
Abstract
While cysteine (CysSH) is known to be exported into the extracellular space, its biological significance is not well understood. The present study examined the movement of extracellular CysSH using stable isotope-labeled cystine (CysSSCys), which is transported into cells and reduced to CysSH. Exposure of HepG2 cells to 100 µM stable isotope-labeled CysSSCys resulted in 70 µM labeled CysSH in cell medium 1 h after CysSSCys exposure. When the cell medium was collected and incubated with either hydrogen peroxide (H2O2) or atmospheric electrophiles, such as 1,2-naphthoquinone, 1,4-naphthoquinone and 1,4-benzoquinone, CysSH in the cell medium was almost completely consumed. In contrast, extracellular levels of CysSH were unaltered during exposure of HepG2 cells to H2O2 for up to 2 h, suggesting redox cycling of CysSSCys/CysSH in the cell system. Experiments with and without changing cell medium containing CysSH from HepG2 cells revealed that oxidative and electrophilic modifications of cellular proteins, caused by exposure to H2O2 and 1,2-naphthoquinone, were significantly repressed by CysSH in the medium. We also examined participation of enzymes and/or antioxidants in intracellular reduction of CysSSCys to CysSH. These results provide new findings that extracellular CysSH derived from CysSSCys plays a role in the regulation of oxidative and electrophilic stress.
GRAPHICAL ABTSRACT
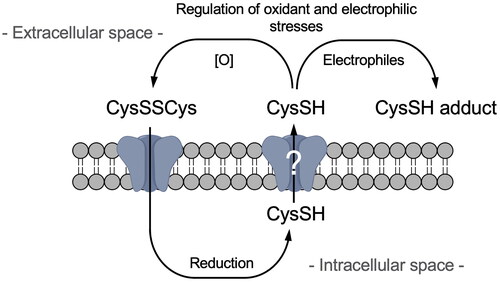
Introducton
Cysteine (CysSH) is an amino acid with a thiol group and is used to synthesize the tripeptide glutathione (GSH), known to have antioxidant capability. A variety of cells are able to export CysSH into the extracellular environment, resulting in low intracellular levels of CysSH compared with those of GSH, whereas extracellular concentrations of CysSH are higher than those of GSH [Citation1,Citation2]. These steady state concentration differences have raised questions regarding the biological significance of the extracellular CysSH.
Several lines of evidence have indicated that reactive sulfur species, such as CysSH persulfide (CysSSH), exhibit high antioxidant capability [Citation3,Citation4]. Although it was previously reported that CysSSH is secreted into extracellular space under basal conditions [Citation5,Citation6], we subsequently showed that a variety of cells (e.g. HepG2 cells, HEK293 cells, primary mouse hepatocytes) are capable of exporting excess CysSSH into the extracellular space via cystine (CysSSCys)-dependent antiporters under conditions of sulfur stress [Citation7]. However, we recently found that extracellular CysSH levels were about 200-fold of that of CysSSH in A431 cells, and that the atmospheric electrophiles 1,2-naphthoquinone (1,2-NQ), 1,4-NQ and 1,4-benzoquinone (1,4-BQ) readily reacted with CysSH, forming CysSH adducts, whereas the reactivity of these electrophiles with CysSSH was relatively low [Citation8].
These findings suggest that extracellular CysSH, and not CysSSH, may act as a negative modulator of electrophilic stress. In the current study, HepG2 cells were exposed to stable isotope-labeled CysSSCys and extracellular CysSH levels and covalent and oxidative modifications of cellular proteins were monitored to address role of extracellular CysSH under electrophilic or oxidative stress. We also examined whether cellular enzymes and/or antioxidants could contribute to the reduction of CysSSCys to CysSH. The results showed that intracellular CysSH secreted to the extracellular space acts as a nucleophile to regulate oxidants and electrophiles prior to the potential uptake of such xenobiotics into cells, demonstrating for the first time an important role of CysSH in protecting cells exposed to oxidants and electrophiles.
Materials and methods
Materials
1,2-NQ (96.5% purity determined by gas chromatography), glutathione (GSH) and streptavidin-peroxidase were obtained from Sigma-Aldrich (St Louis, MO, USA). 1,4-NQ (98% purity determined by gas chromatography), 1,4-BQ (99.1% purity determined by gas chromatography) and dihydrolipoic acid (DHLA) were purchased from Tokyo Chemical Industry (Tokyo, Japan). Hydrogen peroxidase (H2O2) and dansyl chloride were purchased from Wako (Osaka, Japan). L-Cysteine-13C3-15N-d7.DCl.D2O was obtained from Taiyo Nippon Sanso (Tokyo, Japan). Hank’s balanced salt solution (HBSS) was obtained from Thermo Fisher Scientific (Walthan, MA, USA). Biotin-conjugated dimedone derivative (DCP-Bio1) was purchased from Merk Millipore (Darmstadt, Germany). β-(4-hydroxyphenyl)ethyl iodoacetamide (HPE-IAM) was obtained from Chem-Impex International (Wood Dale, IL, USA). β-Nicotinamide-adenine dinucleotide phosphate reduced form tetrasodium salt (NADPH) was obtained from Oriental Yeast (Tokyo, Japan). L-Cysteine, L-cystine dihydrochloride, β-nicotinamide adenine dinucleotide disodium salt (NADH) and L(+)-ascorbic acid were obtained from Nacalai Tesque (Kyoto, Japan). Coenzyme Q10 (CoQ10) was purchased from Funakoshi (Tokyo, Japan). Polyclonal antibodies against 1,2-NQ were prepared as reported previously [Citation9]. All other reagents and chemicals were of the highest grade available.
Synthesis of stable isotope-labeled CysSSCys
L-Cysteine-13C3-15N-d7.DCl.D2O (2.5 mM) was incubated in 0.2 M borate buffer (pH 9.5) at 37 °C for 72 h. After checking that there is no remaining cysteine in the reaction mixture, aliquots of the reaction mixture were incubated with 5 mM dansyl chloride in 0.1 M borate buffer (pH 9.5) containing 40% acetonitrile at 25 °C for 18 h to quantify the produced stable isotope-labeled CysSSCys by using an Acquity UPLC-MSE system (Waters, MA, USA).
Ultra-performance liquid chromatography-mass spectrometry
UPLC-MSE analysis was performed using an Acquity UPLC system (Waters, MA, USA) equipped with an Acquity UPLC ethylene-bridged hybrid C18 column (2.1 mm × 50 mm i.d., 1.7 μm) maintained at 35 °C. Mobile phases A (0.1% v/v formic acid) and B (100% v/v acetonitrile with 0.1% v/v formic acid) were linearly mixed at a flow rate of 0.3 mL/min using the following gradient system: 5% v/v B with a linear increase over 11 min to 95% v/v B. The total running time, including the initial conditioning of the column, was 15 min, and the injection volume was 10 μL. The eluted compounds were transferred to a 190–500 nm photodiode array detector (Waters) and the electrospray source of a Synapt high-definition mass spectrometer (Waters). Electron spray ionization (ESI) was used, with a capillary voltage of 2.5 kV, sampling cone voltage of 40 V, and transfer cone voltages of 4 V. Low (6 eV) or elevated (incremented from 20 to 35 eV) collision energies were used to generate either the intact precursor ions (low energy) or product ions (elevated energy). The electrospray source temperature was 100 °C, and the detector was operated in the positive-ion mode. Data were acquired in the m/z = 50–1000 range using an independent reference spray via LockSpray interference with leucine enkephalin [M + H] + (m/z = 556.27) as the lock mass to ensure accuracy and reproducibility. Data were analyzed using MassLynx version 4.1 software (Waters).
Cell culture
HepG2 cells obtained from RIKEN Cell Bank (Tsukuba, Japan) were cultured in Minimum Essential Medium α (Wako) containing 10% fetal bovine serum, antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin), and 2 mM L-alanyl-L-glutamine (Invitrogen, Carlsbad, CA, USA) in an incubator supplemented with 5% CO2 at 37 °C.
Determination of intracellular and extracellular CysSH and its related compounds
Liquid chromatography-ESI-tandem mass spectrometry (LC-ESI-MS/MS) and the HPE-IAM probe were used to determine the concentration of intracellular and extracellular CysSH and its related compounds as previously reported [Citation10,Citation11]. Conditioned medium was collected and incubated with 5 mM HPE-IAM at 37 °C for 30 min to yield β-(4-hydroxyphenyl)ethyl acetamide (HPE-AM) adducts for extracellular CysSH and its related compounds. Cells were washed with ice-cold phosphate buffered saline (PBS) and collected by scraping into PBS, then pelleted by centrifugation (9000 × g, 5 min, 4 °C). Using an ultrasonic disruptor (UD-201; Tomy, Tokyo, Japan), cell pellets were homogenized in 100 μL ice-cold methanol containing 1 mM HPE-IAM, then the lysates were incubated at 37 °C for 30 min to yield HPE-AM adducts. To determine the sulfur nucleophile concentration, aliquots containing HPE-AM adducts were diluted with 0.1% (v/v) formic acid containing known amounts of isotope-labeled internal standards, then LC/MS/MS analysis was performed using an EVOQ Qube triple quadrupole mass spectrometer (Bruker, Billerica, MA, USA) coupled to an Advance ultra-high-performance liquid chromatography system (Bruker).
Western blot analysis
HepG2 cells were washed twice with PBS and then lysed in 2% sodium dodecyl sulfate (SDS) solution. After the collected cells were heated to 95 °C for 20 min, protein concentrations were determined using a bicinchoninic acid protein assay kit (Nacalai Tesque) before 2-mercaptoethanol was added to each sample. Cellular proteins were separated using SDS polyacrylamide gel electrophoresis (SDS-PAGE) with 10% w/v polyacrylamide gels and electro-transferred onto polyvinyl difluoride membranes at 2 mA/cm2 for 1 h, following the method of Kyhse-Andersen [Citation12]. Membranes were blocked with 5% w/v skim milk at 25 °C for 1 h and then incubated with primary antibodies overnight at 4 °C. The membranes were washed and then incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. Immunoreactive bands were visualized using enhanced chemiluminescence reagent (Chemi-Lumi One L, Nacalai) and FUSION FX7.EDGE (Vilber Bio Imaging, Marne-la-Vallée, France).
Detection of oxidative modifications of cellular proteins
HepG2 cells in 35 mm dishes were washed twice with ice-cold PBS and then collected in RIPA buffer [50 mM tris-HCl (pH 6.8), 150 mM sodium chloride, 1% v/v NP40, and 0.5% v/v sodium deoxycholic acid] containing 1 mM DCP-Bio1 [Citation13], 200 U/mL catalase (Sigma-Aldrich), 10 mM iodoacetamide (Wako), and protease inhibitor cocktail [150 μl per plate (Nacalai Tesque)]. Lysates were incubated on ice for 1 h and then centrifuged at 14,000 × g for 10 min. Excess unreacted compounds were removed by an ultrafiltration device with a 3 kDa molecular weight cutoff (Merk Millipore) and subjected to SDS-PAGE. Immunoblotting was performed against biotin using an HRP-conjugated antibody.
Enzyme preparation
HepG2 cells in 35 mm dishes were washed twice with ice-cold PBS and then collected in RIPA buffer supplemented with a protease inhibitor cocktail. Lysates were homogenized using an ultrasonic disruptor and then centrifuged at 13,000 × g for 5 min. The high and low-molecular-weight fractions of the resulting cell lysates were obtained using an ultrafiltration device with a 3 kDa molecular weight cutoff.
siRNA transfection
Short interfering RNAs (siRNAs) against thioredoxin (Trx)-related protein 14 (TRP14; Guide: 5′-AGUUUAUUGAUACUAGAACtt-3′, Passenger: 5′-GUUCUAGUAUCAAUAAACUtt-3′), thioredoxin (Trx)(Guide: 5′-UAGACUAAUUCAUUAAUGGtt-3′’, Passenger: 5′-CCAUUAAUGAAUUAGUCUAtt-3′) and Trx-related protein 32 (TRP32; Guide: 5′-UAAUAAGCAGCUGUUCAUCtt-3′’, Passenger: 5′-GAUGAACAGCUGCUUAUUAtt-3′) were purchased from BEX (Tokyo, Japan). Control siRNAs (catalog no. 1022076) were purchased from Qiagen (Valencia, CA, USA). Transient transfection of siRNAs was performed in serum-free medium using the Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific) with slight modification, according to the manufacturer’s protocol. Briefly, cells were grown to approximately 40% confluence and then 6 μL of siRNA duplex (20 μM) and 6 μL of RNAiMAX reagent were both mixed with OPTI-MEM (Invitrogen) in separate tubes. Before addition to the cells, the siRNA and transfection reagent solutions were mixed together and incubated for 20 min at room temperature to allow the formation of complexes.
Statistical analysis
All data are presented as the mean ± SE of at least three independent experiments. Statistical significance was assessed using one- or two-way ANOVA, followed by Tukey or Sidak’s multiple comparison tests using GraphPad Prism version 9.5.0 software, and p < 0.05 was considered significant.
Results
Interaction of secreted extracellular CysSH with oxidative/electrophilic stress agents
As shown in , stable isotope-labeled CysSSCys (100 µM added to media) was taken up by HepG2 cells and reduced to CysSH in a time-dependent manner, resulting in 70 µM stable isotope-labeled CysSH in the media at 1 h after CysSSCys exposure. In contrast, the extracellular concentration of CysSSH was only 0.20 µM (Figure S1). The high CysSH concentration suggested that it may be involved in responses to oxidative or electrophilic stress.
Figure 1. Intracellular and extracellular cysteine (CysSH) content after exposure to stable isotope-labeled cystine and the consumption of extracellular cysteine by hydrogen peroxide or electrophiles in HepG2 cells. (A) Intracellular CysSH content. Cells were exposed to stable isotope-labeled cystine (100 µM) in Hank’s balanced salt solution (HBSS) for 0.5 or 1 h, and the cells were collected with methanol containing 1 mM β-(4-hydroxyphenyl)ethyl iodoacetamide (HPE-IAM), followed by liquid chromatography-ESI-tandem mass spectrometry (LC-ESI-MS/MS) analysis as described in the Methods section. (B) Extracellular CysSH content. Cells were exposed to stable isotope-labeled cystine (100 µM) in HBSS for 0.5 or 1 h, and the conditioned medium was collected and reacted with 5 mM HPE-IAM for 30 min at 37 °C, followed by LC-ESI-MS/MS analysis described in the Methods section. (C) Cells were exposed to stable isotope-labeled cystine (100 µM) in HBSS for 1 h, and the conditioned medium was collected and reacted with 5 mM HPE-IAM for 30 min at 37 °C after incubation with 50 µM of H2O2, 1,2-naphthoquinone (1,2-NQ), 1,4-NQ or 1,4-benzoquinone (1,4-BQ) for 1 h at 37 °C, followed by LC-ESI-MS/MS analysis as described in the Methods section. **p < 0.01 compared with control.
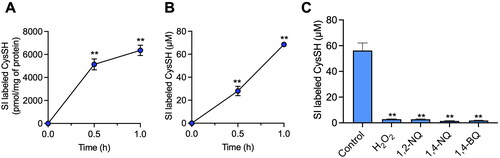
To address this possibility, HepG2 cells were incubated for 1 h with either H2O2 or the atmospheric electrophiles 1,2-NQ, 1,4-NQ and 1,4-BQ. The stable isotope-labeled CysSH in cell medium was consumed by these agents (), and a separate experiment confirmed that the reaction product of H2O2 and CysSH was CysSSCys, noting Michael adducts of electrophiles were also found in a previous similar study8.
Role of extracellular CysSH in regulation of oxidative/electrophilic stress in HepG2 cells
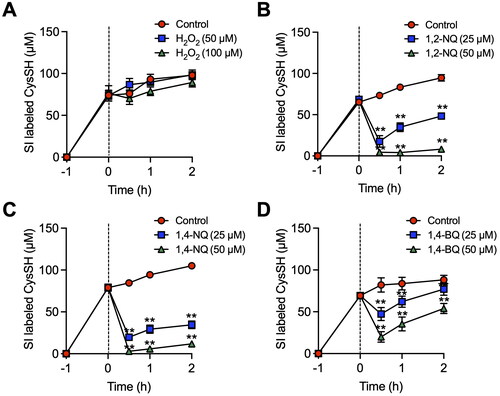
While almost complete oxidation of isotope-labeled CysSH in HepG2 cell medium occurred following H2O2 exposure (), extracellular CysSH derived from CysSSCys levels were unaltered (), indicating that the process was due to redox cycling between stable isotope-labeled CysSSCys and CysSH in the cells. In contrast, addition of 1,2-NQ, 1,4-NQ and 1,4-BQ to the HepG2 culture system caused a concentration-dependent consumption of isotope-labeled CysSH (), consistent with their covalent binding to CysSH.
Figure 2. Alteration of extracellular cysteine (CysSH) content after exposure to stable isotope-labeled cystine in HepG2 cells. (A). Cells were pre-incubated with stable isotope-labeled cystine (100 µM) in Hank’s balanced salt solution (HBSS) for 1 h and then exposed to H2O2 (50 or 100 µM) for 0.5, 1 or 2 h. (B). Cells were pre-incubated with stable isotope-labeled cystine (100 µM) in HBSS for 1 h and then exposed to 1,2-naphthoquinone (1,2-NQ, 50 or 100 µM) for 0.5, 1 or 2 h. (C). Cells were pre-incubated with stable isotope-labeled cystine (100 µM) in HBSS for 1 h and then exposed to 1,4-NQ (50 or 100 µM) for 0.5, 1 or 2 h. (D). Cells were pre-incubated with stable isotope-labeled cystine (100 µM) in HBSS for 1 h and then exposed to 1,4-benzoquinone (1,4-BQ, 50 or 100 µM) for 0.5, 1 or 2 h. The conditioned medium was collected and reacted with 5 mM β-(4-hydroxyphenyl)ethyl iodoacetamide for 30 min at 37 °C, followed by liquid chromatography-ESI-tandem mass spectrometry analysis as described in the Methods section. **p < 0.01 compared with control.
Effect of extracellular CysSH derived from CysSSCys on oxidative and covalent modifications of cellular proteins
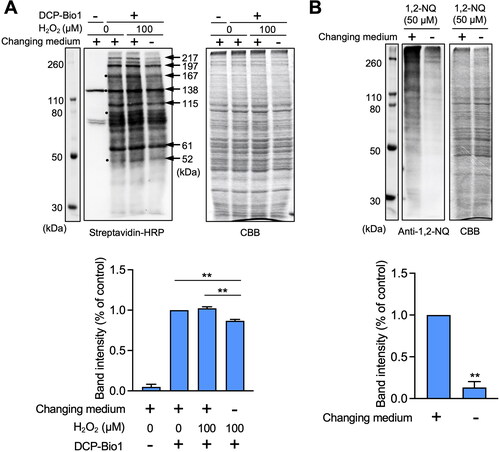
If extracellular CysSH traps H2O2 or atmospheric electrophiles during exposure of HepG2 cells to isotope-labeled CysSSCys, then the oxidative and electrophilic modifications of cellular proteins caused by these agents should be repressed. To confirm this hypothesis, we examined protein modification under conditions of oxidative or electrophilic stress. Oxidative modification of cellular proteins, determined by the dimedone assay for CysSOH [Citation13], indicated numerous protein thiols underwent oxidation to sulfenic acid (-SOH) even under basal conditions (western blot analysis, ), suggesting that a variety of low pKa protein thiols were oxidized as previously described [Citation14]. This observation was also in agreement with our previous finding that numerous protein thiols underwent oxidation under basal conditions in A431 cells [Citation15]. An experiment with culture medium without extracellular CysSH derived from CysSSCys, followed by exposure of HepG2 cells to H2O2, indicated that some cellular proteins were further oxidized, as shown by black circles (). However, this protein oxidation was significantly lower in cell medium containing extracellular CysSH derived from CysSSCys, as shown by the arrows (). Exposure of the cells to 1,2-NQ resulted in extensive modification of cellular proteins covalently bound to 1,2-NQ, shown by western blot analysis (), modifications which were significantly diminished in the presence of extracellular CysSH derived from CysSSCys ().
Figure 3. Effect of extracellular cysteine derived from cystine on oxidative and covalent modifications of cellular proteins in HepG2 cells. (A) Cells were pre-incubated with stable isotope-labeled cystine (100 µM) in Hank’s balanced salt solution for 1 h and then added with or without H2O2 (100 µM) for 1 h. Total cell lysates were prepared with RIPA buffer with or without 1 mM biotin-conjugated dimedone derivative (DCP-Bio1), and subjected to western blot analysis using streptavidin-HRP or SDS-PAGE with Coomassie brilliant blue (CBB) staining. The bar graph represents quantitative results of the upper bands of western blots. Each value represents the mean ± SE of three independent experiments. **p < 0.01 compared with no changing of medium with H2O2 exposure. (B) Cells were pre-incubated with stable isotope-labeled cystine (100 µM) in Hank’s balanced salt solution for 1 h and then incubated with 1,2-naphthoquinone (1,2-NQ, 50 µM) for 1 h. Total cell lysates prepared with RIPA buffer were subjected to western blot analysis using an antibody against 1.2-NQ, or SDS-PAGE with CBB staining. The bar graph represents quantitative results of the upper bands of western blots. Each value represents the mean ± SE of three independent experiments. **p < 0.01 compared with control.
Contribution of enzymes and low molecular chemicals to reduction of CysSSCys to CysSH
We next explored whether enzymes and/or intracellular antioxidants were involved in the reduction of CysSSCys in HepG2 cells. As shown in , heat treatment of HepG2 cell lysate at 95 °C for 10 min reduced the reduction of CysSH by 60%, suggesting that enzyme and antioxidants both participate in the reduction. Incubation of stable-isotope labeled CysSSCys with the high molecular weight fraction in the absence of low molecular weight components, such as antioxidants in the presence of either NAD(P)H, CoQ10 or ascorbic acid, indicated that these antioxidants were not a predominant factor for formation of stable isotope-labeled CysSH (). In contrast, GSH and DHLA were able to reduce stable isotope-labeled CysSSCys to CysSH with minimal protein requirement, because heat treatment had no effect on the reduction (). Interestingly, for the low molecular weight components in HepG2 cells the reduction of CysSSCys to CysSH was almost completely heat-labile (). We therefore investigated the thermal sensitivity of the production of CysSH mediated by GSH and DHLA. As shown in , the DHLA-induced reduction of CysSSCys to CysSH was significantly repressed by heat treatment, unlike the GSH effect. Arnér and his associates previously reported that a thioredoxin-related protein of 14 kDa (TRP14), a member of thioredoxin family, partly plays a role in the reduction of CysSSCys to CysSH in HEK293 cells [Citation16]. However, knockdown of thioredoxin, TRP14 and TRP32 did not block the reduction of stable isotope-labeled CysSSCys to CysSH in HepG2 cells ().
Figure 4. Contribution of enzymes and low molecular chemicals to the reduction of cystine to cysteine (CysSH) in vitro. (A) CysSH production from cystine. High molecular weight fraction (HMWF) prepared from HepG2 cells (0.4 mg/mL) was incubated with stable isotope-labeled cystine (100 µM), with or without the indicated antioxidants (each 100 µM) or heat treatment for 10 min at 37 °C. The resulting solution was incubated with 5 mM β-(4-hydroxyphenyl)ethyl iodoacetamide (HPE-IAM) for 30 min at 37 °C, followed by liquid chromatography-ESI-tandem mass spectrometry analysis as described in the Methods section. (B) Effect of heat on production of CysSH from cystine by the low molecular weight fraction (LMWF). The LMWF prepared from HepG2 cells with or without heat treatment (95 °C for 10 min) was incubated with stable isotope-labeled cystine (100 µM). The resulting solution was incubated with 5 mM HPE-IAM for 30 min at 37 °C, followed by liquid chromatography-ESI-tandem mass spectrometry analysis as described in the Methods section. **p < 0.01 compared with control. CoQ10; coenzyme Q10, DHLA; dihydrolipoic acid, GSH; glutathione, NADPH; β-nicotinamide-adenine dinucleotide phosphate.
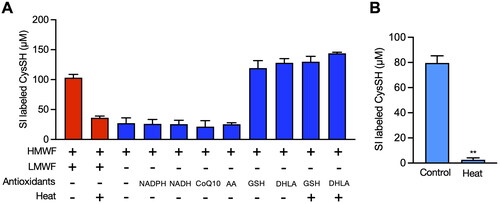
Figure 5. Effect of knockdown of thioredoxin (Trx), Trx-related protein 14 (TRP14) and TRP32 on the production of cysteine (CysSH) from cystine in HepG2 cells. (A) Knockdown of Trx, TRP14 and TRP32. Cells were transfected with control, Trx, TRP14 and TRP32 siRNAs for 48 h, and then total cell lysates were subjected to western blot analysis with the indicated antibodies. (B) Intracellular CysSH content after exposure to stable isotope-labeled cystine. Control siRNA-, Trx siRNA-, TRP14 siRNA- and TRP32 siRNA-transfected cells (48 h) were exposed to stable isotope-labeled cystine (100 µM) in Hank’s balanced salt solution for 0.5, 1 or 2 h, and then cells were collected with methanol containing 1 mM β-(4-hydroxyphenyl)ethyl iodoacetamide (HPE-IAM), followed by liquid chromatography-ESI-tandem mass spectrometry analysis as described in the Methods section. (C) Extracellular CysSH content after exposure to stable isotope-labeled cystine. Control siRNA-, Trx siRNA-, TRP14 siRNA- and TRP32 siRNA-transfected cells (48 h) were exposed to stable isotope-labeled cystine (100 µM) in Hank’s balanced salt solution for 0.5, 1 or 2 h, and the conditioned medium was collected and reacted with 5 mM HPE-IAM for 30 min at 37 °C, followed by liquid chromatography-ESI-tandem mass spectrometry analysis described in the Methods section.
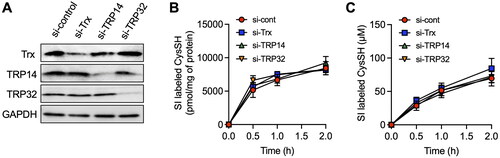
Table 1. Chemical conversion of cystine to cysteine (CysSH) by glutathione (GSH) or dihydrolipoic acid (DHLA).
Discussion
The present study indicates that a considerable amount of CysSH (0.07 µmol) is released from HepG2 cells following 1 h exposure to stable isotope-labeled CysSSCys (0.10 µmol) (). This implies that CysSH produced from CysSSCys in HepG2 cells is transported to the extracellular space for a specific function. Consistent with this notion, the current results suggest that Cys is involved in the reduction of oxidized proteins and modification of reactive electrophiles. The results show:
CysSH in cell medium collected at 1 h after exposure of HepG2 cells to stable isotope-labeled CysSSCys readily reacted with H2O2 and also atmospheric electrophiles, such as 1,2-NQ, 1,4-NQ and 1,4-BQ, resulting in the almost complete consumption of stable isotope-labeled CysSH (). This finding suggests that the movement of intracellular CysSH to the extracellular environment may serve to repress oxidative and electrophilic stress. GSH is an abundant low molecular weight nucleophile found at an intracellular concentration greater than 1 mM [Citation17], whereas CysSH is a minor nucleophile in cells. Conversely, extracellular GSH levels are much lower than their intracellular concentration.
Although extracellular concentrations of CysSH are known to be higher than those of GSH [Citation2], there have been limited studies regarding the biological role of extracellular CysSH. We have studied adaptive and protective responses against electrophilic stress and reported that exposure of cultured cells to xenobiotic electrophiles activates a variety of cellular redox signal transduction pathways, at low doses through covalent modification of negative regulator proteins with low pKa values, and at high doses by disruption of signaling via nonselective and extensive modification of cellular protein thiols [Citation18]. We also found that reactive sulfur species (hydrogen sulfide, CysSSH, glutathione persulfide and their polysulfides) are capable of capturing xenobiotic electrophiles, leading to production of sulfur adducts with little electrophilicity [Citation19–21]. In addition to these observations, we also found that CysSSH is excreted into extracellular spaces under basal condition [Citation5,Citation6].
While these findings suggested that CysSSH may regulate the reactivity of xenobiotic electrophiles prior to potential uptake by cells, the present study with stable isotope-labeled CysSSCys clearly indicated that the intracellular level of CysSH was 1200 times higher than that of CysSSH at 1 h following CysSSCys exposure ( and Figure S1). In addition, our previous study indicated that atmospheric quinones readily reacted with CysSH, but not CysSSH, thereby forming CysSH adducts [Citation8].
Overall, we conclude that extracellular CysSH derived from CysSSCys, not GSH and CysSSH, is a critical low molecular weight chemical to regulate electrophilic stress.
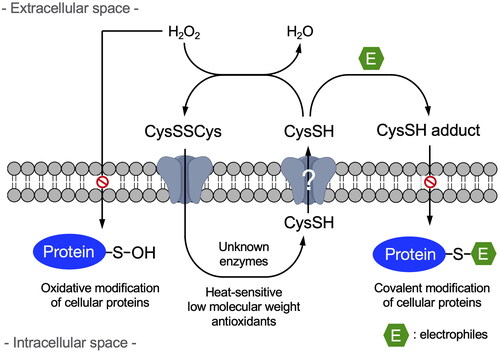
Since it is well established that CysSH causes a 2-electron reduction reaction with H2O2, forming CysSSCys and H2O2 [Citation22], we examined whether or not H2O2 interacts with extracellular CysSH derived from CysSSCys. This study showed that CysSH in the medium of HepG2 cells was completely consumed by H2O2, () whereas extracellular CysSH levels were unaffected in the presence of HepG2 cells exposed to stable isotope-labeled CysSSCys (). The changes in CysSH levels, with and without HepG2 cells, probably reflects the redox reaction between CysSSCys and CysSH. This potential system is interesting, because even if CysSH was oxidized to CysSSCys under extracellular oxidative stress, CysSSCys could be taken up by CysSSCys-dependent antiporters, such as SLC7A11 [Citation7,Citation23], and then undergo reduction to CysSH in the HepG2 cells (). Subsequently, the intracellularly formed CysSH can be transported into the extracellular space. In contrast, extracellular CysSH derived from CysSSCys was consumed by the addition of atmospheric electrophiles in a concentration dependent manner (). It is likely that atmospheric electrophiles trap extracellular CysSH, resulting in the production of associated CysSH adducts [Citation8], not allowing CysSH to be reused. In other words, exposure to atmospheric electrophiles at high doses may deplete extracellular CysSH, and thereby potentially disrupt redox homeostasis in cells.
Figure 6. Scheme for the negative regulation of oxidative and electrophilic stress by extracellular cysteine.
The present study has emphasized the importance of extracellular CysSH derived from CysSSCys in oxidative and electrophilic modifications of cellular protein. Further investigation of the reduction of CysSSCys in HepG2 cells revealed that enzymes and also low molecular weight antioxidants, such as GSH and DHLA, but not NAD(P)H, CoQ10 and ascorbic acid, participate in the reduction (). Of particular interest, the current experiments suggested that the intracellular reduction of CysSSCys to CysSH mediated by low molecular weight antioxidant in HepG2 cells was mainly due to dithiol compounds, like DHLA, because the GSH-dependent reduction was heat-stable (). Finally, it should be noted that NADPH-dependent enzymes do not catalyze the reduction of CysSSCys. While TRP14 was reported to play a role in the reduction of CysSSCys to CysSH in HEK293 cells [Citation16], the present study found that thioredoxin, TRP14 and TRP32 were not the predominant enzymes catalyzing the reduction of CysSSCys in HepG2 cells (). Further study is required to determine the relevant enzyme(s).
Disclosurer statement
There are no conflicts of interest.
Supplemental Material
Download MS Word (90.7 KB)Acknowledgments
We thank Charles Allan, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Additional information
Funding
References
- Wu G, Fang YZ, Yang S, et al. Glutathione metabolism and its implications for health. J Nutr. 2004;134(3):489–492. doi: 10.1093/jn/134.3.489.
- Anderson CL, Iyer SS, Ziegler TR, et al. Control of extracellular cysteine/cystine redox state by HT-29 cells is independent of cellular glutathione. Am J Physiol Regul Integr Comp Physiol. 2007;293:1069–1075.
- Shinkai Y, Kumagai Y. Sulfane sulfur in toxicology: a novel defense system against electrophilic stress. Toxicol Sci. 2019;170(1):3–9. doi: 10.1093/toxsci/kfz091.
- Fukuto JM, Ignarro LJ, Nagy P, et al. Biological hydropersulfides and related polysulfides – a new concept and perspective in redox biology. FEBS Lett. 2018;592(12):2140–2152. doi: 10.1002/1873-3468.13090.
- Lin J, Akiyama M, Bica I, et al. The uptake and release of polysulfur cysteine species by cells: physiological and toxicological implications. Chem Res Toxicol. 2019;32(3):447–455. doi: 10.1021/acs.chemrestox.8b00340.
- Henderson CF, Bica I, Long FT, et al. Cysteine trisulfide protects E. coli from electrophile-induced death through the generation of cysteine hydropersulfide. Chem Res Toxicol. 2020;33(2):678–686. doi: 10.1021/acs.chemrestox.9b00494.
- Akiyama M, Unoki T, Aoki H, et al. Cystine-dependent antiporters buffer against excess intracellular reactive sulfur species-induced stress. Redox Biol. 2022;57:102514.
- Shinkai Y, Onose Y, Akiyama M, et al. Capture of electrophilic quinones in the extracellular space: evidence for a phase zero reaction. Chem Res Toxicol. 2023;36(1):23–31. doi: 10.1021/acs.chemrestox.2c00223.
- Miura T, Kumagai Y. Immunochemical method to detect proteins that undergo selective modification by 1,2-naphthoquinone derived from naphthalene through metabolic activation. J Toxicol Sci. 2010;35(6):843–852. doi: 10.2131/jts.35.843.
- Akiyama M, Unoki T, Shinkai Y, et al. Environmental electrophile-mediated toxicity in mice lacking Nrf2, CSE, or both. Environ Health Perspect. 2019;127(6):67002. doi: 10.1289/EHP4949.
- Akaike T, Ida T, Wei FY, et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat Commun. 2017;8(1):1177. doi: 10.1038/s41467-017-01311-y.
- Kyhse-Andersen J. Electroblotting of multiple gels: a simple apparatus without buffer tank for rapid transfer of proteins from polycrylamide to nitrocellulose. J Biochem Biophys Methods. 1984;10(3-4):203–209. doi: 10.1016/0165-022x(84)90040-x.
- Poole LB, Klomsiri C, Knaggs SA, et al. Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug Chem. 2007;18(6):2004–2017. doi: 10.1021/bc700257a.
- Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–C868.
- Dóka É, Ida T, Dagnell M, et al. Control of protein function through oxidation and reduction of persulfidated states. Sci Adv. 2020;6(1):eaax8358. doi: 10.1126/sciadv.aax8358.
- Pader I, Sengupta R, Cebula M, et al. Thioredoxin-related protein of 14 kDa is an efficient L-cystine reductase and S-denitrosylase. Proc Natl Acad Sci USA. 2014;111(19):6964–6969. doi: 10.1073/pnas.1317320111.
- Meister A. Selective modification of glutathione metabolism. Science. 1983;220(4596):472–477. doi: 10.1126/science.6836290.
- Kumagai Y, Abiko Y. Environmental electrophiles: protein adducts, modulation of redox signaling, and interaction with persulfides/polysulfides. Chem Res Toxicol. 2017;30(1):203–219. doi: 10.1021/acs.chemrestox.6b00326.
- Yoshida E, Toyama T, Shinkai Y, et al. Enzyme in mammalian cells. Chem Res Toxicol. 2011;24(10):1633–1635. doi: 10.1021/tx200394g.
- Akiyama M, Shinkai Y, Unoki T, et al. The capture of cadmium by reactive polysulfides attenuates cadmium-induced adaptive responses and hepatotoxicity. Chem Res Toxicol. 2017;30(12):2209–2217. doi: 10.1021/acs.chemrestox.7b00278.
- Abiko Y, Sha L, Shinkai Y, et al. 1,4-Naphthoquinone activates the HSP90/HSF1 pathway through the S-arylation of HSP90 in A431 cells: negative regulation of the redox signal transduction pathway by persulfides/polysulfides. Free Radic Biol Med. 2017;104:118–128. doi: 10.1016/j.freeradbiomed.2016.12.047.
- Ohtsu I, Wiriyathanawudhiwong N, Morigasaki S, et al. The L-cysteine/L-cystine shuttle system provides reducing equivalents to the periplasm in Escherichia coli. J Biol Chem. 2010;285(23):17479–17487. doi: 10.1074/jbc.M109.081356.
- Sato H, Tamba M, Ishii T, et al. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274(17):11455–11458. doi: 10.1074/jbc.274.17.11455.