Abstract
Etoposide-loaded nanoparticles were prepared using nanoprecipitation and emulsion solvent evaporation techniques using polylactide-co-glycolic acid and poly(ε -caprolactone) in presence of Pluronic F68, respectively. Effect of formulation variables like stabilizer concentration, amount of polymer, and drug was studied. These parameters were found to affect particle size, zeta potential, drug content, and entrapment efficiency of nanoparticles. The methods produced nanoparticles with good entrapment efficiency of around 80%. Recovery of nanoparticles was as high as 95% and drug content was around 1.5%. Increase in lactide content decreased the release of etoposide in vitro and poly(ε -caprolactone) nanoparticles retarded etoposide release for 48 hr. The results show the suitability of polylactide-co-glycolic acid and poly(ε -caprolactone) nanoparticles as potential carriers for controlled delivery of etoposide.
Keywords:
In the recent years, significant effort has been devoted to develop nanotechnology for drug delivery since it offers a suitable means of delivering small as well as high molecular weight drugs, proteins, peptides, or genes to cells and tissue. Nanoparticles represent very promising delivery systems for the encapsulation and effective delivery of poorly soluble drugs. Optimization of existing techniques in combination with new methods using biodegradable polymeric carriers enabled them to emerge as suitable delivery systems with increased acceptance and potential (CitationKumar and Kumar 2001). Nanoparticles of biodegradable polymers may also provide an alternative solution for oral delivery of anticancer drugs across the gastrointestinal barrier due to their extremely small size and their appropriate surface coating to escape from the recognition by P 450/P-gp (CitationBrigger, Dubernet, and Couvreur 2002). Considerable progress has been made in the preparation of well-characterized nanoparticle formulations loaded with a variety of anticancer agents. The use of biodegradable materials avoids the problem related to the removal of the delivery device after drug depletion. Moreover, biodegradable matrices can provide a further control of release rates by combining the diffusive mechanism with polymer degradation (CitationBala, Hariharan, and Kumar 2004). Various biodegradable polymers have been explored as carriers of drugs to increase the therapeutic benefit and reduce side effects. Among these polymers, polylactide-co-glycolide copolymers and poly(ε -caprolactone) have generated interest because of their excellent biocompatibility and biodegradability (CitationSoppimath et al. 2001; CitationBudhian, Siegel, and Winey 2005; CitationBudhian, Siegel, and Winey 2007; CitationGomez-Gaete et al. 2007; CitationHe, Horn, and Hussain 2007).
Etoposide is an established drug used in the first-line treatment of various malignancies like lymphoma, leukemia, liver cancer, lung cancer etc (CitationO'Dwyer et al. 1985). It has highly variable and erratic bioavailability with intra- and intersubject variation. Poor solubility and degradation in the acidic pH are attributed to the poor bioavailability of etoposide. There are formulation problems associated with available products of etoposide like precipitation of etoposide during the dilution of intravenous formulation and hypotension due to the presence of higher amount of excipients used to solubilize etoposide (CitationShah, Chen, and Chow 1989). The present study was taken with an objective to prepare and characterize etoposide-loaded nanoparticles using PLGA-copolymers with different lactide and glycolide ratio and poly(ε -caprolactone). Influence of different formulation parameters like stabilizer concentration, amount of polymer, and etoposide was studied.
MATERIALS AND METHODS
Materials
Polylactide-co-glycolide 50/50 (PLGA 50/50, molecular weight of 10,000), poly(ε -caprolactone) (PCL) (molecular weight of 40,000), and Pluronic F 68 (F68) were procured from Sigma-Aldrich Chemicals, USA. PLGA 75/25 (Purasorb PDLGA® 75/25, molecular weight of 10,000) and PLGA 85/15 (Purasorb PDLGA® 85/15, molecular weight of 10,000) were generously gifted by Purac, The Netherlands. Etoposide was gift from Dabur research foundation, Sahibabad, India. Triple distilled water was used in the preparation of nanoparticles. All other materials were of analytical grade (Spectrochem, Mumbai, India) and used as received.
Preparation of Nanoparticles
Nanoparticles were prepared by nanoprecipitation (CitationFessi et al. 1989; CitationPeltonen et al. 2002) and emulsion solvent evaporation methods (CitationLeroux et al. 1995) using PLGA polymers and PCL, respectively. These methods were modified according to present requirement. The optimized formulation was prepared using nanoprecipitation method as follows: polymer (50 mg) and etoposide (5 mg) were dissolved in acetone. Dichloromethane was used to dissolve PCL and etoposide in case of emulsion solvent evaporation method. Resulting organic phase was added slowly under moderate magnetic stirring into triple distilled water (TDW) containing F68 as stabilizer (1.0% w/v). This aqueous phase immediately turns milky with formation of nanoparticle dispersion. The organic solvent was then removed by evaporating under magnetic stirring or under reduced pressure at 35°C for approximately 1 hr (Rotavapor, Buchi, Switzerland). Entire dispersion was centrifuged at 14,000 rpm at 25°C for 10 min (Cooling Compufuge, Remi, Mumbai) in three cycles. Supernatant was analyzed for free drug content and the sediment constituting nanoparticles was freeze-dried. For freeze-drying, prefreezing of samples was done at –20°C for 20 hr, then the flasks were connected to freeze-drier (Maxi Dry Lyo, Heto, Germany) under vacuum (1 mbar, –110°C). The process was continued till free-flowing powder was obtained.
Different formulation variables like polymer amount (25, 50, 100, and 200 mg), concentration of stabilizer (0.5, 0.75, 1.0, and 2.0% w/v), and etoposide amount (2.5, 5, 10, and 20 mg) were varied and the effect on size, zeta potential, and entrapment efficiency was studied. Only one parameter was changed in each series of experiments. Drug free nanoparticles were prepared by the same methods without the addition of etoposide and characterized.
Characterization
Average particle diameter, polydispersity index, and surface potential for each batch of prepared nanoparticles were determined by using Zetasizer 3000HS (Malvern Instruments, UK). Values reported are the mean and standard deviation of three batches of nanoparticles.
Morphological examination was done using transmission electron microscopy (Morgagni, Philips, Netherlands) following negative staining with phosphotungstic acid (0.5%). Samples were also examined using atomic force microscopy (Nanoscope II, USA).
To characterize physical state of formulations after encapsulation, differential scanning calorimetry (DSC) study of etoposide, polymers, and drug-loaded nanoparticles was performed using DSC-60 (Shimadzu, Japan). About 2.5 mg of sample was weighed and the aluminum pans were crimped and thermograms were recorded at a temperature increase rate of 10°C per minute from 30 to 300°C. Exothermic peaks were directed upward.
Drug Content and Encapsulation Efficiency
Freeze-dried powder of nanoparticles was weighed and dissolved in acetonitrile. Etoposide was estimated by UV spectrophotometer at 240 nm using combination of acetonitrile (ACN): TDW (1:1) as medium. To determine entrapment efficiency of nanoparticles, sediment formed after centrifugation was dissolved in acetonitrile and analyzed for etoposide. Drug content and encapsulation efficiency were calculated by following equations:
In Vitro Drug Release
Release of etoposide from prepared nanoparticles was studied using dialysis bag diffusion method (CitationLevy and Benita 1990). Two milliliters of the nanodispersions were placed in the dialysis bag (Spectrapor, Cut off 12,500 Da), sealed, and dropped in a beaker containing 200 mL phosphate buffer (pH 7.4). The entire system was kept at 37 ± 2°C with continuous stirring at 100 rpm. Samples were withdrawn from the receptor media at predetermined time intervals and replaced by fresh buffer. The amount of etoposide in the samples was determined by (high performance liquid chromatography) HPLC method.
For HPLC (Jasco, Japan) analysis, a reverse phase Lichrocart® RP 18 column (250 × 4.6 mm i.d., pore size 5 μ m, Merck) was used. Mobile phase is a mixture of ammonium acetate buffer (pH 6.0) : methanol (50:50 v/v). Injection volume was 50 μ l. Flow rate was adjusted to 1 mL/min (PU-1580 model intelligent pumps) and the wavelength was set to 240 nm (UV-1575 model intelligent UV/visible detector). Samples were injected using autosampler (AS-1559 model intelligent sampler) after suitable dilution with mobile phase and the chromatograms were analyzed using Borwin software (Jasco, Japan) provided with the system.
RESULTS AND DISCUSSION
The choice of the particular method of encapsulation is determined by solubility characteristics of the polymer and drug. In the present study, nanoprecipitation method for PLGA-copolymers and solvent evaporation technique for PCL were adapted because these are best suitable for encapsulation of insoluble drugs like etoposide. The particle size, polydispersity index, and encapsulation efficiency have been optimized by changing stabilizer concentration, polymer, and etoposide amount. Nanoparticle formation, in methods used here, can be explained in terms of interfacial turbulence and the diffusion processes between two unequilibrated liquid phases. Mechanism of nanoparticle formation can be described on the basis of solvent–polymer, water–solvent, and water–polymer interactions (CitationChorny et al. 2002). These methods allowed preparation of biodegradable nanoparticles with high drug-loading capacity in an effective and reproducible manner.
Volume of organic solvent was an important factor and it also controls the size of nanoparticles formed (CitationPeltonen et al. 2002). After preliminary study, volume of solvent was decided to 10 mL. An increase in the volume of the aqueous phase (5–25 mL) led to decrease in nanoparticle size to certain extent. Above 25 mL of aqueous phase volume, there was no effect on nanoparticle formation. Aqueous phase volume, thus, was optimized and maintained as 25 mL for all the preparations.
Characterization
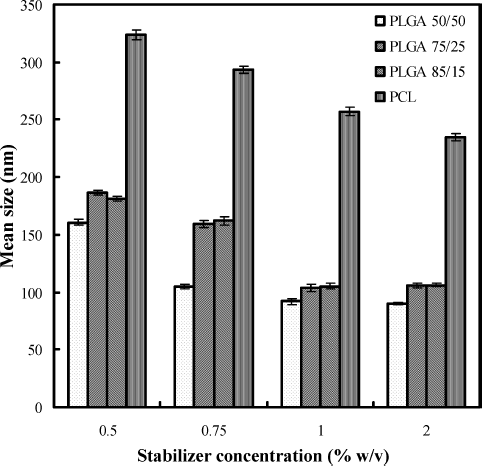
Nanoprecipitation and emulsion solvent evaporation methods produced fine dispersions without any agglomeration. In case of formulations prepared with PLGA-copolymers, particle size found to vary depending on type of PLGA polymer and stabilizer used. It was found that change in composition of lactide and glycolide content in PLGA polymers influenced size of the nanoparticles. For optimized formulation, containing stabilizer 1.0%, PLGA 50/50 produced nanoparticles of 91.8 ± 0.74 nm, whereas at same stabilizer concentrations, PLGA 75/25 and PLGA 85/15 produced nanoparticles of size 103.7 ± 0.18 nm and 105.1 ± 0.38 nm, respectively. PCL leads to the formation of bigger particles (257.2 ± 0.96 nm) when compared with size of nanoparticles prepared by PLGA-copolymers. On the basis of preliminary studies, amount of drug, amount of polymer, and concentration of stabilizer were optimized to 5 mg, 50 mg, and 1.0% w/v. Formulation characters of the optimized formulations prepared with PLGA-copolymers and PCL are given in . Size distribution profiles of formulations prepared with PLGA 50/50 and PCL are given in and , respectively. They show the narrow size distribution of the particles. Transmission electron microscopy photographs of nanoparticles show that all the nanoparticles are round, smooth without showing any agglomeration (), and dispersion does not contain detectable free drug crystals. Atomic force microscopy images ( and ) show that the surface of the particles is smooth and they are spherical. Percent recovery of the formulations prepared with these polymers was very good and was in the range of 85.99–96.71. Entrapment efficiency ranging from 57 to 80% was obtained for the formulations, and the percent drug content was low for all batches with a maximum of around 1.45 for PLGA 85/15 and PCL.
FIG. 1. Size distribution profile of formulation prepared with (A) PLGA 50/50 and (B) PCL.
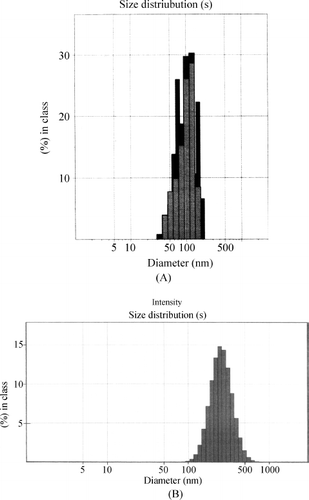
FIG. 2. Transmission electron microscopy image of formulation prepared with (A) PLGA 50/50 (×24,000), (B) PLGA 75/25 (×20,000), (C) PLGA 85/15 (×21,000) and (D) PCL (×24,000).
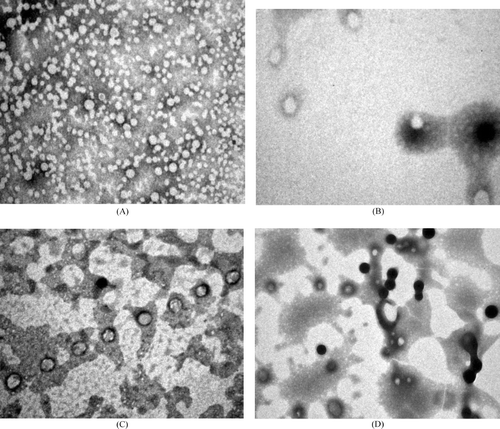
FIG. 3. Atomic force microscopy image of formulation prepared with (A) PLGA 50/50 and (B) PCL.
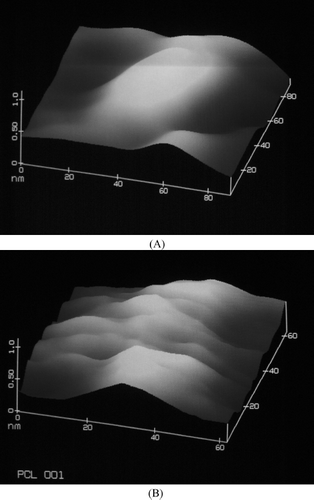
TABLE 1 Formulation characters for the optimized nanoparticle formulations
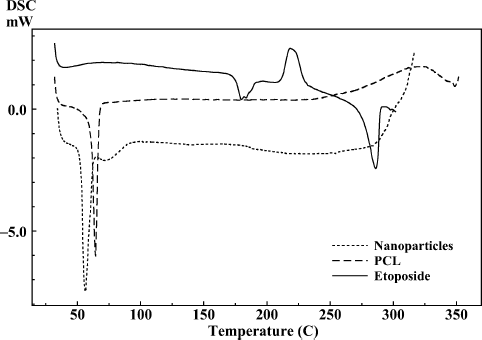
DSC thermograms of drug, polymers, and drug-loaded freeze-dried PLGA 50/50 and PCL nanoparticles are shown in and . DSC thermogram of the drug-loaded nanoparticles prepared with PLGA 50/50 showed glass transition temperature corresponding to PLGA 50/50 at 43.61°C () whereas thermogram of drug-loaded nanoparticle formulation prepared by PCL () showed endothermic melting peak of PCL at 58.84°C. The results show that when nanoparticles were prepared with PLGA 50/50 and PCL polymers, the encapsulated drug did not interact with the polymers and etoposide was encapsulated in the nanoparticle (CitationEspuelas et al. 1997). Absence of drug peak can be attributed to complete encapsulation of etoposide or there might be very less adsorbed drug on the surface of nanoparticles.
FIG. 4. Comparative DSC thermograms of etoposide, PLGA 50/50, and nanoparticles.
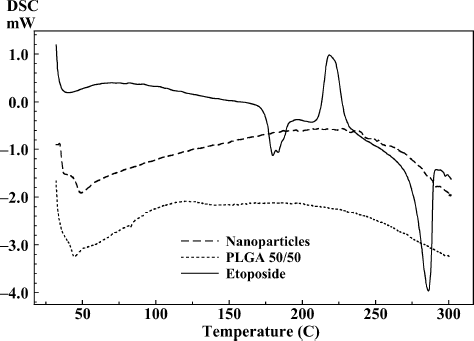
FIG. 5. Comparative DSC thermograms of etoposide, PCL, and nanoparticles.
Effect of Formulation Variables
Stabilizer Concentration
Increase in concentration of F68 from 0.5 to 1.0% w/v made significant decrease in size of nanoparticles prepared with all the polymers in the study. Above 1.0% w/v of F68, there was no significant decrease in size of the particles. Therefore, 1.0% w/v of F68 was considered as optimum for the preparation of nanoparticles and used for optimized products. The effect of stabilizer concentration on the mean size of particles is shown in . An optimum concentration of stabilizer leads to a reduced size of nanoparticles. Insufficient amount of stabilizer is unable to cover the dispersed nanoparticles completely and fails in stabilizing and causes aggregation leading to larger nanoparticles (CitationFeng and Huang 2001). In the solvent evaporation and nanoprecipitation methods, formation and stabilization of particles are crucial factors. The amount of surfactant plays an important role in the protection of particles because it can avoid the agglomeration of particles. There was no difference in size of the particles as the concentration of stabilizer increased to 2% w/v indicating excess stabilizer. Further increase in concentration of stabilizer increased particle size because of the viscosity of the aqueous phase due to higher stabilizer concentration at specific stirring speed (CitationMurakami et al. 1999). Polydispersity index values of the formulations prepared are less than 0.9 for all formulations prepared in the study indicating narrow and homogenous size distribution. Particles prepared with F68 as stabilizer have significant negative surface charge ranging around −33.38 to −18.91 mV for PLGA-copolymers and −33.14 to −23.62 mV for PCL. It was observed that increase in stabilizer concentration changed the zeta potential toward positive value.
FIG. 6. Effect of stabilizer concentration on mean size of particles.
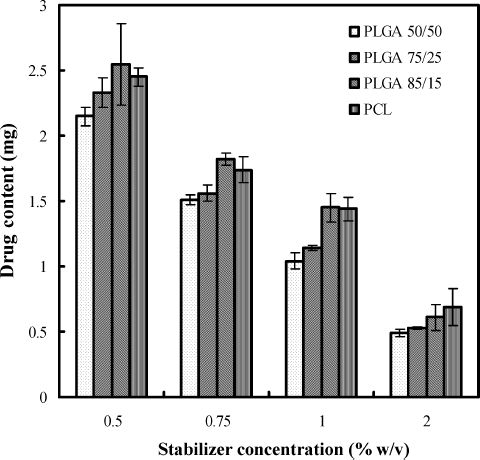
Decrease in drug content was observed with increase in stabilizer concentration. The effect of stabilizer on the drug content of the formulations is shown in . Increase in stabilizer concentration upto 1.0% w/v did not influence entrapment efficiency of etoposide. However, slight decrease in entrapment efficiency was obtained when the stabilizer concentration increased to 2.0% w/v. When the concentration of stabilizer is increased, it helps in solubilizing drug in the aqueous phase. Due to this, when organic solvent is added to the aqueous phase, more amount of etoposide was soluble in the aqueous phase and also helps in leaching of drug from nanoparticles.
FIG. 7. Effect of stabilizer concentration on drug content of formulations.
Polymer Content
An increase in the polymer amount affected the morphology, particle size, polydispersity, zeta potential, drug content, and encapsulation efficiency of the nanoparticles. Initially on increase from 25 to 50 mg, there was no significant increase in mean size of the particles but polydispersity index was slightly increased. Nanoparticles prepared with 25 and 50 mg of polymer formed milky dispersions without any agglomerates. For formulations with PLGA-copolymers, increase in amount to 100 mg increased the size (167.8 and 235.4 nm for 100 and 200 mg, respectively, for PLGA 50/50) and there was formation of small agglomerates. But in formulations prepared with PCL, agglomeration started with 100 mg and when increased to 200 mg, agglomeration was very high leading to precipitation of polymer into large flakes. This is because aqueous phase containing stabilizer was not able to hold this amount of polymer and making to precipitate into agglomerates. Increase in particle size was observed with an increase in amount of polymers by other authors also (CitationOgawa et al. 1988; CitationQuintanar-Guerrero et al. 1996; CitationMurakami et al. 1999; CitationKwon et al. 2001; CitationChorny et al. 2002; CitationBudhian, Siegel, and Winey 2007). Increase in the polymer concentration led to increase in viscosity of the organic phase, and this resulted in poor dispersibility of the organic phase into the aqueous phase. Coarse dispersions were obtained at higher polymer concentrations, which lead to the formation of bigger particles during the diffusion process. This can also be due to the insufficient amount of stabilizer present in the aqueous phase for that particular polymer amount. Entrapment efficiency was influenced by the amount of polymer present in the formulation. When polymer amount was increased from 25 to 200 mg, there was increase in entrapment efficiency for batches prepared with PLGA-copolymers and PCL. For batches prepared with PCL, entrapment efficiency was increased with polymer content from 25 to 50 mg. Increasing content of polymer above 50 mg leads to precipitation and aggregate formation. Therefore, entrapment efficiency, drug content, and recovery for these preparations were not determined.
Amount of Drug
Maintaining a constant initial mass of polymers (50 mg), the mass of etoposide was varied between 2.5 and 20 mg. It was observed that the increase in the amount of etoposide from 2.5 to 10 mg increased the nanoparticle mean diameter from 82.7 to 92.4 nm for PLGA 50/50 and 221.4 to 255.7 nm for PCL. The reason might be, when less amount of etoposide was taken initially, smaller particles were produced after evaporation of organic solvent, whereas if the amount of etoposide added initially is increased, the mean particle size increases because of the high solid content after evaporation. The encapsulation efficiency was increased for all batches when etoposide amount was increased from 2.5 to 10 mg, beyond which there was no effect. This could be due to inadequate amount of polymer present in the system and was not sufficient to entrap the drug inside the matrix.
In Vitro Drug Release
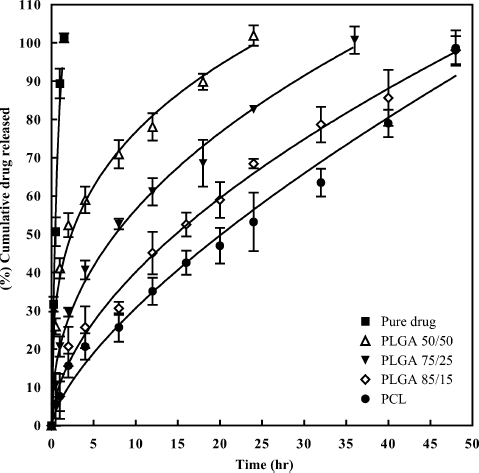
In vitro diffusion profile of pure etoposide from the dialysis bag is given in and shows complete diffusion within 1.5 hr. The cumulative in vitro release profiles of nanoparticle formulations prepared with PLGA-copolymers and PCL with F68 as stabilizer are given in . There was initial burst release of around 41.08% in the first hour for PLGA 50/50, and this rapid initial release may be attributed to the large fraction of drug that was adsorbed or exposed on the surface of the nanospheres. Other scientists also reported the same for different drugs (CitationGomez-Gaete et al. 2007). The small size of the nanoparticles is a major factor, which influences the release rate. The particles have a high surface area; therefore, some amount of the drug will be at or near the particle surface and can be immediately released. Furthermore, the diffusion distances present in the particles are small, which allow drug trapped in the core to rapidly diffuse out and also for the release medium to diffuse in. Release was decreased for batches prepared with PLGA 75/25 (20.65%) and PLGA 85/15 (7.65%) in the first hour of study. Release was extended for 24 hr for PLGA 50/50 and it extended to 36 hr and 48 hr for PLGA 75/25 and PLGA 85/15, respectively. This decrease in release was due to increase in lactide content of the polymer and higher hydrophobic character. Nanoparticles prepared with PCL extended etoposide release up to 48 hr with release around 7.65% in the first hour of study. This indicates that less amount of etoposide was adsorbed or present on the surface of nanoparticles. Maximum amount of etoposide was present in the matrix along with PCL and, therefore, release of etoposide was slow from these formulations. When the in vitro release profiles of formulations prepared with PCL were compared with those prepared with PLGA-copolymers, maximum control in release was observed for PCL formulations. This retardation in release is due to more hydrophobic character of PCL than PLGA copolymers. The most crystalline and hydrophobic polymers exhibit the slowest degradation rate (CitationCoffin and McGinity 1992; CitationLemoine et al. 1996). As etoposide is also hydrophobic in nature, release from the PCL nanoparticle matrix was very less. It was reported by other scientists that release could be extended from these polymers for about months because of slow degradation of polymers in the release medium (CitationLamprecht et al. 2000). In the present study, release was extended only up to 48 hr. This might be due to the low molecular weight of the polymers used in the study.
FIG. 8. In vitro release profiles of pure drug and nanoparticle formulations prepared with PLGA copolymers and PCL.
All the in vitro release data were fitted into various mathematical models (zero-order, first order, Higuchi, Hixson–Crowell, Weibull models) to know the release kinetics, and among all the models used, data were best fitted into Peppas model. Akaike's information criterion (AIC) (CitationYamaoka, Nakagawa, and Uno 1978) and mean sum of squared residuals (MSSR) were calculated for all models and were found to be less for Peppas model indicating best fit. All the kinetic parameters for Peppas model (k, R2, MSSR, n, and AIC) are presented in . Value of “n” in Peppas model indicates that the mechanism of release of etoposide follows anomalous transport. All the polymers are biodegradable and their degradation depends on the molecular weight of the polymer and external medium. Release of drug from these polymers was mainly because of degradation/erosion of polymer.
TABLE 2 Release kinetics for in vitro release of etoposide from nanoparticles using Peppas model
CONCLUSIONS
Etoposide-loaded nanoparticles were prepared successfully using nanoprecipitation method and solvent evaporation method in the presence of F68 as stabilizer. These methods are easy, reproducible, and they produced nanoparticles with narrow size distribution and good entrapment efficiency. Drug content of nanoparticles was found to be low and affected by different formulation variables. Use of different biodegradable polymers produced etoposide-loaded nanoparticles with low polydispersity index with high entrapment efficiency of etoposide. Change in the concentration of stabilizer, polymer, and amount of etoposide found to vary size, polydispersity, drug content, and entrapment efficiency of the prepared nanoparticles. By varying the proportion and nature of polymer, nanoparticles can be made for intended drug release. Release from nanoparticles prepared with PLGA copolymers decreased with increase in lactide content in the polymer. In vitro release of etoposide was controlled for 48 hr from formulations prepared with PLGA 85/15, PCL individually. These studies help optimize the formulation achieving higher efficacy with an improved safety profile of polymer-based nanoparticle formulation. These results justify further investigation of the suitability of these nanoparticles for application in the controlled delivery and/or targeting of etoposide. These formulations can be an alternative to improve the stability of drug with probable enhancement in absorption and bioavailability of the etoposide.
The authors thank the Council of Scientific and Industrial Research (CSIR, New Delhi) for providing scholarship. The authors are grateful to Indian Institute of Chemical Technology (IICT, Hyderabad), All India Institute of Medical Sciences (AIIMS, New Delhi), and Central Electronics Engineering Research Institute (CEERI, Pilani) for extending their help in analyzing samples by Zeta Sizer,Transmission Electron Microscopy, and Atomic Force Microscopy.
Related Research Data
REFERENCES
- I. Bala, S. Hariharan, and M. N. Kumar. (2004). PLGA nanoparticles in drug delivery: the state of the art. Crit. Rev. Ther. Drug Carrier. Syst. 21:387–422.
- I. Brigger, C. Dubernet, and P. Couvreur. (2002). Nanoparticles in cancer therapy and diagnosis. Adv. Drug Del. Rev. 54:631–651.
- A. Budhian, S. J. Siegel, and K. I. Winey. (2005). Production of haloperidol-loaded PLGA nanoparticles for extended controlled drug release of haloperidol. J. Microencapsul. 22:773–785.
- A. Budhian, S. J. Siegel, and K. I. Winey. (2007). Haloperidol-loaded PLGA nanoparticles: systematic study of particle size and drug content. Int. J. Pharm. 336:367–375.
- M. Chorny, I. Fishbein, H. D. Danenberg, and G. Golomb. (2002). Lipophilic drug loaded nanospheres prepared by nanoprecipitation: effect of formulation variables on size, drug recovery and release kinetics. J. Control. Release 83:389–400.
- M. D. Coffin, and J. W. McGinity. (1992). Biodegradable pseudolatexes: the chemical stability of poly (D,L-lactide) and poly (ε -caprolactone) nanoparticles in aqueous media. Pharm. Res. 9:200–205.
- M. S. Espuelas, P. Legrand, J. M. Irache, C. Gamazo, A. M. Orecchioni, J. P. Devissaguet, and P. Ygartua. (1997). Poly (ε -caprolacton) nanospheres as an alternative way to reduce amphotericin B toxicity. Int. J. Pharm. 158:19–27.
- S.-S. Feng, and G. Huang. (2001). Effects of emulsifiers on the controlled release of paclitaxel (Taxol®) from nanospheres of biodegradable polymers. J. Control. Release 71:53–69.
- H. Fessi, F. Puisieux, J. P. Devissaguet, N. Ammoury, and S. Benita. (1989). Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 55:R1–R4.
- C. Gomez-Gaete, N. Tsapis, M. Besnard, A. Bochot, and E. Fattal. (2007). Encapsulation of dexamethasone into biodegradable polymeric nanoparticles. Int. J. Pharm. 331:153–159.
- W. He, S. W. Horn, and M. D. Hussain. (2007). Improved bioavailability of orally administered mifepristone from PLGA nanoparticles. Int. J. Pharm. 334:173–178.
- M. N. Kumar, and N. Kumar. (2001). Polymeric controlled drug-delivery systems: perspective issues and opportunities. Drug Dev. Ind. Pharm. 27:1–30.
- H.-Y. Kwon, J.-Y. Lee, S.-W. Choi, Y. Jang, and J.-H. Kim. (2001). Preparation of PLGA nanoparticles containing estrogen by emulsification-diffusion method. Colloids Surf. A: Physicochem. Eng. Aspects 182:123–130.
- A. Lamprecht, N. Ubrich, M. Hombreiro Perez, C. M. Lehr, M. Hoffman, and P. Maincent. (2000). Influences of process parameters on nanoparticle preparation performed by a double emulsion pressure homogenization technique. Int. J. Pharm. 196:177–182.
- D. Lemoine, C. Francois, F. Kedzierewicz, V. Preat, M. Hoffman, and P. Maincent. (1996). Stability study of nanoparticles of poly (ε -caprolactone), poly (D,L-lactide) and poly(D,L-lactide-co-glycolide). Biomaterials 17:2191–2197.
- J. C. Leroux, E. Allemann, E. Doelker, and R. Gurny. (1995). New approach for the preparation of nanoparticles by an emulsification diffusion method. Eur. J. Pharm. Biopharm. 41:14–18.
- M. Y. Levy, and S. Benita. (1990). Drug release from submicronized o/w emulsion: a new in vitro kinetic evaluation model. Int. J. Pharm. 66:29–37.
- H. Murakami, M. Kobayashi, H. Takeuchi, and Y. Kawashima. (1999). Preparation of poly (dl-lactide-co-glycolide) nanoparticles by modified spontaneous emulsification solvent diffusion method. Int. J. Pharm. 187:143–152.
- P. J. O'Dwyer, B. Leyland-Jones, M. T. Alonso, S. Marsoni, and R. E. Wittes. (1985). Etoposide (VP-16–213). Current status of an active anticancer drug. N. Engl. J. Med. 312:692–700.
- Y. Ogawa, M. Yamamoto, H. Okada, T. Yashiki, and T. Shimamoto. (1988). A new technique to efficiently entrap leuprolide acetate into microcapsules of polylactic acid or copoly(lactic/glycolic) acid. Chem. Pharm. Bull. 36:1095–1103.
- L. Peltonen, P. Koistinen, M. Karjalainen, A. Hakkinen, and J. Hirvonen. (2002). The effect of cosolvents on the formulation of nanoparticles from low-molecular-weight poly(l)lactide. AAPS Pharm. Sci. Tech. 3:E32.
- D. Quintanar-Guerrero, H. Fessi, E. Allemann, and E. Doelker. (1996). Influence of stabilizing agents and preparative variables on the formation of poly (d,l-lactic acid) nanoparticles by an emulsification-diffusion technique. Int. J. Pharm. 143:133–141.
- J. C. Shah, J. R. Chen, and D. Chow. (1989). Preformulation study of etoposide: identification of physicochemical characteristics responsible for the low and erratic oral bioavailability of etoposide. Pharm. Res. 6:408–412.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni, and W. E. Rudzinski. (2001). Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 70:1–20.
- K. Yamaoka, T. Nakagawa, and T. Uno. (1978). Application of Akaike's information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J. Pharmacokinet. Biopharm. 6:165–175.