Abstract
The objective of this study was to design an in situ biodegradable polymer implant controlled-release drug delivery system, using novel combinations of co-solvents and a model polypeptide, calcitonin (CT), and to assess the release of drug as a function of these co-solvents. Formulations were prepared by dissolving/ suspending CT polypeptide in poly-(lactic acid) (PLA) polymer solutions/suspensions containing combinations of a hydrophobic (benzyl benzoate, BB) and a hydrophilic (benzyl alcohol, BA) solvent. The CT-PLA mixtures were each injected into test tubes containing phosphate buffered saline solution to form the in situ implant and sampling was conducted over a 28-day period. The samples were analyzed for drug content using a modified Lowry protein assay procedure. Cumulative drug release demonstrated a rank-order correlation depending on the amount of the hydrophobic (BB) and hydrophilic (BA) solvents within each system. Increasing the amounts of the hydrophobic solvent, BB, in formulations demonstrated a 1.2–4.4-fold increase in CT release. Stability studies of all formulations over a 4-month period showed progressive increase in degradation of the CT polypeptide, especially at 37°C, but a slower degradation pattern prevailed at 4° and 20°C. Differential scanning calorimetric studies revealed a homogenous mixture of drug in the polymer matrix. Overall, these studies demonstrated the feasibility of designing controlled release systems capable of releasing a polypeptide drug as a function of influence of different co-solvent combinations.
The past two decades have seen a dramatic increase in the use of synthetic biodegradable polymers to design controlled-release drug products (Davis and Ilum Citation1988). Advantages offered by these drug delivery systems include reduced frequency of dosing, prolonged drug release periods, higher bioavailability, improved patient compliance and comfort, and low in vivo fluctuation of drug concentrations (Yolles, Eldridge, and Woodland Citation1971). More recently, attention has focused on the development of in situ biodegradable injectable systems whereby the drug can be dissolved or suspended in a biodegradable polymer such as polylactide (PLA) or copolymer of polylactic/glycolic acids (PLGA) dissolved in a nontoxic, physiologically acceptable solvent(s) (Dunn et al. Citation1994; Eliaz and Kost Citation2000; Chandrashekar and Udupa Citation1996) that upon injection forms a gel-like mass releasing drug over a period of time. The in situ biodegradable polymer injectable system has several advantages over the more traditional polymer microsphere drug delivery systems (Jain et al. Citation2000). These include less number of processing steps, production as suspensions or solutions that do not need mixing, less complex method of manufacture, easier to use, higher dose of drug delivered and ability to inject with smaller diameter (25–28 gauge) injection needles compared with microsphere preparations (21–23 gauge). Additionally, the in situ implants demonstrate less tissue irritation than microspheres and exhibit low initial burst release due to a low surface area to volume ratio (Cleland Citation2001).
The process by which these implants form in situ is as follows: when the drug-polymer mixture is injected into the body in liquid form, the biocompatible solvent dissipates into surrounding tissue and water comes in contact with the preparation. This leads to phase separation whereby a gel-like mass is formed due to coagulation of the polymer, hence forming an implant in situ. Drug is released from these implants over a period of time by a combination of diffusion of the drug and erosion of the polymers (Jain Citation2000).
Most technology for developing polymer drug delivery systems use organic solvents such as acetone, ethyl acetate, dichloromethane, dioxane, hexane, tetrahydrofuran, propylene glycol, and 2-pyrrolidone (Matschke et al. Citation2002). Specifically, for implants formed in situ, a broad range of solvents have been investigated including N-methyl-2-pyrrolidone (NMP) (Dunn et al. Citation1994), dimethyl sulfoxide (DMSO), triacetin, benzyl benzoate, glycofurol and glycerol formal (Bleiberg et al. Citation1993; Graham, Brudbeck, and McHush Citation1999; Chern and Zingermann Citation1999). Of these, the most preferred solvents are NMP and DMSO because of their pharmaceutical precedence (Dunn et al. Citation1997). However, the use of NMP as an excipient in parenteral pharmaceutical technology recently has come under close scrutiny by the European regulatory authorities because it has shown to have significant toxicity such as neurotoxicity, nongenotoxic cancerogenicity, or teratogenicity (ICH Topic Q3C 2000). Both NMP and DMSO have extensive toxicity data documented for oral, intraperitoneal, and intravenous administration of these solvents, but not for subcutaneous or intramuscular use (Royals et al. Citation1999). However, in a Krantz et al. (Citation2001) study, PLA or PLGA was dissolved in NMP, DMSO, or 2-pyrrolidone and injected intramuscularly into Sprague-Dawley rats. It was shown that all these solvents are myotoxic and can damage muscles.
Chandrashekar and Udupa (Citation1996) and Singh et al. (Citation1997) tried propylene glycol and triacetin, respectively. However, triacetin suffers from the same problems of NMP and DMSO and propylene glycol is not recommended due to its hemolytic potential (Cherng-Chyi Fu et al. Citation1998). Glycofurol was utilized as a solvent to deliver soluble necrosis receptor factor from an in situ forming PLGA implants in vivo (Eliaz et al. Citation2000). Glycofurol, although used in parenteral products (Crowther, Pillingi, and Owen Citation1997), has very little toxicological data available. A composition of a 75/25 PLGA in glycerol formal (hydrophilic) and triacetin (lipophilic) solvent combination was used to formulate a veterinary drug product (Chern and Zingermann Citation1999) and blood levels were observed over a period of 12 months subsequent to subcutaneous administration.
The objective of this study was to design an in situ biodegradable polymer implant drug delivery system for the controlled-release of a model polypeptide drug, calcitonin (CT), using combinations of a physiologically acceptable hydrophobic and hydrophilic co-solvent systems, benzyl benzoate (BB) and benzyl alcohol (BA). Both these solvents have a well-established track record as safe for parenteral use and meet the criteria for excipients in parenteral administration (Moltu et al. Citation2000). Five formulations containing various proportions of the co-solvent system were prepared to assess the differences in release of CT polypeptide drug over a period of 28 days. Studies were conducted with differential scanning calorimetry (DSC) to characterize the implant formulations further. Stability of CT polypeptide in co-solvent formulations was assessed over a 4-month period at different temperatures. CT was chosen as a model polypeptide drug because previous studies have demonstrated its usefulness as a model for polymer-based drug delivery systems (Prabhu, Sullivan, and Betageri 2002).
MATERIALS AND METHODS
Bovine CT drug, polylactic acid (PLA), BB and BA solvents were purchased from Sigma Chemicals (St. Louis, MO, USA). Phosphate buffer salts was obtained from Fisher Scientific (Fairlawn, NJ, USA). All chemicals were used as received.
Preparation of PLA Solutions Using Co-Solvents
PLA polymers were dissolved or suspended in (A) BB solvent alone (100:0) or a mixture of BB and BA solvents of various concentrations (B) 80:20 (C) 60:40, (D) 50:50, and (E) 20:80, respectively, using a novel approach that bypassed the application of direct heat. An ultrasonifier (Branson Sonifier 250) was used whereby the PLA polymer was suspended in each of the co-solvent systems and sonicated for an average of 45 sec resulting in a clear polymer solution.
Preparation of In Situ Implants Containing CT
Weighed amounts of CT polypeptide (2.50 mg/formulation) was dissolved or suspended in 3 ml of each of the PLA solutions (Formulations A–E) and stirred for a few minutes. To verify the formation of biodegradable implants in situ, the formed solution/suspension mixture was injected into 2 ml of phosphate buffer solution (PBS, pH 7.4). The resultant effect was the precipitation of polymer and formation of a gel matrix entrapping the drug thus forming an in situ implant. “Blank” in situ implants were prepared using the same procedure but containing no CT polypeptide. All samples were prepared in triplicate.
Physical Characterization Studies
Thermal Analysis
Differential scanning calorimetry (DSC) curves were recorded on a scanning calorimeter with a thermal analysis data system (Mettler 80, TA 2000, Toledo, OH, USA). The instrument was calibrated using indium as the standard. Samples (2 mg) were heated in sealed aluminum pans at a scanning rate of 10°C/min from 0 to 300°C under a nitrogen purge using an empty aluminum pan as reference.
Measurement of Encapsulation Efficiency
The amount of CT polypeptide loaded in the in situ implants was determined by direct recovery of the polypeptide from the polymer. The implants were dissolved into a tube containing 5 ml of methylene chloride, and subsequently extracted into 5 ml of water. The supernatant was collected and analyzed for polypeptide content using the Lowry assay procedure (Lowry et al. Citation1951). The protein-loading efficiency was calculated using the following formula:
where, Mactual is the amount of CT in each sample and Mtheoretical is the theoretical amount calculated from quantity added during the manufacturing process.
In Vitro CT Release Studies from In Situ Implants
Tubes containing the in situ implants of CT (as prepared above) were placed in a water bath/shaker and temperature was maintained at 37 ± 0.5°C. At predetermined intervals, 2 ml of the supernatant was withdrawn and replaced with fresh PBS medium. Sampling was conducted for a period of 28 days. All samples were subjected to CT analysis using a modified Lowry assay procedure at a spectrophotometric wavelength of 750 nm.
Stability Studies
CT polypeptide (300 μ g/ml) was added to each of the PLA solutions (Formulations A–E) and all formulations were stored for a period of up to 4 months at 4°, 20°, and 37°C in glass tubes. Samples were analyzed using a modified Lowry assay at 1-, 2-, and 4-month intervals to assess the stability of CT polypeptide.
RESULTS AND DISCUSSION
The choice of solvents plays an important role in the design of injectable drug delivery systems (Matschke et al. Citation2002). The ideal qualities of solvent(s) include safety, nontoxicity, stability, biocompatibility, and meeting all required guidelines for parenteral administration. As per the above qualities, the BB/BA mixtures proved to be ideal candidates for our proposed project. BB (hydrophobic) and BA (hydrophilic) solvents at different concentrations demonstrated a rank-order release pattern of CT over a 28-day period (). The release depended on the ratio of hydrophobic to hydrophilic solvent components, whereby increasing proportions of BB resulted in an increase in the rate of drug release from the implants and vice versa. As seen from , the cumulative release of CT was fastest from Formulation A with over 1000 μg of the drug released within 7 days, 1700 μg in 14 days, 2100 μg in 21 days, and 2200 μg toward the end of the run (28 days). Considering that the nature of the solvent (BB) in the formulation was hydrophobic, we expected to see a slower release profile compared with formulations containing higher amounts of hydrophilic solvents. This is because of the potential effect of the hydrophobic solvent to encapsulate the drug more efficiently and prevent it from leaving the gel when introduced into the aqueous medium. However, as observed from , the drug release rate was much higher from Formulation A than for other formulations. In Formulation A, the PLA polymer, for the most part, remains in solution whereas the polypeptide drug is suspended in the hydrophobic solvent medium.
FIG. 1 Cumulative release (in μg) of calcitonin (CT) from PLA in situ implants in different co-solvent combinations.
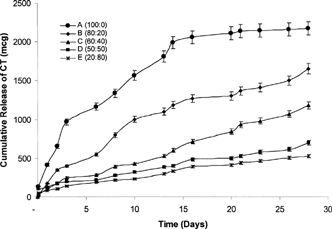
We postulated that the PLA at the surface of the gel implant precipitates out upon introduction into the aqueous medium forming a thin outer coat of the gel mass but essentially leaves the remaining PLA in solution along with the suspended drug. Thus, the only barrier for the CT polypeptide drug to overcome is that of a less rigid, lower density outer PLA coat that allows the highly water soluble drug to escape rapidly as it crosses the barrier to come in contact with the aqueous surrounding.
Formulations B–E followed a rank-order correlation depending upon the concentration of BB and BA solvents within each system, but all showed slower release than Formulation A. Formulation B exhibited significantly slower release kinetics compared with Formulation A with only ∼500 μg being released at 7 days, 1100 μg at 14, 1300 μg at 21, and 1600 μg at 28 days. Similarly, the release of drug from Formulations C–E slowed even further with approximately 1100, 600, and 500 μg total drug being released by the end of the run, respectively. This effect seemingly demonstrates a direct correlation to the amount of hydrophilic solvent in the formulation.
Our observations suggest that the CT dissipates out of the hydrophilic component leaving less drug to be entrapped within the mostly hydrophobic gel implant which accounts for their lower release rate overall. As the hydrophilic component of the formulation is increased, the formation of the gel matrix causes the release of drug to be slowed further into the aqueous medium. However, the expected burst release as a result of drug dissipating into the aqueous medium also was not evident. Other studies conducted in our laboratory using testosterone as the model drug produced similar results (data not shown). Overall, a 1.2–4.4-fold increase in the release of polypeptide was observed as the content of the hydrophobic solvent increased and hydrophilic solvent decreased.
Formulation A showed a burst effect in the release of drug during the initial 24 hr whereas release from Formulations B–E were more muted during the same time period. The presence of the hydrophilic solvent, regardless of proportion, appears to play a role in controlling this phenomenon.
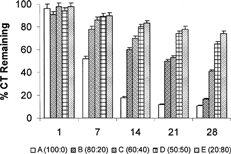
shows the percentage activity remaining in each formulation after the 28-day cycle. A comparison of CT remaining in the implant at day 1 and day 28 revealed a most dramatic drop for Formulation A at 88% whereas Formulation E only exhibited a drop of 20%. Formulations B–D droped by 64, 44, and 24%, respectively, reinforcing the trend established in . Overall, Formulation B offered the most gradual drop in activity over the time period of the study suggesting that such a formulation could be the most ideally suited in terms of release of the CT polypeptide drug.
FIG. 2 Percent CT remaining in the PLA in situ implants at 1, 7, 14, 21, and 28 days.
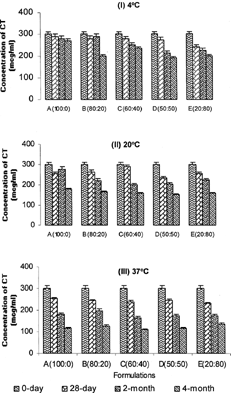
A 4-month long stability study of Formulations (A–E) at 3 different temperatures (4°, 20°, and 37°C) revealed a progressive increase in degradation of the polypeptide at 37°C compared with 4° and 20°C (). On average, ≈30% of the polypeptide degraded at the end of 4 months at 37°C compared with 12% decline at 4°C and 20% decline at 20°C. However, across Formulations (A–E) at any given temperature (4°, 20°, or 37°C), there was no significant difference (p > 0.05) in degradation of the CT polypeptide when subjected to different concentrations of the hydrophobic and hydrophilic solvents. This suggests exposure to different solvents has no effect on the stability of the polypeptide. Overall, the degradation patterns suggested that CT is not likely to remain stable in solution over prolonged periods of time (i.e., > 4 months).
FIG. 3 Storage stability of CT formulations over a 4-month period at (I) 4°, (II) 20°, and (III) 37°C.
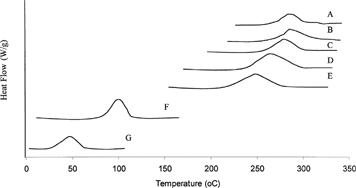
DSC thermograms () showed single endothermal peaks for CT, PLA, and drug/polymer mixed in different solvent combinations. The endothermal melting peak temperature for PLA was measured at 50°C, CT at 109°C, and CT implant Formulations (A) 100:0, (B) 80:20, (C) 60:40, (D) 50:50, and (E) 20:80 at 283°C, 280°C, 275°C, 262°C, and 249°C, respectively. The marked shift in peak temperatures from the original endothermal peaks of CT and PLA suggest that the drug is homogenously dispersed in the polymer matrix and the increasing amount of hydrophobic solvent, BB, accounts for the proportionate increase in peak temperatures. As the content of hydrophilic solvent (BA) increases and hydrophobic solvent (BB) decreases, the peak temperatures demonstrate a shift accordingly. However, the peak heights of solvent combinations were not significantly different from each other, but all were lower than the peak height for CT drug. The DSC curves for the solvent combinations clearly demonstrated the influence of increasing/decreasing hydrophobic solvent content on the in situ implants and the observations made here were consistent with the in vitro dissolution behavior of the implant systems.
FIG. 4 DSC thermograms of (A) BB only, (B) 80:20, (C) 60:40, (D) 50:50, and (E) 20:80 (BB:BA combinations) containing (F) CT and (G) PLA polymer.
Encapsulation efficiency of in situ implants was calculated to be ∼70% on average. Despite the low surface to volume ratio occupied by the gel implants, the lower encapsulation in implants was understandable due to the possible dilution effect caused by the lag time between the injection and formation of the gel matrix (Jain Citation2000). However, in general, the encapsulation was much higher in implants than expected.
CONCLUSIONS
The use of BB/BA co-solvent systems and their effect on the release of CT demonstrated that the composition and content of solvents definitely affected the release of polypeptide drugs from these controlled release delivery systems. Specifically, the higher the hydrophobic solvent content, higher the rate of drug release was observed and vice versa. Stability data suggested that long-term storage of CT in solution may not be feasible. Overall, the study demonstrated the feasibility of incorporating a polypeptide drug in an organic co-solvent system that has relevance in future design of in situ implant systems with predictable release kinetics.
The authors thank the Western University Research Committee for awarding a student fellowship to L.P. Tran through which this project was initiated and for additional intramural funds to complete the work.
REFERENCES
- Bleiberg B., Beers T. R., Persson M., Miles J. Metabolism of triacetin-derived acetate in dogs. Am. J. Clin. Nutr. 1993; 58: 908–911, [PUBMED], [INFOTRIEVE], [CSA]
- Chandrashekhar G., Udupa N. Biodegradable injectable implants for long term drug delivery using poly (lactic-co-glycolic) acid copolymers. J. Pharm. Pharmacol. 1996; 48: 669–674, [CSA]
- Chern R. T., Zingermann J. R. Liquid polymeric compositions for controlled release of bioactive substances. 1999, WO Publications Number 9947073
- Cherng-Chyi Fu R., Lidgate D. M., Whatley J. L., McCullough T. The biocompatibility of parenteral vehicles in vivo/in vitro screening comparison and effect of excipients on hemolysis. J. Parent. Sci. Technol. 1998; 41(5)164–168, [CSA]
- Cleland J. L. Injectable gels for local and systemic delivery of proteins. Abstract #0221, Controlled Release Society Annual meeting, San Diego, CA., 2001
- Crowther M. A., Pilling A., Owen K. The evaluation of glycofurols as a vehicle for use in toxicity studies. Hum. Exp. Toxicol. 1997; 16: 406, [CSA]
- Davis S. S., Ilum L. Polymeric microspheres as drug carriers. Biomaterials 1988; 9: 111–115, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Dunn R. L., English J. P., Cowsar D. R., Vanderbelt D. D. Biodegradable in situ forming implants and methods of producing the same. U.S. Patent 5 278 201 11, 1994
- Dunn R. L., Tipton A. J., Southard G. L., Rogers J. A. Biodegradable polymer composition. U. S. Patent 5 599 552, 1997
- Eastell R. Treatment of menopausal osteoporosis. N. Eng. J. Med. 1998; 338: 736, [CSA], [CROSSREF]
- Eliaz R. E., Kost J. Characterization of a polymeric PLGA—injectable implant delivery system for the controlled release of proteins. J. Biomed. Mat. Res. 2000; 50: 388–396, [CSA], [CROSSREF]
- Eliaz R. E., Wallach D., Kost J. Delivery of soluble tumor necrosis factor receptor from in-situ forming PLGA implants: in vivo. Pharm. Res. 2000; 17(12)1546–1550, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Graham D., Brodbeck K. J., McHugh A. J. Phase inversion dynamics of PLGA solutions related to drug delivery. J. Contrd. Rel. 1999; 58: 233–245, [CSA], [CROSSREF]
- ICH Topic Q3C (M). Maintenance of note for guidance on impurities: residual solvents (CPMP, ICH/283/95). European Agency for the Evaluation of Medical Products, London, UK., 2000
- Jain R. A., Rhodes C. T., Railkar A. M., Malick A. W., Shah N. A. Controlled release of drugs from injectable in-situ formed biodegradable PLGA microspheres: effect of formulation variables. Eur. J. Pharm. Biopharm. 2000; 50: 257–262, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Jain R. A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000; 21: 2475–2490, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Kranz H., Brazeau G. A., Napaporn J., Martin R. L., Millard W., Bodmeier R. Myotoxicity studies of injectable biodegradable in-situ forming drug delivery system. Int. J. Pharm. 2001; 212: 11–18, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. Protein measurement with folin phenol agent. J. Biol. Chem. 1951; 193: 265, [PUBMED], [INFOTRIEVE], [CSA]
- Matschke C., Isele U., Hoogevest P. V., Fahr A. Sustained release injectables formed in-situ and their potential use for veterinary products. J. Control. Rel. 2002; 85: 1–15, [CSA], [CROSSREF]
- Moltu F., Laurent A., Rufenacht D. A., Doelker E. Organic solvent for pharmaceutical parenterals and embolic liquids: a review of toxicity data. J. Parent. Sci. Technol. 2000; 54(6)457–469, [CSA]
- Royals M. A., Fujuta S. M., Yewey G. L., Rodriguez J., Schultheiss P. C., Dunn R. L. Biocompatibility of a biodegradable in-situ forming implant system in rhesus monkeys. J. Biomed. Mater. Res. 1999; 45: 231–239, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Singh U. V., Udupa N., Kamath R., Umadevi P. Enhanced anti-tumor efficacy of methotrexate poly (lactide-co-glycolide) acid injectable gel implants in mice bearing sarcoma—180. Pharm. Sci. 1997; 3: 133–136, [CSA]
- Yolles S., Eldridge J. E., Woodland J. H. R. Sustained delivery of drugs from polymer drug mixtures. Polymer News 1971; 1: 9–12, [CSA]