Abstract
Colloidal gold, a sol comprised of nanoparticles of Au0, has been used as a therapeutic for the treatment of cancer as well as an indicator for immunodiagnostics. However, the use of these gold nanoparticles for in vivo drug delivery has never been described. This communication outlines the development of a colloidal gold (cAu) nanoparticle vector that targets the delivery of tumor necrosis factor (TNF) to a solid tumor growing in mice.
The optimal vector, designated PT-cAu-TNF, consists of molecules of thiol-derivatized PEG (PT) and recombinant human TNF that are directly bound onto the surface of the gold nanoparticles. Following intravenous administration, PT-cAu-TNF rapidly accumulates in MC-38 colon carcinoma tumors and shows little to no accumulation in the livers, spleens (i.e., the RES) or other healthy organs of the animals. The tumor accumulation was evidenced by a marked change in the color of the tumor as it acquired the bright red/purple color of the colloidal gold sol and was coincident with the active and tumor-specific sequestration of TNF. Finally, PT-cAu-TNF was less toxic and more effective in reducing tumor burden than native TNF since maximal antitumor responses were achieved at lower doses of drug.
INTRODUCTION
Targeting cancer therapeutics to solid tumors is facilitated by particle delivery systems capable of escaping phagocytic clearance by the reticuloendothelial system (RES; Papisov Citation1998; Moghimi and Patel Citation1998; Woodle Citation1998). Under ideal conditions such delivery systems preferentially extravasate the tumor vasculature and accumulate within the tumor microenvironment (Nafayasu et al. Citation1999; Maruyama et al. Citation1999). By design, a particle delivery system capable of sequestering a cancer drug solely within a tumor may also reduce the accumulation of the drug in healthy organs (Papisov Citation1998; Moghimi and Patel Citation1998; Woodle Citation1998; Nafayasu et al. Citation1999; Maruyama et al. Citation1999). Consequently, these delivery systems may increase the relative efficacy or safety of a cancer therapy, and thus serve to increase the drug's therapeutic index.
The field of particle-based drug delivery is currently focused on two chemically distinct colloidal particles, liposomes and biodegradable polymers (Müller et al. Citation2000; Jain et al. Citation1998; Rafferty et al. Citation1996; Ogawa Citation1997; Maruyama et al. Citation1998). Both delivery systems encapsulate the active drug. The drug is released from the particle as it lyses, in the case of lipsomes, or disintegrates, as described for biodegradable polymers.
Colloidal gold nanoparticles represent a completely novel technology in the field of particle-based tumor-targeted drug delivery. The synthesis of these particles was first reported by Michael Faraday, who, in Citation1857, described the chemical process for the production of nanosized particles of Au0 from gold chloride and sodium citrate (Faraday Citation1857). In the 1950s the discovery that these particles could bind protein biologics without altering their activity paved the way for their use in handheld immunodiagnostics and in histopathology (Chandler et al. Citation2001). Of particular relevance to this communication is the use of radioactive colloidal gold nanoparticles, made from Au198, for the treatment of liver cancer and sarcoma (Rubin and Levitt Citation1964; Root et al. Citation1954). Intravenous administration of these nanoparticles resulted in drug-associated toxicities due to radiation exposure. However, no demonstrable toxicities were noted from the particles themselves. More recently, gold nanoparticles have been assembled into scaffolds for use in DNA diagnostics and biosensors (Mirkin et al. Citation1996).
In toto, colloidal gold nanoparticles represent a versatile biomedical platform. In this report we describe the formulation of these gold nanoparticles for the targeted delivery of the potent yet highly toxic anticancer protein biologic, tumor necrosis factor (TNF), to a solid tumor. TNF has been shown to have antitumor action, but its associated systemic toxicities have stymied its use as a systemic cancer treatment (Schiller Citation1991; Furman Citation1993; Taguchi 1998; Creaven Citation1987). Initial formulations of the vector, manufactured by binding only TNF to the particles, were less toxic than native TNF and effective in reducing tumor burden in a murine model. Subsequent studies revealed that the safety of this vector was primarily due to its rapid uptake and clearance in the RES. This vector was reformulated to include molecules of thiol-derivatized polyethylene glycol (PEG-THIOL) that were bound with molecules of TNF on the gold nanoparticles surface. The new vector, PT-cAu-TNF, avoids detection and clearance by the RES, and actively and specifically sequesters TNF within a solid tumor. The altered biodistribution correlated to improvements in drug safety and dose-to-dose efficacy of TNF treatment in our murine tumor model.
The studies described below provide the proof of concept for the use of the colloidal gold nanoparticles in tumor-targeted drug delivery. The data support the continued development of this delivery system for potential clinical use and warrant new research on the use of these nanoparticles to develop novel tumor-targeting drug therapies.
MATERIALS AND METHODS: PREPARATION OF COLLOIDAL GOLD NANOPARTICLES
One of the most widely used methods for manufacturing colloidal gold nanoparticles involves the reduction of chloroauric acid (Au+3, HAuCl4) to neutral gold (Au0) by agents such as sodium citrate (Frens Citation1972). We adapted the method described by Horisberger (Citation1979) to produce 33 nm colloidal gold particles. This method, which is both simple and scalable, provides for the large-scale production of the colloidal gold nanoparticles required for the in vivo assessment of the colloidal-gold–TNF vectors.
Large (8 L) batches of colloidal gold were manufactured using a reflux apparatus that heats and mixes the reactants during particle synthesis (Kontes Glass, Vineland, NJ, USA). The reactants, a 4% gold chloride solution (23.03% stock; OMG, South Plainfield, NJ, USA) and a 1% sodium citrate solution (wt/wt; J.T. Baker Company, Paris, KY, USA) were made in deionized H2O (DIH2O). Particle synthesis was initiated by heating 8 L of DI H2O to a rolling boil under reflux. Subsequently, 20 ml of the 4% gold chloride solution was added through one port in the apparatus. The solution was brought to a boil and kept under reflux during the addition of 320 ml of the sodium citrate solution.
The addition of sodium citrate to the gold chloride initiates a series of well-described reduction reactions characterized by changes in the color of the initial gold chloride solution (Frens Citation1972; Horisberger Citation1979). After particle synthesis, the sol was cooled to room temperature, filtered through a 0.22 μ nitrocellulose filter, and stored at room temperature (RT) until use.
Characterization of the Colloidal Gold Particles
Particle size was determined by three techniques: TEM, dynamic light scatter, and differential centrifugal sedimentation. Electron micrographs were used to physically measure the size of the colloidal gold particles. Dynamic light scattering (DLS; Brookhaven Instruments Corporation, Holtsville, NY), determines particle size by measuring the angles at which an incident light beam is scattered as a function of Brownian motion of the colloidal gold particles. Differential centrifugal sedimentation (DCS; Disc Centrifuge; CPS Instruments, Inc., Stuart, FL), measures particle size by determining the time required for the colloidal gold particles to traverse a sucrose density gradient created in a disc centrifuge. Both DLS and DCS methods use calibrated particle reference standards to estimate the size of the colloidal gold preparation. The studies reported in this communication were performed with gold particles with an estimated diameter of 32.6 ± 3 nm.
Finally, the zeta potential of the particles was determined at the various stages of vector development using a Brookhaven Zeta Potential Analyzer (Brookhaven Instrument Corp., Holtsville, NY).
Binding TNF to Colloidal Gold: Determination of the pH Binding Optimum
The colloidal gold nanoparticles formed by the above process remain in suspension by their mutual electrostatic repulsion generated and maintained by a net negative charge on their surface. Cations present in salt solutions negate this charge repulsion and cause these “naked particles” to agglomerate and eventually precipitate out of solution. Binding of proteins or other stabilizing agents to the particles' surface maintains the sol state by blocking the salt-induced precipitation of the colloidal gold particles.
The binding of proteins to colloidal gold nanoparticles is dependent on the pH of the colloidal gold and the protein solutions. To determine the optimal pH for binding TNF to the colloidal gold particles, the pH of 2 ml aliquots of 33 nm colloidal gold sol was adjusted, as measured on pH strips, from pH 5 to 11 using 1N NaOH. TNF (Knoll Pharmaceuticals, Mount Olive, NJ; provided by the Surgery Branch, NCI, Bethesda, MD; repurified to homogeneity) was reconstituted in DIH2O to a concentration of 1 mg/ml and further diluted to 100 μg/ml in 3 mM TRIS base pH 8. One hundred microliters of this TNF stock was added to the seven aliquots of pH-adjusted colloidal gold and incubated for 15 min. Following this incubation, 100 μl of a 10% NaCl solution was added to each of the aliquots to induce particle precipitation. The pH binding optimum was defined as the pH that allowed TNF to bind to the colloidal gold particles to prevent their precipitation by salt, as described above.
Mechanism of Binding
To investigate the mechanism of the TNF/gold interaction we attempted to release the cytokine from the particles' surface using chemical agents known to disrupt specific chemical bonds and interactions. For example, to determine if the TNF/gold interaction occurred through the formation of dative covalent bonds, 30 μg of colloidal gold bound TNF was incubated with 10 ml of a solution containing 1 μg/ml dithiolthreitol (DTT; made in 1X PBS; Sigma Chemical Company, St. Louis, MO). Similarly, ionic and hydrophobic interactions were assessed by treating 30 μg of the colloidal-gold–bound TNF with either 3 M MgCl2 solution or a PBS solution containing 1% Tween 20, respectively.
Following a 30 min treatment, 1 ml of the various samples was collected and centrifuged at 7,500 rpms for 15 min. A 10 μl sample of the supernatant was added to 990 μl of EIA assay diluent (provided as part of a commercial EIA kit for TNF measurement; CytImmune Sciences, Inc., College Park, MD, USA). The remainder of the supernatant was removed by aspiration and the colloidal gold pellet was resuspended to its original volume in PEG 1450/DIH2O, pH 8. Ten microliters of the resuspended pellet were added to 990 μl of EIA assay diluent. Both the reconstituted pellet and supernatant solutions were serially diluted and analyzed for TNF concentration by EIA (see above).
Saturation Binding Studies
Based on the data obtained from the pH binding study, the pH of a 33 nm colloidal gold sol was adjusted to pH 8 with 1 N NaOH. The sol was divided into 1 ml aliquots to which increasing amounts of TNF were added. After binding for 15 min the samples were centrifuged at 7,500 rpm for 15 min, and the resultant colloidal gold pellets and supernatants were analyzed by EIA as described above.
Development of the cAu-TNF and the PT-cAu-TNF Vectors
Two electrostatically stable vectors were generated for in vivo analysis. The first vector, designated cAu-TNF, was developed by binding 4 μg of TNF per ml of colloidal gold sol. The cAu-TNF vector was shown to be completely stable when exposed to salt or heparanized mouse whole blood. Although this vector was safer than native TNF and was effective at reducing tumor burden, it was rapidly cleared by the RES (see below). The addition of hydrophyllic blockers, such as CARBOWAX 20M (Fisher Scientific, Pittsburg, PA, USA), PEG-8000 (Sigma Chemical Company, St. Louis, MO, USA), or Tetronic 908 (BASF, Mount Olive, NJ, USA) did not block the vector's uptake and clearance by the RES.
Our second approach re-engineered the cAu-TNF vector by binding molecules of TNF and thiol-derivatized PEG on the same particle of colloidal gold. To develop this vector required that both TNF and PEG-THIOL reactants share the available binding sites on the colloidal gold nanoparticle and that each particle was uniformly bound with the same relative amounts of the TNF and PEG-THIOL reactants.
We developed an apparatus to maximize the interaction between the colloidal gold nanoparticles with the TNF reactant during the binding process. The apparatus consists of two large dewars used to hold the TNF and the colloidal gold sol. The dewars are connected to a single T-connector, which reduces the large (2–4 L) volumes of the dewar system to the 100 μl volume of the small mixing chamber in the T-connector. Once the dewars were loaded with their respective solutions (i.e., pH-adjusted colloidal gold sol and TNF at 1 μg/ml) the solutions were physically drawn into the T-connector by a single peristaltic pump at a flow rate of 1 L/min. An in-line vortex (Cole-Palmer Instrument Co., Vernon Hills, IL, USA) was placed immediately downstream of the T-connector to vigorously mix the colloidal gold particles with TNF solutions as they exited the T-connector. The resultant mixture was collected in a flask and allowed to stir for 15 min followed by the addition of the PEG-THIOL (15 μg/ml added as a 10X concentrate in DIH2O; Sun BIO Corp, Walnut Creek, CA, USA). After an additional 15 min BSA (5% v/v at 200 μg/ml in DIH2O) was added to the solution.
Colloidal-gold–bound TNF in each preparation was separated from free TNF by diafiltration through a 50,000 MWCO BIOMAX diafiltration cartridge (Millipore Corporation, Chicago, IL, USA). An aliquot of the permeate (i.e., free TNF) was removed and set aside for TNF determination. The retentate, which contained the TNF-bound colloidal gold, was sterile filtered through a 0.22 μ filter, and a 10 μl aliquot was taken for TNF analysis. The remainder of the retentate was frozen at −80°C for storage or lyophilization. Once the TNF concentration on the colloidal gold vector was determined, a solution of native TNF was prepared in 3 mM Tris as the control for the in vivo studies.
To demonstrate that TNF and the PEG remained as separate entities while binding to the colloidal gold nanoparticles, the above procedure was repeated in the absence of the colloidal gold particles. TNF and 20 kD PEG-THIOL alone were added to 3 mM TRIS solution at the same binding ratios as described above. Individual solutions containing either TNF or PEG-THIOL alone were used as controls. Samples for SDS-PAGE (under denaturing but not reducing conditions) were collected immediately after mixing and 30 min later. The various samples were electrophoresed in separate minigels. TNF was detected on one gel by with Coomassie Blue staining, while the detection of PEG was done using barium iodide as described by Kurfurst (Citation1992).
Tumor Cell Implantation
C57/BL6 mice, implanted with the colon carcinoma tumor cells, MC-38, were used as the model to compare the safety and efficacy of native and colloidal-gold–bound TNF preparations. This animal protocol was reviewed and approved by the University of Maryland, College Park, IACUC committee (protocol number R-2001-3). C57/BL6 mice were implanted with 105 MC-38 tumor cells in one site on the ventral surface. The cells were allowed to grow until they formed a tumor measuring 0.5 cm3, as determined by measuring the tumor in three dimensions (L × W × H) using a digital caliper. The height of the tumors was determined by physically grasping the tumor by its base. Tumor volume was calculated using the following equation: Tumor Volume = (π·H·(H2+3·(L+W/2)))/6.
In Vivo Characterization of the Colloidal Gold-TNF Vectors: Visual Detection of the cAu-TNF and PT-cAu-TNF Vectors in RES and MC-38 Tumors in C57/BL6 Mice
Native TNF, cAu-TNF, or PT-cAu-TNF formulations were generated as described above. These studies were done in separate cohorts and thus depending on the study, 5–24 μg of native TNF, cAu-TNF, or PT-cAu-TNF formulations were intravenously injected, through the tail vein, of MC-38 tumor-burdened C57/BL6 mice. One group of mice treated with either the cAu-TNF or PT-cAu-TNF vector was sacrificed 5 hr after the injection. The livers and spleens from these mice were collected and photographed to document the presence of the colloidal gold vector. Additional photographic studies were done using a second group of mice treated with either the cAu-TNF or PT-cAu-TNF vectors. The latter two groups were used to document the accumulation of the gold vector in the tumor following an injection of the cAu-TNF or PT-cAu-TNF vectors. Finally, studies were done to compare the tumor gold accumulation following the administration of the PT-cAu-TNF vector or its control vector PT-cAu-MSA.
Pharmacokinetic and BioDistribution Studies
To compare the pharmacokinetic and biodistribution profiles of the different vectors, MC-38 tumor-burdened C57/BL6 mice were injected with identical amounts of native TNF or one of the colloidal-gold–bound formulations. Mice (n = 3–4/group/time point/gold formulation) were bled through the retro-orbital sinus at different times after the injection. The blood was immediately diluted 1:1 with PBS containing 1 mg/ml heparin and frozen at −20°C for batch TNF analysis by enzyme immunoassay (EIA). At selected time points various organs were also collected and flash frozen. To determine intraorgan TNF content, the organs were defrosted and homogenized using a polytron tissue disrupter. Debris was removed by allowing the homogenate to stand on ice for 20 min. The supernatant was analyzed for TNF concentration as described above, as well as for total protein using a commercial protein assay (BioRad, Hercules, CA, USA). Organ TNF concentrations were normalized to total protein.
Dose Escalation, Toxicity, and Efficacy of Native and Colloidal-Gold–Bound TNF in MC-38 Tumor-Burdened Mice C57/BL6 Mice
MC-38 tumor-burdened C57/BL6 mice (n = 4–9/group) were intravenously injected with increasing doses of native TNF, cAu-TNF, or PT-cAu-TNF. All mice were scored in 2 hr intervals after injection using the following toxicity rating scale: 0 = normal activity; 1 = piloerection; 2 = loose stools; 3 = lethargy; 4 = unresponsive; and 5 = death. Animals scoring a 4 on two consecutive observations were sacrificed. Treatment efficacy was determined by monitoring the reduction in tumor volume induced by the various TNF treatments. The measures were compared to the initial tumor volume of each animal in the various groups, as well as to animals receiving saline injections (untreated controls). Tumor measurements were made at various time points after injection. Untreated controls were sacrificed when their tumor volumes were 4 cm3.
RESULTS
Characterization of the Colloidal Gold Nanoparticles
Interrogation of our colloidal gold preparations by TEM (), dual-angle light scattering, and differential centrifugal sedimentation revealed that the size of the particles in the colloidal gold preparations were very close to their theoretical size of 30–34 nm (). The particles were homogenous in size with a mean particle diameter of 32.6 ± 3 nm and a polydispersity measure averaging 0.106 for 8 L scale preparations ().
1 (a) Transmission electron microscopy of the colloidal gold nanoparticles. Colloidal gold nanoparticles were manufactured as described in the Materials and Methods. After filtration 10 ml of the sol was centrifuged at 2500rpms for 2 hr. The colloidal gold pellet was analyzed by TEM using standard techniques. Bar = 33 nm. (b) Determination of the pH binding optimum of TNF to the colloidal gold nanoparticles. To determine the pH for binding TNF to the colloidal gold particles, the pH of 2 ml aliquots of 34 nm colloidal gold sol was adjusted, as measured on pH strips, from pH 5 to 11 using 1N NaOH. TNF (Knoll Pharmaceuticals; provided by the Surgery Branch, NCI; repurified to homogeneity) was reconstituted in DIH2O to a concentration of 1 mg/ml and further diluted to 100 μg/ml in 3 mM TRIS base pH 8. One hundred microliters of this TNF stock was added to the seven aliquots of pH-adjusted colloidal gold and incubated for 15min. Following this incubation, 100 μl of a 10% NaCl solution was added to each of the aliquots to induce particle precipitation. The optimal binding pH was defined as the pH that allowed TNF to bind to the colloidal gold particles while preventing the particles' precipitation by salt (Red arrows). (c) Saturation binding of TNF to the colloidal gold nanoparticles. The pH of the 33 nm colloidal gold sol was adjusted to pH 8 with 1 N NaOH. The sol was divided into 1 ml aliquots to which increasing amounts of TNF were added. After binding for 15 min the samples were centrifuged at 7,500 rpm for 15 min, and the resultant colloidal gold pellets and supernatants were analyzed by a quantitative EIA.
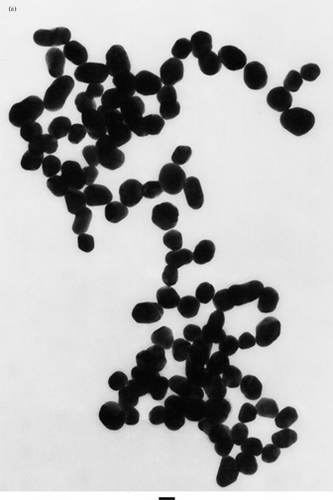
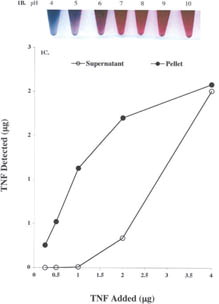
Characteristics of the colloidal gold TNF vector: Colloidal gold particle size characterization
Binding Studies: Determination of pH Binding Optimum and Mechanism of Binding
We determined that the optimal pH for binding TNF to colloidal gold was between 8.0–10.0 (). At lower pH values, the addition of the TNF caused the particles to agglomerate, while adjusting the pH of the sol above 10 resulted in the generation of unstable vector preparations, since the addition of large quantities of NaOH oftentimes precipitated the gold particles before the addition of protein.
The ability of DTT to release 88% of the TNF bound to the colloidal gold particles suggests that the TNF interacts with the gold surface by the formation of dative covalent bonds (Hermanson Citation1996; Nuzzo and Allara Citation1983; ). Conversely, incubating the vector in high ionic strength (i.e., 3 M MgCl2) or detergent-based buffers caused negligible release of TNF from the vector, suggesting that ionic and hydrophobic interactions play a minimal role in the binding of TNF to the colloidal gold nanoparticle ().
Characteristics of the colloidal gold TNF vector: Mechanism of TNF binding to colloidal gold nanoparticles
The change in the colloidal gold nanoparticles' surface charge during the binding of TNF and other stabilizers is shown in . The data presented show that the “naked” colloidal gold particles have an intrinsic negative surface charge of approximately −38 mV which increases to −54 mV as the pH of the sol is adjusted towards the pH binding optimum (). The stepwise binding of TNF and PEG-THIOL neutralized surface charge to a measured value of −34 and −2 mV, respectively.
Characteristics of the colloidal gold TNF vector: Analysis of zeta potential during different stages of PT-cAu-TNF vector manufacture
Saturation Binding of TNF to the Colloidal Gold Particles
The binding of TNF to the colloidal gold nanoparticles exhibited saturation kinetics. As shown in , at 0.5 μg of TNF/ml of gold sol virtually all the TNF was bound to the colloidal gold particles, with an insignificant amount (less than 1%) present as free TNF in the supernatant. As the mass of TNF added to the gold sol is increased, more of the protein bound the gold nanoparticles until all of the available binding sites were occupied with TNF. Saturation of the colloidal gold particles occurred at a binding concentration of 4 μg/ml ().
In a final characterization study, we observed that TNF and PEG-THIOL do not interact by crosslinking with each other during binding to the colloidal gold particle. In brief, we observed no co-migration of either entity as determined by SDS-PAGE analysis, suggesting that TNF and PEG-THIOL independently bind to the colloidal gold nanoparticle (data not shown).
In Vivo Characterization of the Colloidal Gold TNF Vector: Visual Detection of the cAu-TNF and PT-cAu-TNF Vectors in RES and MC-38 Tumors in C57/BL6 Mice
One of the advantages of the colloidal gold nanoparticle delivery system is the relative ease of tracking the vector once it is injected. Although visualization of individual particles requires sophisticated techniques, the accumulation of the particles localized en masse in a particular organ is facilitated by the color of the vector itself. The physical state of the particles can be determined by the color the particles reflect. For example, in a completely stabilized monodispersed state, 33 nm colloidal gold nanoparticles appear red-burgundy in color, while in a precipitated state the particles appear black.
We used these color characteristics to track the location of both the cAu-TNF and the PT-cAu-TNF vectors after their injection into MC-38 tumor-burdened C57/BL6 mice. As shown in , the cAu-TNF vector is sequestered within the livers and spleens as evidenced by the dramatic change in the color of these organs. This uptake was rapid, and oftentimes occurred within 5 min after intravenous injection of the cAu-TNF vector. The fact that the color of the organs was not different from the black precipitates formed when naked colloidal gold particles are exposed to salt suggested that the cAu-TNF vector was destabilized and precipitated during its sequestration in the liver. Finally, the gold staining in the liver and spleen did not dissipate with time since these organs remained black for a month after treatment (data not shown). Interestingly during this period of observation the mice behaved normally and did not exhibit signs of distress, suggesting that the sequestration of the particles in the liver did not induce any clinically adverse toxicities. The addition of hydrophyllic blockers such as carbowax, pluronic, and tetronic to the cAu-TNF vector failed to prevent its uptake and clearance in the RES and at times actually resulted in increased vector toxicity.
2 Hepatic and splenic uptake of the cAu-TNF formulation and the PEG-THIOL cAu-TNF formulation (PT-cAu-TNF) compared to an untreated mouse. Two MC-38 tumor-burdened C57/BL6 mice were injected intravenously with the cAu-TNF vector, and another two tumor-burdened C57/BL6 mice were injected with the PT-cAu-TNF formulation. An untreated mouse and the four treated mice were sacrificed 5 hr after injection of 15 μg of the two different cAu formulations, and all five animals were perfused with heparinized saline. The livers and spleens from these animals were collected and photographed using a digitial camera. (a) Liver and spleen from an untreated mouse. (b) Livers and spleens from mice receiving the cAu-TNF vector. (c) Liver and spleen from mice receiving the PT-cAu-TNF vector.
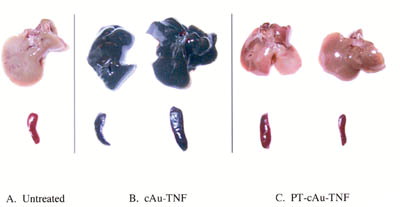
The uptake of the colloidal gold vector by the RES was significantly blunted by the incorporation of PEG-THIOL onto the PT-cAu-TNF vector. The visual data obtained with the PT-cAu-TNF vector clearly show a marked decrease in gold uptake by the livers and spleens since these organs were no longer black with the gold precipitates but appeared similar in color to the untreated controls (). Furthermore, the ability to see the vector allowed us to track its accumulation in and around the MC-38 tumors over time. Unlike the black color of the gold that accumulated in the liver and spleen following cAu-TNF treatment, the color of the gold seen in the tumor following the administration of the PT-cAu-TNF was reddish-purple, suggesting that the gold particles remained in a monodispersed colloidal state during their residence in the circulation and their accumulation in the tumor.
Interestingly, the pattern of PT-cAu-TNF accumulation in and around the tumor site changed with time. As can be seen in , the PT-cAu-TNF was initially (i.e., 0–2 hr) sequestered solely in the tumor. Over time, however, vector staining was apparent on the skin and tissues surrounding the tumor ( at 3 and 5 hr). During the blunt dissection of sacrificed animals' tumors, we observed that the extratumor staining in these animals was restricted to the dermal layer where the tumor cells were initially implanted. No staining was present on the underlying abdominal muscle bed on which the tumor rested. This observation suggests that the peripheral staining may be due to the accumulation of the vector in the blood vessels, possibly new blood vessels, feeding the tumor mass. Whether the staining represents a sequestration of the drug vector in these blood vessels or the passive leakage of the vector from the tumor has not been addressed and will be the focus of future investigations.
3 (a) Accumulation of the PT-cAu-TNF vector in MC-38 tumors over time. A MC-38 tumor-burdened C57/BL6 mouse was intravenously injected with 15 μg of the PT-cAu-TNF vector. The ventral surface of the animal was photographed at the indicated times, showing the color changes of the tumor over 5 hr. Red arrows show tumor uptake of the vector; blue arrows show accumulation of the vector in the tissues surrounding the tumor. (b) Comparative accumulation of the native TNF and PT-cAu-TNF vectors in MC-38 tumors. Fifteen micrograms of the native TNF and the PT-cAu-TNF vectors were intravenously injected into MC-38 tumor-burdened C57/BL6 mice. At specific time points the MC-38 tumors in these mice were photographed to document the vectors' accumulation. Tumor staining indicated by red arrows, and extratumor staining indicated by blue arrows. (c) Accumulation of a PEG-THIOL–stabilized colloidal gold-MSA vector and the PT-cAu-TNF vector in MC-38 tumors. Fifteen micrograms of the PT-cAu-TNF vector and an equal volume of the PT-cAu-MSA vector were intravenously injected into MC-38 tumor burdened mice. A wavelength scan of each preparation verified that the preparations had identical colloidal gold particle concentrations. The time course of each vector's accumulation in MC-38 tumors was photographed.
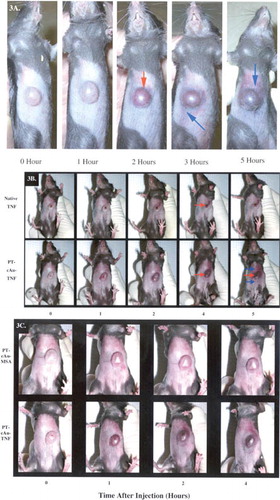
To determine whether the tumors' color change was due to a hemorrhagic response caused by TNF we compared the tumors of mice receiving a 15 μg injection of native TNF with those receiving the same dose of the PT-cAu-TNF vector. As can be seen in , mice treated with native TNF began to show tumor eschar formation, which typically follows intravenous administration of TNF. The tumor eschar seen with native TNF treatment was clearly different from the tumor color change observed following PT-cAu-TNF treatment (, respectively). Additionally, tumor-bearing mice treated with a control PT-cAu-MSA (murine serum albumin) vector exhibited a similar tumor staining as those treated with PT-cAu-TNF, albeit the color change occurred at a much slower rate and was of lower intensity than that observed with PT-cAu-TNF (). Nevertheless, the similarity in tumor color suggested that the colloidal gold particles and not the TNF were responsible for the observed changes.
Pharmacokinetic Analysis
Pharmacokinetic data supported the visual evidence observed on the distribution of the cAu-TNF and PT-cAu-TNF vectors. Animals treated with the cAu-TNF preparation exhibited significantly lower blood levels than animals injected with native TNF () at all time points studied. For example, Cmax values were 50–80% lower in animals treated with the cAu-TNF drug compared to animals treated with native TNF. The suppressed TNF blood levels persisted throughout the time course studied.
4 (a) The pharmacokinetic profiles of native TNF and the cAu-TNF vector in MC-38 tumor-burdened C57/BL6 mice. MC-38 tumor-burdened mice (n = 3/group/time point) were intravenously injected with 10 μg of either native TNF or the cAu-TNF vector. At the indicated time points the mice were anesthetized and bled through the retro-orbital sinus, and the blood was diluted 1:1 with PBS containing 1 mg/ml heparin. TNF concentrations were determined using an EIA. Data are presented as the mean ± SEM blood concentration from three mice per time point.*ast; p < 0.05 cAu-TNF versus native. (b) The pharmacokinetic profiles of native TNF and the PT-cAu-TNF vector in MC-38 tumor-burdened C57/BL6 mice. Mice were bled through the retro-orbital sinus at 5, 180, and 360 min after the injection, and the blood samples were analyzed as described above. Data are presented as the mean ± SEM blood TNF concentration from 3 mice/time point. Δ p < 0.1 * p < 0.05 versus native group.
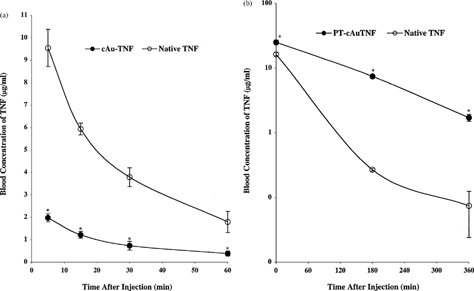
Conversely, PT-cAu-TNF increased the overall residence time for TNF in the circulation (). Unlike the suppressed Cmax values described above, mice treated with PT-cAu-TNF had nearly identical initial blood levels of TNF as mice treated with native TNF. However, 3 and 6 hr after injection, mice treated with PT-cAu-TNF had approximately 20-fold higher blood levels of TNF than mice receiving native TNF.
The pharmacokinetic data are consistent with the observations made on the visual distribution of the various gold TNF vectors. The rapid (i.e., 5 min) uptake and clearance of the cAu-TNF vector by the RES is consistent with the lower Cmax measured at that time. In turn, blocking the RES uptake and clearance of the gold particle, using the PT-cAu-TNF vector, increased the residence time of the TNF in the circulation.
TNF Bio-Distribution Analysis
cAu-TNF was only marginally effective at increasing the amount of TNF delivered to the tumor. For example, 8 hr after administering an intravenous injection of either native TNF or cAu-TNF, the intratumor TNF concentrations were 2.5 times higher in the cAu-TNF group than in the group treated with native TNF (182 ± 50.3 pg/mg protein vs. 63.7 ± 31 pg/mg protein for the cAu-TNF–treated mice and the native TNF-treated mice, respectively).
The data presented in show that the PT-cAu-TNF vector actively sequestered TNF in the MC-38 tumors. Five minutes after intravenous injection, similar intratumor TNF levels were measured in the mice treated with native TNF or the PT-cAu-TNF vector (1.1 ng/mg protein versus 0.82 ng/mg protein, respectively). For animals treated with native TNF, the amount of TNF in the tumor remained the same over the 6 hr time course (). In contrast, animals treated with the PT-cAu-TNF vector exhibited a significant increase in the intratumor TNF concentration over time. Intratumor TNF concentrations increased from a baseline value of 0.8 ng of TNF/mg protein at 5 min to 6.0–7.0 ng of TNF/mg of protein at 3 hr. These elevated values remained constant for an additional 3 hr ().
5 (a) Intratumor TNF accumulation following an intravenous injection of the PT-cAu-TNF. Mice were sacrificed 5, 180, and 360 min after the injection of 15 μg of native TNF or PT-cAu-TNF. The tumors were removed, homogenized, and analyzed for TNF concentrations and total protein as described in the Material and Methods section. Data are presented as the mean ± SEM of tumor TNF concentration, expressed in ng TNF/mg of total protein, from 3 mice/time point/treatment group. *p < 0.05 versus native; p < 0.05 versus 5 min time point for PT-cAu-TNF treatment. Time-to-time comparison NS for native. Organ TNF distribution analysis from MC-38 tumor-burdened C57/BL6 mice receiving either (b) PT-cAu-TNF or (c) native TNF. Livers, lung and brains from the animals were processed and analyzed for TNF and protein concentrations as described in the Material and Methods section. Data are presented as the mean ± SEM of intraorgan TNF concentration from 3 mice/time point/formulation injected.
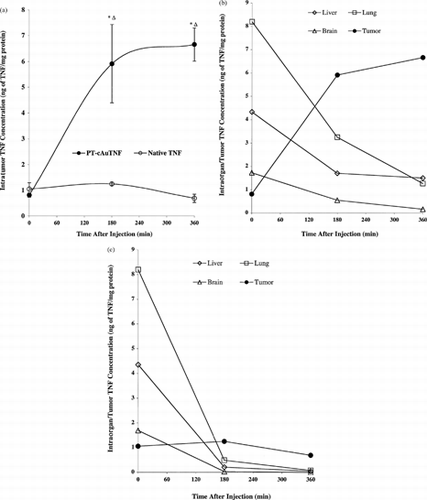
The accumulation of the PT-cAu-TNF vector in the MC-38 tumor mass was an active process. This hypothesis is supported by two sets of observations. First, the intratumor TNF concentrations measured in animals treated with PT-cAu-TNF increased as a function of time, while over the same time course TNF levels in the blood and other organs isolated from these animals fell (). This pattern was also observed in animals treated with native TNF. Second, the pattern of TNF distribution in the healthy organs such as the liver, lung, and brain reflected the exponential decay profile observed in the blood, which was also observed in animals treated with native TNF. Consequently, the accumulation of TNF resulting from the administration of the PT-cAu-TNF vector is specific to the tumor () and not other organs.
Comparison of the Antitumor Efficacy of Native TNF, cAu-TNF, and PT-cAu-TNF: Dose Escalation Studies
The toxicity of native TNF was dose-dependent. Five micrograms of native TNF/mouse caused piloerection and diarrhea within 1–2 hr of injection. With increasing doses of native TNF more severe toxicities were observed. At a dose of 15 μg of TNF/mouse 30–50% of the animals were “cold to the touch” and unresponsive, and ultimately died within 24 hr. At a dose of 24 μg all of the mice died.
The antitumor efficacy of the two lead colloidal gold TNF vectors was tested in C57/BL6 mice implanted with MC-38 colon carcinoma cells. Both the cAu-TNF and PT-cAu-TNF vectors exhibited significant antitumor efficacy in this model. However, the efficacy of the colloidal gold vectors was related to the accumulation of TNF in the MC-38 tumors.
In our first series of efficacy studies we observed that neither 6 nor 12 μg of cAu-TNF improved the efficacy of TNF (compared on a dose-to-dose basis with native TNF) in reducing tumor burden (). However, 24 μg of cAu-TNF did induce significant tumor reduction. Furthermore, this vector was also safer than native TNF since 12 and 24 μg of native TNF caused 25% and 100% of the animals to die, respectively. None of the mice receiving the same dose of TNF, as part of the cAu-TNF vector formulation, became sick or died. These safety and efficacy data are consistent with the vectors' uptake by the RES and with its marginal ability to improve the delivery of TNF to the MC-38 tumors. Thus the improved safety seen with this vector may be directly related to the rapid clearance of TNF observed in the PK studies. Yet the vectors' ability to deliver the remainder of the TNF in the tumor may account for its antitumor efficacy. Similar observations have been described for other colloidal carriers.
6 (a) Comparison of the antitumor efficacy of native TNF and the cAu-TNF vector in MC-38 tumor-burdened C57/BL6 mice. MC-38–tumored C57/BL6 mice (n = 4/group/dose) were intravenously injected with increasing doses of native TNF or the cAu-TNF vector. The antitumor responses for the various treatment groups were measured by determining three dimensional (L × W × H) tumor measurements 10 days after treatment. Data are presented as the mean ± SEM tumor volume (in cm3) for each group. †All animals receiving the 24 μg native TNF treatment died within 24 hr of treatment. *p ≤ 0.05 versus untreated controls (b) Antitumor efficacy of native TNF and the PT-cAu-TNF vector: effect of PEG-THIOL on antitumor efficacy. MC-38 tumor-burdened C57/BL6 mice were divided into 9 groups with 9 animals/group. One group served as an untreated control group. Two groups were intravenously injected with either 7.5 or 15 μg of native TNF. Two groups were also intravenously injected with either 7.5 or 15 μg of the PT-cAu-TNF vector. Tumor measurements were made on various days after the treatment on animals that survived TNF treatment. Statistical difference between the various groups was determined using a paired t-test. †, p < 0.05 for the 7.5 μg of dose of native TNF versus untreated controls.§, p < 0.05 for the 7.5 μg of dose of the PT-cAu-TNF treatment versus untreated controls and native TNF groups. *, p < 0.05 for the 15 μg of dose of native or both PT-cAu-TNF treatments versus untreated controls and the 7.5 μg of dose native TNF. The 7.5 μg of the PT-cAu-TNF vector was not statistically different from the 15 μg of either native TNF or PT-cAu-TNF treatments.
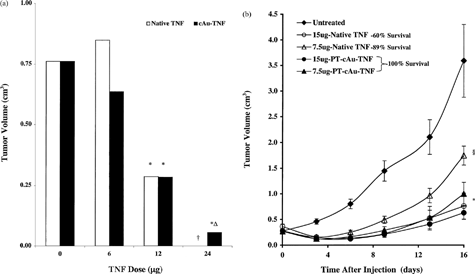
The ability of the PT-cAu-TNF vector to actively sequester TNF within the MC38-tumors supported its ability to improve dose-to-dose safety and efficacy of TNF administration. Similar to the above study we observed that a single injection of native TNF caused dose-dependent toxicity and regression of established MC-38 tumors. While 7.5 and 15 μg of native TNF caused MC-38 tumors to regress by 51 and 79%, respectively, these same doses caused 11 and 40% of the animals to die, respectively. Conversely, none of the animals receiving the same doses of TNF as part of the PT-cAu-TNF formulation died. Nevertheless, both of these PT-cAu-TNF treatments caused the maximal antitumor response observed (72 and 82% inhibition, respectively; ).
DISCUSSION
To achieve tumor-targeted drug delivery nanoparticle systems must address technical and biological concerns that influence their distribution. An early challenge faced by these technologies was their rapid opsinization in the blood and subsequent clearance by the RES. This process proved saturable, since at a high enough dose of nanoparticles, the ability of the RES to clear such particles could be exceeded (Bergqvist et al. Citation1987; Bradfield Citation1980). Once RES saturation was achieved, particles not trapped in the liver or spleen could deliver drugs to solid tumors. The data obtained with the cAu-TNF vector are consistent with these observations. Although a majority of the vector was cleared by the RES, a fraction of the drug was delivered to the tumor.
A more practical approach in addressing RES uptake and clearance was the modification of the nanoparticle surface to include hydrophilic blockers such as PEG as well as block copolymers in the tetronic and pluronic families of surfactants. Indeed, these modifications significantly altered the biodistribution of liposomes and biopolymers, since liver and splenic uptake of these hydrated particles was marginal. Furthermore, these preparations were shown to passively accumulate in solid tumors through extravascation of the leaky vasculature that fed them (Cleland Citation1997; Baban Citation1998; Molema 1999; Moghimi Citation1999; Redhead et al. Citation2001; Illum et al. Citation1987).
Unfortunately, blockers such as polyethylene glycol-based stabilizers (PEGs), including carbowax, as an example, or block copolymers such as poloxamine or poloxamer (Cleland Citation1997; Baban Citation1998; Molema 1999; Redhead et al. Citation2001; Illum et al. Citation1987), failed to alter the biodistribution of the cAu-TNF vector when compared to the cAu-TNF vector alone. One possible explanation for the inability of these blockers to alter the cAu-TNF biodistribution may be due to an inability of the TNF-saturated gold particles to directly bind the hydrophilic blockers. As a result such poorly bound blockers were unable to stop the vectors' uptake by the RES.
Our second strategy sought to stabilize the colloidal gold particle itself rather than the drug-saturated vector. This vector (PT-cAu-TNF) was manufactured by simultaneously binding TNF and PEG-THIOL to the surface of the particle. Both moieties bind to the gold nanoparticle through the formation of a dative covalent bond. For TNF, the sulfhydryl groups present on the cysteine residues presumably form this bond, while the single thiol group present on the PEG-THIOL moiety serves to bind and orient the stabilizer on the surface of the particle. Similar to the PEG moieties on liposomes, PEG-THIOL hydrates the gold nanoparticle by attracting water. Consequently, these hydrophilic “bristles” of PEG-THIOL create a water brush that shields the particle from the RES.
RES avoidance is, however, only one of the challenges that particle-based delivery technologies face in achieving tumor-targeted drug delivery. A second and potentially more significant obstacle may lie in the high interstitial fluid pressure recently described for solid tumors (Netti et al. Citation1999; Stohrer et al. Citation2000; Zhang et al. Citation2000; Rofstad 2002). In brief, high interstitial pressure is thought to interfere with the uptake of cancer therapies by solid tumors (Netti et al. Citation1999; Stohrer et al. Citation2000; Zhang et al. Citation2000). Consequently, passive accumulation of particle systems may only achieve marginal delivery of drugs to tumors.
The PT-cAu-TNF drug vector may in part counter the interstitial fluid pressure in solid tumors by actively accumulating TNF within the tumor mass. We believe that is facilitated by the dual roles played by TNF while bound to the vector. Certainly, the cytokine serves as the therapeutic responsible for tumor regression. However, TNF also serves as a targeting ligand that anchors the vector in and around the tumor.
This hypothesis is supported by the longer time necessary for the control/PT-cAu-MSA vector to sequester in the tumor. In effect, the MSA-based gold vector demonstrates the passive component of the PT-cAu-TNF tumor sequestration, which we believe is similar in action to liposomes. Absent the TNF on the vector's surface, the MSA-based vector passively extravascates the leaky tumor vasculature. In contrast, the presence of TNF on the surface of the PT-cAu-TNF vector anchors the vector within the tumor bed by binding to its receptor. Over time this leads to an accumulation of TNF and the gold in the tumor.
Further facilitating the sequestration is the number of TNF molecules carried by single particle of colloidal gold. Given the particle diameter of 33 nm and the efficient binding of TNF to the particle, we estimate that each particle of colloidal gold binds approximately 400 molecules of TNF. Thus the binding of a single TNF molecule to its receptor may arrest 400 additional molecules of TNF in the tumor, resulting in a significant amplification of active drug in and around the tumor.
Although the fate of colloidal gold particles was not evaluated in the current studies, several preclinical models suggest that the electrostatically stabilized particles are taken up by hepatocytes (Hardonk et al. Citation1985; Renaud et al. Citation1989), not Kupffer cells, excreted into the bile and expelled from the body in feces (Hardonk et al. Citation1985; Renaud et al. Citation1989). Two key factors influence the clearance of gold particles. First, smaller colloidal gold particles stabilized with either a protein or a polymer were preferentially taken up by the hepatocytes and ultimately excreted into the bile and eliminated in the feces (Hardonk et al. Citation1985; Renaud et al. Citation1989). Secondly, blocking Kupffer cell activity with gadolinium chloride also increased the fraction of particles cleared by the hepatocytes (Renaud et al. Citation1989). The size and RES-avoiding properties of PT-cAu-TNF vector support similar mechanisms for clearance of the particles.
Limiting the distribution of TNF to solid tumors has been shown to significantly increase the therapeutic index of the drug (Lejeune Citation1995; Fraker et al. Citation1996; Alexander et al. Citation1998). Clearly, the dramatic improvements seen in the efficacy of TNF in the isolated limb perfusion studies suggest that sequestering TNF within a solid tumor not only improves the safety of the drug but also vastly improves the efficacy of combination therapies, such as treatment with chemotherapeutics. In part these effects may be due to the ability of TNF to directly reduce the interstitial fluid pressure (Kristensen et al. Citation1996; Nedrebo et al. 1998). Alternatively, TNF is known to induce direct antitumor effects on a variety of tumor phenotypes as well as the newly formed blood vessels feeding the tumor. Finally, systemic administration of PT-cAu-TNF may also potentiate the immunotherapeutic effects of TNF by sensitizing immune cells to recognize and destroy cancer cells. In effect, the PT-cAu-TNF may be an early stimulus in the anticancer immune response, which may be enhanced by additional immunotherapies. In toto, tumor sequestration of TNF, as shown with the PT-cAu-TNF vector, may induce one or multiple mechanisms to effect a significant antitumor response. Future studies will clarify the mechanism of action of PT-cAu-TNF and its clinical potential.
The data described in this report represent the first description of the use of the colloidal gold nanoparticles for tumor-targeted drug delivery. Future efforts will focus on identifying the mechanism and site of action for the vector and determining the general applicability of the vector to treat primary and metastatic tumors in preclinical models. Additional studies are focused on expanding the repertoire of drugs to deliver with this novel nanoparticle vector as well as determining if the vector is capable of delivering multiple therapies on a single particle.
We thank Drs. H. Richard Alexander and Steven K. Libutti, Surgery Branch, National Cancer Institute, NIH for providing TNF for these experiments, for their collegial support and encouragement throughout the course of these studies, and for reviewing this manuscript. This work was supported by an ATP Grant (Grant #99-01-1101) from The National Institute of Standards and Technology.
REFERENCES
- Alexander H. R., Bartlett D. L., Libutti S. K., Fraker D. L., Moser T., Rosenberg S. A.. 1998. Isolated hepatic perfusion with tumor necrosis factor and melphalan for unresectable cancers confined to the liver. J. Clin. Oncol.. 16: 1479–1489. [PUBMED], [INFOTRIEVE]
- Baban D. F.. 1998. Control of tumour vascular permeability. Ad. Drug Delivery Rev.. 34: 109–119. [CROSSREF]
- Bergqvist L., Sundberg R., Ryden S., Strand S. E.. 1987. The “critical colloid dose” in studies of reticuloendothelial function. J. Nucl. Med.. 28: 1424–1429. [PUBMED], [INFOTRIEVE]
- Bradfield J. W.. 1980. A new look at reticuloendothelial blockade. Br. J. Exp. Pathol.. 61: 617–623. [PUBMED], [INFOTRIEVE]
- Chandler J., Robinson N., Whiting K.. 2001. Handling false signals in gold-based tests. I.V.D. Technol.. 72, 2001. 34–35
- Cleland J. L.. 1997. Protein delivery from biodegradable microspheres. In Protein Delivery: Physical Systems, Sanders, Hendren, 1–43, New York, Plenum Press
- Creaven P. J., Plager J. E., Dupere S., Huben R. P., Takite H., Mittleman A., Proefrock A.. 1987. Phase I clinical trial of recombinant human tumor necrosis factor. Cancer Chemother. Pharmacol.. 20: 137–144. [PUBMED], [INFOTRIEVE]
- Faraday M.. 1857. Experimental relations of gold (and other metals) to light. Philos. Trans. R. Soc. London. 14: 145–181
- Fraker D. L., Alexander H. R., Andrich M., Rosenberg S. A.. 1996. Treatment of patients with melanoma of the extremity using hyperthermic isolated limb perfusion with melphalan, tumor necrosis factor, and interferon gamma: results of a tumor necrosis factor dose-escalation study. J. Clin. Oncol.. 14: 479–489. [PUBMED], [INFOTRIEVE]
- Frens G.. 1972. Controlled nucleation for the regulation of particle size in monodisperse gold suspensions. Nature Phys. Sci.. 241: 20–22
- Furman W. I., Strother D., McClain K., Bell B., Leventhol B., Pratt C. B.. 1993. Phase I clinical trial of recombinant human tumor necrosis factor in children with refractory solid tumors: A pediatric oncology group study. J. Clin. Oncol.. 11: 2205–2210. [PUBMED], [INFOTRIEVE]
- Hardonk M. J., Harms G., Koudstaal J.. 1985. Zonal heterogeneity of rat hepatocytes in the vivo uptake of 17 nm colloidal gold granules. Histochemistry. 83: 473–477. [PUBMED], [INFOTRIEVE]
- Hermanson G. T.. 1996; Bioconjugate Techniques.. Academic Press, Inc., San Diego, California
- Horisberger M.. 1979. Evaluation of colloidal gold as a cytochemical marker for transmission and scanning electron microscopy. Biol. Cellulaire. 36: 253–258
- Illum L., Davis S. S., Muller R. H., Mak E., West P.. 1987. The organ distribution and circulation time of intravenously injected colloidal carriers sterically stabilized with a block copolymer—poloxamine 908. Life Sci.. 40: 367–374. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Jain R., Shah N. H., Malick A. W., Rhodes C. T.. 1998. Controlled drug delivery by biodegradable poly(ester) devices: different preparative approaches. Drug Dev. Indust. Pharm.. 24: 703–727
- Kristensen C. A., Nozue M., Boucher Y., Jain R. K.. 1996. Reduction of interstitial fluid pressure after TNF-alpha treatment of three human melanoma xenografts. Br. J. Cancer. 74: 533–536. [PUBMED], [INFOTRIEVE]
- Kurfurst M.. 1992. Detection and molecular weight determination of polyethylene glycol-modified hirudin by staining after sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anal. Biochem.. 200: 244–248. [PUBMED], [INFOTRIEVE]
- Lejeune F. J.. 1995. High dose recombinant tumour necrosis factor (rTNFα) administered by isolation perfusion for advanced tumors of the limbs: a model for biochemotherapy of cancer. Eur. J. Cancer. 3: 1009–1016. [CROSSREF]
- Maruyama K., Takizawa T., Takahashi N., Tagawa T., Nagaike K., Iwatsuru M.. 1998. Targeting efficiency of PEG-immunoliposome-conjugated antibodies at PEG terminals. Ad. Drug Delivery Rev.. 24: 235–242. [CROSSREF]
- Maruyama K., Ishida O., Takizawa T., Moribe K.. 1999. Possibility of active targeting to tumor tissues with liposomes. Ad. Drug Delivery Rev.. 40: 89–102. [CROSSREF]
- Mirkin C. A., Letsinger R. L., Mucic R. C., Storhoff J. J.. 1996. A DNA based method for rationally assembling nanoparticles into macroscopic materials. Nature. 382: 607–609. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Moghimi S. M.. 1999. Re-establishing the long circulatory behaviour of poloxamine-coated particles after repeated intravenous administration: applications in cancer drug delivery and imaging. Biochim. Biophys. Acta. 1472: 399–403. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Moghimi S. M., Patel H. M.. 1998. Serum-mediated recognition of liposomes by phagocytic cells of the reticuloendothelial system—the concept of tissue specificity. Ad. Drug Delivery Rev.. 32: 45–60. [CROSSREF]
- Molema G., de, Leij L. F. M. H., Meijer D. K. F.. 1997. Tumor vascular endothelium: barrier or target in tumor directed drug delivery and immunotherapy. Pharm. Res.. 14: 2–38. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Müller R. H., Mäder K., Gohla S.. 2000. Solid lipid nanoparticles (SLN) for controlled drug delivery—a review of the state of the art. Eur. J. Pharm. Biopharm.. 50: 161–177. [CROSSREF]
- Nafayasu A., Uchiyama K., Kiwada H.. 1999. The size of liposomes: a factor, which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs. Ad. Drug. 40: 75–87
- Nedrebo T., Berg A., Reed R. K.. 1999. Effect of tumor necrosis factor-α, IL-1-β, and IL-6 on interstitial fluid pressure in rat skin. Am. J. Physiol.. 277. Heart Circ. Physiol.. 46: H1857–H1862. [PUBMED], [INFOTRIEVE]
- Netti P. A., Hamberg L. M., Babich J. W., Kierstia D., Graham W., Hunter G. J., Wolf G. L., Fischnan A., Boucher Y., Jain R. K.. 1999. Enhancement of fluid filtration across tumor vessels: implication for delivery of macromolecules. Proc. Natl. Acad. Sci. USA. 96: 3137–3142. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Nuzzo R. G., Allara D. L.. 1983. Adsorption of Bifunctional Organic Disulfides on Gold Surfaces. J. Am. Chem. Soc.. 105: 4481–4483
- Ogawa Y.. 1997. Injectable microcapsules prepared with biodegradable poly(α-hydroxy) acids for prolonged release of drugs. J. Biomater. Sci. Polymer Edn.. 8: 391–409
- Papisov M. I.. 1998. Theoretical considerations of RES-avoiding liposomes: molecular mechanisms and chemistry of liposome interactions. Ad. Drug Delivery Rev.. 32: 119–138. [CROSSREF]
- Rafferty D. E., Elfaki M. G., Montgomery P. C.. 1996. Preparation and characterization of a biodegradable microparticle antigen/cytokine delivery system. Vaccine. 14: 532–538. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Redhead H. M., Davis S. S., Illum L.. 2001. Drug delivery in poly(lactide-co-glycolide) nanoparticles surface modified with poloxamer 407 and poloxamine 908: in vitro characterisation and in vivo evaluation. J. Control Release. 70: 353–363. [CROSSREF], [PUBMED], [INFOTRIEVE]
- Renaud G., Hamilton R. L., Havel R.. 1989. Hepatic metabolism of colloidal gold-low-density lipoprotein complexes in the rat: evidence for bulk excretion of lyosomal contents into bile. Hepatology. 9: 380–392. [PUBMED], [INFOTRIEVE]
- Root S. W., Andrews G. A., Knieseley R. M., Tyor M. P.. 1954. The distribution and radiation effects of intravenously administered colloidal Au 198 in man. Cancer. 7: 856–866. [PUBMED], [INFOTRIEVE]
- Rubin P., Levitt S. H.. 1964. The response of disseminated reticulum cell sarcoma to the intravenous injection of colloidal radioactive gold. J. Nuclear Med.. 5: 581–594
- Schiller J. H., Storer B. E., Witt P. L., Alberti D., Tombes M. M. B., Arzoomanian R., Proctor R. A., McCarthy D., Brown R. R., Voss S. D., Remick S. C., Grem J. L., Borden E. C., Trump D. L.. 1991. Biological and clinical effects of intravenous tumor necrosis factor-a administered three times weekly. Cancer Res.. 51: 1651–1658. [PUBMED], [INFOTRIEVE]
- Stohrer M., Boucher Y., Stangassinger M., Jain R. K.. 2000. Oncotic pressure in solid tumors is elevated. Cancer Res.. 60: 4251–4255. [PUBMED], [INFOTRIEVE]
- Taguchi T.. 1988. Phase I study of recombinant human tumor necrosis factor (rHuTNF-PT-050). Cancer Detect. Prev.. 12: 561–572. [PUBMED], [INFOTRIEVE]
- Woodle M. C.. 1998. Controlling liposome blood clearance by surface grafted polymers. Ad. Drug Delivery Rev.. 32: 139–152. [CROSSREF]
- Zhang X. Y., Luck J., Derhirst M. W., Fan Yuan F.. 2000. Interstitial hydraulic conductivity in a fibrosarcoma. Am. J. Physiol. Heart Circ. Physiol.. 279: H2726–H2734. [PUBMED], [INFOTRIEVE]