Abstract
Acyclovir, a selective antiherpes virus agent, was loaded in the hollow microspheres to improve bioavailability and patient compliance by prolonging the residence time in the gastrointestinal tract. The hollow microspheres of acyclovir were prepared by solvent evaporation diffusion method using Eudragit S 100 as a controlled polymer. We found that the process conditions that provided the high % yield of the hollow microspheres were the use of 5:8:2 of dichloromethane: ethanol: isopropanol as a solvent system and stirring at 300 rpm for 60 min. The size of the microspheres prepared from different ratios of acyclovir and Eudragit S 100 was 159–218 μm. When the drug-to-polymer ratio was increased, the size and percent drug content increased. The highest percent drug entrapment was obtained at the ratio of 600 mg acyclovir: 1 g Eudragit S 100. The hollow microspheres tended to float over 0.1 M hydrochloric acid containing 0.02% Tween 20 solution for 24 hr. The rate of acyclovir released from the microspheres was generally low in simulated gastric fluid without enzyme due to the low permeability of the polymer. However, in phosphate buffer pH 6.8, the drug release increased as the drug load increased due to the swelling property of the polymer. In simulated intestinal fluids without enzymes, the polymer completely dissolved resulting in instant release of the drug in this medium.
Controlled drug delivery systems (DDS) are aimed primarily to achieve more predictable and increase bioavailability of drugs; however, the development process is generally precluded by several physiological difficulties, such as an inability to restrain and localize the DDS within desired regions of the gastrointestinal (GI) tract and highly variable nature of gastric-emptying process. The emptying process can last from a few minutes to 12 hr depending on physiological state of the subject and design of pharmaceutical formulation. This variability may lead to unpredictable bioavailability and time to achieve peak plasma levels because the majority of drugs are preferentially absorbed in upper part of the small intestine (duodenum and jejunum) (CitationRouge et al. 1996; CitationSingh and Kim 2000). Furthermore, the relatively brief gastric emptying time in humans, which normally averages 2 to 3 hr through the major absorption zone (stomach and upper region of the intestine), can result in incomplete drug release from DDS leading to diminished efficacy of the administered of dose (CitationRubestein et al. 1989).
Therefore, control of placement of DDS in a specific region of the GI tract and prolonged gastric retention time offers numerous advantages, especially for drugs exhibiting an absorption window in the GI tract or drugs with a stability problem, resulting in increase of drug release and drug absorption in specific region. These considerations have led to the development of oral controlled release (CR) dosage forms possessing gastric retention capabilities.
During the past decade, many studies have been performed concerning the sustained release dosage forms of drug, which can prolong the gastric emptying time (GET) such as swelling-type dosage forms, bioadhesive systems, modified-shape systems, and high-density formulations. One attempt to overcome this problem is to extend the GI transit time of devices, by using floating dosage forms that have specific density lower than that of gastric fluids (1.004 g/cm3) and remain buoyant in the stomach contents. The various buoyant preparations include hollow microspheres (microballoons), granules, powders, capsules, tablets (pills), and laminated films (CitationDeshpande et al. 1996, Citation1997; CitationChien, 1997; CitationWhiteheas et al. 1998; CitationDressman 2000; CitationSingh and Kim 2000).
Many floating systems are dominated by a single-unit formulation, such as hydrodynamically balanced systems. However, one disadvantage of this formulation is that a device fails to float for an extended period of time in the GI tract so that less than the desired amount of drug may be adsorbed. As a result, it may cause high variability in bioavailability. In contrast, a multiple-unit floating system, which distributes widely throughout the GI tract, provides a longer lasting and more reliable release of drugs (CitationDeshpande et al. 1996, Citation1997; CitationWhiteheas et al. 1998; CitationSingh and Kim 2000).
The polymer microspheres are multiple-unit systems, which can be prepared by emulsion solvent diffusion method (CitationMalamataris and Avgerinos 1990; CitationKawashima et al. 1991, Citation1992, Citation1993; CitationThanoo et al. 1993; CitationCrotts and Park 1995; CitationJayanthi et al. 1995; CitationScholes 1998; CitationLee et al. 1999, Citation2000; CitationEl-Kamel et al. 2001; CitationSoppimath et al. 2001). This method has been the most widely used since it is known to be simple and able to be produced in a small batch size (CitationSwarbrick and Baylan 1994; CitationQuintanar-Guerrero et al. 1998a, Citation1998b; CitationScholes 1998; CitationMathiowitz 1999). In this method, dispersion of drug in the solution of polymer in organic solvent is stabilized in an external phase containing surfactant stabilizer. Polyvinyl alcohol (PVA) is most widely used as a surfactant stabilizer and appears to most effective for formation of microparticles (CitationMathiowitz 1999). In final stage, the microspheres are normally obtained after organic solvent evaporation and solidification of polymer droplets.
Acyclovir [9-(2-hydroxyethoxymethyl) guanine] is an acyclic nucleoside analogue of guanosine that is a potent and selective antiviral agent. Acyclovir has a relatively short plasma half-life (3 hr). When orally administered, it is slowly and scarcely absorbed from the gastrointestinal tract. The plasma concentration reaches its therapeutic level in 1.5 to 2 hr. The estimated total bioavailability of acyclovir is between 15% and 30% and decreases with increasing dose (CitationJohn and Deboral 1989; CitationAntona et al. 1994). Acyclovir is almost completely unionized and has the maximum solubility (2.5 mg/ml) at pH 7.0. Various sustained release dosage forms of acyclovir had been successfully developed, including bioadhesive microspheres for ophthalmic administration (CitationGenta et al. 1997; CitationDe Jalon et al. 2001), biodegradable microspheres for intravitreal administration (CitationConti et al. 1997; CitationHerrero-Vanrell et al. 2000), liposomes for ocular delivery system (CitationLaw and Hung 1998; CitationLaw et al. 2000), and controlled release capsules for ophthalmic administration (CitationTu et al. 2001).
Only one study reported the development of oral sustained release of acyclovir, which was designed as magnetic depot tablets dosage form. In such a system, the gastric retention time could be prolonged; however, the magnetic device could cause some problem of mucus irritation (CitationGroning et al. 1998). In this study, the hollow microspheres were developed to extend the gastric retention time of acyclovir. The hollow microspheres that can float on gastric fluids would provide more time for acyclovir absorption at the absorption zone of the intestinal region. It is expected that as the fraction of the hollow microspheres gradually pass down to the intestinal region where they are soluble, the loaded drug starts to dissolve and is absorbed into the systemic circulation. As a result, a sustained release formulation of this drug may be achieved and the drug can be efficiently delivered thereby improving absolute bioavailabilities, reducing the frequency of the drug administration, and raising patient compliance.
Eudragit S 100 is an anionic copolymerization product of methacrylic acid and methyl methacrylate and preferably used as a sustained release polymer (CitationHarris and Ghebre-Sellassie 1997; CitationLehmann 1997; CitationKibbe 2000). It is a low density polymer (0.83–0.85 g/cm3) and soluble in intestinal fluid (pH > 7). Dichloromethane (methylene chloride) is the most widely used organic solvent because it possesses high volatility (boiling point of 41°C) and can be evaporated during production, resulting in a round cavity inside microspheres. The microspheres with a round cavity, called hollow microspheres, yield low density microspheres that can float in GI fluids for longer times (CitationMathiowitz 1999; CitationSoppimath et al. 2001). In addition, the release of a drug can be controlled by using the suitable ratios of polymer to drug and the mixing conditions including equipment geometry, speed, and time.
The aim of our study was to develope multiple-unit hollow microspheres of acyclovir to prolong the release of the drug. The microspheres were prepared by o/w (oil-in-water) emulsion solvent diffusion technique, using Eudragit S 100 as a controlled release polymer. PVA was used as a surfactant stabilizer and the mixture of dichloromethane, ethanol, and isopropanol was an organic solvent in the systems for production of hollow microspheres. The effects of polymer to drug ratios on the release characteristics and physiochemical properties of the microspheres were investigated.
MATERIALS AND METHODS
Acyclovir was obtained from Resfar (Italy). An enteric polymer used was Eudragit S 100 (Rohm GmbH, Germany) and polyvinyl alcohol (Sigma, USA) was used as a surfactant stabilizer. Dichloromethane, isopropanol, and methanol were purchased from Lab-Scan Analytical Science (Ireland) and ethanol was received from Carlo Erba (USA). Distilled water was used throughout the experiments. All chemicals were pharmaceutical grade and used without further modification.
Process Conditions for Hollow Microspheres
The hollow microspheres without drug were prepared by o/w emulsion solvent evaporation diffusion method modified from the methods previously reported (CitationKawashima et al. 1992, Citation1993; CitationCrotts and Park 1995; CitationLee et al. 1999, Citation2000; CitationEl-Kamel et al. 2001; CitationSoppimath et al., 2001). Eudragit S 100 was used as a controlled release polymer. Ethanol, isopropanol, and dichloromethane were organic solvents of the system. Water containing 0.6 % w/v polyvinyl alcohol as a surfactant stabilizer was an aqueous phase. Briefly, the polymer solution was prepared using 1 g of Eudragit S 100 and 15 ml of the polymer solvents by dissolving Eudragit S 100 in the mixture of ethanol and isopropanol, and dichloromethane was added afterward. The polymer solution was mixed with 1000 ml external phase containing 0.6% w/v polyvinyl alcohol and the mixture was continuously stirred at 25 ± 0.5°C.
The formed microspheres were harvested by vacuum filtration and washed three times with 100 ml of diluted hydrochloric acid (0.01 M HCl) and 100 ml of distilled water, respectively. Finally, the harvested microspheres were dried at 50°C for 12 hr. The process variables that may affect the microsphere preparation were investigated, including polymer solvent ratio (dichloromethane: ethanol: isopropanol at 5:8:2 and 5:10:0), stirring speed (250 and 300 rpm), and stirring time (15 min, 30 min, and 1 hr).
Hollow Microspheres of Acyclovir
Hollow microspheres of acyclovir were prepared by the method described above and using 1 g of Eudragit S 100 and different amounts (100, 200, 400, 600 and 800 mg) of acyclovir. In this method, acyclovir powder was added to dichloromethane before adding to the polymer solution of ethanol and isopropanol, and subsequently mixing with 1000 ml external phase (0.6% polyvinyl alcohol in water). The process conditions (i.e., solution ratio and the stirring speed and time) that provided highest percent yields as studied previously were used in the preparation of acyclovir microspheres.
Evaluation of Hollow Microspheres of Acyclovir
Particle Size and Distribution
The particle size distribution of the microspheres was determined by a laser diffraction particle size analyzer (Mastersizer S long bed, Marvern, UK). First, 1 g of hollow microspheres was floated in 200 ml of 0.02% Tween 20 in aqueous solution and stirred at 37 ± 0.5°C. Second, particle size distribution was obtained when a laser light passed through the microspheres and then diffracted the intensity in an angular distribution. The data obtained were evaluated using volume distribution diameter (d) values of 10%, 50%, and 90% and Span values. The Span value is a statistical parameter useful to evaluate the particle size distribution and calculated applying the following equation:
Quantitative Analysis of Acyclovir Content in Hollow Microspheres
The hollow microspheres equivalent to acyclovir 30 mg were accurately weighed and dissolved in 100 ml of 0.1 N sodium hydroxide solution. The solution was stirred by a magnetic stirrer for 4 hr. Next 1 ml of the solution was transferred to 50 ml volumetric flask and diluted with 0.1 N sodium hydroxide solution to the volume. An absorbance of the solution was assayed by UV spectrophotometer (Spectronic 601, Melton Roy, USA) at wavelength 254 nm, using 0.1 N sodium hydroxide solution as a blank. An amount of acyclovir was calculated from the calibration curve. The analysis was performed in triplicate. The percentages of production yield of the hollow microspheres, percentages of drug content, percentages of theoretical content, and percentages of drug entrapment were calculated from the following equations:
Floating Ability
First 50 mg of the hollow microspheres were placed in 50 ml beaker. Second, 20 ml of 0.1 M HCl containing 0.02% Tween 20 were added and stirred with a magnetic stirrer at 37 ± 0.5°C by using a controlled temperature water bath. Floating microparticles were collected at 1, 2, 4, 8, 12, and 24 hr. The experiments were performed in triplicate and the percentage floating of the hollow microspheres was calculated from the following equation:
Microscopic Studies
The microscopic appearance of the hollow microspheres was observed under optical and scanning electron microscopes (Jeol JSM-5410 LV, Tokyo, Japan). The scanning electron microscope photomicrographs were taken at 15 kV in various magnifications appropriate to each formulation.
Differential Scanning Calorimetry (DSC)
The thermal properties of hollow microspheres, acyclovir powder, and Eudragit S 100 were evaluated by differential scanning calorimetry (Perkin-Elmer DSC 7, USA). Thermograms of acyclovir, polymer (Eudragit S 100), and hollow microspheres were obtained with 5°C/min of heating rate between the temperature range of 50°C to 280°C.
Powder X-Ray Diffraction
The X-ray diffraction analysis for pure drug, polymer, and hollow microspheres was performed by using power X-ray diffractometer (Jeol JDX-8030, Tokyo, Japan). The X-ray diffraction patterns were recorded using Ni-filtered Cu-radiation generated at 45 kV and 35 mA as X-ray source. The measurement conditions were 0.04 degree-step angles, 1.5 sec/count time over range of 5° to 45°(2θ).
Fourier Transform Infrared (FTIR) Spectroscopy
The IR spectra of the hollow microspheres obtained from Fourier-transform infrared spectroscope (Perkin-Elmer 1760x, USA) were compared with those of acyclovir crystal, Eudragit S 100 powder, and their physical mixture (1:1). The potassium bromide (KBr) disc was prepared and scanned from 4000 to 400 cm− 1.
In Vitro Drug Release
The dissolution of acyclovir from the hollow microspheres was carried out using USP Dissolution Apparatus II, paddle method at 37 ± 0.5°C for 24 hr. Dissolution media were 900 ml of simulated gastric fluid, simulated intestinal fluid, and phosphate buffer pH 6.8. A weight of the hollow microspheres corresponding to 1 g of acyclovir was dispersed in each medium. The speed of stirrer was maintained at 100 rpm. At an appropriate interval, 5 ml of samples were withdrawn and replaced with an equivalent volume of fresh dissolution medium to maintain the volume of dissolution medium to be constant. The sample solution was filtrated through 0.45 μ m cellulose acetate membrane and analyzed for the concentration of acyclovir by UV spectrophotometer at wavelength 254 nm. The amounts of acyclovir released were calculated from the calibration curve of the same dissolution medium.
Data Analysis
The data of two sets were evaluated by pair t-test, and p values of less than 0.05 (p < 0.05) were considered to be a significant difference. For more than two sets of the data, comparison was performed by LSD, a type of one way analysis of variance (ANOVA) test, and p values of less than 0.05 (p < 0.05) were considered to be a significant difference.
RESULTS AND DISCUSSION
Effect of Process Variables
Although the solvent evaporation process is conceptually simple, a large number of process variables exist that can affect the nature of the obtained product (CitationMathiowitz 1999). In this study, effects of process variables, i.e., solvent ratio, stirring rate, and speed, on the yield and particle size of the hollow microspheres without drug were first studied by using 1 g of Eudragit S 100 to prepare the hollow microspheres. The effects of solvent ratio on the percentages of production yield and the size of the hollow microspheres are shown in . When the ethanol and dichloromethane with or without isopropanol were used, the percentages of production yields of microspheres were 95.04% and 91.79%, respectively. Average sizes of microspheres using 5:8:2 and 5:10:0 dichloromethane: ethanol: isopropanol were 154 μ m and 156 μ m, respectively. The two solvent systems yielded the microspheres with an equal size (p > 0.05); however, higher percentage of production yield was obtained when using 5:8:2 dichloromethane: ethanol: isopropanol (p < 0.05). Since isopropanol diffused into the aqueous phase slower than ethanol, an addition of isopropanol provided more time for droplet formation and hardening of the hollow microspheres. Thus the yield of microspheres was improved (CitationLee et al. 1999, Citation2000).
TABLE 1 Effects of process variables on percentage of production yield and size of the hollow microspheres without drug prepared by using 1 g of Eudragit S 100 (n = 3)
The effects of stirring speed on the percentages of production yield of the hollow microspheres also are shown in . When the stirring speed was increased from 250 to 300 rpm, the mean particles size of microspheres decreased from 202 μ m to 155 μ m whereas the percentages of production yield of the hollow microspheres increased from 91.04% to 94.01%. Therefore, an increase in stirring rate decreased the droplet size of the microspheres. This result was similar to the study by CitationLee et al. (1999), which reported that the particle size of ketoprofen-loaded hollow microspheres reduced with higher agitation speed.
The effects of stirring time on the percentages of production yield of the hollow microspheres can be observed in . When the hollow microspheres were prepared at 15, 30, and 60 min stirring time, the total production yield were 90.25%, 92.57%, and 94.56%, respectively. The mean particle sizes of microspheres prepared by using 15, 30, and 60 min stirring time were 162 μ m, 158 μ m, and 163 μ m, respectively. The longer stirring time provided enough time for the solvents to diffuse and thus improved the yield of the hollow microspheres. Therefore, the highest percentage of production yield of the hollow microspheres was obtained when using 60 min stirring time.
From this investigation, we found that optimized conditions for the preparation of the hollow microspheres were the use of 5:8:2 dichloromethane: ethanol: isopropanol as a solvent system, 300 rpm stirring speed, and 60 min stirring time.
Evaluation of the Hollow Microspheres of Acyclovir
Particle Size
The mean diameters of the obtained hollow microspheres of acyclovir are presented in . Theoretically, the size of the hollow microspheres increases when drug-to-polymer ratio increases due to the higher viscosity of the internal phase when increasing the drug concentration (CitationMalamataris and Avgerinos 1990). As expected, when acyclovir was increased from 100, 200, 400, 600, and 800 mg per 1 g of Eudragit S 100, the size of microspheres increased from 159 μ m, 186 μ m, 194 μ m, 201 μ m, and 217 μ m, respectively. This result was in agreement with the previous discussion. In addition, the narrow size distribution of all microspheres was indicated by the low span values, except for the microspheres produced by using 100 mg acyclovir.
TABLE 2 Particle size analysis of the hollow microspheres prepared by using different amounts of acyclovir
Quantitative Analysis of Acyclovir Content and Yield
Total production yield, drug content, and entrapment efficiency of the hollow microspheres prepared at different drug-to-polymer ratios are presented in . The total percentage yields of hollow microspheres prepared using 100, 200, 400, 600, and 800 mg of acyclovir and 1 g of Eudragit S 100 and determined by weighing after drying were 93.39%, 87.99%, 78.99%, 76.25%, and 66.11%, respectively. The percentage yield decreased with an increase in drug-to-polymer ratio. This effect agreed with that reported by CitationEl-Kamel et al. (2001), which indicated that the percentage yield of ketoprofen hollow microspheres was reduced with increased of ketoprofen content. However, the percentages of drug content of 100, 200, 400, 600, and 800 mg were 6.93%, 13.39%, 24.31%, 37.35%, and 37.85%, respectively. It can be seen that the percentage of drug content increased rapidly with an increase in acyclovir content from 100 mg to 400 mg, and then slowly increased from 600 mg to 800 mg.
TABLE 3 Total production yield, drug content, theoretical content, and drug entrapment of acyclovir hollow microspheres
Furthermore, the percentages of the entrapment efficiencies of 100, 200, 400, 600, and 800 mg acyclovir were 76.24%, 80.32%, 85.12%, 99.57%, and 85.17%, respectively. The percentage of drug entrapment also increased with an increase in acyclovir content except for the formulation of highest amount of acyclovir used (800 mg). In the last formulation, the percentage of entrapment efficiency was reduced as compared with microspheres prepared by using 600 mg acyclovir. This resulted from insufficient polymer for entrapment of increased amount of the drug. Therefore, the optimum condition of the drug and polymer ratio was 600 mg acyclovir: 1 g Eudragit S 100 which provided the highest percentages of drug entrapment and production yield.
Floating Ability
The floating test was carried out to investigate the floating ability of the prepared microspheres. The microspheres were spread over the surface of an acid medium and the fraction of the microspheres settled down as a function of time was quantified. As shown in , the percentage of all hollow microspheres floating on the 0.1 M HCl containing 0.02 % Tween 20 was reduced almost linearly from initial time to 24 hr. The result also showed a tendency that an increase in acyclovir content resulted in shorter floating time. This may be because the high concentration of drug in the microspheres increased the density of microspheres and reduced the floating time. However, more than 50% of the hollow microspheres of all formulations were able to float for longer than 24 hr.
FIG. 1. Percentages of floating ability from different formulations of the hollow microspheres floated in the 0.1 M hydrochloric acid solution containing 0.02% Tween 20.
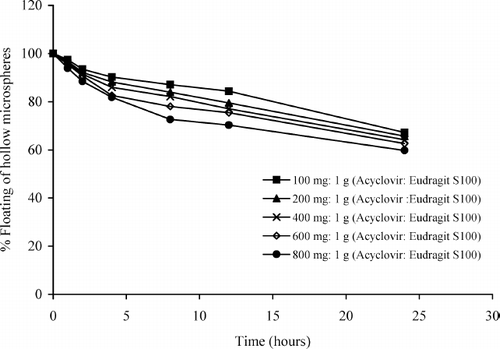
Therefore, all hollow microspheres showed an ability to float on acid medium up to 24 hr which their fraction decreased almost linearly up to 24 h. From the results, we assumed that the microspheres of acyclovir could be floated in gastric fluid and the fraction of these microspheres could gradually pass down to the intestinal region for extended duration.
Morphology
The scanning electron micrographs of the hollow microspheres of various amounts of acyclovir are shown in . We can see that all microspheres were spherical in shape without pores on the surface. An internal round cavity in the hollow microspheres formed during the solvent evaporation diffusion process could be evidently seen. Although the surfaces of the hollow microspheres were relatively smooth, needle-like drug crystals were clearly observed. The drug crystals were found in greater extent on the surface when acyclovir content was increased (400, 600, and 800 mg acyclovir).
FIG. 2. Scanning electron micrographs of the microspheres prepared by using acyclovir:Eudragit S 100 at (1) 100 mg:1 g; (2) 200 mg:1 g; (3) 400 mg:1 g; (4) 600 mg:1 g; and (5) 800 mg:1 g, showing (a) spherical shape particle, (b) hollow structure, and (c) drug crystal on the surface.

Differential Scanning Calorimetry
The DSC thermograms of the microspheres, powdered Eudragit S 100, and powdered acyclovir are shown in . Eudragit S 100 () did not show any endothermic peak, as similar to that reported by CitationEl-Kamel et al. (2001). However, the DSC thermogram of acyclovir () showed a small endothermic peak at 171°C, followed by a sharp melting peak at 242°C. The DSC thermogram obtained from this study slightly differed from the studies by CitationGenta et al. (1997) and CitationKristl et al. (1996) indicating that the DSC curve of acyclovir exhibited a small melting endothermic peak at approximately 170°C and a sharp melting peak at 252°C. This result may be due to the differences in experimental conditions and instruments.
FIG. 3. The DSC thermogram of the hollow microspheres prepared by using acyclovir:Eudragit S 100 at (a) 100 mg:1 g; (b) 200 mg:1 g; (c) 400 mg:1 g; (d) 600 mg:1 g; and (e) 800 mg:1 g, compared with the DSC thermograms of (f) Eudragit S 100 powder and (g) acyclovir crystal.
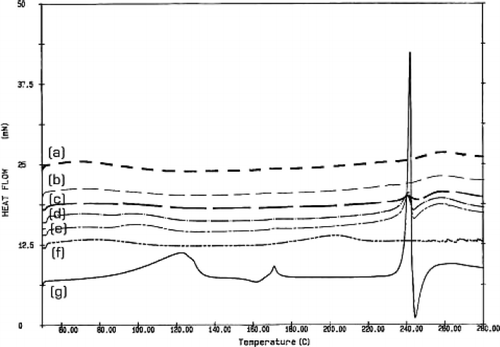
The DSC thermograms of the microspheres prepared by using 400, 600, and 800 mg of acyclovir () showed a small endothermic peak at 242°C, similar to that of acyclovir powder. Therefore, we believed that acyclovir would disperse as drug crystal in polymer matrix, as confirmed by observing drug crystal on outer shell in the scanning electron micrographs presented in . For the hollow microspheres produced from 100 and 200 mg of acyclovir, their DSC thermograms () did not show the endothermic peak of acyclovir at 242°C. This may be because acyclovir in the polymer matrix was relatively low so that its endothermic peak could not be detected in the DSC thermograms. However, the drug could be a crystalline characteristic in the polymer matrix. The results agreed well with the observation from scanning electron micrograph (), which showed that the surface of the microspheres prepared using 100 and 200 mg acyclovir was smooth and drug particles were hardly seen on the surface.
Powder X-Ray Diffraction
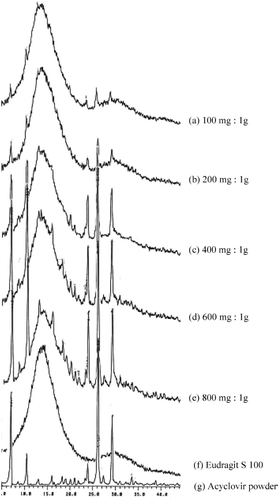
The X-ray powder diffractometry is widely used for identification of solid phases. The powder X-ray pattern of every crystalline form of a compound is unique (CitationBrittan 2000). In this study, the X-ray patterns of the pure drug and the polymer are shown in . The X-ray pattern of Eudragit S 100 showed a halo pattern () indicating its existence in amorphous form, whereas that of acyclovir exhibited crystalline characteristics, as shown in identical to that reported by CitationGenta et al. (1997). The X-ray patterns of microspheres prepared by using different amounts of acyclovir can be seen in ), which showed a combined pattern of those of the polymer and the pure drug. The intensity of these diffraction peaks increased with increasing the ratio of drug-to-polymer in microspheres. These results indicated that acyclovir was present as drug crystal in the polymer matrix.
FIG. 4. The X-ray patterns of the hollow microspheres prepared using acyclovir:Eudragit S 100 at (a) 100 mg:1 g; (b) 200 mg:1 g; (c) 400 mg:1 g; (d) 600 mg:1 g; and (e) 800 mg:1 g, compared with the X-ray patterns of (f) Eudragit S 100 powder and (g) acyclovir crystal.
FTIR Spectroscopy
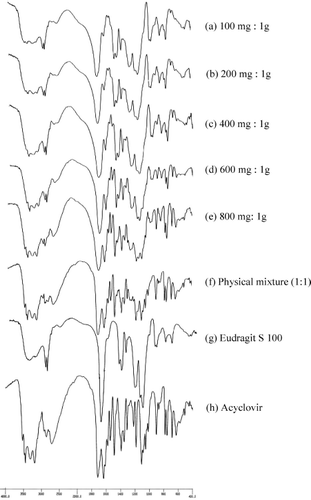
The FTIR spectroscopy has been known as a tool for drug identification. The spectra of the hollow microspheres prepared by using different amount of acyclovir were compared with those of Eudragit S 100 powder, acyclovir powder, and the physical mixture (1:1). The FTIR spectra of Eudragit S 100 powder and acyclovir powder are presented in , respectively. The spectrum of Eudragit S 100 showed its principle peaks at wavenumbers 2954, 1733, 1265, 1194, and 1156 cm− 1 and that of acyclovir had peaks at wavenumbers 3442, 3186, 2713, 1714, 1633, 1486, 1390, 902, 781, 753, and 683 cm− 1. Comparing the spectra of the hollow microspheres and the physical mixture of 1:1 acyclovir (), the IR spectra of the hollow microspheres were identical with those of Eudragit S 100 and acyclovir powders. The results indicated no interaction between Eudragit S 100 and acyclovir.
FIG. 5. The FTIR spectra of the hollow microspheres prepared by using acyclovir: Eudragit S 100 at (a) 100 mg:1 g; (b) 200 mg:1 g; (c) 400 mg:1 g; (d) 600 mg:1 g; and (e) 800 mg:1 g, compared with the FTIR spectra of (f) physical mixture (1:1); (g) Eudragit S 100 and (h) acyclovir.
In Vitro Drug Release Study
The dissolution of the hollow microspheres prepared with different amounts of acyclovir was studied in pH 1.2 simulated gastric fluid (SGF) without enzyme, pH 6.8 phosphate buffer, and pH 7.2 simulated intestinal fluid (SIF). The dissolution profiles of acyclovir hollow microspheres prepared by using different amounts of acyclovir (100, 200, 400, 600, and 800 mg) in SGF (pH 1.2) are shown in . The release of drug from the hollow microspheres was generally low (no more than 41 %) in SGF. This is due to the fact that Eudragit S 100 has a low permeability and is insoluble in acid medium because it has high intermolecular attraction (hydrogen bonding) between the hydroxyl groups of carboxylic moiety and oxygen of ester groups increases the degree of compactness of the polymer and decreases its porosity and permeability (CitationEl-Kamel et al. 2001).
FIG. 6. Dissolution profiles of the hollow microspheres containing different ratios of acyclovir:Eudragit S 100 in simulated gastric fluid pH 1.2.
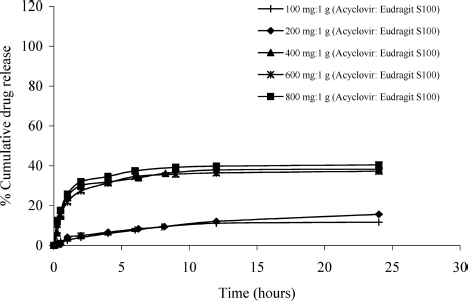
In this study, the cumulative drug release of the hollow microspheres containing 100 and 200 mg acyclovir were found to be only 11.62% and 15.59%, respectively, after 24 hr. In those formulations, the microspheres were nearly coated with polymer and the slow drug release was achieved, due to the difficultly of drug diffusion through the polymer matrix. However, the hollow microspheres containing higher amounts of acyclovir (400, 600, and 800 mg) had 38.24%, 39.01%, and 40.46% of cumulative release, respectively, after 24 hr. In such formulations, the drug was in higher concentration in the microspheres and the drug crystal on the surface of these hollow microspheres resulted in higher release of drug into the acid medium.
The dissolution profiles of acyclovir hollow microspheres prepared by using different amounts of acyclovir (100, 200, 400, 600, and 800 mg) in phosphate buffer (pH 6.8) are shown in . After 24 hr, the release of drug from the hollow microspheres containing 100, 200, 400, 600, and 800 mg acyclovir reached 10.30%, 32.62%, 65.18%, 87.51%, and 91.15%, respectively. In this pH medium, Eudragit S 100 swelled and the drug could diffuse through the highly permeable polymer easier than in the acid medium. Moreover, acyclovir dissolved in this pH more than in acid medium and exited mostly in unionized form. As a result, the percentages of cumulative drug release from all formulations increased except for the hollow microspheres containing 100 mg acyclovir, which showed similar percentage of cumulative drug release as that in SGF.
FIG. 7. Dissolution profiles of the hollow microspheres containing different ratios of acyclovir:Eudragit S 100 in phosphate buffer pH 6.8.
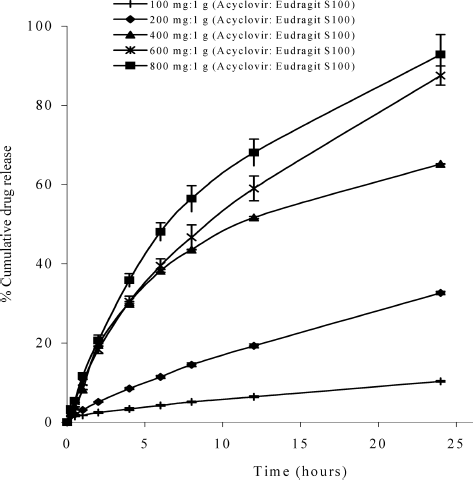
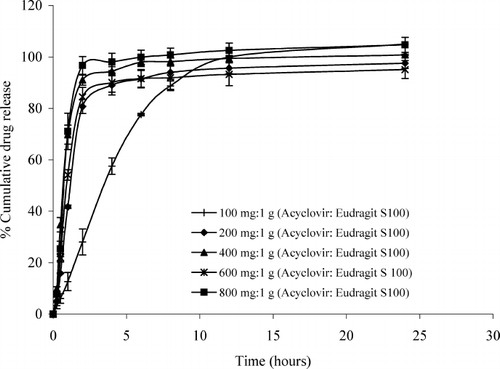
The dissolution profiles of acyclovir hollow microspheres prepared by using different amounts of acyclovir (100, 200, 400, 600, and 800 mg) in SIF (pH 7.2) are shown in . The release of drug from the hollow microspheres containing 100, 200, 400, 600, and 800 mg acyclovir reached 104.99%, 97.59%, 100.76%, 99.39%, and 104.69%, respectively, after 24 hr. In this medium, the polymer completely dissolved; therefore, the drug readily released from the hollow microspheres.
FIG. 8. Dissolution profiles of the hollow microspheres containing different ratios of acyclovir:Eudragit S 100 in simulated intestinal fluid pH 7.2.
CONCLUSION
The hollow microspheres of acyclovir prepared by emulsion solvent evaporation using optimal process conditions (5:8:2 dichloromethane: ethanol: isopropanol as a solvent system, 300 rpm stirring speed, and 60 min stirring time) could be obtained successfully. The hollow microspheres had a size range between 159–218 μ m. All hollow microspheres floated on acid medium up to 24 hr and the fraction of them was almost linearly reduced up to 24 hr, indicating a tendency to float in the stomach and to increase the GI retention time. The SEM micrographs revealed hollow structure of the microspheres, and the DSC, X-ray diffraction, and FTIR results indicated that acyclovir dispersed in crystalline form in the matrix of Eudragit S 100 without drug-polymer interaction. The microspheres showed a low release rate of drug in SGF (pH 1.2) due to the low permeability of the polymer. In phosphate buffer pH 6.8, the polymer swelled and the drug could diffuse into the medium resulting in an increase in the percentages of cumulative drug release. In SIF (pH 7.2), the polymer was completely dissolved and the drug freely released into the medium.
In summary, our results led to the conclusion that the hollow microspheres of acyclovir developed in this study could be floated in the acid medium with linearly reduction during 24 hr. As the fraction of the microspheres passed down the higher pH region (pH 6.8), the drug release depended on the drug loaded, and all drug released completely when the microspheres reached the pH 7.2 region.
The authors are grateful for financial support received from Mahidol University Research Grant.
REFERENCES
- J. W. Antona, F. Diana, and L. G. Karen. (1994). Acyclovir: a reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic efficiency. Drugs 47:153–205.
- H. G. Brittan. ( Ed.). (2000). Physical Characterization of Pharmaceutical Solids, vol. 70, 1–424. New York: Marcel Dekker.
- Y. Chien. (1997). Oral drug delivery and delivery systems. In Novel Drug Delivery Systems, ed. Y. Chien., 139–196. New York: Marcel Dekker.
- B. Conti, C. Bucolo, C. Giannavola, G. Puglisi, P. Giunchedi, and U. Conte. (1997). Biodegradable microspheres for the intravitreal administration of acyclovir: in vitro/in vivo evaluation. Eur. J. Pharm. Sci. 5:287–293.
- G. Crotts, and T. G. Park. (1995). Preparation of porous and nonporous biodegradable polymeric hollow microspheres. J. Control. Rel. 35:91–105.
- E. G. De Jalon, N. J. Blanco-Prieto, P. Ygartua, and S. Santoyo. (2001). Topical application of acycovir-loaded microparticles: quantification of the drug in porcine skin layers. J. Control. Rel. 75:191–197.
- A. A. Deshpande, C. T. Rhoes, N. H. Shah, and A. W. Malick. (1996). Controlled-release drug delivery systems for prolonged gastric residence: an overview. Drug Dev. Ind. Pharm. 22:531–539.
- A. A. Deshpande, N. H. Shah, C. T. Rhodes, and W. Malick. (1997). Development of a novel controlled-release system for gastric retention. Pharm. Res. 14:815–819.
- J. B. Dressman. (2000). Gastrointestinal variables. In Oral Absorption: Prediction and Assessment. J. B. Dressman, and H. Lennerias., 1–17. New York: Marcel Dekker.
- A. H. El-Kamel, M. S. Sokar, S. S. Gamal, and V. F. Naggar. (2001). Preparation and evaluation of ketoprofen oral delivery system. Int. J. Pharm. 220:13–21.
- I. Genta, B. Conti, P. Perugini, F. Pavanetto, A. Spadaro, and G. Puglosi. (1997). Bioahesive microspheres for ophthalmic administration of acylclovir. J. Pharm. Pharmacol. 49:737–742.
- R. Groning, M. Berntgen, and M. Georgarakis. (1998). Acyclovir serum concentrations following peroral administration of magnetic depot tablets and the influence of extacorporal magnets to control gastrointestinal transit. Eur. J. Pharm. Biopharm. 46:285–291.
- M. R. Harris, and I. Ghebre-Sellassie. (1997). Aqueous polymeric coating for modified release oral dosage forms. In Aqueous Polymeric Coating for Pharmaceutical Dosage Forms. J. Mcginity., pp.81–100. New York: Marcel Dekker.
- R. Herrero-Vanrell, L. Ramiez, A. Fernandez-Carballido, and M. F. Refojo. (2000). Biodegradable PLGA microspheres loaded with ganciclovir for intraocular administration, encapsulation technique, in vitro release profiles and sensitization process. Pharm. Res. 17:1323–1328.
- G. Jayanthi, S. B. Jayaswal, and A. K. Srivastave. (1995). Formulation and evaluation of terfenadine microballoons for oral controlled release. Pharmazie 50:769–770.
- J. O. John, and M.-C. R. Deboral. (1989). Acyclovir: an update review of its antiviral activity. Drugs 37:233–309.
- Y. Kawashima, T. Iwamoto, T. Niwa, H. Takeuchi, and T. Hino. (1993). Role of the solvent-diffusion-rate modifier in a new emulsion solvent diffusion method for preparation of ketoprofen microspheres. J. Microencapsul. 10:329–340.
- Y. Kawashima, T. Niwa, H. Takeuchi, T. Hino, and Y. Itoh. (1991). Preparation of multiple unit hollow microspheres (microballlons) with acrylic resin containing trailast and their drug release characteristics (in vitro) and floating behavior (in vivo). J. Control. Rel. 16:279–290.
- Y. Kawashima, T. Niwa, H. Takeuchi, T. Hino, and Y. Itoh. (1992). Hollow microsphees for use as a floating controlled drug delivery system in stomach. J. Pharm. Sci. 81:135–140.
- A. H. Kibbe. ( Ed.). (2000). Handbook of Pharmaceutical Excipients, 3rd ed., London: American Pharmaceutical Association and Pharmaceutical Press.
- A. Kristl, S. Srcic, F. Vrecer, B. Sustar, and D. Vojnovic. (1996). Polymorphism and pseudomorphism: influencing the dissolution properties of the quanine derivative acyclovir. J. Control. Rel. 39:231–235.
- S. L. Law, K. J. Huang, and C. H. Chiang. (2000). Acyclovir-containing liposomes for potential ocular delivery cornea penetration and absorption. J. Control. Rel. 63:135–140.
- S. L. Law, and H. Y. Hung. (1998). Properties of acyclovir-containing liposomes for potential ocular delivery. Int. J. Pharm. 16:253–239.
- J. H. Lee, T. G. Park, and H. K. Choi. (1999). Development of oral drug delivery system using floating microspheres. J. Microencapsul. 16:715–729.
- J. H. Lee, T. G. Park, and H. K. Choi. (2000). Effect of formulation and process variables on the characteristics of microspheres for water-soluble drugs prepared by w/o/o double emulsion solvent diffusion method. Int. J. Pharm. 196:75–83.
- K. O. Lehmann. (1997). Chemistry and application properties of polymethacrylate coating systems. In Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms, ed. J. W. Mcginity., 101–176. New York: Marcel Dekker.
- S. Malamataris, and A. Avgerinos. (1990). Controlled release indomethacin microspheres prepared by using emulsion solvent diffusion technique. Int. J. Pharm. 62:105–111.
- E. Mathiowitz. (1999). Encyclopedia of Pharmaceutical Technology, vol. 2, 493–546. New York: Marcel Dekker.
- D. Quintanar-Guerrero, E. Allemann, E. Doelker, and H. Fessi. (1998a). Preparation and characterization of nanocapsules from preformed polymers by a new process based on emulsification-diffusion technique. Pharm. Res. 15:1056–1062.
- D. Quintanar-Guerrero, E. Allemann, H. Fessi, and E. Doelker. (1998b). Preparation techniques and mechanisms of formation of biodegradable nanoparticles from preformed polymers. Drug Dev. Ind. Pharm. 24:1113–1128.
- N. Rouge, P. Buri, and E. Doelker. (1996). Drug absorption sites in the gastrointestinal tract and dosage forms for site-specific delivery. Int. J. Pharm 136:117–139.
- A. Rubestein, H. Vincent, K. Li, P. Gruber, and J. R. Robinson. (1989). Gastrointestinal physiological variables affecting the performance of oral sustained release dosage forms. In Oral Sustained Release: Design and Evaluation. ed. A. Yacobi, and E. H. Walega. 125–156. New York: Pergamon Press.
- S. Scholes., (1998). Production technique for biodegradable microspheres. In Controlled Drug Delivery: Challenge and Strategies, ed. 89–91. New York: American Chemical Society Press.
- B. N. Singh, and K. H. Kim. (2000). Floating drug delivery systems: an approach to oral controlled drug delivery via gastric retention. J. Control. Rel. 63:235–259.
- K. S. Soppimath, A. R. Kulkarni, and T. M. Aminabhavi. (2001). Development of hollow microspheres as floating controlled-release systems for cardiovascular drugs: preparation and release characteristics. Drug Dev. Ind. Pharm. 27:507–515.
- J. Swarbrick, and J. C. Baylan. ( Eds.). (1994). Enclyclopedia of Pharmceutical Technology, vol. 10, 1–29. New York: Marcel Dekker.
- B. C. Thanoo, M. C. Sunny, and A. Jayakrishnan. (1993). Oral sustained release drug delivery systems using polycarbonate microspheres capable of floating on the gastric fluid. J. Pharm. Pharmacol. 45:21–24.
- J. Tu, L. Wang, J. Yang, H. Fei, and X. Li. (2001). Formation and pharmacokinetic studies of acyclovir controlled release capsule. Drug Dev. Ind. Pharm. 27:687–692.
- L. Whiteheas, J. T. Fell, J. H. Collett, H. L. Sharma, and A. M. Smith. (1998). Floating dosage forms: an in vivo study demonstrating prolonged gastric retention. J. Control. Rel. 55:3–12.