Abstract
The purpose of this study was to develope and characterize a micellar formulations of N-{[(2-hydroxy-5- nitrophenyl)amino]carbonothioyl}-3,5-dimethylbenzamide (DM-PIT-1)—a new small molecule non-lipid antagonist of phopshotidylinositol-3.4.5-triphopshate and inhibitor of the PI3-kinase pathway. Micelle-forming PEG2000-PE was used to solubilize DM-PIT-1. To improve the specificity of the micellar DM-PIT-1, cancer-targeting anti-nucleosomal mAb2C5 antibodies as well as Tumor necrosis factor- Related Apoptosis-Inducing Ligand (TRAIL) were attached to the surface of polymeric micelles. DM-PIT-1 was effectively incorporated (> 70%) into 14–16 nm micelles, which had a negative surface zeta potential of 4–5 mV. Micellar DM-PIT-1 demonstrated high in vitro cytotoxicity against various cancer cells. An improved potency of the dual-activity DM-PIT-1/TRAIL combination nanoparticles in inducing death of TRAIL-resistant cancer cells was shown. Efficacy of the TRAIL therapy was enhanced by combining it with the 2C5 antibody cancer-targeted micellar form of DM-PIT-1. In conclusion, DM-PIT-1 micellar preparations can be used for targeted combination therapy against TRAIL-resistant cancers.
Introduction
We have recently identified a small molecule N-{[(2- hydroxy-5-nitrophenyl)amino] carbonothioyl}- 3, 5-dimethylbenzamide (DM-PIT-1), the first-in-class small molecule non-lipid inhibitor of PIP3/PH domain interaction, demonstrating high apoptotic activity, especially against PTEN-deficient cancer cells (Skidan et al. Citation2008b). Despite this selectivity and high ability to induce the apoptosis in cancer cells both in vitro and in vivo, suppress the migration of cancer and endothelial cell in vitro, and inhibit metastatic process and tumor angiogenesis in vivo (Skidan et al. Citation2007), therapeutic potential of DM-PIT-1 may be limited because of its very poor aqueous solubility.
One of the techniques commonly used to overcome the problem of poor solubility of new compounds is to increase its solubility by chemical modifications (Safavy et al. Citation2007). Another standard approach is the use of a water-miscible co-solvent (Scripture, Figg, and Sparreboom Citation2005; Kawakami et al. Citation2006; Rajebahadur et al. Citation2006). However, chemical modifications of small molecules often result in lost biological activity, while co-solvents are usually not physiological inert and provoke a variety of toxic side-effects (Masini et al. Citation1985; Gelderblom et al. Citation2001; van Zuylen et al. Citation2001). An alternative approach suggests using various colloidal drug delivery systems providing improved drug solubilization and stability (Mallick et al. Citation2007).
In this regard, polymeric micelles have emerged as an attractive colloidal nanosized drug delivery system (Torchilin Citation2007). Micelles prepared from conjugates of polyethylene glycol (PEG) and diacyllipids, such as phosphatidylethanolamine (PE), are of particular interest because the use of lipid moieties as hydrophobic blocks forming the micelle core allows for an efficient solubilization of poorly soluble substances and provides aqueous stability to the micelles (Lukyanov et al. Citation2002). Small size of PEG-PE-micelles facilitates their passive targeting into different pathological tissues, such as tumors, via the enhanced permeability and retention (EPR) effect (Kwon et al. Citation1995; Maeda Citation2001). Active targeting of such drug-loaded micelles can be achieved using various specific ligands, such as peptides (Sethuraman and Bae Citation2007), proteins (Lee et al. Citation2007), folic acid (Yuan et al. Citation2008), and monoclonal antibodies (Elbayoumi et al. Citation2007), attached to the micelle surface via reactive groups, such as p-nitrophenylcarbonyl group (Torchilin et al. Citation2001, Citation2003), incorporated into the micelle corona.
Therefore, we hypothesized that the cytotoxicity against cancer cells of DM-PIT-1 will be significantly enhanced by its entrapment into PEG-PE-based polymeric micelles, which, if needed, can be additionally modified with tumor cell-specific targeting ligand. DM-PIT-1-loaded PEG-PE micelles may also be used in combination with other anticancer agents to achieve higher efficacy. In this study, we have also investigated the possibility of the combined action onto cancer cells of the micellar DM-PIT-1 and Tumor necrosis factor-Related Apoptosis-Inducing Ligand (TRAIL, a cytokine of the TNFα family). TRAIL was chosen for its promising specific tumoricidal activity (Kelley and Ashkenazi Citation2004; Pei et al. Citation2004) due to the selective upregulation of TRAIL-dependent apoptotic signaling in the transformed, but not in normal cells (Pan et al. Citation1997a,Citationb; Sheridan et al. Citation1997). However, the development of the TRAIL-based anticancer therapeutics for human use revealed two serious limitations. First, TRAIL was found to be toxic towards normal human liver cells (Jo et al. Citation2000), necessitating the development of strategies to target it more specifically to cancer cells. Second, different types of tumor cells have developed multiple ways to evade TRAIL-mediated toxicity through the upregulation of intracellular antiapoptotic signaling (Guo et al. Citation2002; Ballestrero et al. Citation2003; Chawla-Sarkar et al. Citation2004).
In the present study, we have also investigated the cytotoxic activity of DM-PIT-1-loaded PEG-PE micelles modified with the monoclonal antibody 2C5 (mAb 2C5), specifically recognizing a broad variety of cancer cells via the cancer cell surface-bound nucleosomes released from the apoptotically dying neighboring cancer cells (Iakoubov and Torchilin Citation1997). With all this in mind, we have prepared DM-PIT-1-loaded PEG-PE-based micelles additionally conjugated with human recombinant soluble TRAIL and/or cancer-specific mAb 2C5 and investigated their effect on TRAIL-sensitive and TRAIL-resistant human and murine cancer cell lines.
Materials and methods
Materials
1,2-Disteratoyl-sn-glycero-3-phosphoethanolamine- N-[methoxy(polyethylene-glycol)-2000] (PEG-PE), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and phosphatidyl-ethanolamine lissamine rhodamine B (Rh-PE) were purchased from Avanti Polar Lipids (Birmingham, AL) and used without further purification. Triethylamine (TEA) was from Sigma-Aldrich Chemical Co. (St. Louis, MO). Polyethyleneglycol-diphenyl carbonate (PEG-(NPC)2, MW 3400, was from New SunBio (Orinda, CA). Purified monoclonal antibody 2C5 (mAb 2C5) was prepared by Harlan Bioproducts for Science (Indianapolis, IN) using the hybridoma cell line from our laboratory. Recombinant His6-tagged human TRAIL was produced in E.coli and purified using Ni2+ chromatography as previously described (Pan et al. Citation1997a,Citationb). The bacterial expression vector was a generous gift of Dr V. Dixit (Genentech). Polyvinylidene difluoride dialysis membranes with molecular weight cut-off sizes of 100 and 250 kDa were from Spectrum Laboratories Inc. (USA). Cell culture media and supplements were from CellGro (Kansas City, MO). All other reagents and buffer solution components were analytical grade preparations. Distilled and deionized water was used in all experiments. Human glioblastoma cell line U-87 MG (ATCC, HTB-14, TRAIL-resistant), murine melanoma cell line B-16, F1 (ATCC, CRL-6323), and murine metastatic breast cancer cells 4T1 (ATCC, CRL-2539) were purchased from the American Type Culture Collection (Manassas, VA).
Preparation and characterization of DM-PIT-1-loaded PEG-PE micelles
Synthesis of pNP-PEG3400-PE
pNP-PEG-PE was synthesized according to the earlier published procedure (Torchilin et al. Citation2001). Briefly, 800 mg of PEG-(NPC)2 was dissolved in chloroform to obtain a 200 mg/ml solution. A further 1.3 ml DOPE (25 mg/ml) was added into the solution. After further addition of 25 μl of TEA, the sample was incubated overnight at room temperature (RT) with stirring under argon. A rotary evaporator was used for the removal of organic solvents. A water bath at RT was used for the formation of pNP-PEG-PE micelles in 0.001 M HCl. Subsequently, a Sepharose CL-4B column (2.5 × 50 cm) was used to separate the micelles from the unbound PEG and free pNP using 0.001 M HCl as an eluent. Pooled fractions containing pNP-PEG-PE were freeze-dried. Finally, pNP-PEG-PE micelles was extracted with chloroform and stored as a chloroform solution at −80°C.
Preparation of DM-PIT-1-loaded PEG-PE micelles
PEG-PE was dissolved in chloroform at 20 mg/ml. A DM-PIT-1 stock solution at 0.5 mg/ml was obtained by dissolving the DM-PIT-1 in acetonitrile. Two milliliters of DM-PIT-1 stock solution was added to 1.25 ml of a chloroform solution of PEG-PE. The organic solvents were removed by the rotary evaporation to form a thin film of drug/micelle material mixture. This film was further dried under high vacuum overnight to remove any traces of remaining solvent. To form the micelles, the film was re-hydrated in a 10 mM HEPES buffer saline, pH 7.4, and sonicated for 10 min. The non-entrapped, precipitated DM-PIT-1 was removed by filtration through a 0.22 μm filter (Fisher Scientific, USA).
Preparation of the surface-modified PEG-PE micelles
Two milliliters from the stock solution of DM-PIT-1 (0.5 mg/ml) were added to a mixture of PEG-PE and p NP-PEG-PE (95:5 molar ratio) in chloroform and DM-PIT-1-loaded PEG-PE-based micelles prepared as above. One milliliter of a 12 μM solution of mAb 2C5 or TRAIL (1 mg/ml) in PBS buffer, pH 7.4, was added to 1 ml (for the attachment of mAb 2C5 antibody) or 2 ml (for the attachment of TRAIL) of drug-loaded pNP-PEG-PE-containing micelles at the total PEG-PE concentration of 2.2 mM. The pH of the final mixture was adjusted to 8.5–8.8. The incubation for 3 hr at RT resulted in a sufficient mAb 2C5 or TRAIL binding and simultaneous hydrolysis of non-reacted pNP groups. The mAb2C5-antibody- or TRAIL-containing micelles were separated from the unbound mAb mAb2C5 or TRAIL by the dialysis against 10 mM HEPES-buffered saline, pH 7.4, using polyvinylidene difluoride dialysis membranes (MWCO 250 and 100 kDa, respectively, Spectrum Laboratories, Inc. CA). Control micelles were prepared using the same lipid components and in the same concentration as DM-PIT-1-loaded micelles but contained no DM-PIT-1.
The immunological activity of the micelle-attached mAb 2C5 was estimated by the standard enzyme-linked immunosorbent assay (ELISA) using the water-soluble fraction of calf thymus nucleohistone (Worthington Biochemical, Lakewood, USA) as an antigen and horseradish peroxidase/anti-mouse IgG conjugate (ICN Biomedical, Aurora, USA) as a secondary antibody. Anti-mouse IgG/peroxidase conjugate was quantified by the degradation of diaminobenzidine supplied as a ready-for-use solution (Neogen, Lexington, USA). The intensity of the color developed was analyzed by an ELISA reader, Labsystems Multiscan MCC/340 (Labsystems and Life Sciences International, Helsinki, Finland).
The association of TRAIL with the surface of polymeric micelles was confirmed by dissolving extensively dialyzed micelles in SDS loading buffer (50 mM Tris HCl, pH 6.8, 100 mM DTT, 2% SDS, 12.5% glycerol, 0.1% bromophenol blue), and SDS gel-electrophoresis, followed by Coomassie Blue staining (GelCode Blue reagent, Pierce, Rockford, USA).
Quantification of DM-PIT-1 in micelles by HPLC
The amount of DM-PIT-1 in the micellar preparations was determined by the isocratic reverse-phase HPLC (Hitachi, Elite La Chrome, Tokyo, Japan). Before HPLC analyses, the DM-PIT-1-loaded micelles were destroyed by acetonitrile and then centrifuged at 14,000 g for 5 min and the amount of DM-PIT-1 was determined. The analysis was performed on a C-18 column (Richard Scientific, Novato, USA) with the photodiode-array detection (L-2455, Hitachi, Elite La Chrome, Tokyo, Japan). The elution was performed with an isocratic mobile phase consisting of acetonitrile and water (70:30) with 0.1% formic acid (v/v). The flow rate was 1 ml/min and the total run time was 10 min. DM-PIT-1 were detected by the UV absorbance (320 nm). Detection and quantification limits of DM-PIT-1 in acetonitrile were below 5 and 10 ng/mL, respectively. The calibration curve studied in the range of 10 to 1000 ng/mL was linear, R2 = 0.9994. The retention time for DM-PIT-1 was 6.3 min. All samples were analyzed in duplicate. Drug fraction in micelles (wt%) was calculated as the amount of DM-PIT-1 in micelles divided by the amount of the polymer used in micelle preparation.
Micelle properties
The mean size of different micelles was measured by the dynamic light scattering (DLS) with a scattering angle of 90° at 25°C using a N4 Plus Submicron Particle System (Coulter Corporation, Miami, FL). The micelle suspensions were diluted with a 10 mM HEPES buffer saline, pH 7.4, until a concentration providing a light scattering intensity of 5 × 104 to 1 × 106 counts was achieved. The measurements were done in triplicate.
Micelle surface charge analysis was performed using a Zeta Phase Analysis Light Scattering (PALS) UltraSensitive Zeta Potential Analyzer instrument (Brookhaven Instruments, Holtsville, USA). Each sample of micelle suspension was diluted with deionized distilled water to have the signal intensity within the limits required by the instrument. The measurements were done in triplicate.
The stability of the micelles was monitored by the changes in particle size in the micellar samples during the storage period of 72 hr and 1 week at 4°C. To test the stability of micelles in the presence of blood serum components, DM-PIT-1-loaded micelles were incubated with 0.2% bovine serum albumin (BSA) at the PEG-PE concentration of 4 mM for 96 hr at RT. The samples were diluted with HEPES-buffered saline, pH 7.4, and analyzed for the changes in the micelle size as above.
Cell viability experiments
Human U87MG glioblastoma cells were plated at a density of 1 × 104 cells/well in 96-well plates (Corning, Inc., Corning, USA). Cells were cultured in DMEM10 medium (supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin) and maintained at 37°C in a 5% CO2 atmosphere. After the overnight incubation, the medium was replaced with medium containing formulated DM-PIT-1 at concentrations ranging from 1 to 200 μg/ml of DM-PIT-1. Then the cells were incubated at 37°C, 5% CO2 for an additional 24, 48, or 72 hr. The cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) method (CellTiter96®AQueous One Solution Cell Proliferation Assay, Promega, Madison, USA), or using the CellTiter-Glo ATP Viability assay (Promega, Madison, USA) according to manufacturer’s instructions.
Statistical analysis
The results were expressed as a mean ± standard deviation (SD). Statistical analysis was performed using the Student’s t-test, with a p-value of less than 0.05 considered statistically significant.
Results and discussion
Preparation and characterization of micellar DM-PIT-1
DM-PIT-1-loaded PEG-PE micelles were prepared using the thin-film hydration method. The incorporation of DM-PIT-1 into these polymeric micelles significantly increases its solubility. If free DM-PIT-1 precipitates from the aqueous solution already at ~ 30 μM, the micellar drug stays in solution even at the concentration of above 4 mM.
For the detection and quantification of DM-PIT-1 in solubilized/micellar forms we used the reversed-phase HPLC method. The calibration curve for the quantification of DM-PIT-1 was linear over standard DM-PIT-1 concentrations of 10 to 1000 ng/ml with the orrelation coefficient R2 = 0.999. The retention time of DM-PIT-1 was approximately 6.3 min (). This HPLC method is a simple and straightforward way to determine DM-PIT-1 concentration in micellar formulations.
Figure 1. Reverse phase HPLC profile of N-{[(2-hydroxy-5-nitrophenyl)amino]carbonothioyl}-3,5-dimethylbenzamide (DM-PIT-1) in polymeric micelles made of PEG-PE. Analysis was performed on a C-18 column with photodiode-array detection. DM-PIT-1 was detected by the UV absorbance (320 nm). The retention time for DM-PIT-1 was approximately 6.3 min.
![Figure 1. Reverse phase HPLC profile of N-{[(2-hydroxy-5-nitrophenyl)amino]carbonothioyl}-3,5-dimethylbenzamide (DM-PIT-1) in polymeric micelles made of PEG-PE. Analysis was performed on a C-18 column with photodiode-array detection. DM-PIT-1 was detected by the UV absorbance (320 nm). The retention time for DM-PIT-1 was approximately 6.3 min.](/cms/asset/283482d3-f2c5-4ef9-9fb7-c0fc8f2e68e6/idrd_a_351963_f0001_b.gif)
We have obtained several different preparations of DM-PIT-1-loaded PEG-PE-based micelles including those modified with mAb 2C5 or TRAIL. Some properties of these preparations are presented in . As one can see from , loading with DM-PIT-1 slightly increases the size of plain PEG-PE micelles, while the further attachment of mAb 2C5 or TRAIL to drug-loaded micelles does not have any effect on the micelle size: all DM-PIT-1-loaded micelles had a size of ca. 14 to 16 nm and a narrow size distribution. All PEG-PE-based micelles have a slightly negative surface zeta potential of 4 to 5 mV. All DM-PIT-1-loaded micelles contained approximately 1 to 2 wt% of the drug.
Table 1. Some properties of micellar formulations.
DM-PIT-1-loaded PEG-PE micelles were stable in aqueous dispersions exhibiting no aggregation for up to 1 week. Micelle incubation in 0.2% BSA at 4°C for 96 hr also did not provoke any changes in micelle size, i.e. no micelle aggregation/destabilization occurs ().
Table 2. Stability of DM-PIT-1-loaded micelles upon storage.
The latter result is in good agreement with earlier findings, which showed that PEG-PE micelles were stable in the presence of blood serum components (Roby, Erdogan, and Torchilin Citation2006).
mAb 2C5- and TRAIL-modified DM-PIT-1-loaded micelles
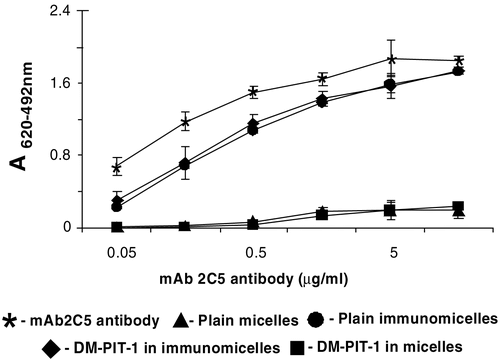
The ELISA experiments clearly demonstrated that mAb 2C5 attached to the surface of DM-PIT-1-loaded micelles to a major extent retains its specific activity required for the successful targeting of drug-loaded micelles to cancer cells ().
Figure 2. ELISA results for different micellar preparations compared to the non-modified mAb 2C5. Data represent the mean ± SD of triplicate samples.
The same coupling chemistry as was used for attaching mAb 2C5 to PEG-PE micelles via pNP groups allowed for the successful coupling of TRAIL with drug-loaded micelles. This association of TRAIL with PEG-PE micelles was determined by the standard gel-electrophoresis procedures. The amount of TRAIL attached to the surface of polymeric micelles was approximately 7% of initially added. As shown in , the intense TRAIL band in TRAIL-modified PEG-PE micelles confirms good association of the protein with micelles.
Figure 3. Gel-electrophoresis of TRAIL preparations. (1) TRAIL attached to micelles via pNP-PEG-PE moities, (2) soluble TRAIL (control). Ten microliters of TRAIL-modified PEG-PE micelles were loaded on 12% SDS-PAGE and stained using GelCode® Blue Stain Reagent. Control lane contains 50 ng of the recombinant TRAIL used for conjugation.
Cytotoxicity of micellar DM-PIT-1 alone or in combination with TRAIL
The cytotoxic action of DM-PIT-1-loaded PEG-PE micelles on murine B-16 melanoma and 4T1 metastatic breast carcinoma cells after different incubation time with different concentration of micellar preparations (as DM-PIT-1) is shown in . It could be clearly seen that micellar DM-PIT-1 effectively kills both cancer cells at concentrations around 100 μM. Taking into account the fact that the aqueous solubility of free DM-PIT-1 is under 30 μM, only the micellar form of the drug can be really effective. Naturally, the efficacy of drug actions increases with the increase in the incubation time.
Figure 4. In vitro cytotoxicity of micellar DM-PIT-1 against murine B-16 melanoma (left) and 4T1 metastatic breast cancer cells (right). Cancer cells were incubated with micellar DM-PIT-1 for 24, 48, and 72 hr. Data represent the mean ± SD of three experiments and are expressed relative to untreated controls.
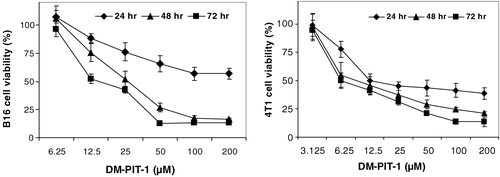
Other tested cell lines, such as human prostate carcinoma LNCaP, breast adenocarcinoma MCF-7, glioblastoma U87MG, and breast carcinoma MBA-MD-231, demonstrate similar sensitivity towards the micellar DM-PIT-1 (data not shown).
In order to check if the cytotoxicity of DM-PIT-1 in PEG-PE micelles can be further improved by combining it with TRAIL (TRAIL-modified DM-PIT-1-loaded PEG-PE micelles), TRAIL-resistant U87MG cells have been used. The data presented in show that, while drug-free PEG-PE micelles modified with TRAIL are not cytotoxic for U87MG cells and DM-PIT-1-loaded PEG-PE micelles demonstrate cytotoxicity within the earlier established range, the additional attachment of TRAIL to drug-loaded micelles results in the synergistic effect and leads to significantly enhanced cell death. The viability of U87MG cells was significantly lower in all groups treated with TRAIL-modified DM-PIT-1-loaded PEG-PE micelles than in groups treated with the plain DM-PIT-1-loaded micelles groups (p < 0.01).
Figure 5. Toxicity of the micellar DM-PIT-1 and its combination with TRAIL against TRAIL-resistant U87MG cells. Cells were treated with indicated amounts (in μM) of plain micelles modified with TRAIL (1); micellar DM-PIT-1 (2), or micellar DM-PIT-1 modified with TRAIL (3). The amount of lipid in all wells was kept constant at 2.5 mg/ml by adding empty micelles. Cell viability was determined after 24 hr using the MTT assay. Data represent the mean ± SD of three independent experiments and are expressed relative to the untreated control (* p < 0.05, ** p < 0.01).
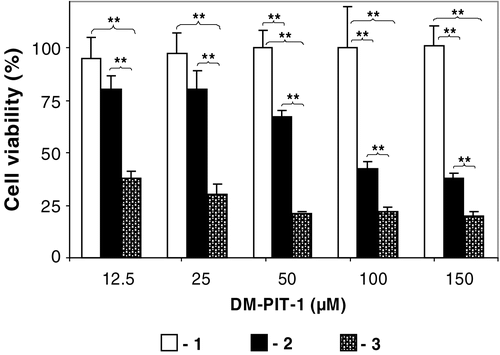
These data show that the combination of DM-PIT-1 and TRAIL in one micellar preparation provides a convenient method for their simultaneous delivery to cancer cells. These results offer a new approach that should be beneficial for a broad range of both TRAIL-sensitive and TRAIL-resistant cancers.
Drug-loaded micelles modified with mAb 2C5
We have also investigated if the modification of DM-PIT-1- loaded PEG-PE micelles with cancer cell- specific mAb 2C5 can still further enhance their cytotoxicity. The data presented in clearly show that the attachment of mAb 2C5 to DM-PIT-1-loaded micelles significantly increased the cytotoxicity of the micellar DM-PIT-1 over the whole investigated DM-PIT-1 dose range (p < 0.05).
Figure 6. (A) The cytotoxicity towards U87MG cells of mAb 2C5-modified DM-PIT-1-loaded PEG-PE micelles (1) compared to mAb 2C5-free preparation (2); (B) Same as in (A) but in the presence of the soluble TRAIL at 50 ng/ml. Cell viability was determined using the MTT assay. Viability of cells in the presence of plain micelles, immunomicelles, and TRAIL was observed at comparable concentrations of drug-loaded particles. Data represent the mean ± SD of three independent experiments and are expressed relative to the untreated control (* p < 0.05, ** p < 0.01).
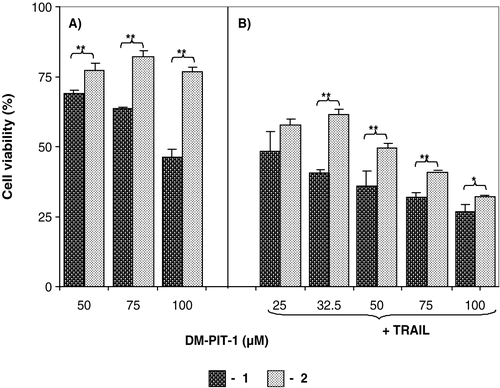
The increase in the efficacy of immunomicellar DM-PIT-1 in immunomicelles is explained by the higher quantity of DM-PIT-1-containing micelles associated with the cells surface because of specific interaction of mAb 2C5 antibody with cancer cells. These results are in good agreement with earlier published data on the increased cytotoxicity of mAb 2C5-modified drug-loaded micelles against cancer cells (Roby et al. Citation2006; Elbayoumi et al. Citation2007; Skidan et al. Citation2008).
Interestingly, it looks like the use of DM-PIT-1 can overcome TRAIL resistance in resistant cells (U87MG cells), since the addition of a soluble TRAIL into the system quite significantly increases the cytotoxicity of both mAb 2C5-modified and non-modified DM-PIT-1-loaded PEG-PE micelles (still the maximum effect being achieved in the case of mAb 2C5 targeted immunomicelles), see Figure 6B.
In summary, a novel proapoptotic agent poorly soluble in water, DM-PIT-1, was successfully formulated into PEG-PE-based micelles and demonstrated high in vitro cytotoxicity against various cancer cells. This cytotoxicity can be still further increased by additional modification of DM-PIT-1-loaded micelles with TRAIL or with cancer-specific monoclonal antibody. Such preparations can be used for targeted combination therapy against TRAIL-resistant cancers. Testing these preparations in vivo is the subject of our current research.
Acknowledgment
This study was supported in part by the NIH grant R01-EB01961 to Vladimir Torchilin.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ballestrero A, Montemurro F, Gonella R, Capaldi A, Danova M, Friedman D, Puglisi M, Aglietta M, Patrone F 2003. Dose-dense vinorelbine and paclitaxel with granulocyte colony-stimulating factor in metastatic breast cancer patients: anti- tumor activity and peripheral blood progenitor cell mobilization capability. Breast Cancer Res Treat. 82, 185–190.
- Chawla-Sarkar M, Bae SI, Reu FJ, Jacobs BS, Lindner DJ, Borden EC 2004. Downregulation of Bcl-2, FLIP or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 11, 915–923.
- Elbayoumi TA, Pabba S, Roby A, Torchilin VP 2007. Antinucleosome antibody-modified liposomes and lipid-core micelles for tumor-targeted delivery of therapeutic and diagnostic agents. J. Liposome Res. 17, 1–14.
- Gelderblom H, Verweij J, Nooter K, Sparreboom A 2001. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 37, 1590–1598.
- Guo F, Nimmanapalli R, Paranawithana S, Wittman S, Griffin D, Bali P, O’Bryan E, Fumero C, Wang HG, Bhalla K 2002. Ectopic overexpression of second mitochondria- derived activator of caspases (Smac/DIABLO) or cotreatment with N-terminus of Smac/DIABLO peptide potentiates epothilone B derivative-(BMS 247550) and Apo-2L/TRAIL-induced apoptosis. Blood 99, 3419–3426.
- Iakoubov LZ, Torchilin VP 1997. A novel class of antitumor antibodies: Nucleosome-restricted antinuclear autoantibodies (ANA) from healthy aged nonautoimmune mice. Oncol. Res. 9, 439–446.
- Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR, Strom SC 2000. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat. Med. 6, 564–567.
- Kawakami K, Oda N, Miyoshi K, Funaki T, Ida Y 2006. Solubilization behavior of a poorly soluble drug under combined use of surfactants and cosolvents. Eur. J. Pharm. Sci. 28, 7–14.
- Kelley SK, Ashkenazi A 2004. Targeting death receptors in cancer with Apo2L/TRAIL. Curr. Opin. Pharmacol. 4, 333–339.
- Kwon GS, Naito M, Yokoyama M, Okano T, Sakurai Y, Kataoka K 1995. Physical entrapment of adriamycin in AB block copolymer micelles. Pharm. Res. 12, 192–195.
- Lee H, Hu M, Reilly RM, Allen C 2007. Apoptotic epidermal growth factor (EGF)-conjugated block copolymer micelles as a nanotechnology platform for targeted combination therapy. Mol. Pharm. 4, 769–781.
- Lukyanov AN, Gao Z, Mazzola L, Torchilin V 2002. Polyethylene glycol-diacyllipid micelles demonstrate increased accumulation in subcutaneous tumors in mice. Pharm. Res. 19, 1424–9.
- Maeda H 2001. The enhanced permeability retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv. Enzyme Regul. 41, 189–207.
- Mallick S, Pattnaik S, Swain K, De PK 2007. Current perspectives of solubilization: potential for improved bioavailability. Drug Dev. Ind. Pharm. 33, 865–873.
- Masini E, Planchenault J, Pezziardi F, Gautier P, Gagnol JP 1985. Histamine-releasing properties of Polysorbate 80 in vitro and in vivo: correlation with its hypotensive action in the dog Agents Actions 16, 470–477.
- Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM 1997a. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277, 815–818.
- Pan G, O’Rourke K, Chinnaiyan A, Gentz R, Ebner R, Ni J, Dixit VM 1997b. The receptor for the cytotoxic ligand TRAIL. Science 276, 111–113.
- Pei Z, Chu L, Zou W, Zhang Z, Qiu S, Qi R, Gu J, Qian C, Liu X 2004. An oncolytic adenoviral vector of Smac increases antitumor activity of TRAIL against HCC in human cells and in mice. Hepatology 39, 1371–1381.
- Rajebahadur M, Zia H, Nues A, Lee C 2006. Mechanistic study of solubility enhancement of nifedipine using vitamin E TPGS or solutol HS-15. Drug Deliv. 13, 201–206.
- Roby A, Erdogan S, Torchilin V 2006. Solubilization of poorly soluble PDT agent, meso-tetraphenylporphin, in plain or immunotargeted PEG-PE micelles results in dramatically improved cancer cell killing in vitro. Eur. J. Pharm. Biopharm. 62, 235–240.
- Safavy A, Raisch K, Mantena S, Sanford L, Sham S, Krishna N, Bonner J 2007. Design and development of water-soluble curcumin conjugates as potential anticancer agents. 50, 6284–6288.
- Scripture C, Figg WD, Sparreboom A 2005. Paclitaxel chemotherapy: from empiricism to a mechanism-based formulation strategy. Ther. Clin. Risk Manag. 1, 107–114.
- Sethuraman V, Bae YH 2007. TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J. Contr. Rel. 118, 216–224.
- Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, Goddard AD, Godowski P, Ashkenazi A 1997. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277, 818–821.
- Skidan I, Degterev A, Torchilin V 2007. Micellar form of novel poorly soluble proapoptotic agent, Dimethyl-PIT. 2007. 34th Annual CRS meeting, Long Beach, CA, July 7–11, 2007, Abstract #247.
- Skidan I, Dholakia P, Torchilin V 2008a. Photodynamic therapy of experimental B-16 melanoma in mice with tumor-targeted 5,10,15,20-tetraphenylporphin-loaded PEG-PE micelles. J. Drug Target. 16, 486–493.
- Skidan I, Dholakia P, Degterev A, Torchilin V 2008b. PEG-PE micelles with novel poorly-soluble proapoptotic agent: activity in vivo. 35th Annual CRS meeting, New York, July 12–16, Abstract #648.
- Torchilin V 2007. Micellar nanocarriers: pharmaceutical perspectives. Pharm. Res. 24, 1–16.
- Torchilin V, Levchenko T, Lukyanov A, Khaw B, Klibanov A, Rammohan R, Samokhin G, Whiteman K 2001. p-Nitrophenylcarbonyl-PEG-PE-liposomes: fast and simple attachment of specific ligands, including monoclonal antibodies, to distal ends of PEG chains via p-nitrophenylcarbonyl groups. Biochim. Biophys. Acta 1511, 397–411.
- Torchilin V, Lukyanov A, Gao Z, Papahadjopoulos-Sternberg B 2003. Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proc. Natl. Acad. Sci. USA 100, 6039–6044.
- van Zuylen L, Karlsson MO, Verweij J, Brouwer E, de Bruijn P, Nooter K, Stoter G, Sparreboom A 2001. Pharmacokinetic modeling of paclitaxel encapsulation in Cremophor EL micelles. Cancer Chemother. Pharmacol. 47, 309–318.
- Yuan H, Miao J, Du YZ, You J, Hu FQ, Zeng S 2008. Cellular uptake of solid lipid nanoparticles and cytotoxicity of encapsulated paclitaxel in A549 cancer cells. Int. J. Pharm. 348, 137–145.