Abstract
A redispersible dry emulsion (DE) formulation of atorvastatin calcium (AC) was developed to enhance the in vitro dissolution of AC, thereby increasing its gastrointestinal absorption. The spray-drying technology was used where Plurol Oleique CC 497 was chosen as the oil phase. Effects of carriers, surfactants, and homogenizers on the characteristics of DE containing AC were systematically investigated. The final formulation consisted of dextrin and Poloxamer 188 as carrier and surfactant, respectively, and was homogenized by a high pressure homogenizer before spray drying. The in vitro release of AC from the optimized DE was significantly higher than that of pure AC powder (76% vs. 30% at 24 hr). The in vitro intestinal absorption of AC from the DE formulation was 0.77 μg/cm2 at 2 hr, which was a 2.33-fold increase compared to the pure unformulated AC powder. These results suggest that the oral dry emulsion formulation could improve the intestinal absorption of AC.
Introduction
Statin drugs are effective inhibitors of hydroxy- methylglutaryl-coenzyme A reductase (HMG-CoA), the rate-limiting enzyme associated with the de novo synthesis of cholesterol (Witztum Citation1996). These agents are known to decrease the low-density lipoprotein cholesterol (LDL-C) and triglycerides (TG) levels while increasing that of the high-density lipoprotein cholesterol (HDL-C) in plasma, thereby reducing the risk factor for coronary heart diseases (CHD) (Gordon et al. Citation1989; Pedersen et al. Citation1998b; Ballantyne et al. Citation1999). Atorvastatin calcium (AC), a commonly used form of atorvastatin, is insoluble in aqueous solutions of pH 4 and below, while being very slightly soluble in water and in pH 7.4 phosphate buffer. It is classified by the biopharmaceutical classification system (BCS) as a Class 2 drug (Amidon et al. Citation1995; Kasim et al. Citation2004; Wu and Benet Citation2005) which is often reported to have low bioavailability owing to the insufficient dissolution process in the gastrointestinal tract. In fact, the bioavailability of the commercially available tablet formulation of AC, Lipitor®, is about 14% (Kearney et al. Citation1993).
Formulation strategies to improve the oral bioavailability of Class 2 drugs include increasing the dissolution rate (Okonogi et al. Citation1997) and solubilizing the drug in the gastrointestinal tract (Bhargava and Agrawal Citation2008). Emulsions have been reported to be efficient for improving the dissolution rate and increasing bioavailability of poorly water-soluble drugs (Tarr and Yalkowsky Citation1989). However, creaming, flocculation, coalescence, and phase separation are often observed in emulsions, giving rise to stability problems (Floyd Citation1999; Welin-Berger and Bergenstahl Citation2000).
Dry emulsions (DE) could serve as an alternative to overcome the disadvantages of the conventional emulsions (Molina and Cadorniga Citation1995; Shively and Thompson Citation1995). DE formulations are typically prepared from O/W emulsions containing a soluble or an insoluble solid carrier in the aqueous phase by spray drying (Takeuchi et al. Citation1992a, Citation1992b; Pedersen et al. Citation1998a), lyophilization (Corveleyn and Remon Citation1998a, Citation1998b), or evaporation (Shively and Dec Citation1994). Solid carriers include gelatin (Nakamoto et al. Citation1975), lactose (Pedersen et al. Citation1998a; Heinzelmann and Franke Citation1999), maltodextrin (Corveleyn and Remon Citation1999; Heinzelmann and Franke Citation1999), mannitol (Molina and Cadorniga Citation1995), povidone (Shively Citation1993a, Citation1993b), and sucrose (Shively and Thompson Citation1995; Porter et al. Citation1996).
Herein, we report on the development of a new DE type formulation of atorvastatin calcium (AC) which resulted in enhanced solubility and improved stability. The spray drying technology was utilized for preparation followed by a systematic characterization. Moreover, in vitro dissolution and in vitro intestinal permeability studies were performed to optimize the formulation.
Materials and methods
Materials
Atorvastatin calcium (crystalline form) was purchased from Beijing HuaFeng United Technology Co. Ltd (Beijing, China). Labrafil WL 2609 BS, Labrafil M 2125 CS, Lauroglycol FCC, Labrafac Lipophile WL 1349, Capryol 90, Labrafil M 1944 CS, Peceol, Lauroglycol, and Maisine 35-1 were received as gifts from Gattefossé Co. (Saint Priest Cedex, France) while Plurol Oleique CC 497 was purchased from Gattefossé Co. (Saint Priest Cedex, France). Dextrin from corn, soybean oil, mineral oil, castor oil, isopropyl myristate (IPM), and sesame oil were obtained from Sigma-Aldrich Co. (St. Louis, MO). Lactose monohydrate, Tween 80, and Poloxamer 188, Cremophor EL were purchased from Kanto Chemical Co. Inc (Tokyo, Japan), Tokyo Kasei Kogyo Co. Ltd (Tokyo, Japan), and BASF Co. (Ludwigshafen, Germany), respectively. Acetonitrile and methanol were HPLC grade and supplied from Fisher Scientific Korea Ltd. (Seoul, Korea). Sodium dodecylsulfate (SDS) was purchased from Tokyo Kasei Kogyo Co. Ltd (Tokyo, Japan). All other chemicals and solvents were of analytical grade.
Solubility of AC in oils
The solubility of AC was determined in various oils including Labrafil WL 2609 BS, Labrafil M 2125 CS, Lauroglycol FCC, Labrafac Lipophile WL 1349, Capryol 90, Labrafil M 1944 CS, Peceol, Lauroglycol, Maisine 35-1, Plurol Oleique CC 497, soy bean oil, mineral oil, and castor oil. Excess AC was added into 1 ml of each oil in the centrifugal tube and mixed (100 rpm) in a shaking incubator (Jeio-Tech, Seoul, Korea) at 25°C for 48 hr. The solution was centrifuged at 13,200 rpm for 5 min to remove the excess AC, and then the concentration of AC in the supernatant was measured by HPLC after appropriate dilution with methanol.
Preparation of dry emulsion (DE)
Based on the result of the solubility study (, Plurol Oleique CC 497 was selected as the oil phase. The effect of solid carriers (i.e. lactose monohydrate, HPMC, and dextrin) and surfactants (i.e. Tween 80, Cremophor EL, and Poloxamer 188) and homogenizing conditions (i.e. basic homogenizer and high pressure homogenizer) were systematically investigated in mono factorial design (). An accurately measured amount (430 mg) of AC was dissolved in 25 g Plurol Oleique CC 497 and mixed with 100 ml of water phase containing various surfactants (4%, w/v). The mixture was dispersed by magnetic stirring for 30 min and the resulting coarse emulsion was mixed with 150 ml of solid carrier solution containing 25 g of various solid carrier (Plurol Oleique:carrier = 1:1, w/w) by magnetic stirring for 15 min. Further homogenizing either by Ultra-Turrax® T25 Basic Homogenizer or by EmulsiFlex-C5 high pressure homogenizer and spray drying using the Büchi mini spray dryer (Büchi, Switzerland) proceeded under the following conditions: inlet temperature at 120°C, aspiration of 100%, drying air flow at 800 Nl/hr, and feeding rate of the emulsion at 5 ml/min. The resulting dry emulsion powder was stored in well-closed vials at room temperature.
Table 1. Solubility of AC in various oils.
Table 2. Formulation of dry emulsion of atorvastatin calcium.
Characterization of dry emulsion (DE)
Droplet size and distribution measurement
The reconstituted emulsions were obtained from dispersing DE powder into distilled water. The droplet size and distribution of emulsions before spray drying and reconstituted emulsions were determined using a NICOMP 370 Submicron Particle Sizer (Particle Sizing System, Santa Barbara, CA). The SPAN value was employed to express the width of the droplet size distribution which was defined as:
SPAN = [d(v, 0.9) − d(v, 0.1)]/d(v, 0.5)
where d(v, 0.1), d(v, 0.5), and d(v, 0.9) were 10%; 50%, and 90% volumetric diameters.
Scanning electron microscopy (SEM)
The surface morphology of the DE powder was observed by scanning electron microscopy (JSM-5310LV, JEOL, Tokyo, Japan) at an accelerating voltage of 20 KV. Samples were mounted on a double-faced adhesive tape and sputtered with platinum for 250 sec before scanning electron microscopy.
Powder X-ray diffraction (pXRD)
DE powder was observed for X-ray pattern using a D5005 powder X-ray diffractometer (Bruker, Karlsruhe, Germany) with CuKα radiation (λ = 1.54,056 å) generated from a copper source operating at a voltage of 40 KV and a current of 40 mA. The test samples were packed into 0.5 mm deep graphite sample holders. The samples were scanned over the range of 3–50° 2θ at a scan rate of 5°/min.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) measurements were carried out with a DSC-Q1000 modulated differential scanning calorimeter (TA Instruments Inc., UK). About 3–5 mg DE samples were placed in hermetic aluminum pans with lid and clamp sealed. The samples were heated from 30 to 400°C at a rate of 10°C/min. An empty aluminum pan was used as a reference.
In vitro dissolution test
In vitro dissolution tests were performed using a dissolution tester (Electrolab, Bombay, India) following the USP paddle method. AC powder or reconstituted DEs equivalent to 10 mg of AC was placed in the dialysis bag (Spectra/Por® membranes, MWCO: 12000, Spectrum Medical Industries Inc. TX) located in dissolution media (900 ml of distilled water) containing 0.2% (w/v) of sodium dodecylsulfate (SDS) at 37 ± 0.5°C and was stirred with a rotating paddle at 50 rpm. Aliquots of dissolution media (5 ml) were withdrawn at predetermined time intervals for 24 hr, and were replaced with equal volumes of the fresh media.
In vitro intestinal absorption study using the Ussing chamber
Male Sprague-Dawley rats (250–300 g, Dae-Han Biolink, Daejeon, Korea) were used to perform the in vitro permeation study. All rats were provided with food (SamYang Company, Seoul, Korea) and water ad libitum, and maintained in a light-controlled room (light: 07:00–19:00, dark: 19:00–07:00) kept at a temperature of 22 ± 2°C and a relative humidity of 55 ± 5% (Animal Center for Pharmaceutical Research, College of Pharmacy, Seoul National University, Seoul, Korea). The experimental protocols involving animal study were approved by the Animal Care and Use Committee of the College of Pharmacy, Seoul National University, according to the National Institutes of Health Guidelines (NIH publication #85-23, revised in 1985).
Animals were fasted for 2 days prior to the experiment and the intestinal tissue was excised after being humanely sacrificed by ketamine. The freshly excised small intestine was immediately rinsed twice with ice-cold phosphate buffered saline (PBS, pH 7.4) and placed in ice-cold PBS under continuous oxygenation with O2/CO2 (95%/5%) bubbling. The specimen was cut open along the mesenteric border with blunt-end scissors and divided into 1 cm segments. The resulting intestinal sheets were mounted as flat sheets between two acrylic half-cells and they were joined to form the complete Ussing chamber. The tissue pieces were bathed on both sides with 1 ml of PBS (pH 7.4) at 37°C controlled by a water-jacketed reservoir. The PBS was also bubbled with an O2/CO2 (95%/5%) mixture gas to circulate fluid in each chamber at 37°C. After 30 min equilibration, 1 ml of the AC suspension or reconstituted dry emulsion containing equal amount of AC (1.72 mg as AC) was infused into the mucosal side of the chamber while 1 ml of fresh PBS (37°C) was added in the serosal side. At predetermined time intervals, 0.5 ml of samples were withdrawn from the serosal side and the same volume of fresh PBS at 37°C was refilled immediately.
Physical stability of dry emulsion
The physical stability of DE powder in well-sealed containers was tested at 40°C with 75% relative humidity in a climatic chamber (EYELA, Tokyo, Japan) and ambient temperature, respectively. The X-ray diffraction and DSC of DE powder were examined and its morphology observed by SEM for 3 months.
HPLC analysis of AC
The concentrations of AC were quantified by HPLC with a UV detector, as previously described (Shen and Zhong Citation2006). HPLC system consisted of a pump (Waters 515), an automatic injector (Waters 717 plus autosampler), and a UV detector set at 248 nm (Waters 2487, Waters Corporation, Milford, MA). An analytical column (LiChrospher® 100 RP-18, 5 μm, Darmstadt, Germany) was eluted with a mobile phase consisting of acetonitrile and 0.05% acetic acid solution (65:35, v/v) at a flow rate of 1.0 ml/min.
Statistical analysis
A value of p less than 0.05 was considered to be statistically significant when t-test was performed between the two means for the unpaired data. All data are expressed as the mean ± standard deviation.
Results and discussions
Formulation of dry emulsion (DE)
To select a suitable oil phase for the DE formulation, the solubility of AC in various oils was determined, as shown in . AC has a solubility ranging from 0.022 mg/ml in mineral oil, 3.21 mg/ml in Labrafil WL 2609 BS, to 16.24 mg/ml in Plurol Oleique CC497. Based on the solubility study, Plurol Oleique CC497 was chosen as the oil phase and the formulation was designed as shown in .
All freeze-dried DE formulations containing AC could be reconstituted by dispersing them in distilled water. Droplet size of each formulation both in liquid emulsion before spray drying and in reconstituted emulsion after spray drying showed a mono-modal distribution (data not shown). The mean droplet size and SPAN values of each formulation listed in shows that the droplet size and distribution were affected by carriers, surfactants, and homogenizing conditions. The mean droplet size of Rx3, where dextrin was used as the carrier, was relatively smaller than that of Rx1 and Rx2. This may be due to lactose monohydrate’s sensitivity to moisture causing coalescence after spray drying.
Table 3. Droplet size and distribution of the dry emulsions.
HPMC seemed to increase the viscosity of the emulsion before spray drying, thereby increasing the droplet size during the spray drying process. Compared to Tween 80 (Rx3) and Cremophore EL (Rx4), Poloxamer 188 (Rx5) resulted in the increase of the mean droplet size of DE, although the reason is not clear from this study. However, homogenizing conditions seemed to exert the most significant effect on the mean droplet size, as shown in Rx5 and Rx6 (). The use of a high pressure homogenizer as in the case for Rx6 resulted in the smallest droplet size. Rx6 could therefore show better gastrointestinal absorption of the drug compared to others since reports have shown that the reduction of droplet size of emulsions enhance the gastrointestinal absorption of poorly water-soluble drugs (Tarr and Yalkowsky Citation1989).
Scanning electron micrographs of the AC powder and freshly prepared DE formulations containing AC are shown in . As expected from the results of the droplet size study, the morphology of Rx1 and Rx2 powder showed agglomeration during the preparation of the SEM samples due to lactose monohydrate and HPMC, respectively. On the other hand, Rx3 and Rx4 powder showed well separated particles, although with wrinkles and deep dents on the surface, which are probably due to the liquid surfactants Tween 80 and Cremophor EL that can induce the shrinkage of particles during the spray drying process. Rx5 and Rx6 powder were well separated spherical particles with smooth surfaces, which was consistent with the report by Elversson and Millqvist-Fureby (Citation2006). Thus, Rx6 formulation where dextrin and Poloxamer 188 were used as carrier and surfactant, respectively, was chosen for further evaluation studies, and compared with the Rx2 and Rx5 formulations.
Figure 1. Scanning electron micrographs of pure AC powder and six freshly prepared DE formulations containing AC. Rx5’ and Rx6’ indicate the Rx5 and Rx6 DE stored at 40°C with 75% relative humidity for 3 months. The composition of every formulation is listed in .

Powder X-ray diffraction (pXRD)
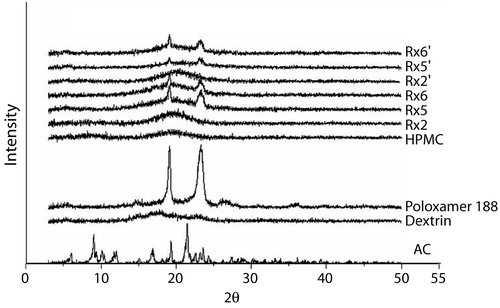
Powder X-ray diffraction diffractograms of pure AC powder, Rx2, Rx5, and Rx6 formulations are shown in . Distinct peaks found in the diffractogram indicate that the AC powder is present in crystalline form. The spectrum of Poloxamer 188 also showed a distinct peak pattern. In the Rx5 and Rx6 formulations, Poloxamer 188 still seemed to remain, while the diffraction peaks corresponding to AC crystalline disappeared, indicating that AC was in the amorphous state.
Figure 2. Powder X-ray diffractograms of three freshly prepared DE formulations containing AC (Rx2, Rx5, and Rx6). Rx2’, Rx5’, and Rx6’ indicate the DE stored at 40°C with 75% relative humidity for 3 months. The bottom four diffractograms are HPMC, Polaxamer 188, dextrin, and pure AC powder, for comparison.
Differential scanning calorimetry (DSC)
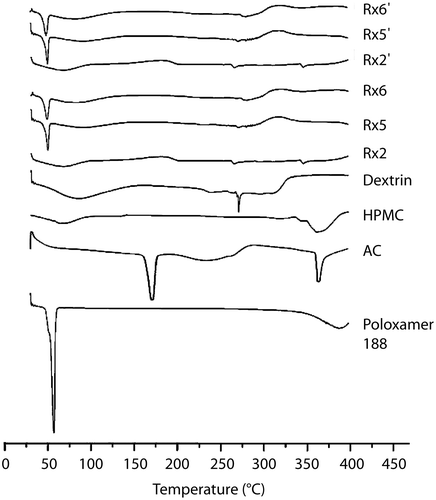
DSC thermograms obtained from AC powder, Rx2, Rx5, and Rx6 formulations are shown in . An endothermic peak near 171°C, which indicates the melting point of the AC powder, disappeared in the thermograms of the formulations. Thus, no crystalline form of AC seems to be present in those formulations, which is consistent with the data from the X-ray diffraction studies.
Figure 3. DSC thermograms of three freshly prepared DE formulations containing AC (Rx2, Rx5, and Rx6). Rx2’, Rx5’, and Rx6’ indicate the DE stored at 40°C with 75% relative humidity for 3 months. DSC of dextrin, HPMC, pure AC powder, and Polaxamer 188 were also shown for comparison.
In vitro dissolution test
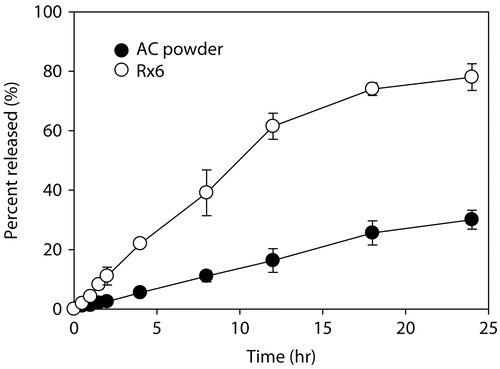
The dissolution profiles of the Rx6 formulation and pure AC powder into 0.2% SDS solution are shown in . Release of AC from the Rx6 formulation was significantly faster than that from the pure AC powder. Also, the amount of AC released for 24 hr was higher from Rx6 than that of the AC powder (i.e. 76% vs. 30%, respectively). Initial burst release was not observed for DE, indicating that AC was dispersed in the oil phase and was not crystallized on the surface even after spray-drying, as confirmed from the results of the X-ray diffraction and DSC study.
Figure 4. Dissolution profiles of AC from pure AC powder (•) and formulation Rx6 (ο) at 37.0 ± 0.5°C for 24 h. Each data represents the mean ± SD of three determinations.
In vitro intestinal absorption study by Ussing chamber
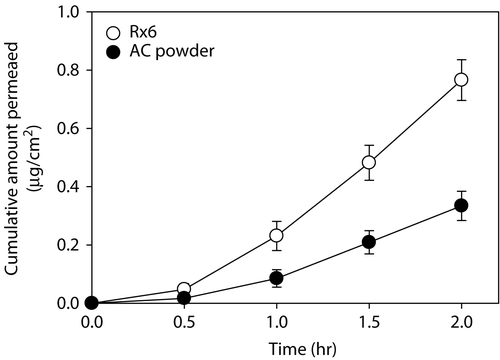
The intestinal absorption of class 2 drugs could be increased by enhancing the dissolution rate, thereby increasing the bioavailability (Grove et al. Citation2006; Ahmed and Aboul-Einien Citation2007). Based on previous reports, the feasibility of the DE formulation as a drug delivery system which can enhance the intestinal absorption of AC was investigated in vitro by using the Ussing chamber. As shown in , the amount of AC permeated through the rat intestine from the Rx6 formulation was significantly higher than that from pure AC powder at 2 h (i.e. 0.77 μg/cm2 vs. 0.33 μg/cm2, p < 0.05). As mentioned above, the Rx6 formulation consisted of dextrin and Poloxamer 188 as carrier and surfactant, respectively, and was homogenized by high pressure homogenizer before spray drying. Well homogenized emulsion with smaller droplet size and narrow distribution was obtained through the high pressure homogenizer, which may be the reason for improved permeation of AC in the Rx6 formulation (Itoh and Matsui Citation2002).
Figure 5. In vitro intestinal absorption profile of AC from AC powder (•) and formulation Rx6 (ο). Each data point represents the mean ± SD of three determinations.
Physical stability of dry emulsion
Rx5’ and Rx6’ in Figure 1 are SEM images for Rx5 and Rx6 formulations after being stored at 40°C with 75% relative humidity for 3 months, respectively. The results showed that the outer structures of Rx5 and Rx6 formulations remained unchanged after storage. That is, well-separated spherical particles with smooth surface were observed in the formulations. The X-ray diffraction and DSC results also support that the inner properties of the dry emulsion of the Rx5 and Rx6 formulations did not significantly change at the above-mentioned storage conditions. However, when Tween 80 was used as surfactant and dextrin as carrier (Rx3), the dry emulsion powder melted into a sticky liquid under the same storage conditions (data not shown). Even though the dry emulsion consisting of Tween 80 and HPMC (Rx2) kept the solid form after storage in the above condition for 3 months, the SEM results showed that the particles were sticky together and there was no clear boundary between the particles (data not shown).
Conclusion
In the present study, DE containing AC was successfully prepared by the spray-drying method. Carriers and surfactants affected the outer morphology and stability of the DE formulations when Plurol Oleique CC 497 was used as an oil phase. At the same time, the homogenizing method influenced the droplet size and distribution of the emulsion, which seemed to be the most critical factor affecting the in vitro dissolution and intestinal absorption of AC from the DE. In conclusion, dry emulsion formulation of AC could be a feasible approach for enhancing the dissolution rate, thereby increasing the bioavailability of AC after oral administration.
Acknowledgment
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ahmed IS, Aboul-Einien MH 2007. In vitro and in vivo evaluation of a fast-disintegrating lyophilized dry emulsion tablet containing griseofuvin. Eur. J. Pharm. Sci. 32, 58–68.
- Amidon GL Lennernas H Shah VP, Crison JR 1995. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12, 413–420.
- Ballantyne CM Herd JA Ferlic LL Dunn JK Farmer JA Jones PH Schein JR, GottoJr. AM 1999. Influence of low HDL on progression of coronary artery disease and response to fluvastatin therapy. Circulation 99, 736–743.
- Bhargava S, Agrawal GP 2008. Preparation & characterization of solid inclusion complex of cefpodoxime proxetil with beta-cyclodextrin. Curr. Drug Deliv. 5, 1–6.
- Corveleyn S, Remon JP 1998a. Formulation of a lyophilized dry emulsion tablet for the delivery of poorly soluble drugs. Int. J. Pharm. 166, 65–74.
- Corveleyn S, Remon JP 1998b. Bioavailability of hydrochlorothiazide: conventional versus freeze-dried tablets. Int. J. Pharm. 173, 149–155.
- Corveleyn S, Remon JP 1999. Stability of freeze-dried tablets at different relative humidities. Drug Dev. Ind. Pharm. 25, 1005–1013.
- Elversson J, Millqvist-Fureby A 2006. In situ coating-An approach for particle modification and encapsulation of proteins during spray-drying. Int. J. Pharm. 323, 52–63.
- Floyd AG 1999. Top ten considerations in the development of parenteral emulsions. Pharm. Sci. Technol. To. 2, 134–143.
- Gordon DJ Probstfield JL Garrison RJ Neaton JD, Castelli WP Knoke JD JacobsJr. DR Bangdiwala S, Tyroler HA 1989. High-density lipoprowith lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 79, 8–15.
- Grove M Müllertz A, Nielsen JL, Pedersen GP 2006. Bioavailability of seocalcitol II: Development and characterization of self-microemulsifying drug delivery system (SMEDDS) for oral administration containing medium and long chain triglycerides. Eur. J. Pharm. Sci. 28, 233–242.
- Heinzelmann K, Franke K 1999. Using freezing and drying techniques of emulsions for the microencapsulation of fish oil to improve oxidation stability. Colloids Surf. B 12, 223–229.
- Itoh K, Matsui S 2002. Improvement of physicochemical properties of N-4472 Part II: characterization of N-4472 microemulsion and the enhanced oral absorption. Int. J. Pharm. 246, 75–83.
- Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennernas H, Hussain AS, Junginger HE, Stavchansky SA, Midha KK, Shah VP, Amidon GL 2004. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 1, 85–96.
- Kearney AS, Crawford LF, Mehta SC, Radebaugh GW 1993. The interconversion kinetics, equilibrium, and solubilities of the lactone and hydrocyacid forms of the HMG-CoA reductase inhibitor. CI-981. Pharm. Res. 10, 1461–1465.
- Molina C, Cadorniga R 1995. Physical stability of lyophilized and sterilized emulsions. (S.T.P.) Pharma Pratiques 5, 63–72.
- Nakamoto Y, Hashida M, Muranishi S, Sezaki H 1975. Studies on pharmaceutical modification of anticancer agents. Enhanced delivery of bleomycin into lymph by emulsions and drying emulsions. Chem. Pharm. Bull. 23, 3125–3131.
- Okonogi S, Oguchi T, Yonemochi E, Puttipipatkhachorn S, Yamamoto K 1997. Improved dissolution of ofloxacin via solid dispersion, Int. J. Pharm. 156, 175–180.
- Pedersen GP, Fäldt P, Bergenståhl B, Kristensen HG 1998a. Solid state characterisation of a dry emulsion: a potential drug delivery system. Int. J. Pharm. 171, 257–270.
- Pedersen TR, Olsson AG, Faergeman O, Kjekshus J, Wedel H, Berg K, Wilhelmsen L, Haghfelt T, Thorgeirsson G, Pyörälä K, Miettinen T, Christophersen B, Tobert JA, Musliner TA, Cook TJ 1998b. Lipoprotein changes and reduction in the incidence of major coronary heart disease events in the Scandinavian Simvastatin Survival Study (4S). Circulation 97, 1453–1460.
- Porter CJH, Charman SA, Williams RD, Bakalova MV, Charman WN 1996. Evaluation of emulsifiable glasses for the oral administration of cyclosporin in beagle dogs. Int. J. Pharm. 141, 227–237.
- Shen H, Zhong M 2006. Preparation and evaluation of self-microemulsifying drug delivery system (SMEDDS) containing atorvastatin. J. Pharm. Pharmacol. 58, 1183–1191.
- Shively ML 1993a. Characterization of oil-in-water emulsions prepared from solid-state emulsions: effect of matrix and oil phase. Pharm. Res. 10, 1153–1156.
- Shively ML 1993b. Droplet size distribution within oil-inwater emulsions prepared from solid state dispersions. J. Colloid Interface Sci. 155, 66–69.
- Shively ML, Dec SF 1994. Solid-state emulsions: evaluation by 1H and 13C solid-state nuclear magnetic resonance. Pharm. Res. 11, 1301–1305.
- Shively ML, Thompson DC 1995. Oral bioavailability of vancomycin solid-state emulsions. Int. J. Pharm. 117, 119–122.
- Takeuchi H, Sasaki H, Niwa T, Hino T, Kawashima Y, Uesugi K, Ozawa H 1992a. Design of redispersible dry emulsion as an advanced dosage form of oily drug (vitamin E nicotinate) by spray-drying technique. Drug Dev. Ind. Pharm. 18, 919–937.
- Takeuchi H, Sasaki H, Niwa T, Hino T, Kawashima Y, Uesugi K, Ozawa H 1992b. Improvement of photostability of ubidecarenone in the formulation of a novel powdered dosage form termed redispersible dry emulsion. Int. J. Pharm. 86, 25–33.
- Tarr BD, Yalkowsky SH 1989. Enhancement intestinal absorption of cyclosporine in rats through the reduction of emulsion droplet size. Pharm. Res. 6, 40–43.
- Welin-Berger K, Bergenståhl B 2000. Inhibition of Ostwald ripening in local anesthetic emulsions by using hydrophobic excipients in the disperse phase. Int. J. Pharm. 200, 249–260.
- Witztum, J. L. 1996. Drugs used in the treatment of hyperlipoproteinemias. In: The pharmacological Basis of Therapeutics, 9th ed, eds Ruddon JG, Limbird LE, Molinoff PB, Ruddon RW, 875–897. New York, McGraw-Hill.
- Wu CY, Benet LZ 2005. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm. Res. 22, 11–23.