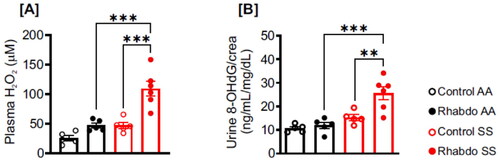
Rhabdo-induced oxidative stress is amplified in SCD mice
Plasma H2O2 level was insignificant in control AA, rhabdo AA, and control SS groups (). However, plasma H2O2 level was significantly elevated in glycerol-treated SS mice (). Similarly, the urinary levels of 8-OHdG, an oxidative DNA damage product were comparable in control AA, rhabdo AA, and control SS groups (). By contrast, 8-OHdG level in rhabdo SS mice was significantly higher ().
Figure 4. Rhabdo induces oxidative stress in SS mice. (A) Plasma H2O2 and (B) urinary 8-OHdG in control and rhabdo AA and SS mice (one-way ANOVA, with Holm-Šídák’s posthoc test).
TEMPOL mitigates rhabdo-induced AKI and tubular injury in SS mice
Delayed FITC-sinistrin clearance indicated a reduction in GFR of glycerol-treated AA and SS mice when compared with respective controls (). However, glycerol-induced GFR reduction was more significant in the SS mice (). Urine NGAL, plasma L-FABP, and IL-18, which are early biomarkers of AKI, were similar in control AA, rhabdo AA, and control SS groups (). These indices were significantly increased in glycerol-treated SS mice (). Pretreatment of SS mice with TEMPOL protected against GFR reduction and urine NGAL, plasma L-FABP, and IL-18 increases in glycerol-treated SS mice (). Histological counts for tubular damage exemplified by nuclear loss, brush border damage, cast formation, and hypernucleation were higher only in glycerol-treated SS mice, an effect attenuated in TEMPOL-pretreated mice ().
Figure 5. Rhabdo-induced oxidative stress aggravates AKI parameters in SS mice. (A) GFR, (B) urinary NGAL, (C) plasma L-FABP, and (D) plasma IL-18 in control AA and SS, rhabdo AA and SS, and TEMPOL + rhabdo SS groups (one-way ANOVA, with Holm-Šídák’s posthoc test).
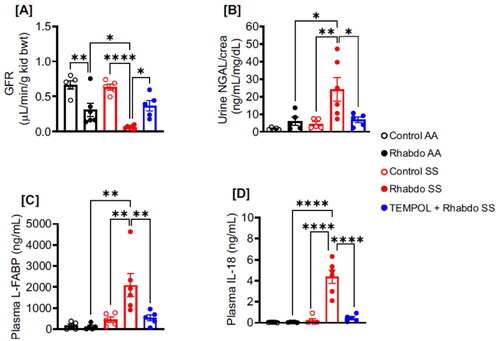
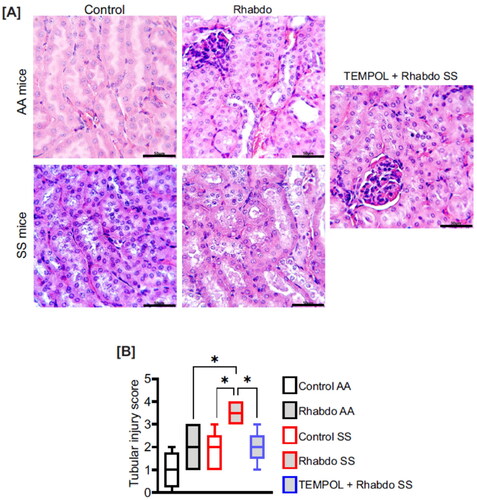
Figure 6. Rhabdo-induced oxidative stress causes significant renal tubular damage in SS mice. (A) Representative H&E-stained kidney sections and (B) box & whiskers plot of tubular injury score in control AA (n = 4) and SS (n = 5), rhabdo AA (n = 5) and SS (n = 6), and TEMPOL + rhabdo SS (n = 5) groups. (one-way ANOVA, with Holm-Šídák’s posthoc test); scale bar = 50 µm.
Discussion
The findings in this study suggest that skeletal muscle damage is more lethal in sickle cell mice than in non-sickling counterparts. All AA mice survived up to 48h post glycerol injection. By contrast, SS exhibited 100% mortality 8–24 h after intramuscular glycerol injection. Hence, the protocol for experiments in this paper, for both AA and SS mice, was adjusted to 5h, where both groups had 100% survival. Increased iron accumulation leading to ROS formation is consistent with other studies [Citation30,Citation31]. Oxidative stress observed in the SS mice was associated with AKI. Hence, to recover the SS mice kidneys from AKI, we scavenged ROS using TEMPOL, which mitigated kidney insufficiency and tubular injury. Therefore, we provide novel data indicating that a shorter duration of rhabdo is sufficient to cause significant kidney injury in SS mice, notably in renal tubular cells driven by oxidative stress caused by renal iron accumulation.
The classical laboratory tests for rhabdo are elevated blood creatine kinase and urine myoglobin [Citation13–16]. The present study found that although basal creatine kinase and urine myoglobin levels in control AA and SS mice were the same, glycerol-induced increases in these markers were ∼ twofold higher in SS mice, suggesting that glycerol-induced rhabdo is aggravated in the SS mice. Since AKI depends on hydration status during rhabdo, an experimental model of glycerol-induced myoglobinuric AKI involves prior water deprivation. Dehydration may increase the build-up of myoglobin as it impairs the ability to get rid of the protein efficiently. Dehydration can also worsen skeletal muscle damage [Citation32]. Impaired urine concentration is common in SCD, which increases the risk of cellular dehydration [Citation7,Citation33]. Muscle function and energetics have also been shown to be altered in SCD [Citation34,Citation35]. Hence, potential contributing factors to amplified plasma creatine kinase levels and urine myoglobin in the rhabdo SS mice may include exacerbated dehydration and impaired muscle function.
Since SS mice and humans experience chronic hemolysis, releasing free heme and iron into the bloodstream, it is unsurprising to observe high basal plasma heme and iron levels in the mice [Citation36,Citation37]. Myoglobin, an approximately 17 kDa iron- and oxygen-binding protein, is filtered in kidney glomeruli and reabsorbed in the proximal tubules by endocytic receptors megalin and cubilin [Citation38]. An acidic environment drives oxyradical-producing ferrihemate dissociation from myoglobin [Citation15]. Hence, a combination of hemolysis and increased heme and iron released from myoglobin may contribute to rhabdo-induced increases in plasma heme and renal iron levels. Ferritin is a major regulator of intracellular iron [Citation39,Citation40]. An iron storage protein, ferritin also catalyzes the oxidation of Fe2+ to Fe3+ [Citation39,Citation40]. Vertebrate ferritin molecules exist in 2 distinctive subunits: light chain (ferritin LC) and heavy chain (ferritin HC) [Citation39,Citation40]. Unlike ferritin HC, ferritin LC lacks ferroxidase activity [Citation39,Citation40]. Renal iron accumulation increases tissue ferritin levels [Citation41], consistent with the findings of this study. The upregulation of ferritin HC in the SS mice may be due to increased ferroxidase activity.
Although the proximal tubule reabsorbs filtered iron, excessive renal iron loading contributes to AKI and CKD, of many provenances [Citation42,Citation43]. Iron nephrotoxicity is primarily mediated by increased oxidative cellular injury, driven mainly by the Fenton reaction, whereby Fe3+ reacts with H2O2 to generate highly toxic oxyradicals [Citation44]. Although basal plasma H2O2 and urinary 8-OHdG (a biomarker of oxidative DNA damage) were similar in AA mice after 5 h of glycerol injection, these parameters were significantly increased in SS mice. The findings of this study are consistent with previous reports on the significant contribution of ROS-induced DNA damage on renal tubular injury [Citation45,Citation46].
GFR was reduced by rhabdo in both AA and SS mice, but the reduction was more severe in SS mice, indicating a more impaired renal function in the SS mice. Plasma L-FABP and urinary NGAL are associated with the severity of the renal tubular injury, with NGAL being able to detect renal tubular damage as early as 2 h after injury [Citation47–49]. There is also evidence that plasma L-FABP plays a cytoprotective role by mitigating H2O2-induced oxidative stress [Citation49]. Plasma IL-18, as a proinflammatory cytokine, is both a mediator and biomarker of early AKI [Citation50,Citation51]. Only in the rhabdo SS mice were the urinary and plasma biomarkers of AKI elevated. Histopathological analysis suggested significant renal tubular cell damage occurred only in the rhabdo SS mice. These findings illustrate the heightened sensitivity of the SS mice to rhabdo-induced AKI and the severity of renal function impairment and tubular injury associated with renal iron accumulation and oxidative stress in rhabdo SS mice.
The antioxidant properties of TEMPOL are attributed to its superoxide dismutase mimetic activity and ability to facilitate H2O2 metabolism and inhibit Fenton reactions [Citation52,Citation53] TEMPOL injected intraperitoneally to mice was tolerated up to 275 mg/kg and alleviated whole-body radiation injury [Citation54]. At 100 mg/kg, TEMPOL provided significant protection against ROS-dependent AKI in cisplatin-treated mice [Citation55]. We show here that pretreatment with the same dose of TEMPOL ameliorated rhabdo-induced AKI in SS mice. The critical point of this study was to focus on the early phase of AKI. Hence, our study did not determine whether TEMPOL attenuates mortality in SS mice exposed to a longer duration of glycerol injection. However, this question requires further investigation.
This study suggests that rhabdo can cause significant renal damage in as little as five hours in SS mice. We attribute this heightened level of injury to oxidative stress facilitated by iron accumulation in the kidneys. Previous studies have suggested that military personnel and athletes with SCT are at a greater risk of exercise-induced rhabdo, the mechanisms of which are debatable [Citation17–22]. Despite the high prevalence of AKI and CKD in SCD and its association with substantial morbidity and mortality, the available treatment options remain limited. Further studies are necessary to investigate whether pharmacologically neutralizing free radicals is a valuable approach to preventing or managing rhabdo-induced early AKI in SCD humans.
Authors’ contributions
A.A. conceived and designed research. J.D.W., R.K., and J.M.A. performed experiments. F.P. performed histological analysis. A.A. and J.D.W. analyzed and interpreted results, prepared figures, and drafted the manuscript.
Acknowledgments
The authors would like to acknowledge the UAB-UCSD O'Brien Core Center for Acute Kidney Injury Research for assistance with independent creatinine analysis.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. doi: 10.1016/S0140-6736(10)61029-X.
- Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376(16):1561–1573. doi: 10.1056/nejmra1510865.
- Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev Pathol. 2019;14(1):263–292. doi: 10.1146/annurev-pathmechdis-012418-012838.
- (WHO), W. H. O. Sickle-cell anaemia - Report by the Secretariat. 2006.
- DeBaun MR, Ghafuri DL, Rodeghier M, et al. Decreased median survival of adults with sickle cell disease after adjusting for left truncation bias: a pooled analysis. Blood. 2019;133(6):615–617. doi: 10.1182/blood-2018-10-880575.
- McClellan AC, Luthi J-C, Lynch JR, et al. High one year mortality in adults with sickle cell disease and end-stage renal disease. Br J Haematol. 2012;159(3):360–367. doi: 10.1111/bjh.12024.
- Nath KA, Hebbel RP. Sickle cell disease: renal manifestations and mechanisms. Nat Rev Nephrol. 2015;11(3):161–171. doi: 10.1038/nrneph.2015.8.
- Ataga KI, Saraf SL, Derebail VK. The nephropathy of sickle cell trait and sickle cell disease. Nat Rev Nephrol. 2022;18(6):361–377. doi: 10.1038/s41581-022-00540-9.
- Steinberg MH. Sickle cell anemia, the first molecular disease: overview of molecular etiology, pathophysiology, and therapeutic approaches. Sci World J. 2008;8:1295–1324. doi: 10.1100/tsw.2008.157.
- Wesson DE. The initiation and progression of sickle cell nephropathy. Kidney Int. 2002;61(6):2277–2286. doi: 10.1046/j.1523-1755.2002.00363.x.
- Ware RE, Rees RC, Sarnaik SA, et al. Renal function in infants with sickle cell anemia: baseline data from the BABY HUG trial. J Pediatr. 2010;156(1):66–70.e1. doi: 10.1016/j.jpeds.2009.06.060.
- Tejani A, Phadke K, Adamson O, et al. Renal lesions in sickle cell nephropathy in children. Nephron. 1985;39(4):352–355. doi: 10.1159/000183404.
- Torres PA, Helmstetter JA, Kaye AM, et al. Rhabdomyolysis: pathogenesis, diagnosis, and treatment. Ochsner J. 2015;15(1):58–69.
- Knochel JP. Rhabdomyolysis and myoglobinuria. Annu Rev Med. 1982;33(1):435–443. doi: 10.1146/annurev.me.33.020182.002251.
- Efstratiadis G, et al. Rhabdomyolysis updated. Hippokratia. 2007;11:129–137.
- Bosch X, Poch E, Grau JM. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009;361(1):62–72. doi: 10.1056/NEJMra0801327.
- Harrelson GL, Fincher AL, Robinson JB. Acute exertional rhabdomyolysis and its relationship to sickle cell trait. J Athl Train. 1995;30(4):309–312.
- O'Connor FG, Bergeron MF, Cantrell J, et al. ACSM and CHAMP summit on sickle cell trait: mitigating risks for warfighters and athletes. Med Sci Sports Exerc. 2012;44(11):2045–2056. doi: 10.1249/MSS.0b013e31826851c2.
- Nelson DA, Deuster PA, Carter R, et al. Sickle cell trait, rhabdomyolysis, and mortality among U.S. Army soldiers. N Engl J Med. 2016;375(5):435–442. doi: 10.1056/NEJMoa1516257.
- Kerle KK, Nishimura KD. Exertional collapse and sudden death associated with sickle cell trait. Mil Med. 1996;161(12):766–767.
- Quattrone RD, Eichner ER, Beutler A, et al. Exercise collapse associated with sickle cell trait (ECAST): case report and literature review. Curr Sports Med Rep. 2015;14(2):110–116. doi: 10.1249/jsr.0000000000000137.
- Buchanan BK, Siebert DM, Zigman Suchsland ML, et al. Sudden death associated with sickle cell trait before and after mandatory screening. Sports Health. 2020;12(3):241–245. doi: 10.1177/1941738120915690.
- Sklar AH, Perez JC, Harp RJ, et al. Acute renal failure in sickle cell anemia. Int J Artif Organs. 1990;13(6):347–351.
- Meneghini R. Iron homeostasis, oxidative stress, and DNA damage. Free Radic Biol Med. 1997;23(5):783–792. doi: 10.1016/s0891-5849(97)00016-6.
- Wu L-C, Sun C-W, Ryan TM, et al. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood. 2006;108(4):1183–1188. doi: 10.1182/blood-2006-02-004812.
- Ryan TM, Ciavatta DJ, Townes TM. Knockout-transgenic mouse model of sickle cell disease. Science. 1997;278(5339):873–876. doi: 10.1126/science.278.5339.873.
- Schock-Kusch D, Xie Q, Shulhevich Y, et al. Transcutaneous assessment of renal function in conscious rats with a device for measuring FITC-sinistrin disappearance curves. Kidney Int. 2011;79(11):1254–1258. doi: 10.1038/ki.2011.31.
- Pais GM, Chang J, Liu J, et al. A translational rat model to assess glomerular function changes with vancomycin. Int J Antimicrob Agents. 2022;59(5):106583. doi: 10.1016/j.ijantimicag.2022.106583.
- Fanous MS, Afolabi JM, Michael OS, et al. Transdermal measurement of glomerular filtration rate in mechanically ventilated piglets. J Vis Exp. 2022;(187):e64413. doi: 10.3791/64413.
- Galaris D, Barbouti A, Pantopoulos K. Iron homeostasis and oxidative stress: an intimate relationship. Biochim Biophys Acta Mol Cell Res. 2019;1866(12):118535. doi: 10.1016/j.bbamcr.2019.118535.
- Galaris D, Pantopoulos K. Oxidative stress and iron homeostasis: mechanistic and health aspects. Crit Rev Clin Lab Sci. 2008;45(1):1–23. doi: 10.1080/10408360701713104.
- Ozkan I, Ibrahim CH. Dehydration, skeletal muscle damage and inflammation before the competitions among the elite wrestlers. J Phys Ther Sci. 2016;28(1):162–168. doi: 10.1589/jpts.28.162.
- Brugnara C. Sickle cell dehydration: pathophysiology and therapeutic applications. Clin Hemorheol Microcirc. 2018;68(2-3):187–204. doi: 10.3233/ch-189007.
- Merlet AN, Chatel B, Hourdé C, et al. How sickle cell disease impairs skeletal muscle function: implications in daily life. Med Sci Sports Exerc. 2019;51(1):4–11. doi: 10.1249/mss.0000000000001757.
- Chatel B, Hourdé C, Gondin J, et al. Impaired muscle force production and higher fatigability in a mouse model of sickle cell disease. Blood Cells Mol Dis. 2017;63:37–44. doi: 10.1016/j.bcmd.2017.01.004.
- Nguyen J, Abdulla F, Chen C, et al. Phenotypic characterization the townes sickle mice. Blood. 2014;124(21):4916–4916. doi: 10.1182/blood.V124.21.4916.4916.
- Schein A, Enriquez C, Coates TD, et al. Magnetic resonance detection of kidney iron deposition in sickle cell disease: a marker of chronic hemolysis. J Magn Reson Imaging. 2008;28(3):698–704. doi: 10.1002/jmri.21490.
- Gburek J, Birn H, Verroust PJ, et al. Renal uptake of myoglobin is mediated by the endocytic receptors megalin and cubilin. Am J Physiol Renal Physiol. 2003;285(3):F451–458. doi: 10.1152/ajprenal.00062.2003.
- Honarmand Ebrahimi K, Hagedoorn P-L, Hagen WR. Unity in the biochemistry of the iron-storage proteins ferritin and bacterioferritin. Chem Rev. 2015;115(1):295–326. doi: 10.1021/cr5004908.
- Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta. 1996;1275(3):161–203. doi: 10.1016/0005-2728(96)00022-9.
- Zager RA, Johnson AC, Hanson SY. Parenteral iron nephrotoxicity: potential mechanisms and consequences. Kidney Int. 2004;66(1):144–156. doi: 10.1111/j.1523-1755.2004.00716.x.
- van Raaij S, van Swelm R, Bouman K, et al. Tubular iron deposition and iron handling proteins in human healthy kidney and chronic kidney disease. Sci Rep. 2018;8(1):9353. doi: 10.1038/s41598-018-27107-8.
- Walker VJ, Agarwal A. Targeting iron homeostasis in acute kidney injury. Semin Nephrol. 2016;36(1):62–70. doi: 10.1016/j.semnephrol.2016.01.003.
- Leaf DE, Swinkels DW. Catalytic iron and acute kidney injury. Am J Physiol Renal Physiol. 2016;311(5):F871–f876. doi: 10.1152/ajprenal.00388.2016.
- Yan M, Tang C, Ma Z, et al. DNA damage response in nephrotoxic and ischemic kidney injury. Toxicol Appl Pharmacol. 2016;313:104–108. doi: 10.1016/j.taap.2016.10.022.
- Wang P, Ouyang J, Jia Z, et al. Roles of DNA damage in renal tubular epithelial cells injury. Front Physiol. 2023;14:1162546. doi: 10.3389/fphys.2023.1162546.
- Zeng X-F, Li J-M, Tan Y, et al. Performance of urinary NGAL and L-FABP in predicting acute kidney injury and subsequent renal recovery: a cohort study based on major surgeries. Clin Chem Lab Med. 2014;52(5):671–678. doi: 10.1515/cclm-2013-0823.
- Mishra J, Dent C, Tarabishi R, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005;365(9466):1231–1238. doi: 10.1016/S0140-6736(05)74811-X.
- Kashani K, Cheungpasitporn W, Ronco C. Biomarkers of acute kidney injury: the pathway from discovery to clinical adoption. Clin Chem Lab Med. 2017;55(8):1074–1089. doi: 10.1515/cclm-2016-0973.
- Wu H, Craft ML, Wang P, et al. IL-18 contributes to renal damage after ischemia-reperfusion. J Am Soc Nephrol. 2008;19(12):2331–2341. doi: 10.1681/ASN.2008020170.
- Gonzalez F, Vincent F. Biomarkers for acute kidney injury in critically ill patients. Minerva Anestesiol. 2012;78(12):1394–1403.
- Goralska M, Holley B, McGahan MC. The effects of tempol on ferritin synthesis and Fe metabolism in lens epithelial cells. Biochim Biophys Acta. 2000;1497(1):51–60. doi: 10.1016/s0167-4889(00)00038-0.
- Soule BP, Hyodo F, Matsumoto K-I, et al. The chemistry and biology of nitroxide compounds. Free Radic Biol Med. 2007;42(11):1632–1650. doi: 10.1016/j.freeradbiomed.2007.02.030.
- Hahn SM, Tochner Z, Krishna CM, et al. Tempol, a stable free radical, is a novel murine radiation protector. Cancer Res. 1992;52(7):1750–1753.
- Soni H, Kaminski D, Gangaraju R, et al. Cisplatin-induced oxidative stress stimulates renal fas ligand shedding. Ren Fail. 2018;40(1):314–322. doi: 10.1080/0886022x.2018.1456938.