Abstract
Liposomes-encapsulated indomethacin/cyclodextrins (IMC/ CD) inclusion complexes were prepared. The characteristics and pharmacokinetics of the combined system were investigated. The high drug entrapment values of 2.38 ± 0.16 μg/mg and 2.48 ± 0.12 μ g/mg for liposomes-encapsulated IMC/ β-CD and IMC/HP-β-CD inclusion complexes were achieved, as only 1.60 ± 0.09 μ g/mg for conventional liposomes. Encapsulating IMC/CD inclusion complexes into liposomes resulted in a slow release of drug. Following intravenous administration, both liposomes-encapsulated inclusion complexes showed significantly improved AUC0 − ∞ compared with that of conventional liposomes (p < 0.05). After intramuscular administration, Cmax has been increased to 5.21 ± 1.14 μ g · ml−1 and 6.02 ± 1.22 μg · ml− 1 for liposomes-encapsulated IMC/ β -CD and IMC/HP-β -CD inclusion complexes, respectively, whereas only 2.43 ± 0.69 μg · ml− 1 for liposomes-encapsulated free drug (p < 0.01).
In the past few decades, drug delivery with liposomes as carrier systems provided options and opportunities for designing site specific drug therapy. Liposomal systems can delivery drugs to their activity site with a predetermined rate of clearance and distribution (Allen Citation1998; Banerjee Citation2001; Sihorkar and Vyas Citation2001). Both hydrophilic drugs and lipophilic drugs can be encapsulated in the inner aqueous phase or in lipids bilayers. However, with a limited hydrophobic domain, the carriers can accommodate only a limited amount of hydrophobic drug and the drug/lipid ratio may be especially low for drugs that have a low affinity for the lipids (McCormack and Gregoriadis Citation1994a). Consequently, the administration of a large amount of carrier may be required to reach a therapeutic concentration (Perkins et al., Citation2000). Moreover, the stability of liposomes may be problematic because of the interaction between lipophilic drugs and lipid bilayers (Torchilin, Citation1985; Takino et al., Citation1994).
Cyclodextrins (CD) are well known for their ability to solubilize poorly water-soluble drugs. CD can accommodate water-insoluble drugs in their cavities to form soluble inclusion complexes. The inclusion complexes can increase solubility and dissolution rate, decrease volatility, reduce irritability, and increase stability and pharmacological activity (Uekama Citation2004; Davis and Brewster Citation2004; Loftsson et al. Citation2005). In vivo, rapid dissociation of drug/CD inclusion complexes takes place either because of dilution or because other blood components displace the included drug. The released drug is then metabolized as free drug while the cyclodextrins moiety is excreted through the kidneys. Therefore, CD have no any ability to target drugs to biological sites (McCormack and Gregoriadis Citation1994b; Duchene, Ponchel, and Wouessidjewe Citation1999). The disadvantage of inclusion complexes may result in the failure of increasing drug therapeutic efficacy (Michaelis et al. Citation2005).
To circumvent the problems associated with liposomes or cyclodextrin inclusion complexes in a single system, together with combining their individual advantage, the concept of joining liposomes and cyclodextrin complexes of lipophilic drugs and forming drugs-in-cyclodextrinsin-liposomes formulations was proposed first by McCormack and Gregoriadis (Citation1994a, Citation1994b). The complementary carrier could be prepared by dehydration-rehydration (Kirby and Gregoriadis Citation1984) or spray-drying method (Skalko, Pavelic, and Becirevic Citation2000). This novel drug delivery system has been proved to augment entrapment efficiency (Fatouros, Hatzidimitriou and Antimisiaris Citation2001), and improve drug stability (Loukas, Jayasekera and Gregoriadis Citation1995a; Loukas, Vyza, and Valiraki Citation1998).
However, up to now, there has been little literature concerning the pharmacokinetic profiles of liposomes-encapsulated drug/CD inclusion complexes. It is well-known that encapsulation drug in liposomes often increases therapeutic efficiency due to the better pharmacokinetic profiles (Phan et al. Citation1997; Immordino et al. Citation2003; Gabizon, Shmeeda, and Barenholz Citation2003). Thus, whether the effect can be altered further by liposomes-encapsulated drug/CD inclusion complexes is worthy of being investigated.
The major aim of our work was to compare the in vivo pharmacokinetic parameters of liposomes-encapsulated inclusion complexes as well as free drugs. For this purpose, we used indomethacin (IMC) as a lipophilic drug model. IMC, a nonsteroid anti-inflammatory drug, also is a potent cerebral arteriolar vasoconstrictor. Clinically, the intravenous IMC in the treatment of patent ductus arteriosus in preterm infants is widely used (Vandekerckhove, Macrae and Slavik Citation2005; Watanabe et al. Citation2003). Recently, CitationKim, Lee, and Kang (2005) reported that entrapment of IMC into nanospheres exhibited a significant potential for sustained release of drug and showed slow clearance of IMC following intravenous administration. In the present study, we prepared liposomes-encapsulated IMC/β -cyclodextrin (β -CD) inclusion complexes and IMC/hydroxypropyl-β -cyclodextrin (HP-β -CD) inclusion complexes. The drug loading efficiency and in vitro release of different liposomes formulations were examined. To evaluate the in vivo behavior of liposomes-encapsulated IMC as free drug or inclusion complexes, the pharmacokinetic study of different formulations was performed on rabbits by intravenous and intramuscular administration.
MATERIALS AND METHODS
Indomethacin was purchased from NingBo Pharmaceutical Corp. (China). β -CD, and HP-β -CD (average degree of substitution 3) were obtained from Zhengjia Chemical Corp. (NingBo, China). Soybean phosphatidulcholine (SPC) came from Lipoid (Ludwigshafen, Germany) and cholesterol (Chol) was obtained from Sigma (St. Louis MO, USA). All other reagents and solvents used were of analytical grade.
Phase Solubility Studies
Phase solubility studies were carried out first to select an appropriate stoichiometric ratio of CD to IMC, according to Higuchi and Connors (Citation1965). Excess amounts of drug were added to 5 mL of phosphate buffer saline (PBS, pH 7.4) containing β -CD or HP-β -CD (0.001–0.015 M concentration range) in 10 mL tubes and shaken at 25 ± 0.5°C. At equilibrium after 3 days, suspensions were filtered by 0.45 μ m pore size filters. An aliquot from each tube was appropriately diluted with 50% isopropanol and assayed spectrophotometrically at 320 nm to evaluate the amount of (IMC) dissolved. Each experiment was carried out in triplicate. The phase solubility diagrams were obtained by plotting the mean solubility versus CD concentrations. The stoichiometry and apparent binding constants (KC) of the IMC/CD inclusion complexes were calculated from the slope and intercept of the straight lines of the phase solubility diagrams according to the following equation
where S0 is the solubility of the drug in the absence of CD.
Preparation of IMC/CD Inclusion Complexes
The IMC/CD inclusion complexes were prepared by freeze-drying method. Briefly, IMC (100 mg) and β -CD (317 mg) or HP-β -CD (385 mg) with 1:1 molar ratio (according to the results of phase solubility) were accurately weighted and dissolved in 50 mL of 60°C distilled water. The mixture was stirred on magnetic stirrer for 2.5 h at room temperature and avoided light, then filtered through a 0.45 μm membrane filter unit. The filtrates were freeze-dried to yield amorphous yellow powder. The content of IDN in inclusion complexes was determined by ultraviolet spectrophotometer.
Differential Thermal Analysis (DTA)
Thermal analysis of IMC, CD, physical mixture, and IDN/CD inclusion complexes were performed using a CDR-4P DTA (Shanghai Balance Instrufactory). All samples used were in solid state and were heated at a 10°C min−1 scanning rate between 50–400°C under atmosphere.
Infrared Spectroscopy (IR)
The IR spectra of IMC, CD, physical mixture, and IDN/CD inclusion complexes were taken from dried samples prepared by the potassium bromide disk method and scanned for absorbance from 500 to 4000 per cm. The instrument (Vector 22, Bruker) was operated under a dry air purge.
Entrapment of Inclusion Complexes in Liposomes
IMC/CD inclusion complexes were encapsulated by dehydration-rehydration method (Kirby and Gregoriadis Citation1984). SPC and Chol at weight ratio of 2:1 were placed in a pear-shaped flask and dissolved in diethyl ether. The organic solvent was completely evaporated in a rotary evaporator to form a thin film of lipids, then hydrated with PBS (pH 7.4) containing IMC/CD inclusion complexes. The multilamellar vesicles (MLVs) were obtained by homogenizing using JY92-II ultrosonic cells fragmentation instrument (XinZhi Institute, NingBo, China). After appropriately diluting with distilled water, the multilamellar dehydration-rehydration vesicles were formed by freeze-drying (LGJO.5-II Freeze Dryer, China) and rehydrated the freeze-drying product with 0.9%NaCl solution. To obtain liposmoes encapsulated free IMC, the equivalent amount of IMC was dissolved in diethyl ether and prepared by thin-film method and freeze-drying.
Encapsulation Efficiency
Nanosep® OD030C33 centrifuge tube (Pall Biosciences Lab USA) with 30 kD cut-off was used to separate the unentrapped IMC/CD inclusion complexes in liposomes. The liposomes suspensions were centrifuged at 15 000 g for 10 min at 4°C (Hereaus Instruments) and additional PBS was added to wash. After repetition of filtration and centrifugation three times, the filtrates were combined and the amount of unentrapped value was measured by ultraviolet spectrophotometer. The encapsulation efficiency was calculated from this value with respect to the initial IMC/CD inclusion complexes.
Loading Efficiency
The loading efficiency of liposomes-encapsulated free IMC or IMC/CD inclusion complexes were compared. The different liposomes were centrifuged at 50 000 g for 2 hr to remove the unentrapped drug or inclusion complexes. The pellets were then resuspended in 0.9%NaCl solution and finally freeze-dried. For determining the amount of IMC in the liposomes, the lyophilized products were accurately weighted, 0.5 ml of 10% Triton X-100 was added, and vortex-mixed. The volume was made up to 10 ml with 50% isopropanol and determined by HPLC.
In Vitro Drug Release
Evaluation of the release of IMC from the liposomes was performed with a dialysis method. First, l mL of the separated liposomes was placed into the dialysis bag with 30 kD cut-off and then transferred into 15 mL PBS (pH 7.4) at 37°C. Second, the solution in the dialysis bag was stirred at 100 rmp· min− 1. Then, at fixed time intervals, samples of 3 mL were withdrawn and the same volume of fresh PBS were replaced. After appropriate dilution with 50% isopropanol, samples were analyzed with ultraviolet spectrophotometer.
Pharmacokinetic Experiment Design
New Zealand rabbits weighting 2.0 ± 0.2 kg were obtained from a licensed supplier (Experimental Animal Centre, Zhejiang University). Then 12 hr before the experiment, the animals were fasted but had free access to water. Each animal received a single dose (equivalent to 3.5 mg IMC/kg body weight) of liposomes-encapsulated IMC or IMC/CD inclusion complexes by intravenous or intramuscular administration. At scheduled times, the blood samples were collected from the ear marginal vein and put into Eppendorf tubes with heparin as anticoagulant, then centrifuged at 3500 rpm for 10 min. Serum was collected and kept at −20°C before determination. Pharmacokinetic parameters were obtained by 3p97 program.
Assay of IMC Serum Concentration
First, 0.5 mL of serum sample, 0.1 mL of PBS (pH 5.8), and 3 mL of ethyl acetate were vortex-mixed in a 10 mL glass tube for 5 min. Second after centrifugation at 3500 rpm for 5 min, the organic phase was precisely aspirated. The remainder was extracted by addition of 3 mL of ethyl acetate. The two organic phases were mixed and evaporated to dryness under a gentle stream of nitrogen at 50°C in a water bath. Then, the dried residue was dissolved in mobile phase and analyzed by an established HPLC method. The analytical column was Diamonsil™C18 (5 μm, 150 × 4.6 mm ID, Dikma Technologies, DALIAN). The mobile phase consisted of 75% methanol and 25% phosphoric acid solution (0.34%) and was delivered at the flow rate of 1 mL/min. Next, 20 μ L of sample was injected into HPLC systems (Agilent 1100 series, USA). IMC was detected at 260 nm wavelength with an ultraviolet detector. Chromatographic data were analyzed using Agilent Chemstation provided by the HPLC system.
RESULTS AND DISCUSSION
Formation of Inclusion Complexes
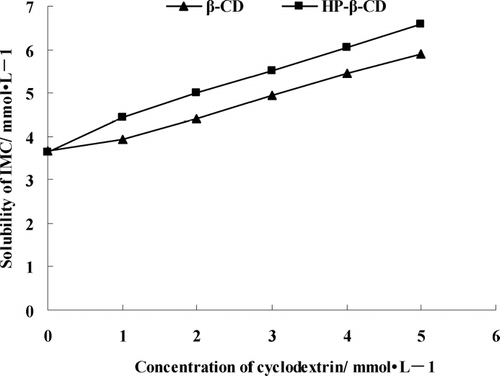
shows the phase solubility diagrams of IMC in different concentrations of the β -CD or HP-β -CD. The solubility of IMC increased linearly as a function of both β -CD and HP-β -CD concentrations, a typical feature of the AL-type complex. These results show the formation of a 1:1 (mol/mol) water-soluble inclusion complexes in solution. The apparent 1:1 stability constant (KC), calculated from the linear plot of the phase solubility, was 243 M−1 for IDN/β -CD and 360 M − 1 for IDN/ HP-β -CD, indicating a similar and moderate interaction between the drug and β -CD or HP-β -CD (Loukas et al. Citation1995b).
FIG. 1 Phase solubility diagrams for indomethacin with increasing concentrations of β -CD and HP-β -CD.
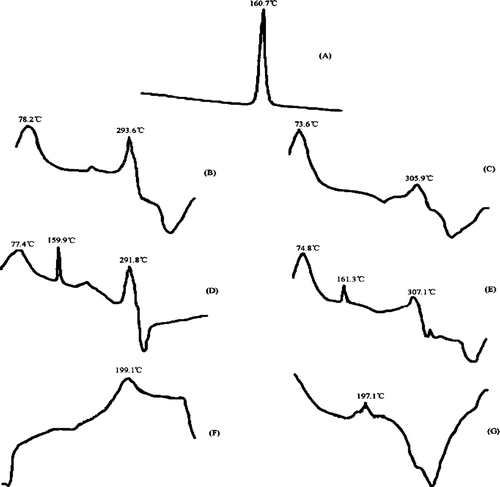
The thermograms of DTA and IR spectra were used to identify the IMC, CD, physical mixtures, and inclusion complexes. The DTA thermograms of IMC exhibited an endothermic peak at 160.7°C, corresponding to its melting point. β -CD or HP-β -CD alone showed a broad endothermic peak at 70–80°C representing its dehydration. The physical mixtures showed the same compositions corresponded to superimposition of the thermograms of the individual compositions. However, for complexes, new endothermic peaks at 199.1°C and 197.1°C for IMC/β -CD and IMC/HP-β -CD complexes appear instead of IMC or CDs endothermic peak, which confirms the formation of true inclusion complexes ().
FIG. 2 DTA thermograms of (A) indomethacin (IMC), (B) β -CD, (C) HP-β -CD, (D) IMC + β -CD (physical mixture), (E) IMC+HP-β -CD (physical mixture), (F) IMC/β -CD inclusion complexes, and (G) IMC/HP-β -CD inclusion complexes.
Similar to DTA, the IR spectra (not shown) gave clear evidence that the formation of IMC/β -CD and IMC/HP-β -CD inclusion complexes prepared under the conditions of this study. Strong absorption peaks were seen at 1690 and 1720 cm−1 for pure IMC, which can be attributed to IMC carbonyl group stretching vibration. In the spectra of the inclusion complexe, this brand was shifted towards shorter frequencies: 1590 and 1680 cm− 1. There was no change in the spectra of physical mixtures.
Encapsulation of IMC/CDs Complexes into Liposomes
It is evident that liposomes passively entrap solutes in their aqueous phase; in general, increasing the concentration of solutes results in greater amounts being entrapped. However, an essential step of preparation in our experiment necessitates freeze-drying, which leads to vesicle fusion and the formation of large liposomes. This can be overcome by the addition of cyclodextrins, that serve as cryoprotecction. However, the encapsulation efficiency could be greatly reduced due to the presence of cyclodextrins. The contradiction can be minimized by adjusting the CDs to lipids ratio and by increasing the volume of suspension prior to freeze-drying (Kirby and Gregoriadis Citation1984). Because excessive dilution of inclusion complexes leads to dissociation (Mesen and Putterman Citation1991), it becomes necessary to optimize preparation parameters.
In this experiment, the factors influencing encapsulation efficiency of IMC/CDs inclusion complexes into liposomes investigated were including lipids concentration, drug-to-lipid ratio, and volume of the solution prior to freeze drying. Through optimization, the optimal concentration of lipids was 30 mg/mL and drug-to-lipid weight ratio was 1:7. The final lyophilized volume was 10 mL. Based on these conditions, 62.4 ± 5.8% and 59.2 ± 4.1% of the encapsulation efficiency were achieved for IMC/β -CD and IMC/HP-β -CD inclusion complexes, respectively.
The loading efficiency of liposomes-encapsulated free IMC, IMC/β -CD, and IMC/HP-β -CD inclusion complexes were 1.60 ± 0.09 μ g/mg, 2.38 ± 0.16 μ g/mg, and 2.48 ± 0.12 μ g/mg, respectively. Significant difference was found between the free drug and inclusion complexes (p < 0.001). The high entrapment ability of IMC in the form of inclusion complexes in comparison to plain drug is an indubitable advantage of this approach. Increasing loading efficiency can be explained to the formation of aqueous inclusion complexes and encapsulation in the aqueous phase of liposomes.
Release Studies
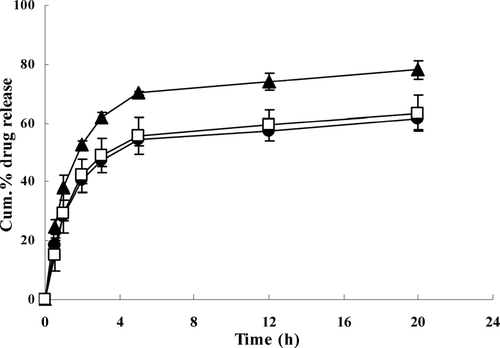
shows the in vitro drug release profiles of liposomes-encapsulated different IMC formulations. As shown, liposomes-encapsulated inclusion complexes result in a slow release compared with conventional liposomes. For liposomes-encapsulated free drug, IMC is directly and rapidly released from the lipid bilayers because hydrophobic drug is mostly entrapped in the domain (Maestrelli et al. Citation2005). However, this was not the case for inclusion complexes. In this situation, two routes may account for the drug release. One is that IMC/CDs inclusion complexes are transported from inner aqueous phase to lipid bilayers, then release intact inclusion complexes. The release rate depends on the diffusion of inclusion complexes. The other way is release of free drug, which is in equilibrium with the inclusion complexes. The release rate also is affected by a stability constant of inclusion complexes. Whether release of whole inclusion complexes or free drug, it had first to overcome the lipidic barrier since inclusion complexes were encapsulated in the inner of aqueous phase of liposomes. Therefore, the release of liposomes-encapsulated inclusion complexes was more slowly. There is no significant difference of release rate between liposomes-encapsulated IMC/β -CD and IMC/ HP-β -CD inclusion complexes because the two inclusion complexes have a similar stability constant, as calculated from phase solubility diagrams. The results of in vitro release deduced that entrapment of inclusion complexes in inner aqueous of liposomes provided a more stable encapsulation of the drug because the interaction between drug and lipid bilayers could be avoided.
FIG. 3 Release profiles of liposomes-encapsulated IMC (▴), IMC/β -CD inclusion complexes (□), and IMC/HP-β -CD inclusion complexes (•) in phosphate buffer saline (pH 7.4). Results are expressed as means ± S.D. of four determinations.
Pharmacokinetic Studies
The assay of IMC concentrations in serum was validated. The intraday and interday precisions were determined by analyzing IMC control samples at three concentrations for 5 times in 1 day or 5 times in 5 consecutive days, respectively. Both the coefficients of variation were below 5%. The results of extraction recovery were all above 80% for three concentrations. A linear calibration plot prepared using peak area of the standard solutions (0.199–9.960 μ g/ml) of IMC with plasma was used to assay the concentration of the drug in the serum samples. and present the mean plasma concentration versus time profiles of IMC after intravenous or intramuscular administration of liposomes-encapsulated IMC or IMC/CDs inclusion complexes. Following intravenous administration, the curves of three preparations were all fitted a two-compartment open model. Pharmacokinetic parameters are summarized in . The values T1/2β and AUC0 − ∞ were 75.4 ± 13.6 min and 390.4 ± 23.4 μg · min· ml−1, respectively, for liposomes-encapsulated IMC; 106.8 ± 16.8 min and 580.8 ± 50.9 μg · min · ml−1, respectively, for liposomes-encapsulated IMC/β -CD, and 122.7 ± 63.1 min and 629.0 ± 150.7 μg · min · ml−1, respectively, for liposomes-encapsulated IMC/HP-β -CD.
FIG. 4 Mean serum concentration-time profiles in rabbits for liposomes-encapsulated IMC (▴), and IMC/β -CD inclusion complexes (□), and IMC/HP-β -CD inclusion complexes (•) after intravenous administration. The vertical bars represent the standard error of mean (n = 4).
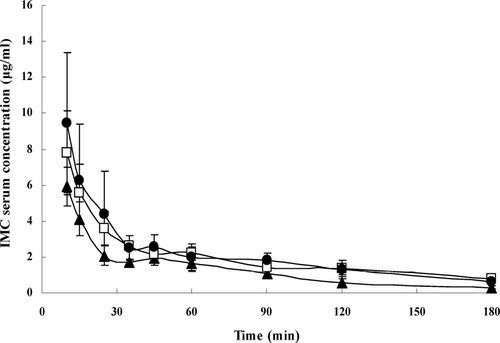
FIG. 5 Mean serum concentration-time profiles in rabbits for liposomes-encapsulated IMC (▴), IMC/β -CD inclusion complexes (□), and IMC/HP-β -CD inclusion complexes (•) after intramuscular administration. The vertical bars represent the standard error of mean (n = 4).
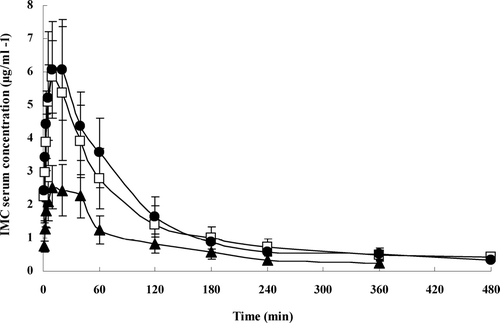
TABLE 1 Pharmacokinetic parameters of liposomes-encapsulated IMC, IMC/β -CD inclusion complexes, and IMC/HP-β -CD inclusion complexes after intravenous administration
Compared with liposomes-encapsulated free IMC, both liposomes-encapsulated inclusion complexes produced much higher AUC0 −∞(p < 0.05). Lipsomes encapsulated IMC/β -CD inclusion complexes also gave significantly longer T1/2β (p < 0.05). Unexpectedly, no significant difference was observed between liposomes-encapsulated IMC/HP-β -CD inclusion complexes and free IMC, probably due to the limited animals. Considering that the entrapping drug inclusion complexes into liposomes results in a slow in vitro release rate, therefore their systematic circulation time prolongs.
lists the pharmacokinetic parameters after intramuscular administration. A one-open compartment was suitably selected to draft the plasma mean concentration versus time profiles. The Cmax has been increased to 5.2 ± 1.1 μg · ml−1 and 6.0 ± 1.2 μg · ml−1 for liposomes-encapsulated IMC/β -CD and IMC/HP-β -CD inclusion complexes, respectively, whereas only 2.4 ± 0.7 μg · ml−1 for liposomes-encapsulated free drug (p < 0.001). Therefore, the AUC0 − ∞ or AUMC0 − ∞ of liposomes-encapsulated inclusion complexes significantly increase (p < 0.01). IMC possesses the characteristics of high protein-bound (up to 99%) (Tayyab et al. Citation1999). As there are many proteins and enzyme in muscular tissues, it is well understood that IMC released from conventional liposomes is easily bound with proteins and gradually degraded. For liposomes-encapsulated inclusion complexes, the simultaneous release of cyclodextrin, which can displace the binding fraction IMC to plasma, promotes the IMC release further. In addition, the formation of inclusion complexes dramatically decreases the drug protein binding and more drug is absorbed from injection site and transported to blood.
TABLE 2 Pharmacokinetic parameters of liposomes-encapsulated IMC, IMC/β -CD inclusion complexes, and IMC/HP-β -CD inclusion complexes after intramuacular administration
CONCLUSION
The possibility of using the combined approach of CD inclusion complexes and liposome entrapment to improve the in vivo pharmacokinetics of hydrophoblic drug has been investigated in the present work. This versatile carrier could overcome the limitation of liposome encapsulated lipophilic drugs with low entrapped efficiency. Using IMC as hydrophobic drug, IMC/β -CD and IMC/HP-β -CD inclusion complexes were entrapped into liposomes by dehydration-rehydration method. Compared with conventional liposomes, entrapping inclusion complexes into liposomes not only increased the IMC loading efficiency, but also altered the in vitro release. Moreover, the present approach could demonstrate feasibility to improve the drug's pharmacokinetic behavior in vivo, better than conventional liposomes. These results revealed that the complementary carrier system could broaden the scope and accessibility of present and future therapeutic agents.
This work was supported by the National Natural Science Foundation of China (No.30371692).
REFERENCES
- Allen T. M. Liposomal drug formulations. Rationale for development and what we can expect for the future. Drug 1998; 56: 747–56
- Banerjee R. Liposomes: applications in medicine. J. Biomater. Appl. 2001; 16: 3–21
- Davis M. E., Brewster M. E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004; 12: 1023–1035
- Duchene D., Ponchel G., Wouessidjewe D. Cyclodextrins in targeting application to nanoparticles. Adv. Drug Del. Rev. 1999; 36: 29–40
- Fatouros D. G., Hatzidimitriou K., Antimisiaris S. G. Liposomes encapsulating predinisolone and predinisolone-cyclodextrin complexes: Comparison of menbranes interity and drug release. Eur. J. Pharm. Sci. 2001; 13: 287–296
- Gabizon A., Shmeeda H., Barenholz Y. Pharmacokinetics of pegylated liposomal doxorubin: Review of animal and human sutides. Clin. Pharmacokin. 2003; 42: 419–436
- Higuchi T., Connors K. A phase solubility technique. Adv. Anal. Chem. Instrum (New York) 1965; 4: 117–211
- Immordino M. L., Brusa P., Arpicco S., Stella B., Dosio F., Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J. Control Rel. 2003; 91: 417–429
- Kim S. Y., Lee Y. M., Kang J. S. Indomethacin-loaded methoxy poly(ethylene glycol)/poly(D,L-lactide) amphiphilic diblock copolymeric nanospheres: Pharmacokinetic and toxicity studies in rodents. J. Biomed. Mater. Res. A 2005; 74: 581–590
- Kirby C., Gregoriadis G. Dehydration–rehydration vesicles (DRV): A new method for high yield drug entrapment in liposomes. Biotechnology 1984; 2: 979–984
- Loftsson T., Jarhi P., Masson M., Jarvinen T. Cyclodextrins in drug delivery 2005; 2: 335–351
- Loukas Y. L., Jayasekera P., Gregoriadis G. Novel liposome based multicomponent systems for the protection of photolabile agents. Int. J. Pharm. 1995a; 117: 85–94
- Loukas Y. L., Vyza A. E., Valiraki A. P., Gregoriadis G. Inclusion complexes and stability studies of organophosphorus insecticide with stability constant. Analyst. 1995b; 120: 533–538
- Loukas Y. L., Vraka V., Gregoriadis G. Drugs, in cylodextrins, in liposomes: A novel approach to the chemical stability of drugs sensitive to hydrolysis. Int. J. Pharm. 1998; 162: 137–142
- Maestrelli F., Rodriguez M. L. G., Rabasco A. M., Mura P. Preparation and characterization of liposomes encapsulating ketoprofen-cyclodextrin complexes for transdermal drug delivery. Int. J. Pharm. 2005; 298: 55–67
- McCormack B., Gregoriadis G. Drugs-in-cyclodextrins-in liposomes: A novel concept in drug delivery. Int. J. Pharm. 1994a; 112: 249–258
- McCormack B., Gregoriadis G. Entrapment of cyclodextrin-drug complexes into liposomes: Potential advantages in drug delivery. J. Drug Target 1994b; 2: 449–454
- Mesen J. L., Putterman P. Pharmacologyical application of a–hydroxypropyl-β -cyclkodextrin. New Trends in Cyclodextrins and Derivatives, D. Duchen. Editionds De Sante, Paris 1991; 369–407
- Michaelis M., Zimmer A., Handjou N., Cinatl J., Cinatl J. J. Increased systemic efficacy of aphidicolin encapsulated in liposomes. Oncol. Rep. 2005; 13: 157–160
- Perkins W. R., Ahmad I., Li X., Hirsh D. J., Masters G. R., Fecko C. J., Lee J., et al. Novel therapeutic nano-particles (lipocores): Trapping poorly water soluble compounds. Int. J. Pharm. 2000; 200: 27–39
- Phan G., Herbet A., Cholet S., Benech H., Deverre J. R., Fattal E. Pharmacokinetics of DTPA entrapped in conventional and long-circulating liposomes of different size for plutonium decorporation. J. Control Rel. 1997; 110: 177–188
- Sihorkar V., Vyas S. P. Potential of polysaccharide anchored lipsomes in drug delivery, targeting and immunization. J. Pharm. Pharm. Sci. 2001; 4: 138–158
- Skalko B. N., Pavelic Z., Becirevic L. M. Liposomes containing drug and cyclodextrin prepared by the one-step spray-drying method. Drug Dev. Ind. Pharm. 2000; 26: 1279–1284
- Takino T., Konishi K., Takakura Y., Hashida M. Long circulating emulsion carrier systems for highly lipophilic drugs. Biol. Pharm. Bull. 1994; 17: 121–125
- Torchilin V. P. Liposomes as targetable drug carriers. Crit. Rev. Ther. Drug Carrier Syst. 1985; 2: 65–115
- Tayyab S., Haq S. K., Aziz S. M. A., Khan M. M., Muzammil S. Effect of lysine modification on the conformation and indomethacin binding properties of human serum albumin. Int. J. Biol. Macro. 1999; 26: 173–180
- Uekama K. Design and evaluation of cyclodextrin-based drug formulation. Chem. Pharm. Bull. 2004; 52: 900–915
- Vandekerckhove K., Macrae D., Slavik Z. Nonsurgical treatment of patent arterial duct in term neonates with congenital heart disease: The role of intravenous indomethacin. Pediatr. Cardiol. 2005; 26: 698–699
- Watanabe K., Tomita H., Ono Y., Yamada O., Kurosaki K., Echigo S. Intravenous indomethacin therapy in infants with a patent ductus arteriosus complicating other congenital heart defects. Circ. J. 2003; 67: 750–752