Abstract
In our work depot delivery systems of celecoxib were developed using multivesicular liposomes. Moreover, the solubility of celecoxib was enhanced by complexing drug with cyclodextrin to overcome the limitation of conventional therapy. The multivesicular liposomes (MVLs) bearing celecoxib-β -cyclodextrin inclusion complex were prepared by reverse phase evaporation method, and multilamellar vesicles (MLVs)-bearing drug complex was prepared by the cast film method. The formulations were characterized for vesicle size, encapsulation efficiency, and in vitro drug release. In vivo performance of multivesicular liposomes bearing celecoxib-β -cyclodextrin inclusion complex was evaluated by assessing anti-inflammatory activity using carrageenan-induced rat paw edema volume method. The results were compared with that of celecoxib-cyclodextrin complex and MLVs containing celecoxib-β -cyclodextrin inclusion complex in equal amounts. Phase solubility studies for the celecoxib-β -cyclodextrin inclusion complex clearly indicated an increase in aqueous solubility of celecoxib with an increase in β –CD concentration. The in vitro release studies reveal that MLVs release more than 80% drug within 48 hr whereas MVL formulations release nearly the same amount of drug in 120 hr. In vivo data reveal that reduction in paw volume with MVL formulation was not rapid and fast, but the effect was maintained for prolonged periods, and even after 24 hr there was 40.7 ± 3.40% reduction in paw volume. MVL formulation showed more sustained and prolonged anti-inflammatory effect compared with plain drug and MLVs. We concluded that multivesicular liposome can be successfully utilized for the sustained delivery of celecoxib.
Celecoxib, a specific cycloxegenase inhibitor (COX-2), is a nonsteroidal anti-inflammatory drug, poorly soluble in water, and widely used as analgesic and for acute and long-term treatment of rheumatoid arthritis and osteoarthritis (Hubbard et al. Citation1996, Simon et al. Citation1998). Celecoxib has serious side-effects on oral administration, such as gastrointestinal (GI) toxicities, gastric mucosal ulceration and hemorrhage due to inhibition of prostaglandin production, that restricts its oral use and makes it a good candidate for transdermal administration. However, due to the excellent barrier function of the skin, there is need to use safe and effective enhancers for improving transdermal absorption of drugs.
Liposomes have been widely used as safe and effective vehicles for topical drug delivery systems since, in spite of the controversial literature about possible vehicle-mediated phenomena of skin uptake (Alvarez-Rom'an Naik et al. Citation2004), in several cases they allowed effective drug penetration and enhanced clinical efficacy (Gregoriadis Citation2000; Verma et al. Citation2003). They have shown great potential as versatile drug delivery systems to deliver numerous bioactives that include proteins, peptides, antineoplastic agents, antibiotics, and antiviral drugs (Ran and Yalkowsky Citation2003; Mumper and Hoffaman Citation2000). Liposome formulations of interferons, peptides, and hormones (calcitonin) have been used successfully as intramuscular depots. However, the conventional liposomes (unilamellar and multilamellar liposomes) have certain limitations such as low entrapment efficiency, stability problems, and release of drugs after a single breach in the external membrane (Lasic and Papadjopulos Citation1995).
These challenges have been met successfully by the use of a new carrier, i.e., a multivesicular liposomal (MVL) drug delivery system (Kim et al. Citation1996; Khatibi, Howell, and McCully Citation1991; Lanston et al. 2003). The MVL systems are characterized by their sui generis structure of multiple nonconcentric aqueous chambers surrounded by a network of lipoidal membranes (Lasic et al. Citation1995). These systems can be differentiated from unilamellar vesicles (ULVs) and MLVs in two major aspects: size and composition. MVL have a size range of 5 to 30 μ m compared with ULVs (1 μ m) and MLVs (1–5 μ m). In addition to the general chemical composition of conventional liposomes, MVLs contain a neutral lipid (triolein, tricaprylin, tributyrin, and tributyrine) as an integral component that is responsible for its unique multivesicular structure (Kim et al. Citation1983; Spector, Zasadzinshi, and Sankaram Citation1996). As MVLs are composed of multiple nonconcentric aqueous chambers, they contain 95% water and therefore provide a unique carrier system for hydrophilic drugs including a variety of therapeutic proteins (Katre et al. Citation1998). The structural characteristics of MVLs make them ideal vehicles for locoregional drug delivery. The MVL particles are manufactured with lipids that are membrane tissue components of the human body; thus they are highly biocompatible and nonimmunogenic (Spector et al. Citation1996). Their unique particle size is large enough to preclude their entry into the capillaries and thus remain at the site of administration.
However, the entrapment of poorly water soluble drugs in the lipid bilayers of liposomal membranes is often limited in terms of drug-to-lipid mass ratio and requires the use of organic solvents (McCormack and Gregoriadis Citation1994; Gregoriadis Citation2000). Furthermore, the carrier functions of liposomes through the skin layers often cannot actually be achieved for lipophilic drugs that, when incorporated in the membrane bilayers rather than entrapped in the aqueous core of the vesicles, are rapidly released from the carriers after administration (Takino et al. Citation1994).
The solubility of the drug was enhanced in the present project by making water soluble complex with β -cyclodextrin. The role of cyclodextrins as possible enhancers of percutaneous absorption of drugs also has been investigated (Loftsson et al. Citation1998; Loftsson and Masson Citation2001). Cyclodextrins may improve dermal absorption of drugs mainly by increasing their thermodynamic activity in the vehicles and favoring their delivery to the skin surface (Loftsson et al. Citation1998; Matsuda and Arima Citation1999; Masson et al. Citation1999). Recently, the entrapment into the aqueous phase of liposomes of lipophilic drugs in the form of water soluble celecoxib-β -cyclodextrin inclusion complex has been proposed as a possible approach for circumventing the problems associated with both systems and combines their relative advantages in a single system. This concept establishes a novel system in drug delivery by joining liposomes and celecoxib-β -cyclodextrin inclusion complex of lipophilic drugs and forming drugs-in-cyclodextrin–bearing liposomes formulations (McCormack and Gregoriadis Citation1994). In the present project, MVLs bearing-celecoxib-β -cyclodextrin inclusion complex were prepared and characterized for their performance in vitro and in vivo.
MATERIALS AND METHODS
Celecoxib was obtained as a generous gift sample from M/s Dr. Reddys laboratories (Hyderabad, India). Hydrogenated soya phosphatidyl choline (HSPC) was supplied as a generous gift sample from Lipoid (Germany). β -cyclodextrin (β -CD), phospatidyl glycerol (PG), cholesterol (CH), dicetyl phosphate (DCP), tributyrin (TB), tricaprylin (TC), triolein (TO), glycine and L-lysine were procured from Sigma Chemical Co. (St. Louis, MO, USA). Diethyl ether and chloroform were procured from Loba Chemicals (Mumbai, India). All other reagents used in this study were of analytical grade. Double distilled water was used throughout the experiments.
Preparation of Celecoxib-β-Cyclodextrin Inclusion Complex
Celecoxib-β -cyclodextrin inclusion complex in molar ratio 1:1 was prepared using the kneading method reported by Reddy et al. (Citation2004) with little modifications. Briefly β -cyclodextrin (1 mM) and double distilled water (1.7 ml) were mixed together in a mortar to make a homogenous paste, celecoxib (1mM) was then added slowly while grinding, a small quantity of ammonium hydroxide (0.25 ml 50% NH4OH) was then added to assist the dissolution of celecoxib. The mixture was then ground for 1 hr with an appropriate quantity of water added to maintain a suitable consistency. The preparation was then dried at 45°C in a rotary evaporator (Buchi R) and then the dried mass was sieved (Retsch, type vibro) and the 50–100 μ m sieve granulometric fraction was used for following studies.
Characterization of Celecoxib-β-Cyclodextrin Inclusion Complex
Phase solubility studies were preformed according to the method reported by Higuchi and Connors (Citation1965). Excess celecoxib was added to 30 ml of purified water (pH 6.8) containing various concentrations of β –CD (0.002 M–0.01 M) in a series of 100 ml volumetric flasks and then the mixture was shaken for 24 hr at room temperature (28 ± 2°C) at 150 rpm. The mixture was then kept aside to achieve equilibrium. These mixtures were then filtered through 0.45 μ m Millipore filter and assayed for celecoxib by measuring the absorbance at 254 nm, against blank solutions. The mixture was shaken until three consecutive samples estimated same amount of drug.
Differential Scanning Calorimetry (DSC) measurements were performed using a Mettler Toledo DSC 821. DSC module was controlled by STAR software (Mettler-Toledo GmbH, Switzerland). Indium standard was used to calibrate the DSC temperature and enthalpy scale. The samples were sealed in 40 μ l aluminum pans, the lids were peered, and the DSC thermograms were recorded at a heating rate of 10°C/min from 50–350°C under nitrogen atmosphere.
Fourier transform infrared (FT-IR) spectra were obtained on a Jasco V5300 (Jasco, Japan) FT-IR system using the KBr disk method and were recorded over the wave number range of 4000 to 400 cm− 1.
Preparation of MVLs
The multivesicular liposomes containing celecoxib-β -cyclodextrin inclusion complex were prepared in two steps using double emulsification technique (Katre et al. Citation1998; Jain et al. Citation2005a) with slight modifications. The first step involved the formulation of water in oil emulsion. A lipid solution consisted of 13.2 mM hydrogenated soya phospatidyl choline, 19.88 mM cholesterol, 2.44 mM neutral oil (TB/TO/TC), and 2.80 mM negatively charged lipid (DCP) in 1 ml mixture of chloroform, and diethyl ether (1:1 v/v) was emulsified with an equal volume of aqueous solution of the celecoxib-β -cyclodextrin inclusion complex (10 mg) by stirring the mixture for 9 min at 40 g. In the second step, this (w/o) emulsion subsequently was emulsified with second aqueous phase containing 1.5% glycine and 40 mM lysine for 15 sec at 80 g, which resulted in w/o/w double emulsion. Organic solvents were removed by flushing nitrogen into the mixture at temperature 30–36°C. For the separation of liposomes from unentrapped material, an equal volume of 5% sucrose (w/v) solution was added to the liposome preparation. The liposomes were pelleted in a clinical centrifuge at 600 g for 5 min. The supernatant was removed and the liposomes were resuspended in 5% sucrose.
MLVs Preparation
MLVs were prepared by cast film method reported by Bangham et al. (Citation1965) with slight modifications. Briefly 13.2 mM hydrogenated soya phospatidyl choline, 19.88 mM cholesterol, and 2.80 mM phosphatidyl glycerol (PG) were dissolved in chloroform and casted a thin film by evaporating the solvent under reduced pressure using rotary evaporator (Buchi-type, York Sci., Bombay). Lipid film was hydrated with (10 ml) saline phosphate buffer (pH 7.4) containing celecoxib-β -cyclodextrin inclusion complex (10 mg) followed by continuous vortexing of the flask for ∼ 1 hr to get multilamilar vesicles (MLV).
In Vitro Characterization
Celecoxib-β -cyclodextrin inclusion complex entrapped MVL formulation and MLVs liposomal formulation were characterized for the following attributes:
Vesicle morphology. The shape and surface topography were visualized by scanning electron microscopy (SEM). The samples of SEM were prepared by lightly sprinkling the lyophilized MVLs on double adhesive tape that was struck to an aluminum stub.
Vesicle size and size distribution of various formulations were determined using laser light diffraction particle size analyzer (CILAS 1064L, France).
Vesicle count/mm3 was determined using hemocytometer (Feinopptik, Blakenburg, Germany) and spreading the diluted vesicle suspension over the slide and counting the number of vesicles in the chambers of hemocytometer. Total number of vesicles/mm3 were calculated using following formula (Jain et al. Citation2005b).
Percent encapsulation efficiency was determined by first centrifugation of the vesicle suspension at 60 g for 30 min. The supernatant liquid was removed and pellet was washed 2–3 times with the phosphate buffer saline (PBS-pH 7.4) and then finally lysed with triton X-100 (0.5% w/v). Methanol was added 1–2-fold followed by mixing to obtain clear solution and analyzed for celecoxib content at 254 nm spectrophotometerically.
In vitro drug release from different formulations was determined using home fabricated Franz diffusion cell and cellophane membrane (MWCO-12,000) in which the donor compartment contained the formulation while the receptor compartment was filled with PBS (pH 7.4). The sink condition was maintained by using 40% v/v PEG-400 in PBS in the receptor compartment and the temperature was maintained at 37± 1°C with the help of a circulating water bath. Samples (1 ml) were withdrawn at appropriate time intervals and compensated with equal quantity of PBS (pH 7.4) containing 40% v/v PEG-400. Samples were filtered through 0.45μ m Millipore filter and drug content was determined spectrophotometrically at 254 nm (Cintra 10 UV-visible spectrophotometer).
Preparation of Skin Samples for In Vitro Studies
Albino rats of either sex (150–200 g) were used in the study. These studies were carried out according to the guidelines of the council for the purpose of control and supervision of experiments on animals, Ministry of Social Justice and Empowerment, Government of India. The rats were killed by an overdose of diethylether anesthesia. Hairs on the abdominal area of the rats were removed by trimming with the clipper followed by shaving with an electrical shaver. Care was taken not to damage the stratum corneum. The hair-free abdominal skin was excised with surgical blade and pair of scissors, and the adhering subcutaneous fat, tissues, and capillaries were removed. The skin was cut in 3 × 3 cm samples for permeation studies.
In Vitro Drug Skin Permeation Studies
The in vitro drug skin permeation across rat skin from different formulations was assessed using Franz diffusion cell (Jain et al. Citation2005b). The PBS (pH 7.4) containing 40% v/v PEG-400 was used as the receptor medium in the diffusion cell. The skin was sandwiched between the receptor compartment and donor compartment in such a way that the dermal portion was continuously bathed with the receptor fluid maintained at 37 ± 1°C by circulating water bath and stratum corneum side exposed to ambient temperature. The content of the receptor fluid was stirred continuously using magnetic stirrer. Samples were withdrawn at different time interval, replaced with the same volume of fresh solution, filtered, and amount of drug was determined spectrophotometrically at 254 nm (Cintra 10 UV-visible spectrophotometer).
In Vivo Performance Studies
Carrageenan induced paw edema method was used to study the in vivo performance of the prepared drug delivery system. Anti-inflammatory activity was determined by measuring change in the volume of inflammed paw, produced by injection of carageenan (0.1 ml of 1% w/v) using plethysmograph. Male albino rats selected for the study were weighed and marks were made on the right hind paw just behind tibia-tarsal junction on each animal, so that every time the paw was dipped in the plethysmograph up to the fixed mark to ensure constant paw volume. Animals were divided into four groups including one control group with each group comprised of 6 animals.
On the basis of in vitro characteristics MVL-3 was chosen for in vivo studies. The formulations MVL-3, MLV-P, and celecoxib-β -cyclodextrin inclusion complex was dispersed in PBS (pH 7.4) and the dose of 10 mg/kg body weight was applied topically to albino rats of respective groups excluding the control animals. The controlled group animals were injected with saline (0.9% NaCl) containing no drug. After 30 min of topical application of formulations, 0.1 ml of 1% w/v carrageenan (in 0.9% normal saline) was injected in the subplanter region of the right hind paws. The initial reading just after injection and subsequent paw volumes was measured up to 8 hr of 1-hr intervals and after 24 hr. The percent inhibition of edema induced by carrageenan was calculated for each group using the following equation:
where Vcontrol = mean edema volume of rats in controlled group and Vtreated = edema volume of each rat in test group.
Statistical Analysis
The results were expressed as ± SD and statistically analyzed using ANOVA (analysis of variance). A probability level of p < 0.05 was considered significant.
RESULTS AND DISCUSSION
Preparation and Characterization of Celecoxib-β-Cyclodextrin Inclusion Complex
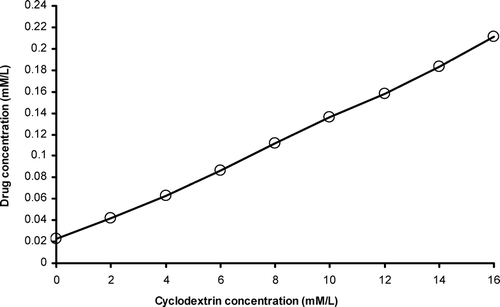
The phase solubility diagram for complex formation between celecoxib and β –CD at room temperature is presented in and clearly indicates an increase in aqueous solubility of celecoxib with an increase in β –CD concentration. The diagram obtained was of AL type according to Higuchi and Connors (Citation1965). Since the straight line had a slope, less than 1, we interpreted that the increase in solubility was observed due to the formation of a 1:1 M complex. The stability constant (KC) of celecoxib and β –CD complex (1:1) was calculated as 218.7 M–1from the linear plot of the phase solubility diagram according to the following equation:
where So is the solubility of drug in the absence of β –CD. This indicates that the celecoxib-β -cyclodextrin inclusion complex at 1:1 ratio is adequately stable.
FIG. 1 Phase solubility diagram of celecoxib with β -cyclodextrin in water at room temperature (28 ± 2°C).
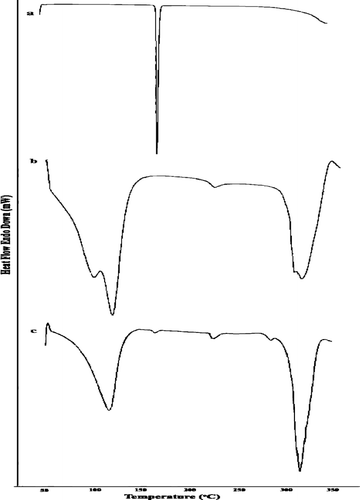
The solid complexes were prepared by the kneading method at 1:1 molar concentrations of drug and β –CD. The thermal behavior of celecoxib-β -cyclodextrin inclusion complex was studied using DSC to confirm the formation of the solid complex. DSC thermograms of celecoxib, cyclodextrin, and celecoxib solid complexes are shown in . The thermogram of celecoxib exhibited an endothermic peak, at 160.54°C corresponding to its melting point. β –CD alone showed a broad endothermic segment at 115.38°C representing a loss of water molecule. The DSC thermogram of the celecoxib-β -cyclodextrin inclusion complex shows the persistence of the endothermic peak of pure drug, giving clear evidence about the formation of complexes.
FIG. 2 DSC thermograms of (a) celecoxib, (b) β -cyclodextrin, and (c) celecoxib-β -cyclodextrin complex.
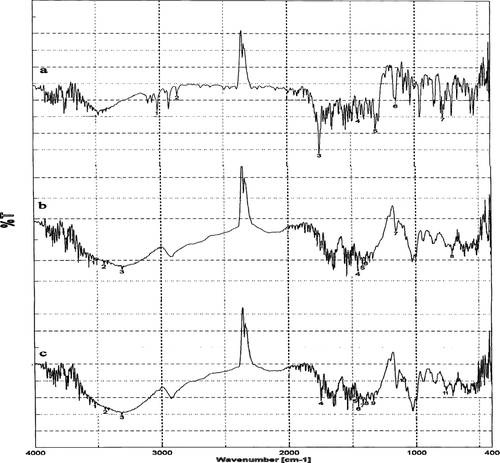
The FT-IR is a powerful technique for the physical characterization of molecules. The vibrational modes of a molecule are used to deduce structural information and also the type of polymorphism existing in drug. IR spectra of β -cyclodextrin gives characteristics peaks on OH stretch : primary OH group (3526 cm− 1), O–H stretch – secondary OH groups (3420 cm− 1), C–H bond in different ways (3296 cm− 1), C–O stretch – sec alcohol (1456 cm− 1), C–H stretch – sec alcohol (1456 cm− 1), C–C stretch in ring (1396 cm− 1), C–O–C ring stretchings (1156 cm− 1), and characteristic vibrations of β –CD (850-1640 cm− 1) The sharp decrease of the intensity of the O–H band at 3420 cm–1in the spectrum of celecoxib-β -cyclodextrin inclusion complex occurred due to total deprotonation of the hydroxyl groups in the complex. Suppression of OH vibrational modes in the 3000–3700 cm–1 region is related to evidence of host–guest interaction as a consequence of complete water release on inclusion ().
FIG. 3 Infrared spectra of (a) celecoxib, (b) β -cyclodextrin, and (c) celecoxib-β -cyclodextrin complex.
Preparation of Multivesicular Liposomes
The multivesicular liposomal formulations containing the celecoxib were prepared by a modified reverse phase evaporation method (w/o/w double emulsification technique) using an ampipathic lipid with a net neutral charge, cholesterol, a triacylglycerol, and a negatively charged lipid. Neutral oil is an integrative structural component of the corners or edges where membranes meet each other and thus stabilizes membrane boundaries (Spector et al. Citation1996). Negatively charged lipids constitute an essential component of MVL structure because they increase the interlamellar distance between successive bilayer of MVL structure, which leads to greater overall captured volume. The presence of charged lipids also reduces the likelihood of aggregation following the formation of MVLs (Kim et al. Citation1983).
Shape and Size of Vesicle
Shape and surface morphology of the prepared MVLs were evaluated by scanning electron microscopy. We observed that the bilayer forms the outermost membrane and the internal space are divided into numerous compartments by bilayer septum, and that most of the MVLs were fairly spherical in shape, and the surface of the particle showed a characteristic smoothness (). The particle size and size distribution of MVL vesicles was determined using lazer diffraction particle size analyzer (Cilas-1604, France). We found that the diameter of MLVs was ∼ 1.32 ± 1.04 μ m whereas MVLs formulations were in the range of 6.52 ± 1.54–10.42 ± 0.44 μ m as shown in .
FIG. 4 Scanning electron microscopic image of multivesicular liposome (MVL-3).

TABLE 1 Composition and characterization of various formulations
Vesicle Count
The number of liposomes/mm3 (vesicle count) was counted by hemocytometer under an optical microscope. We observed that the number of MLVs/mm3 was ∼ 142.33 ± 0.46 × 103 (). But the MVLs counts were less and showed larger size when compared with MLVs. The vesicle count was found inversely related to particle size: as the particle size of MVL increased, the vesicle count was decreased accordingly.
Percent Encapsulation
Percent encapsulation of the celecoxib in various formulations () clearly shows that only 27.68 ± 2.90% of drug encapsulation was observed with plain liposomes, whereas it increases to 62.24 ± 2.42, 70.34 ± 2.78, and 88.24 ± 3.82% in MVL-1, MVL-2, and MVL-3, respectively. The increase in percent encapsulation efficiency may be attributed to their unique structure of the vesicles. These MVL formulations contain numerous nonconcentric aqueous chambers and the aqueous-to-lipid ratio is much higher. These particles contain more than 90% water thereby making technology suitable for encapsulating hydrophilic (water soluble) drugs. The increase in encapsulation efficiency also depends on the fact that the neutral oil forms the part of corner or edges where bilayer membranes meet each other and thus stabilizes the boundaries of inner aqueous compartments. Triolein molecule having a longer acyl chain anchors the lipid bilayer membrane over a larger area thereby increasing the size of inner aqueous compartments, which in turn contributes to higher encapsulation efficiency i.e., 88.24 ± 3.82% (MVL–3).
In Vitro Drug Release
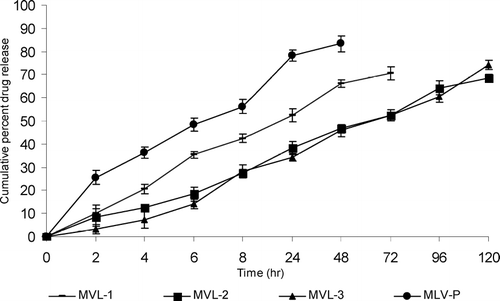
The in vitro release studies reveal that MLVs formulation release more than 80% drug within 48 hr while MVL formulations release nearly same amount of drug in 120 hr. Among the various formulations, MVL-3, consisting of triolein, releases 78.5 ± 2.84 drug within 120 hr. The MVL-2 (tricaprylin) formulation released ∼ 72.4 ± 1.47% of the drug within 120 hr and 83.3 ± 2.26% of drug release was observed with SP MVL-1 (tributyrin) formulation within 72 hr as shown in ().
FIG. 5 In vitro drug release profile of various formulations in PBS pH 7.4. Results are given as mean ± S.D. (n = 3).
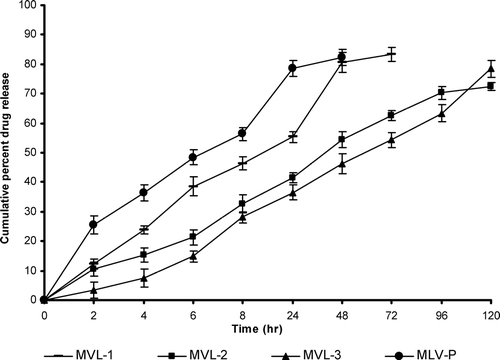
The slow release pattern of the MVL formulations as compared with the plain liposomal formulations may be attributed to the fact that there are more barriers to the diffusion of drug from the MVLs. In MLVs formulation, single breach in the external membrane results in the total release of the internal aqueous contents. But MVL formulations release drug slowly because MVLs consist of numerous aqueous compartments. Thus drug first passes through these compartment and then diffuses into the external surrounding medium. More sustained release pattern of drug from the formulation consisting of triolein could be due to the nature of the neutral oil, i.e., slow release neutral oil while tributyrin and tricaprylin belong to the category of fast release neutral oil.
Two expected mechanisms are responsible for the release of encapsulated drug from MVL. One is the coalescence of the thin film separating the aqueous chambers, and the other mechanism, “compositional ripening,” occurs without film rupture; instead it occurs by diffusion and/or permeation of the encapsulated substances. Thus in vitro release can be used for the relative ranking of the various formulations and it is clear from the above data that MVL formulation exerts a better, sustained, and more controlled in vitro drug release profile than the MLVs formulation.
In Vitro Drug Skin Permeation Studies
In vitro skin permeation studies give valuable information about the product behavior in vivo. The amount of drug permeated indictates the amount of drug available for absorption. The phosphate buffer saline (pH 7.4) containing 40% v/v PEG 400 was used as receptor fluid based on the solubility consideration of celecoxib-β -cyclodextrin inclusion complex and maintaining the sink condition.
In vitro drug skin permeation studies clearly exhibited 83.4 ± 3.43% of celecoxib-β -cyclodextrin inclusion complex penetrated through skin within 48 hr from MLV formulation, whereas MVL formulations released approximately the same amount of drug in 120 hr. MVL–3 formulations, which had triolein and slow release neutral oil, released drug slowly, thus prolonging the duration of drug permeated to 74.2 ± 2.08% until 120 hr. MVL–2 and MVL–1 with tricaprylin and tributyrin, respectively, as neutral oils showed drug permeation to 68.6 ± 1.36% in 120 hr and 70.6 ± 2.88% in 72 hr, respectively (). Thus, on the basis of skin permeation studies, we concluded that although skin permeation rate of MLV formulation was greater than MVL formulation, the MVLs bearing celecoxib:β –cyclodextrin also exhibited good permeation. The biphasic skin permeation pattern of celecoxib:β –cyclodextrin from MVLs was similar to that drug release pattern observed through cellophane membrane.
FIG. 6 In vitro drug skin permeation profile of various formulations. Results are given as mean ± S.D. (n = 3).
In Vivo Performance Evaluation
The MVLs bearing celecoxib-β -cyclodextrin inclusion complex was tested for their in vivo performance by measuring anti-inflammatory activity and comparing with drug-β –CD solution and MLV formulation. Intraperitoneal administration of celecoxib-β –CD solution (10 mg/kg body weight) to albino rats exhibited rapid onset in reduction of paw volume. It reached to 58.3 ± 2.56% reduction in 3 hr and then declined to 20.2 ± 2.24% in 8 hr. The MLVs formulation bearing celecoxib-β -cyclodextrin inclusion complex (10 mg/kg body weight) reduced the maximum paw edema to 50.2 ± 1.34% in 3 hr, which started to decline with time to 36.2 ± 2.74% in 8 hr. The MVLs bearing celecoxib-β -cyclodextrin inclusion complex reduced the maximum paw volume, which was not rapid but the action was maintained for prolonged period of time, and in 6 hr a 68.2 ± 1.68% reduction in paw volume was observed that started decline with time to 64.2 ± 1.56% in 8 hr. Even after 24 hr a 40.7 ± 3.24% reduction in paw volume was observed ().
FIG. 7 Anti–inflammatory activity profile of various formulations in carregeenen-induced rat paw edema. Results are given as mean ± S.D. (n = 6).
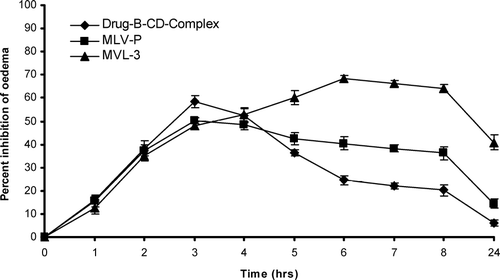
The formulation under study not only decreased the inflammation to the larger magnitude but also sustained this magnitude. Carrageenan-induced paw edema was taken as a prototype of exudative phase of inflammation. The development of oedema has been described as biphasic. The initial phase is attributable to the release of histamine, serotonin, and kinin in the first hour after injection of carrageenan. A mere pronounced second phase is related to the release of prostaglandin-like substances in 2–3 hr. The studies revealed that reduction in paw edema volume was rapid and maximum when celecoxib-β -cyclodextrin inclusion complex solution was administered intraperitoneally; MVL formulation exhibited 40.70 ± 3.24% paw edema reduction for a prolonged period of time i.e., for 24 hr. This could be due to more drug available for the edema reduction and easy elimination from the system in celecoxib-β -cyclodextrin inclusion complex administration, whereas MVL released the drug in a sustained manner for a longer period of time.
CONCLUSION
In the present work we investigated use of the combined approach of cyclodextrin complexation and entrapped this complex in MVLs to increase the skin permeation properties of celecoxib. Drug complexation with β -cyclodextrin results in significant improvement of drug dissolution properties Results of in vivo studies reveal that the MVL formulation showed more sustained and prolonged anti inflammatory effect as compared with celecoxib-β -cyclodextrin inclusion complex and MLVs formulation. MVL could provide a versatile vehicle for controlled and sustained delivery of celecoxib. MVL formulations exhibited extended duration of action. This could be due to the formation of a depot and slow diffusion of drug from the multivesicular liposomes.
ACKNOWLEDGMENTS
The authors extend their gratitude to M/s Lipoid, GmbH, Germany, for supplying HSPC, and one of the authors Mr. Yashwant Gypta is thankful to CSIR, New Delhi, India for providing financial assistance.
REFERENCES
- Alvarez-Rom'an Naik A., Kalia Y. N., Fessi H., Guy R. H. Visualization of skin penetration using confocal laser scanning microscopy. Eur. J. Pharm. Sci. 2004; 58: 301–316
- Bangham A. D., Standish M. M., Watkins J. C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965; 13: 238–252
- Gregoriadis G. Liposome Technology, vols. I–III, 2nd ed. CRC Press, Boca Raton, FL 2000; 367–398
- Higuchi T., Connors K. A. Phase solubility diagram. Adv. Anal. Chem. Instrum. 1965; 4: 117–212
- Hubbard R. C., Koep R. J., Yu S. SC-58635 (celecoxib), a novel COX-2 selective inhibitor, is effective as a treatment for osteoarthritis in a short-term pilot study (abstract). Arthr. Rheum. 1996; 39: S226, (astract 1188)
- Jain S. K., Jain R. K., Chourasia M. K., Jain A. K., Chalasani K. B., Soni V., Jain A. Design and development of multivesicular liposomal depot delivery system for controlled systemic delivery of acyclovir sodium. AAPS PharSciTech. 2005a; 6: 35–40
- Jain S. K., Chourasia M. K., Masuriha R., Soni V., Jain A., Jain N. K., Gupta Y. Solid lipoid nanoparticles bearing flurbiprofen for transdermal delivery. Drug Del. 2005b; 12: 207–215
- Katre N. V., Asherman J., Schaefer H., Hora M. Multivesicular liposome (Depo Foam) technology for the sustained delivery of insulin like growth factor-I (IGF I). J. Pharm. Sci. 1998; 87: 1341–1345
- Khatibi S., Howell S. B., McCully C. Prolongation of action in CSF by encapsulation into multivesicular liposomes. Am. Soc. Clin. Oncol. 1991; 10: 282–286
- Kim S., Turker M. S., Chi E. Y., Sela S., Martin G. M. Preparation of multivesicular liposomes. Biochim. Biophys. Acta 1983; 728: 339–348
- Kim T., Murdane S., Gruber A., Kim S. Sustained-release morphine for epidural analgesia in rats. Anesthesiology. 1996; 85: 331–338
- Kim S., Turker M. S., Chi E. Y., Sela S., Martin G. M. Preparation of multivesicular liposomes. Biochim. Biophys. Acta 1983; 728: 339–348
- Langston M. V., Rampresad M. P., Karali T. T., Galluppi G. R., Katre N. V. Modulation of the sustained delivery of myelopoietin (Leridistim) encapsulated in multivesicular liposomes (DepoFoam). J. Control. Rel. 2003; 89: 87–99
- Lasic D. D., Papadjopoulos D. Liposomes revisited. Science 1995; 267: 1275–1276
- Loftsson T., Masson M. Cyclodextrins in topical drug formulations: theory and practice. Int. J. Pharm. 2001; 225: 15–30
- Loftsson T., Masson M., Sigurdsson H. H., Magmusson P., Legoffic F. Cyclodextrins as co-enhancers in dermal and transdermal drug delivery. Pharmazie 1998; 53: 137–139
- Masson M., Loftsson T., Masson G., Stefansson E. Cyclodextrins as permeation enhancers: some theoretical evaluations and in vitro testing. J. Contr. Rel. 1999; 59: 107–118
- Matsuda H., Arima H. Cyclodextrins in transdermal and rectal delivery. Adv. Drug Del. Rev. 1999; 36: 81–99
- McCormack B., Gregoriadis G. Drugs-in-cyclodextrins-in liposomes: a novel concept in drug delivery. Int. J. Pharm. 1994; 112: 249–258
- Mumper R. J., Hoffman A. S. The stabilization and release of hirudin from liposomes or lipid-assemblies coated with hydrophobically modified dextran. AAPS PharmSciTech. 2000; 1: E3
- Ran Y., Yalkowsky S. H. Halothane, a novel solvent for the preparation of liposomes containing 2-40-amino-30-methylphenul benzothiazole (AMPB), an anticancer drug: a technical note. AAPS PharmSciTech. 2003; 4: E20
- Reddy M. N., Rehana T., Ramkrishna S., Chowdary K. P. R., Diwan P. V. β -cyclodextrin complexes of celecoxib: molecular modeling, characterization and dissoulution studies. AAPS Pharm Sci. 2004; 6: 1–9
- Simon L. S., Lanza F. L., Lipsky P. E. Preliminary study of the safety and efficacy of SC-58635, a novel cyclooxygenase-2 inhibitor. Arthr. Rheum. 1998; 41: 1591–1602
- Spector M. S., Zasadzinshi J. A., Sankaram M. B. Technology of multivesicular liposomes, a model biliquid. Foam. Langmuir 1996; 12: 4704–4708
- Takino T., Konishi K., Takakura Y., Hashida M. Long circulating emulsion carrier systems for highly lipophilic drugs. Biol. Pharm. Bull. 1994; 17: 121–125
- Verma D. D., Verma S., Blume G., Fahr A. Particle size of liposomes influences dermal delivery of substances into skin. Int. J. Pharm. 2003; 258: 141–151