Abstract
To achieve the effective intracellular delivery of siRNA and silence specific genes, various types of conjugates between cell-penetrating peptides (CPPs; Transportan, Penetratin, Tat) and cationic peptides were developed. Uptake, intracellular localization, cytotoxicity, and biological activity of siRNA were significantly dependent on the kind of CPP used and the length of the cationic peptides in the conjugate. Transportan-based conjugates yielded both high internalization of siRNA and strong gene silencing activity, while Penetratin- and Tat-based conjugates did not. These different properties of CPPs emphasize the importance of careful peptide selection and design when attempting the application of CPP technology.
Introduction
Various oligonucleotide derivatives have demonstrated remarkable potential for therapeutic application as gene regulators (CitationPatil et al., 2005). In particular, small interfering RNA (siRNA; chemically synthesized RNA duplexes of 19–23 residues) is widely recognized as a powerful tool for targeted gene silencing by RNA interference (CitationElbashir et al., 2001). The major limitations on the use of siRNA in vitro and in vivo are its poor cellular internalization and chemical instability, and these problems have led to the development of various vehicles (CitationPaula et al., 2007).
Since the discovery of the cell membrane translocation activity of Penetratin (Pen) (CitationDerossi et al., 1994), various cell-penetrating peptides (CPPs), which are usually basic, hydrophobic, and/or amphipathic, have shown great potential for the delivery of bioactive molecules such as proteins, peptides, and nucleic acids both in vitro and in vivo (CitationPujals et al., 2006; CitationPatel et al., 2007). However, the mechanism responsible for the translocation of CPPs and their cargos across the cell membrane remains unclear. It seems likely that both the translocation process and the intracellular trafficking pathway are influenced by various factors, such as the cell type, the physicochemical properties of the cargo, the concentration and type of CPP, and the vehicle linked with the CPP (CitationEl-Andaloussi et al., 2007). Recent studies have suggested that CPPs interact electrostatically with proteoglycans on the cell surface and then are internalized by endocytosis (CitationAbes et al., 2007).
CPPs are likely to be a promising tool for the delivery of siRNA to cells. Vehicles such as liposomes (CitationZhang et al., 2006), dendrimers (CitationKang et al., 2005), and MEND (multifunctional envelope-type nano device) (CitationNakamura et al., 2007) can be covalently bound to CPPs to enhance the cellular uptake of siRNA and the silencing of specific genes.
Some researchers have reported that the formation of covalently bound conjugates between CPPs and siRNA offers another potential delivery strategy. For this strategy to work, one molecule of a CPP [Pen (CitationDavidson et al., 2004; CitationMuratovska and Eccles, 2004; CitationTurner et al., 2007), transportan (CitationMuratovska and Eccles, 2004), or Tat (CitationTurner et al., 2007; CitationChiu et al., 2004)] should be linked to one siRNA molecule. However, the ease with which CPPs bind to siRNA makes this approach difficult to utilize (CitationMeade and Dowdy, 2007). The intra/inter-molecular ionic interaction of basic CPPs with siRNA leads to impairment of translocation because positive charges on the CPPs are crucial for their cellular uptake (CitationMeade and Dowdy, 2007; CitationJiang et al., 2004). This approach is also difficult to employ because it is necessary to synthesize siRNA with a reactive site (e.g. a thiol group) and purify the conjugates from a mixture of siRNA, CPPs, and ionic complexes without denaturizing the siRNA.
Another method that has been proposed involves the formation of non-covalent electrostatic bonds between siRNA and CPPs, including arginine-rich peptides (CitationUnnamalai et al., 2004; CitationWang et al., 2007), MPG peptide (CitationSimeoni et al., 2003) and its analogue (CitationVeldhoen et al., 2006), and a Pen analogue (CitationLundberg et al., 2007). The preparation of such CPP-siRNA complexes is technically simple, because these are formed through electrostatic interaction in an aqueous solution and there is no need for purification or for the synthesis of chemically modified siRNA. Although some researchers have demonstrated successful delivery of siRNA into cells using this method, the non-covalent complexes may reduce the translocation efficiency of CPPs, which must simultaneously perform the two different roles of membrane translocation and stable siRNA retention. In the present study, cationic peptides (CPs) of different lengths were covalently linked to CPPs to serve as preferential siRNA-binding sites that would not interfere with translocation. We selected three of the most extensively investigated CPPs, Transportan10 (TP) (CitationSoomets et al., 2000), Pen (CitationDerossi et al., 1994), and Tat (CitationVivès et al., 1997), and assessed their potential as siRNA carriers after conjugation with CPs.
Materials and methods
Materials
Fluorescein-labeled double-stranded RNA oligomer (Block-iT™ Fluorescence Oligo) was purchased from Invitrogen Co. (Carlsbad, CA). As noted in the manufacturer’s instructions, the sequence of BLOCK-iT™ Fluorescent Oligo is not homologous with any known gene, thus avoiding the induction of any cellular response to introduction of the oligomer itself. SiRNAs designed to target GL3 luciferase mRNA (Control siRNA duplex Firely Luciferase GL3) and designed to minimize sequence homology with any known vertebrate transcript (Stealth™RNAi Negative Control Medium GC Duplex) were purchased from Nippon Gene Co., Ltd. (Tokyo, Japan) and Invitrogen Co., respectively. Synthetic peptides were purchased from Sigma-Aldrich Japan (Tokyo, Japan) and were purified by HPLC with an ODS-80TS column (Tosoh Co., Tokyo, Japan). The sequences of the three CPPs were as follows: TP, AGYLLGKINLKALAALAKKILXC; Pen, RQIKIWFQNRRMKWKKXC; and HIV Tat peptide 48-60 (Tat), GRKKRRQRRRPPQXC. CPs had the following sequences: K4, CKKKK; K6, CKKKKKK; K10, CKKKKKKKKKK; K12, CKKKKKKKKKKKK; and H6K6, CHHHHHHKKKKKK. All of the peptides had a C-terminal free acid. ‘X’ in the sequences means aminohexanoic acid, which was used as a spacer molecule.
Plasmid DNA (pCMV-luciferase) and Lipofectamine™ 2000 (LF2000) were purchased from Promega Co. (Madison, WI) and Invitrogen, respectively.
Synthesis of peptide conjugates
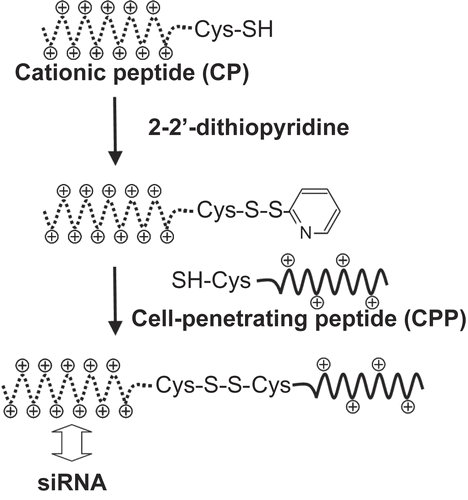
CPPs were conjugated with CPs through a disulfide linkage (). CPs with cystein at the N-terminal (5 mg) were incubated with 2-2’-dithiopyridine (5 mg) in 0.5 ml of a water/acetonitrile mixture (3:1 v/v) for 1 hr at 25°C. After washing with diethyl ether, the activated CPs were purified by HPLC with an ODS-80TS column and then freeze-dried. Next, the activated CPs and CPPs with cystein at the C-terminal were incubated together in water for 1 hr at 25°C. The resulting CPP-CP conjugates were purified by HPLC on an ODS-80TS column and then freeze-dried. The conjugates were finally characterized by MALDI-TOF mass spectrometry.
Figure 1. Synthesis of a conjugate between a cell-penetrating peptide (CPP) and a cationic oligolysine peptide (CP) via a disulfide linkage.
Formation of complexes between siRNA and peptide conjugates
Various amounts of CPP-CP conjugates were mixed with 1.4 μm fluorescein-labeled siRNA in 10 mm phosphate buffer (pH 7.2). After incubation for 30 min at 25°C, the mixture was analyzed by 20% polyacrylamide gel electrophoresis (PAGE) in TBE buffer at 20 mA. Size of complexes was also evaluated. After various amounts of CPP-CP conjugates were mixed with 7.2 μm siRNA in 10 mm phosphate buffer (pH 7.2) and incubated for 30 min at 25°C, the diameter of the complexes was measured by analysis of dynamic light scatter using a Zetasizer Nano ZS (Malvern Instruments Ltd, Worcestershire, UK).
Cell viability assay
FRSK cells (a rat epidermal keratinocyte cell line) were added to a 96-well plate at 2 × 104 cells per well and incubated overnight at 37°C in MEM containing 10% fetal bovine serum (FBS). The cells were washed with phosphate-buffered saline (PBS) and incubated in 100 μl of Opti-MEM I reduced serum medium (Invitrogen). Complexes were formed by mixing various amounts of peptide conjugates and siRNA (Negative control Medium GC Duplex) in phosphate buffer ( 10 mm, pH 7.2), and allowing the mixtures to stand for 30 min at 25°C. Subsequently, FRSK cells were incubated with the complexes for 2 hr at 37°C in Opti-MEM, and cell viability was determined with a Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) based on the metabolism of 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium (WST-8) by living cells.
Internalization of siRNA in cells
The siRNA content of cells after incubation was determined as reported elsewhere (CitationLundberg et al., 2007). FRSK cells were added to a 24-well plate at 12 × 104 cells per well and incubated overnight at 37°C in MEM containing 10% FBS, after which the cells were washed with PBS and incubated in 450 μl of Opti-MEM. Complexes were formed by mixing various amounts of conjugates and fluorescein-labeled siRNA in phosphate buffer ( 10 mm, pH 7.2), after which the mixture was allowed to stand for 30 min at 25°C. The resulting complexes were added to the culture medium and cells were incubated for 2 hr at 37°C, followed by washing with MEM containing FBS and subsequent washing with PBS. Cells were recovered by trypsinization and centrifugation for 5 min at 1000 rpm. After washing the cells with MEM containing FBS and subsequent washing with PBS by centrifugation, the cell pellet was lysed in 300 μl of a 100 mm aqueous solution of NaOH. Next, the lysate was centrifuged for 10 min at 12,000 rpm and the fluorescence intensity of the supernatant was measured by a microplate reader (Wallac 1420 multilabel counter, Perkin Elmer). In addition, the protein content of the lysate was measured by using a DC protein assay kit (BioRad).
The uptake of siRNA by cells incubated in the presence of various endocytosis inhibitors was also determined. Cells were pre-incubated with a number of reagents, including sucrose ( 100 mm), chlorpromazine (30 μm), cytochalasin D (30 μm), nocodazole (33 μm), filipin (5 μg/ml), and colchicine (10 μg/ml), in Opti-MEM for 1 hr at 37°C before addition of the siRNA-conjugate complexes. The cellular content of siRNA was determined according to the method mentioned above.
Fluorescence microscopy
FRSK cells, HTC1 cells (a rat 2-acetylaminofluorene-induced hepatocellular carcinoma cell line) and RAW 264.7 cells (a murine macrophage-like cell line) were used for evaluation of the intracellular localization of siRNA. Cells were added to 8-well chamber slides (Gibco) at 7.5 × 104 cells per well and incubated overnight at 37°C in MEM containing 10% FBS. The cells were washed with PBS and incubated in 450 μl of Opti-MEM. Complexes were formed by mixing the peptide conjugates and fluorescein-labeled siRNA in phosphate buffer ( 10 mm, pH 7.2), after which the mixture was let stand for 30 min at 25°C. After incubation of cells with the resulting complexes for 2 hr at 37°C (final siRNA concentration = 60 nm), the cells were washed twice with MEM containing FBS. The cells were immediately observed by fluorescence microscopy (BZ-9000, Keyence) in MEM without fixation. Cells were also incubated with dextran labeled with tetramethylrhodamine (Mw 10,000, neutral, Molecular Probes) in the presence of complexes, and the internalization of dextran in cells was observed similarly.
Luciferase gene silencing
FRSK cells were added to a 24-well plate at 1.2 × 105 cells per well and incubated overnight at 37°C in MEM containing 10% FBS, after which the cells were washed with PBS and incubated in Opti-MEM. Complexes were formed by mixing conjugates and siRNA (siRNA duplex Firely Luciferase GL3 or Negative control Medium GC Duplex) at a 60-fold molar ratio (peptides/siRNA) in phosphate buffer ( 10 mm, pH 7.2) and allowing the mixture to stand for 30 min at 25°C. The resulting complexes were added to the wells of the plate (final siRNA concentration = 60 nm) and incubated with the cells for 2 hr at 37°C. After washing the cells with MEM containing FBS and subsequent washing with PBS, the cells were incubated in Opti-MEM for 5 hr at 37°C with complexes of LF2000 and pCMV-luciferase plasmid (LF2000-to-plasmid weight ratio = 2.5). After changing the medium to MEM containing FBS, the cells were incubated for a further 18 hr and then luciferase expression in each well was determined by a Luciferase Assay System (Promega) according to the manufacturer’s instructions. LF2000 was also tested as a siRNA carrier instead of the peptide conjugates. The conditions for siRNA delivery were optimized based on the manufacturer’s protocol, and mixing was done at a constant ratio of 2 μg/ml LF2000 to 100 nm siRNA.
Results and discussion
CPPs and CPs were covalently bound through a disulfide linkage between their C- and N-terminal cystein residues, respectively (). The synthesized conjugates were purified by HPLC and characterized by MALDI-TOF mass spectrometry, and conjugates with at least 95% purity were used in this study.
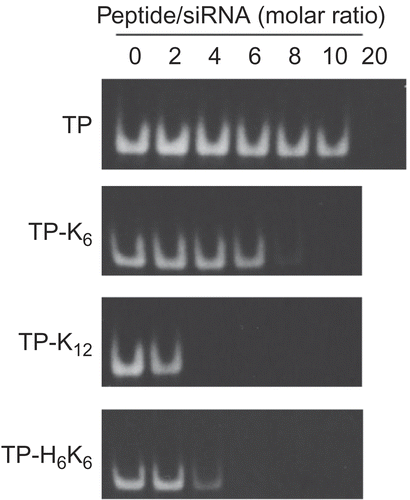
Complex formation between conjugates and fluorescein-labeled siRNA in 10 mm phosphate buffer was evaluated by a gel migration assay (). Detection of siRNA migration on the gels decreased along with an increase in amount of the peptide conjugates added, indicating complex formation between siRNA and each conjugate. The molar ratio between the peptide conjugates and siRNA that was required to achieve complex formation depended on the conjugate, while the charge ratio (positive charges derived from lysine, arginine, and histidine in the conjugates versus negative charges derived from phosphate in siRNA) was almost constant (1~2) for each any conjugate, suggesting the formation of complexes through electrostatic interaction. Despite the presence of the same positive charge on a molecule of TP-H6K6 and one of TP-K12, the former showed a weaker association with siRNA due to the low pKa value (< 6.0) of histidine. Similar results were also obtained with conjugates containing Pen and Tat.
Figure 2. Formation of complexes between TP-CP conjugates and siRNA. Various amounts of TP-CP conjugates and a constant amount of fluorescein-labeled siRNA were mixed in phosphate buffer ( 10 mm, pH 7.2). Migration of siRNA was analyzed by 20% PAGE and fluorescence images of gels were shown.
Formation of complexes between the CPP-CP conjugates and siRNA was also evaluated spectrometrically. Although complexes with most of the peptides underwent precipitation after being let stand for 30 min at 25°C, mixtures that contained Pen-K10, TP-K6, TP-K12, or TP-H6K6 at a 60-fold molar ratio (peptide/siRNA) formed small colloidal particles. The diameter of the complexes formed by adding TP-K12 or Pen-K10 was approximately 200 nm, and that by adding TP-K6 or TP-H6K6 was approximately 600 nm. These results indicated that the complexes had different physicochemical properties, depending on the mixing ratio and the composition of CPPs and CPs.
The cytotoxicity of the siRNA complexes was evaluated by measuring the metabolism of WST-8. Complexes formed by adding the peptides at a 20-fold molar ratio showed no cytotoxicity, and those at a 60-fold molar ratio showed no or slight cytotoxicity (). However, TP-K12 mixed with siRNA at a 60-fold molar ratio had severe cytotoxicity. CitationSaar et al. (2005) have reported that TP peptide shows stronger cytotoxicity than Pen peptide and Tat peptide. Thus, this strong cytotoxicity may be due to both the property of TP and excess positive charge in the conjugate.
Table 1. Cytotoxicity of complexes between peptide conjugates and siRNA.
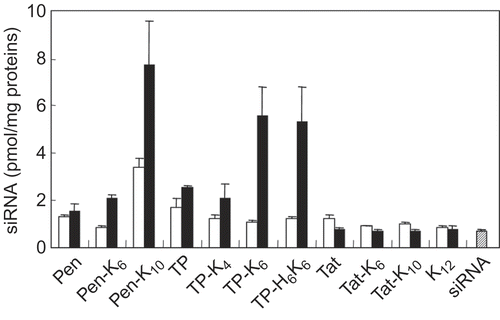
The amount of siRNA internalized by cells was determined by using fluorescein-labeled siRNA. As shown in , CPPs alone (Pen, TP, or Tat) did not enhance the translocation of siRNA into cells. After complex formation with CPP-CP conjugates, the amount of internalized siRNA depended on the kind of CPP, the length of the CPs, and the mixing ratio. It was found that Pen-K10, TP-K6, and TP-H6K6 all significantly enhanced the uptake of siRNA at a 60-fold molar ratio, while Pen-K6, TP-K4, Tat- K6, and Tat-K10 did not. When dextran labeled with tetramethylrhodamine was co-incubated with the complexes, no internalization of dextran in cells was observed. Additionally, K12 peptide also did not enhance the internalization of siRNA. These results indicated that the internalization of siRNA was not due to the potential cytotoxic effect of the complexes at the cell membrane, but due to the CPP properties. Accessibility of the cationic residues on CPPs is crucial for the interaction of these molecules with cells (CitationJiang et al., 2004), and neutralization of the charged CPPs by electrostatic interaction with siRNA reduces the availability of cationic residues, resulting in poor translocation of siRNA (CitationMeade and Dowdy, 2007). The ratio of cationic amino acids (lysine, arginine, and histidine) to all amino acids was 62% (8AA/13AA) for Tat, 44% (7AA/16AA) for Pen, and 19% (4AA/21AA) for TP. Thus, our results indicate that CPPs with a higher cation density needed longer CPs for effective translocation of siRNA. This suggested that the translocation activity of CPPs was preserved by preferential interaction of siRNA with CPs during the process of complex formation. Furthermore, the uptake of siRNA increased dramatically as the mixing ratio was altered from 20-fold to 60-fold, despite the abundant net positive charge of the complexes at both ratios. The complexes with Pen-K10, TP-K6, or TP-H6K6 at a 60-fold molar ratio formed small colloidal particles, while those at a 20-fold molar ratio formed precipitate. Taken together, these results suggested that charged CPP domain was preferentially exposed on the surfaces of the colloidal particles.
Figure 3. Internalization of siRNA complexes formed with various peptide conjugates by FRSK cells. The cells were incubated for 2 hr at 37°C with mixtures of fluorescein-labeled siRNA (final concentration = 60 nm) and various peptides at a 20-fold (open bars) or 60-fold (closed bars) molar ratio (peptide/siRNA). Then cells were recovered by trypsinization and the fluorescence intensity of the cell lysate was measured. Results were normalized by the amount of protein in the cell lysate. Each data point represents the mean ± SD of three wells.
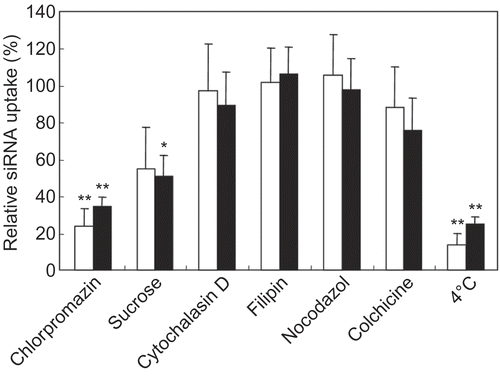
In order to clarify the translocation pathway of the complexes, uptake was investigated in the presence of various endosomal pathway inhibitors (). Cytochalasin D induces the depolymerization of microfilaments involved in phagocytosis and macropinocytosis, nocodazole and colchicine disrupt the microtubules, sucrose and chlorpromazine inhibit clathrin-mediated endocytosis, and filipin disrupts caveolar structure and function. The uptake of siRNA complexes formed with TP-K6 and Pen-K10 showed a decrease when cells were incubated at 4°C, indicating that it was an energy-dependent process. In addition, chlorpromazine and sucrose inhibited the uptake of siRNA complexes formed with any of the conjugates, suggesting that internalization of the complexes occurred via clathrin-mediated endocytosis.
Figure 4. Influence of various endocytosis inhibitors on the uptake of siRNA with peptide conjugates by FRSK cells. The cells were pre-incubated in the presence of each endocytosis inhibitor before addition of the complexes. Then cells were incubated for 2 hr at 37°C with a mixture of fluorescein-labeled siRNA (final concentration = 60 nm) and Pen-K10 (open bars) or TP-K6 (closed bars) at a 60-fold molar ratio (peptide/siRNA). Each data point represents the mean ± SD of three wells. Asterisks (* p < 0.05, ** p < 0.01) indicate values that significantly differ from those measured for cells treated without inhibitors.
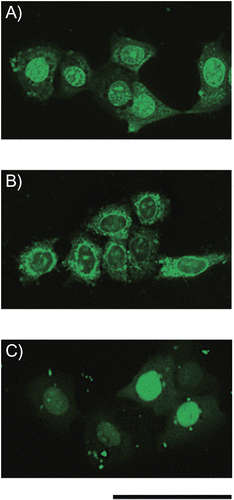
When cells were incubated with complexes containing fluorescein-labeled siRNA and then were observed by fluorescence microscopy without fixation, it was found that the intracellular localization of siRNA was influenced by the type of CPP. SiRNA internalized as complexes with Pen-K10 was localized in the perinuclear region (). On the other hand, siRNA internalized as complexes with TP-K6 was predominantly distributed to the cytoplasm and nuclear envelope (), as was also the case when TP-H6K6 were used (data not shown). LF 2000, a lipid-based carrier, induced the diffuse accumulation of siRNA in the nucleus (). When the localization of siRNA was evaluated using two other cell lines (HTC1 cells and RAW264.7 cells), the results obtained corresponded with those observed using FRSK cells (data not shown).
Figure 5. Fluorescence images of living FRSK cells after treatment with complexes of peptide conjugates and siRNA. A mixture of fluorescein-labeled siRNA (final concentration = 60 nm) with (A) Pen-K10 or (B) TP-K6 at a 60-fold molar ratio (peptide/siRNA) was incubated with the cells for 2 hr at 37°C, after which fluorescence microscopy was done without fixation of the cells. (C) LF 2000 was also tested as a siRNA carrier. Scale bar = 50 μm.
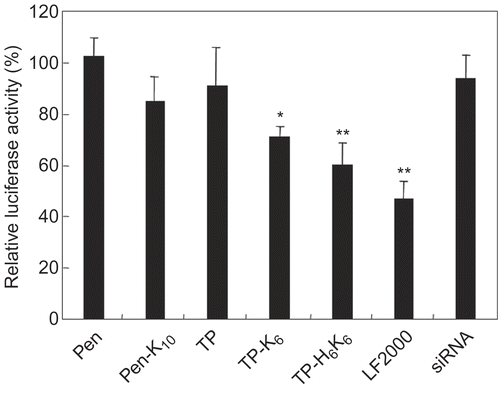
The silencing activity of siRNA complexes formed with CPP-CP conjugates was determined by using an exogenous luciferase gene (). Complexes formed with TP-K6 and TP-H6K6 showed high specific silencing activity, while those with CPP alone and Pen-K10 showed almost no activity.
Figure 6. Luciferase gene silencing by siRNA with peptide conjugates. Mixtures of siRNA (anti-luciferase or negative control siRNA) with various peptide conjugates at a 60-fold molar ratio (peptide/siRNA) were incubated with the cells for 2 hr at 37°C. The final siRNA concentration was 60 nm. Subsequently, the cells were incubated with complexes of LF2000 and pCMV-luciferase plasmid, after which luciferase expression was determined by using a Luciferase Assay System. Silencing of luciferase gene expression is shown as the relative ratio of luciferase activity induced by different siRNA (anti- luciferase siRNA/negative control siRNA). Each data point represents the mean ± SD of three wells. Asterisks (* p < 0.05, ** p < 0.01) indicate values that significantly differ from those measured for cells treated with siRNA.
The biological activity of siRNA is significantly influenced by both its uptake and its localization within cells. A relationship between the intracellular content of siRNA and its biologic effectiveness has already been reported (CitationVeldhoen et al., 2006; CitationMescalchin et al., 2007). In those studies, LF2000 (as well as microinjection and electroporation) induced extremely high siRNA activity, and about 300 copies of siRNA were needed for half maximal target suppression. Furthermore, CitationBerezhna et al. (2006) reported that the intracellular localization of siRNA after translocation with LF2000 was dependent on where the target RNA resides. These results indicate that siRNA complexed with LF2000 easily underwent dissociation in the cytoplasm, where RNA interference is thought to occur predominantly, and subsequently entered the interference machinery. In the present study, TP- and Pen-based conjugates significantly enhanced the internalization of siRNA. However, these conjugates yielded different localization of siRNA in cells and different biological activity. In the case of Pen-based conjugates, Pen-K10 had extremely low silencing activity, and siRNA was localized in both the nucleus and cytoplasm, mainly accumulating in intracellular membranous structures around the nucleus, such as the endoplasmic reticulum and Golgi complex. On the other hand, TP-based conjugates yielded a punctate cytoplasmic distribution of siRNA, suggesting extended residence of siRNA in the endosomes/lysosomes because the translocation pathway was thought to involve clathrin-mediated endocytosis. The biological activity of siRNA internalized with TP-based conjugates was high, but the level was still lower than that achieved with LF2000, probably due to insufficient transfer of siRNA from the endosomes/lysosomes to cytoplasma. Thus, a conjugate that included histidine residues (TP-H6K6) was also used because these residues have the potential to promote escape from the endosomes by lysis (CitationLeng et al., 2007). TP-H6K6 showed the highest silencing activity among the conjugates used, but the activity was almost the same level as TP-K6. This may be due to low lysis ability of the linear peptide, as reported elsewhere (CitationLeng et al., 2005). It may be possible to enhance the activity by optimizing peptide structure as well as to introduce helper molecules such as histidines and fusogenic peptides (CitationWagner et al., 1992). Although it is still unclear why TP- and Pen-based conjugates yielded different patterns of siRNA localization despite uptake via the same route and whether siRNA was dissociated from the conjugates in those intracellular compartments, intracellular localization of siRNA seems to determine its biological activity.
Nevertheless, it was clarified that the utility of CPP-CP conjugates for siRNA delivery were significantly dependent on the kind of CPP and the length of the CP. In addition, among peptide conjugates used in this study, TP-based peptide that included six lysine and six histidine residues was found to be a most available peptide as a siRNA carrier. These findings should help to develop a novel siRNA carrier and activate CPP technology.
Conclusions
Cell-penetrating peptide is an excellent tool for the delivery of various therapeutics because of their ability to cross the cell membrane. However, there have been few studies comparing the efficacy of various cell-penetrating peptides as tools for the delivery of therapeutics, although different peptides may utilize different uptake pathways, show differences in cytotoxicity, and have differing abilities to transport therapeutics. Because the siRNA carriers used in this study only contained CPP-based peptides and siRNA without any effectors, it should be possible to evaluate the efficacy of various CPPs for siRNA delivery. We showed that CPP-CP conjugates markedly enhanced internalization of siRNA in cells. However, the uptake, intracellular localization, cytotoxicity, and biological activity of siRNA were significantly dependent on the kind of CPP used and the length of the CP in the conjugate. Tat modified with K10 exhibited no cytotoxicity, but did not enhance the uptake of siRNA. Pen modified with K10 enhanced the uptake of siRNA, but there was almost no silencing of gene expression. TP-based conjugates yielded high internalization of siRNA and strong silencing activity. The intracellular localization of siRNA varied among TP-based conjugates, Pen-based conjugates, and LF2000. These different properties of CPPs emphasize the importance of careful peptide selection and suggest further design changes for various applications of CPP technology.
Acknowledgment
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Patil SD, Rhodes DG, Burgess DJ. (2005). DNA-based therapeutics and DNA delivery systems: a comprehensive review. AAPS J, 7, E61–77.
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. (2001). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–8.
- Paula DD, Bentley MV, Mahato RI. (2007). Hydrophobization and bioconjugation for enhanced siRNA delivery and targeting. RNA, 13, 431–56.
- Derossi D, Joliot AH, Chassaing G, Prochiantz A. (1994). The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem, 269, 10444–50.
- Pujals S, Fernández-Carneado J, López-Iglesias C, Kogan MJ, Giralt E. (2006). Mechanistic aspects of CPP-mediated intracellular drug delivery: relevance of CPP self-assembly. Biochim Biophys Acta, 1758, 264–79.
- Patel LN, Zaro JL, Shen WC. (2007). Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm Res, 24, 1977–92.
- El-Andaloussi S, Järver P, Johansson HJ, Langel U. (2007). Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: a comparative study. Biochem J, 407, 285–92.
- Abes S, Moulton H, Turner P, Clair P, Richard JP, Iversen P, Gait MJ, Lebleu B. (2007). Peptide-based delivery of nucleic acids: design, mechanism of uptake and applications to splice- correcting oligonucletides. Biochem Soc Trans, 35, 53–5.
- Zhang C, Tang N, Liu X, Liang W, Xu W, Torchilin VP. (2006). siRNA-containing liposomes modified with polyarginine effectively silence the targeted gene. J Contr Rel, 112, 229–39.
- Kang H, DeLong R, Fisher MH, Juliano RL. (2005). Tat-conjugated PAMAM dendrimers as delivery agents for antisense and siRNA oligonucleotides. Pharm Res, 22, 2099–106.
- Nakamura Y, Kogure K, Futaki S, Harashima H. (2007). Octaarginine-modified multifunctional envelope-type nano device for siRNA. J Contr Rel, 119, 360–7.
- Davidson TJ, Harel S, Arboleda VA, Prunell GF, Shelanski ML, Greene LA, Troy CM. (2004). Highly efficient small interfering RNA delivery to primary mammalian neurons induces MicroRNA-like effects before mRNA degradation. J Neurosci, 24, 10040–6.
- Muratovska A, Eccles MR. (2004). Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett, 558, 63–8.
- Turner JJ, Jones S, Fabani MM, Ivanova G, Arzumanov AA, Gait MJ. (2007). RNA targeting with peptide conjugates of oligonucleotides, siRNA and PNA. Blood Cells Molec Dis, 38, 1–7.
- Chiu YL, Ali A, Chu CY, Cao H, Rana TM. (2004). Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem Biol, 11, 1165–75.
- Meade BR, Dowdy SF. (2007). Exogenous siRNA delivery using peptide transduction domains/cell penetrating peptides. Adv Drug Deliv Rev, 59, 134–40.
- Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. (2004). Tumor imaging by means of proteolytic activation of cell- penetrating peptides. Proc Natl Acad Sci USA, 101, 17867–72.
- Unnamalai N, Kang BG, Lee WS. (2004). Cationic oligopeptide- mediated delivery of dsRNA for post-transcriptional gene silencing in plant cells. FEBS Lett, 566, 307–10.
- Wang YH, Hou YW, Lee HJ. (2007). An intracellular delivery method for siRNA by an arginine-rich peptide. J Biochem Biophys Methods, 70, 579–86.
- Simeoni F, Morris MC, Heitz F, Divita G. (2003). Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res, 31, 2717–24.
- Veldhoen S, Laufer SD, Trampe A, Restle T. (2006). Cellular delivery of small interfering RNA by a non-covalently attached cell- penetrating peptide: quantitative analysis of uptake and biological effect. Nucleic Acids Res, 34, 6561–73.
- Lundberg P, El-Andaloussi S, Sütlü T, Johansson H, Langel U. (2007). Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J, 21, 2664–71.
- Soomets U, Lindgren M, Gallet X, Hallbrink M, Elmquist A, Balaspiri L, Zorko M, Pooga M, Brasseur R, Langel U. (2000). Deletion analogues of transportan. Biochim Biophys Acta, 1467, 165–76.
- Vivès E, Brodin P, Lebleu B. (1997). A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem, 272, 16010–7.
- Saar K, Lindgren M, Hansen M, Eiríksdóttir E, Jiang Y, Rosenthal-Aizman K, Sassian M, Langel U. (2005). Cell-penetrating peptides: a comparative membrane toxicity study. Anal Biochem, 345, 55–65.
- Mescalchin A, Detzer A, Wecke M, Overhoff M, Wünsche W, Sczakiel G. (2007). Cellular uptake and intracellular release are major obstacles to the therapeutic application of siRNA: novel options by phosphorothioate-stimulated delivery. Expert Opin Biol Ther, 7, 1531–8.
- Berezhna SY, Supekova L, Supek F, Schultz PG, Deniz AA. (2006). siRNA in human cells selectively localizes to target RNA sites. Proc Natl Acad Sci USA, 103, 7682–7.
- Leng Q, Goldgeier L, Zhu J, Cambell P, Ambulos N, Mixson AJ. (2007). Histidine-lysine peptides as carriers of nucleic acids. Drug News Perspect, 20, 77–86.
- Leng Q, Mixson AJ. (2005). Modified branched peptides with a histidine-rich tail enhance in vitro gene transfection. Nucleic Acids Res, 33, e40.
- Wagner E, Plank C, Zatloukal K, Cotten M, Birnstiel ML. (1992). Influenza virus hemagglutinin HA-2 N-terminal fusogenic peptides augment gene transfer by transferrin-polylysine-DNA complexes: toward a synthetic virus-like gene-transfer vehicle. Proc Natl Acad Sci USA, 89, 7934–8.