Abstract
A 23 full factorial design was employed to evaluate and optimize the drug entrapment efficiency and in vitro drug release from PLGA microparticles encapsulated in a complex crosslinked alginate-pectinate matrix (polysphere). The independent formulation variables included the volume of internal and external phases, and concentration of PLGA. Surface morphology and internal structure of PLGA microparticles and polyspheres were examined by scanning electron microscopy which revealed spherical PLGA microparticles with highly porous surfaces that accounted for the rapid burst effect of this system. Texture analysis was used to profile the matrix resilience, tolerance, and energy absorbed. In vitro drug release was assessed in buffer media on PLGA microparticles and polyspheres. Polyspheres exhibited ideal zero-order release while PLGA microparticles had a burst effect followed by lag phase. Kinetic modeling of in vitro drug release data indicated that formulations were not highly dependent on polymeric erosion as a mechanism for drug release but rather diffusion. A close correlation existed between the matrix tolerance and energy absorbed. Formulations with decreased tolerance absorbed less energy, thus led to rapid surface erosion, lower matrix integrity and hence a burst effect. The converse was true for an increased matrix tolerance, which led to zero-order release supported by superior matrix integrity and a significantly reduced burst effect. The rat subcutaneous model validated in vitro release data and demonstrated that the polyspheres provided flexible yet superior rate-modulated drug delivery.
Rate-modulated drug delivery occurs when a polymer, whether natural or synthetic, hydrophobic or hydrophilic, is combined with a drug in a manner that precisely releases the drug at predetermined intervals. The drug release may be constant or rhythmic over a prolonged period, or it may be triggered in a bioresponsive manner. In either case, the objective behind modulating drug delivery is to achieve a more effective approach in treating biomedical disorders while eliminating the potential for both under- and overdosing. While these advantages can be significant, the potential disadvantages and limitations cannot be ignored (Bendix Citation1998; Vogelson Citation2001; Ruan and Feng Citation2003; Ueng et al. 2004; Pillay et al. Citation2005).
To date, attempts have been made to achieve these desirable characteristics via microencapsulation employing numerous polymers such as thermoplastic polyesters e.g. poly (lactic acid) (PLA) and poly (lactic-co-glycolic acid) (PLGA), and hydrophilic swellable polymers e.g. celluloses (alginate and pection). However, these polymeric systems uniquely possess many disadvantages (Jain Citation2000; Koch et al. Citation2003; Kong and al Moovey Citation2003; Krasaekoopt, Bhandari, and Deeth 2004; Leonard et al. 2004).
Although drug delivery products exist that utilize PLGA, one of the major obstacles that limit their application as microparticulate drug carriers is their initial burst release phase. Consequently, this initial burst effect may become an increasingly tedious challenge to researchers, especially when highly toxic drugs with narrow therapeutic windows are considered for microencapsulation. Furthermore, the consistency of product performance correlates strongly with the physicochemical and physicomechanical properties of PLGA microparticles, which in turn depends on the formulation approach (York Citation2002; Xing et al. Citation2003; You and Pent Citation2005).
In lieu of the aforementioned limitations regarding the use of PLGA microparticles as a drug delivery system, our study focused on developing an effective system to eliminate the initial burst effect and induce zero-order release. This was accomplished by incorporating PLGA microparticles into a platform consisting of a combination of cross-linked alginate and pectinate matrices referred to as “polyspheres”. Our formulation approach endeavored a primary polymeric carrier modified by encapsulation with a second polymer to achieve prolonged release as an implantable device that has the potential to overcome the erratic drug release of native PLGA microparticles. This innovative polysphere formulation is ideal for the treatment or prophylaxis of conditions that require drug delivery over prolonged periods of time and can be fabricated as an implant or tablet matrix.
MATERIALS AND METHODS
PLGA with a copolymer ratio of 85:15 (Mw = 100,000; i.v. 0.72dl/g) (Absorbable Polymers International, Pelham, AL, USA) was used as the hydrophobic biodegradable framework. Medium grade viscosity sodium alginate (200cP for 1%w/v aqueous solution at 20°C) was obtained from FMC Biopolymer (Newark, DE, USA). Zinc sulfate heptahydrate, light paraffin oil, acetonitrile, and petroleum ether were purchased from Fisher Scientific (Norcross, GA, USA). Model drug diclofenac sodium, calcium chloride (anhydrous), and phosphate buffered saline tablets (PBS) (pH 7.4) were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Pectin (28% esterified) was obtained from Herbstreith and Fox KG (Germany). All other chemicals used were of analytical grade.
Preparation of PLGA Microparticles and Polyspheres
PLGA microparticles were prepared using an oil-in-oil (0/0) emulsification-solvent evaporation method adapted from Sweet (Citation2002). Acetonitrile was used as the organic solvent and light mineral oil containing 1%w/v Span 80 as an emulsifier. In accordance with the full factorial design () specific quantitites of PLGA and drug was dissolved in acetonitrile for each formulation, followed by the addition of this solution to the continuous phase. The resulting emulsion was agitated at 400 rpm with a vortex mixer, which allowed solvent evaporation over a 24-hr period. On solvent evaporation, microparticles were filtered, collected and washed with 3× 50 mL volumes of petroleum ether and dried under vacuum.
TABLE 2 23 full factorial design template for the formulation of PLGA microparticles
For encapsulation, sodium alginate (4%w/v) and pectin (4%w/v) was added to 200 mL deionized water. The solution was magnetically stirred to facilitate dissolution of the polymers. Upon dissolution, an optimized PLGA microparticle formulation equivalent to 15 mg of diclofenac sodium was added to the solution. The cross-linking solution was prepared in deionized water (400 mL) by incorporating 2%w/v each of zinc sulfate heptahydrate, zinc gluconate, and calcium chloride. Polyspheres were formed by carefully titrating the polymer solution containing PLGA microparticles, into the cross-linking solution using a 4-channel peristaltic pump set at a rate of 2 mL/min (Labcon, MO, USA) fitted with Tygon® tubing (Fisher Scientific, PA, USA) having a flatended tip of 1/16” diameter opening.
The resultant polyspheres were then agitated in the cross-linking solution for an additional 30 min and thereafter allowed to cure at room temperature (21°C) for 24 hr. The polyspheres were then washed with 3×500 mL volumes of deionized water and dried at room temperature (21°C) for 48 hr. The surface morphology and internal structure of the PLGA microparticles and polyspheres were examined by scanning electron microscopy (SEM), (JEOL 6400, Japan) and high resolution flat bed scanning.
Experimental Design
A 23 full factorial design was developed to optimize the drug entrapment efficiency and in vitro drug release from the PLGA microparticles. The number of experimental runs that related to the tautness of the factorial matrix had to be maximized to ensure that all possible statistical combinations of polymer concentrations and phase volumes were included. This was based on the fact that the system is known to be extremely sensitive to slight changes in formulation parameters. The factor levels for the independent formulation variables (volume of internal and external phases, and concentration of polymer) are listed in .
TABLE 1 Factor levels of independent formulation variables
The factorial matrix was built using compiled Microsoft® Excel 2003 Macros (Add-Ins), namely Essential Regression and Experimental Design Version 2.2 (Gibsonia, PA, USA). The generated matrix required 10 experimental runs that are listed in . The formulations were produced and tested in a random sequence. The interactions response regression model with two centerpoints encompassing 7 linear and main effects terms is shown below:
where the terms in parentheses represent the phase volumes and the concentration of polymer, b0…b6 are the regression coefficients of the system, VE = volume of external phase, VI = volume of internal phase, and CP = concentration of polymer.
Drug Entrapment Efficiency
Drug content within PLGA microparticles was determined by dispersing 25 mg samples in 50 mL of N, N-dimethylformamide (DMF) to facilitate complete dissolution of the drug and polymer. The supernatant was filtered through a 0.45 μm membrane filter (Milli-Q System, Millipore, Bedford, USA) attached to a glass syringe and analyzed by ultraviolet spectroscopy at a wavelength maximum of 290 nm (Hewlett Packard 8453, MD, USA). The quantity of entrapped drug was calculated from a standard curve in DMF and drug entrapment efficiency was calculated using equation 2:
Where DEE = drug entrapment efficiency, AQ = actual quantity of drug entrapped in PLGA microparticles and TQ = theoretical quantity of drug entrapped in PLGA microparticles.
Drug content within polyspheres was determined by placing 200 mg of drug-loaded polyspheres in 500 mL of PBS pH 7.4 at 37°C. The matrices were magnetically stirred to promote swelling and break up of the cross-linked structure and release of diclofenac sodium. The solutions were then vacuum filtered through a 0.45 μm membrane filter and the filtrates were prepared up to 900 mL volumes with PBS pH 7.4. Samples were analyzed with ultraviolet spectroscopy (Hewlett-Packard 8453) at a wavelength maximum of 276 nm. The drug entrapment efficiency was determined using equation 3, a similar empirical relationship stated in equation 2:
where DEE = drug entrapment efficiency, AQ = actual quantity of drug entrapped in polyspheres and TQ = theoretical quantity of drug entrapped in polyspheres.
Textural Profile Analysis of Polyspheres
To determine the matrix resilience, tolerance, and energy absorbed of the polyspheres, a TA.XT plus Texture Analyzer (StableMicroSystems, UK) fitted with a 36 mm cylindrical steel probe and 5 kg loadcell was used. lists the parameter settings. To determine the effect of hydration of the polyspheres on their resilient nature, an orbital shaker operated at 50 rpm (Labline® Instruments, Ill IL, USA) was used to agitate the matrices. Polysphere samples of 300 mg were immersed in 50 mL of PBS (pH 7.4; 37°C). At predetermined time intervals, polyspheres were removed (N = 10) from the medium and analyzed in accordance with the textural parameters listed in using the fully integrated data acquisition and analysis software Texture Exponent® for Windows version 3.2, (StableMicrosystems, UK). Data acquisition was performed at 200 points per second. Typical force-distance and force-time profiles used to determine the responses are depicted in .
TABLE 3 Textural analysis parameter settings
FIG. 1 Typical textural profiles of polyspheres to determine (a) matrix tolerance and energy absorbed and (b) matrix resilience.
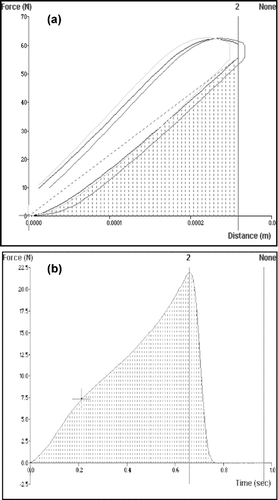
In the matrix tolerance is indicated by the steepness of the upward gradient to the peak force of 40N (namely, between anchors 1 and 2). In general, a steeper gradient indicates greater matrix tolerance. The area under the curve (AUC) of a force-time profile () indicates the energy absorbed by the matrix during analysis. The calculation of matrix resilience, provided by the ratio of the AUC's between anchors 2, 3 and 1, 2 is shown in . An anchor may be defined as a perpendicular drop to the x-axis to divide the area into several boundaries.
Constrained Optimization of PLGA Microparticles
A constrained optimization technique was used to generate the optimum setting for the PLGA microparticles using maximization of drug entrapment as the major optimization objective. In this study, optimization was undertaken using the generalized reduced gradient (GRG2) approach employing Microsoft® Excel 2003 Solver function. Maximization of drug entrapment was proposed as the most favorable parameter (response) for optimization. The following constraint boundaries (mL or mg concentration) for the independent formulation variables were applied:
25 mL ≤ mineral oil ≤ 200 mL
5 mL ≤ acetonitrile ≤ 50 mL
100 mg ≤ PLGA ≤ 2000 mg
The upper limit of 2000 mg of PLGA was feasible in terms of economic viability. The lower limit of 100 mg allowed for uniform microparticles to form. The lower and upper limits of mineral oil and acetonitrile were selected based on the induction of suitable phase separation.
In Vitro Drug Release Studies
In vitro drug release studies were performed on drug-loaded PLGA microparticles and polyspheres. Samples of 300 mg were immersed in 50 mL of PBS (pH 7.4, 37°C) in sealed 100 mL glass bottles and placed in an orbital shaker operated at 50 rpm (Labline® Instruments). Samples of 1 mL buffered media were removed at predetermined time intervals and analyzed by ultraviolet spectroscopy (Hewlett-Packard 8453) at a wavelength maximum of 276 nm. In to maintain sink conditions an equal volume of drug-free buffer was replaced into each bottle.
In Vivo Drug Release Studies
Fully matured 56 adult male rats weighing 300 g were used for in vivo studies. Rats were purchased from Harlan Sprague Dawley, (Indianapolis, Ind., USA). Seven groups of 8 rats per group were used to obtain statistically significant results with three formulations (test groups) each having a blank formulation (control groups). In addition, one formulation contained only diclofenac sodium to serve as a control. Treatment of the various groups were as follows:
Drug solution (diclofenac sodium) [Control]
PLGA microparticles (diclofenac sodium) [Test]
Blank microparticles [Control]
Polyshpheres (diclofenac sodium alone) [Test]
Blank polyspheres [Control]
Polyspheres (diclofenac sodium + PLGA) [Test]
Blank polyspheres [Control]
Each group received a single dose treatment of sterile microparticles or polyspheres implanted subcutaneously in the lower abdominal wall. The rats were anesthetized with halothane and paraffin oil (4% solution). A small incision (3 cm) was made in the lower left flank for implantation of the microparticles and closed with a surgical wound clip. An injection of buprenophrine 0.1 mg/kg subcutaneous was administered to each rat for 3 days after surgery.
Assay Procedure
A saphenous vein technique was used for blood sampling. Briefly, hair was removed from the outer hind limb and the vein was punctured with a 20-gauge needle. Samples of 0.5 mL blood was collected at predetermined time intervals, placed in heparinized tubes, and centrifuged at 4,000 × g for 15 min at 4°C. Plasma samples were then stored at -70°C until further analysis by HPLC.
For the analysis, 0.1 mL of plasma was mixed with 0.4 mL of acetonitrile solution containing ketoprofen (0.16 μ/mL), as an internal standard and centrifuged at 3000 rpm for 10 min to precipitate proteins. The supernatant (0.4 mL) was evaporated under N2(g). The residue was reconstituted in 100/μL of mobile phase and the resulting solution was analyzed by HPLC (Waters, Model 1800) equipped with a Nucleosil C18 column (Alltech, 5/μm, 15 × 0.46 cm i.d.) and PDA detector (Model 969). The mobile phase consisted of acetonitrile and water (adjusted to pH 3 with glacial acetic acid) 50:50 v/v. The eluent was monitored at 280 nm with a flow rate of 1.0 mL/min.
Pharmacokinetic Analysis
Plasma concentration-time curves of diclofenac were evaluated by noncompartmental analysis using WinNonlin® version 5 (Pharsight, Mountain View, CA, USA). Pharmacokinetic parameters such as the peak plasma concentrations (Cmax) and time of their occurrence (tmax) were read directly from the individual plasma concentration-time profiles. The area under the plasma concentration-time curve up to the last quantifiable plasma concentration (AUC) was determined according to the linear trapezoidal rule.
RESULTS AND DISCUSSION
Morphology of PLGA Microparticles
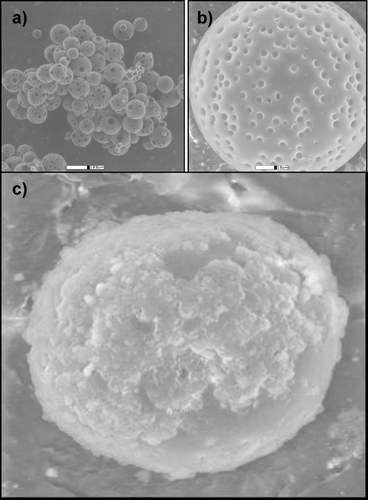
Drug release from PLGA matrices was governed mainly by diffusion through the polymeric matrix. The major mechanism contributing to diffusion and drug release was hydrolysis of PLGA under physiologic conditions. Thus, diffusion of dissolution media into PLGA matrices was a key factor in determining the overall rate of drug release from the PLGA microparticles delivery system. SEM revealed spherical PLGA microparticles with highly porous surfaces ( and ), which may account for the rapid burst effect of this system. depicts a scan of native PLGA microparticles encapsulated within the complex cross-linked alginate-pectinate matrix (polysphere).
FIG. 2 Typical scanning electron micrographs depict the porous surfaces of native PLGA microparticles (a and b) and (c) a PLGA-encapsulated alginate-pectinate polysphere (N = 10).
Regression Analysis
and indicate the DEE and dissolution profiles for the 10 formulations derived from the full factorial design. Repeated forward and backward stepwise regression was used to generate the equations for each response parameter, namely DEE and dissolution after 0.5 hr (Dt0.5) and was fitted to a first-order polynomial model to elucidate both the significant and nonsignificant terms (equations 4 and 5).
where the terms in parentheses represent the phase volumes and the concentration of polymer, b0…b6 are the regression coefficients of the system, VE = volume of external phase, VI = volume of internal phase, and CP = concentration of polymer. The correlation between experimental and predicted data obtained for each of the response parameters in relation to the specific formulations is outlined in .
FIG. 3 Profiles of (a) drug entrapment efficiency and (b) drug release of PLGA microparticles prepared according to the full factorial experimental design.
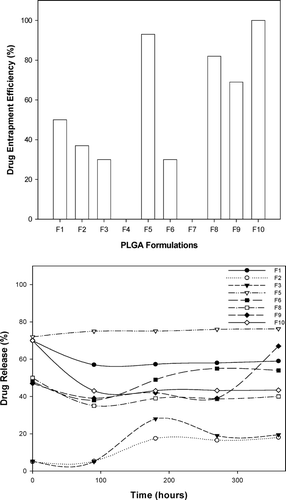
TABLE 4 Correlation between the predicted and experimental DEE and Dt0.5
It is clear that equations 4 and 5 provided a good fit for the experimental data. However, in terms of accuracy of the equations, drug entrapment efficiency was more reliable. This fact is supported by the corresponding values for the regression coefficients, precision indices, and coefficients of variation for each equation as seen in .
TABLE 5 Regression diagnostics for model fitting of DEE and dissolution
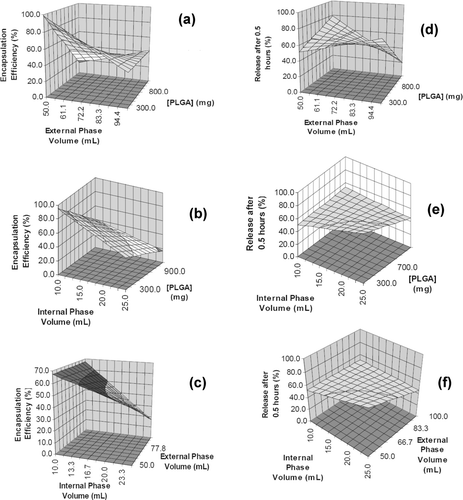
The higher regression coefficient (R) and lower coefficient of variation values indicated that the response model describing the drug entrapment efficiency was more accurate than that describing dissolution after 0.5 hr. In fact, in terms of a 90% confidence interval, only the response model describing drug entrapment efficiency possessed significant coefficients, namely the concentration of polymer and volume of external and internal phases. Based on these significant terms, it was apparent that the PLGA concentration and its interaction with the volume of the external phase had a great impact on the drug entrapment efficiency. Typical response surface plots for the effects of phase volumes and polymer concentration on the drug entrapment efficiency and the phase volumes and polymer concentration on the dissolution after 0.5 hr are shown in .
FIG. 4 Typical surface plots describing drug entrapment efficiency and dissolution after 0.5 hr in terms of: (a) PLGA concentration and external phase volume, (b) PLGA concentration and internal phase volume, (c) external phase volume and internal phase volume, (d) PLGA and external phase volume, (e) PLGA and internal phase volume and (f) internal and external phase volumes.
In Vitro Drug Release
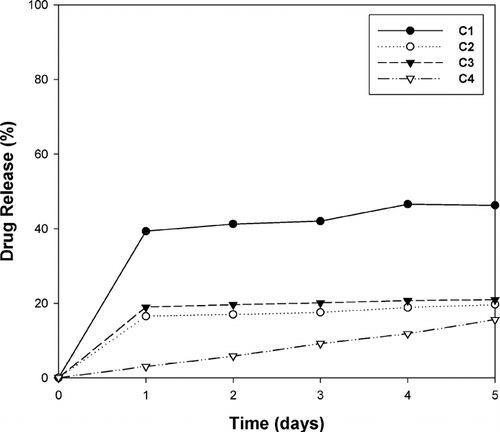
In vitro release studies exhibited significantly different profiles for the various polysphere alginate-pectinate complex formulations (). Polysphere complex C4 exhibited ideal zero-order release (R2 = 0.99), whereas native PLGA (formulation C1) behaved in an expected manner (burst effect followed by a lag phase). To elucidate the dissolution mechanism of polyspheres from a different perspective, we opted to analyze the stress-strain properties of the polysphere complexes.
FIG. 5 In vitro drug release profiles of PLGA microparticles encapsulated within cross-linked alginate-pecintate polyspheres. C1 = PLGA microparticles; C2 = calcium alginate-pectinate polysphere; C3 = zinc-calcium alginate-pectinate polysphere; C4 = calcium-zinc alginate-pectinate polysphere.
Kinetic Modeling of Release Data
Various kinetic models () were used to identify the mechanism involved during drug release from the polysphere formulations using data derived from the release studies (). All least-squares analysis to determine the release mechanism of diclofenac was performed on WinNonlin® Version 5 (Pharsight, USA) using the Guassian-Newton (Levenberg-Hartely) approach. A detailed description of the models and definitions of the pertinent statistical descriptors employed can be found elsewhere (Pillay and Fassihi 1999). indicates the results from kinetic modeling.
TABLE 6 Kinetic analysis of dissolution data
It is evident from that none of the formulations was highly dependent on erosion of polyspheres as a mechanism for drug release (low R2, high AIC, and SBC values). Drug release from the polyspheres was highly dependent on diffusion coupled with matrix relaxation and subsequent swelling, based on a comparison of the Akaike Information Criteria (AIC) and Schwartz Bayesian Criteria (SBC).
Textural Profile Analysis
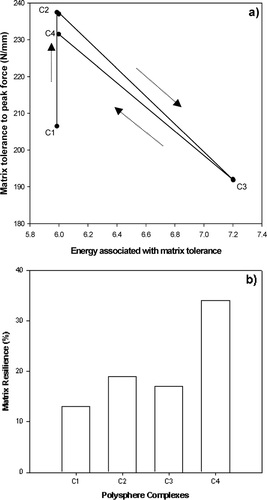
In addition to the physicochemical properties, the degree of complexation significantly altered the physicomechanical behavior of the polyspheres. In an attempt to explain the physicomechanical properties, the polyspheres were subjected to two tests. Initially the tolerance of the polysphere matrices was studied by application of a 40N force. Note that matrix tolerance is an indication of matrix integrity. It was observed that a close correlation existed between matrix tolerance and the energy absorbed to maintain the tolerance in this respect.
reveals that formulation C1 has a decreased tolerance to stress and absorbs less energy. As a result, the matrix integrity was low, thereby leading to rapid surface erosion, incomplete matrix disruption, and hence a burst effect was observed. Formulation C2 revealed an increased tolerance value. Although its energy capacity was not significantly different from formulation C1, the potential of the matrix to rupture was low. Furthermore, this theory was reinforced by the fact that formulation C4 demonstrated an increased tolerance level and energy absorbed (). Thus the maintenance of matrix integrity supported its potential for zero-order release ().
FIG. 6 Profiles of (a) textural analysis of various polyspheres, and (b) the relationship between matrix resilience and the degree of cross-linking after polysphere hydration in PBS pH 7.4. C1 = PLGA microparticles C2 = calcium alginate-pectinate polysphere, C3 = zinc-calcium alginate-pectinate polysphere; C4 = calcium-zinc alginate-pectinate polysphere.
Matrix resilience was assessed on hydrated polyspheres (). A 50% strain force was applied, i.e., the polyspheres were compressed to one-half their originl diameter. At this juncture the polysphere had the capacity to either fracture or return to its original shape and size.
The data in indicate that formulation C1 had a low resilience and consequently this enhanced the driving force behind drug release (). Formulation C2 has a higher resilience than formulation C1 which corresponds to reduced drug release. The matrix resilience of formulation C4 (35%) was significantly higher than formulations C1−3, and correspondingly formulation C4 demonstrated ideal zero-order release (i.e., due to its high resilience the matrix did not rupture and thus no burst effect was observed). Therefore according to the data, the ability of this polysphere formulation (C4) to exhibit only 12% release in 5 days may be a direct result of its increased matrix tolerance and resillience.
In Vivo Drug Release
The initial plasma concentrations of injected diclofenac sodium in pure drug solution were higher compared with those in conventional PLGA microparticles. In particular, in solution, at 1 hr, the plasma concentrations of diclofenac sodium (9–23 μg/mL) were significantly higher than those in PLGA microparticles (3–18 μg/mL). Results indicated that drug in solution may be absorbed faster when compared with conventional PLGA microparticles (). Based on the most fundamental aspects of pharmacokinetics, this was expected. However, at 24 hr the plasma concentrations of diclofenac sodium in solution were not statistically different from those in conventional PLGA microparticles. Superficially, this was not expected since the premise behind PLGA microparticle formulations is to extend and control the release rate, yet from the experimental in vitro data it would have been surprising not to see this phenomenon.
TABLE 7 Pharmacokinetic parameters for pure drug solution and PLGA microparticles
The in vivo study facilitated an understanding of the rate-modulated release properties of diclofenac sodium from polyspheres in the rat subcutaneous model. shows the change of mean plasma concentration of diclofenac sodium after subcutaneous administration of polysphere formulations. lists the pharmacokinetic parameters for the polysphere formulations. Plasma concentration-time curves of diclofenac were evaluated by non-compartmental analysis using WinNonlin® version 5 (Pharsight, USA).
FIG. 7 Plasma concentration-time profile of diclofenac sodium after subcutaneous administration of polyspheres. Each value represents the mean ± S.D. (N = 8).
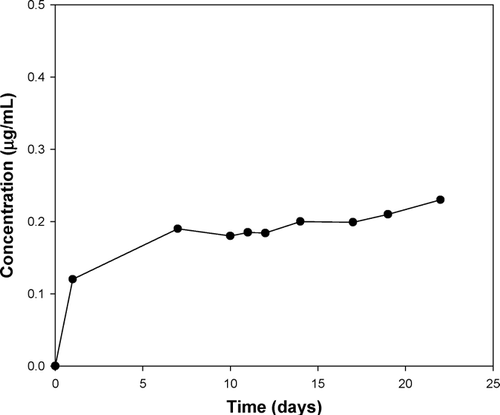
TABLE 8 Pharamockinetic parameters of polysphere formulations
CONCLUSION
The novel cross-linked polyspheres developed in this study proved to provide rate-modulated drug release with a significant reduction in the initial burst effect normally occurring with native PLGA microparticles. Kinetic modeling of polysphere in vitro release data using various relationships indicated that the complexes were not highly dependent on erosion of the polymeric system as mechanism for drug release but rather on diffusion. The application of a 23 full fatorial design served as a useful technique for the characterization and optimization of the PLGA microparticles that were encapsulated into the polyspheres. Multiple regression analysis generated polynomial equations that adequately described the influence of the selected variables at different levels on the responses investigated. Furthermore, polysphere complexes (in particular complex C4) exhibited ideal zero-order in vitro release (R2 = 0.99). From a physicomechanical viewpoint, polyspheres were attractive with regard to applications as matrices for controlled local drug delivery as matrix integrity supported their potential for zero-order release.
REFERENCES
- Bendix D. Chemical synthesis of polylactide and its capacity for medical applications. Polymer Degrad Stab 1998; 59: 129–135
- Jain. The manufacturing techniques of various drug loaded biodegradable poly(lactideo-glycolide) (PLGA) devices. Biomaterials 2000; 21(23)2475–2490
- Koch S., Schwinger C., Kressler J., Heinzen C. H., Rainov N G. Alginate encapsulation of genetially engineered mammalian cells: comparison of production devices, methods and microcapsule characteristics. J. Microencapsul. 2003; 20: 303–316
- Kong H. J., Mooney D. J. The effects of poly(ethyleneimmine) (PEI) molecular weight on reinforcement of alginate hydrogels. Cell Transplant 2003; 12(7)779–85
- Krasaekoopt W., Baandari B., Deeth H. The influene of coating materials on some properties of alginate beads and survivability of microencapsulated probiotic bacteria. Int. Dairy J. 2001; 14: 737–743
- Leonard M., De Boisseson M. R., Hubert P., Dalencon F., Dellacherie E. Hydrophobically modified alginate hydrogels as protein carriers with specific controlled release properties. J. Control, Release. 2001; 98(3)395–405
- Pillay V., Fassihi R. Electrolyte-induced compositional heterogeneity: a novel approach for rate-controlled oral drug delivery. J. Pharm. Sci. 1990; 88: 1140–1148
- Pillay V., Danckwerts M. P., Fassihi R., Muhidinov Z. Novel modulation of drug delivery using binary zich-alginate-pectinate polyspheres for zero-order kinetics over several days: experimental design strategy to elucidate the crosslinking mechanism. Drug Dev. Ind. Pharma. 2005; 31: 191–207
- Ruan G., Feng S. Preparation and characterization of poly(LA)-poly(EG)-poly(LA) (PLAPEG-PLA) microspheres for controlled release of paclitaxel. Biomaterials 2003; 24: 5037–5044
- Sweet J. Formulation and evaluation of controlled release delivery systems for the treatment of alzheimer's disease. AAPSPharmSci. 2002; vol. 4(4), abstract T3204
- Ueng S. W., Yuan I. J., Lee N., Lin S. S., Chan E. C., Weng J. H. In vivo study of biodgradable alginate antibiotic beads in rabbits. J. Orthop. Res. 2001; 22: 592–599
- Vogelson C. T. Advances in drug delivery systems. ACS Publications 2001; 4(4)49–52
- Xing L., Dawei C., Liping X., Rongqing Z. Oral colon-specific drug delivery for bee venom peptide: development of a coated calcium alginate gel beads-entrapped liposome. J. Control Release 2003; 93: 293–300
- York A. Enhanced drug delivery through novel formulation strategies. 2002, Del. CO., Report
- You J. O., Peng C. A. Calcium-alginate nanoparticles for nonviral gene delivery. Tech Proc. NSTI Nanotechnoloy Conf. Trade Show 2005; 1: 270–273