Abstract
DNA vaccination is a promising approach for inducing both humoral and cellular immune responses. The mode of plasmid DNA delivery is critical to make progress in DNA vaccination. Using human papillomavirus type 16 E7 as a model antigen, this study evaluated the effect of peptide-polymer hybrid including PEI600-Tat conjugate as a novel gene delivery system on the potency of antigen-specific immunity in mice model. At ratio of 10:50 PEI-Tat/E7DNA (w/w), both humoral and cellular immune responses were significantly enhanced as compared with E7DNA construct and induced Th1 response. Therefore, this new delivery system could have promising applications in gene therapy.
Introduction
DNA vaccines have been widely used to develop immunity against various pathogens as well as cancer, autoimmune diseases, and allergy. The potential of DNA vaccine to induce an effective immune response is related to the expression levels of the encoded protein in eukaryotic cells. The uptake of naked DNA into a cell is very low in the absence of an efficient delivery agent. Cell-penetrating peptides (CPPs) are a novel class of membrane translocating agents (Citation1–8).
Generally, CPPs consist of less than 30 amino acids, have a net positive charge, and the ability to translocate the plasma membrane and transport a wide variety of cargoes such as proteins, DNAs, toxins, and nanoparticles into the cytoplasm and nucleus (Citation1–8). Interaction of CPP and cargo is either achieved by covalent attachment or by non-covalent complex formation through mainly ionic interactions. In the case of non-covalent complexes, a further assembly of cargo/carrier complexes occurs, leading to the formation of nanoparticles. Prior to the translocation process, the particles attach to the cell surface by ionic interactions of positively charged CPP residues with negatively charged membrane components. Subsequently, complexes are taken up by directly penetrating the cell membrane or by an endocytotic pathway. Recent data suggests that the main uptake route is endocytosis. The CPP must be able to tightly compact and protect DNA, target specific cell-surface receptors, disrupt the endosomal membrane and deliver the DNA cargo to the nucleus (Citation3–8).
One of the best CPPs is the basic domain of human immunodeficiency virus type 1 (HIV-1) Tat protein (Tat49–57: RKKRRQRRR) (Citation9). It can be used for delivery of plasmid DNA into cells without disturbing the stability of the cell membrane and with low toxic effects (Citation9–Citation15). Although Tat peptide is a good carrier, several recent studies have shown that the efficiency of Tat mediated transfection is strongly enhanced with adding chloroquine in culture medium. However, chloroquine co-treatment is not a practical approach for in vivo gene delivery applications (Citation14,Citation15).
It has been confirmed that a type of low molecular weight polymer, so-called polyethylenimine (PEI, mW < 2000 Da), covalently coupled to Tat was able to improve Tat peptide mediated gene delivery as chloroquine (Citation14,Citation15). PEI is the organic macromolecule with the highest cationic-charge-density potential. The amino groups of PEI can be protonated in the acidic endosomal environment, protecting endocytosed DNA from degradation by lysosomal nuclease (Citation14–19).
More advanced polymeric gene delivery systems use macromolecules with a very high cationic charge density that act as endosomal buffering systems, thus suppressing the endosomal enzymes activity and protecting the DNA from degradation (Citation18). Therefore, Tat peptide conjugates with low molecular weight polyethylenimine could be used as the effective non-viral gene delivery vehicles (Citation14,Citation15). Transfection experiments demonstrated that the use of PEI600-Tat conjugates was more effective than the two compounds without chemical conjugation. Furthermore, the newly developed conjugates maintain the desirable property of low cytotoxicity displayed by lower molecular weight PEI polymers and Tat peptides (Citation14,Citation15,Citation18).
In the current study, we chose HPV16E7 as a model antigen for vaccine development because human papillomaviruses (HPVs), particularly HPV16, are associated with most cervical cancers. Also, the HPV oncogenic proteins, E6 and E7, are important in the induction and maintenance of cellular transformation and co-expressed in most HPV-containing cervical cancers. Among many candidate vaccines, DNA vaccines have emerged as an appealing approach to generate antigen-specific immunotherapy because of their simplicity, stability, and safety, in addition to its potent priming of CD8+ T-cell responses (Citation20–23). Naked DNA vaccines do not have the intrinsic ability to amplify or spread in vivo. Therefore, in this study, a new polycationic system was applied based on peptide-polymer hybrid.
Herein, we describe new DNA vaccine using HPV16E7 by means of novel delivery system (PEI600-Tat conjugate) in vivo.
Materials and methods
Large scale preparation of endotoxin-free pcDNA-E7 plasmid
Large-scale purification of pcDNA-E7 plasmid (kindly provided by Professor T. C. Wu, John Hopkins Medical Institutions, USA) was prepared using ion exchange chromatography with an Endofree plasmid Giga kit (QIAGEN, Valencia, CA) according to the manufacturer’s instructions. Plasmid DNA was analysed by agarose gel electrophoresis and quantified by spectrophotometry. Then plasmid was stored in endotoxin-free PBS1X at −20°C until used (Citation24).
Purification of histidine-tagged recombinant HPV16 E7 protein [rE7]
The bacterial pQE30 vector expressing an amino-terminal six histidine-tagged E7 protein has been previously described (Citation24). The E.coli M15 strain was transformed with pQE-E7 and grown at 37°C in LB (Lauri Broth) supplemented with 100 μg/ml ampicillin and 25 μg/ml kanamycin. For protein production, the cultures were grown to an optical density of 0.6–0.8 at 600 nm and protein expression was induced with 1 mM IPTG at 37°C. Protein samples were analyzed by SDS-PAGE in a gel containing 15% (W/V) polyacrylamide followed by staining with Coomassie brilliant blue. Recombinant E7 protein was purified by Fast Protein Liquid Chromatography (FPLC) using Ni-NTA column (Amersham Pharmacia Biotech) under denaturing condition according to the manufacturer’s protocol (QIAGEN). The purified protein fractions were concentrated by ultrafiltration (Amicon) and dialyzed against PBS. The protein concentrations were measured by the BCA procedure (Pierce, Rockford, USA) using BSA as a standard and stored at −70°C, until used.
Synthesis and analysis of PEI600-Tat conjugate
Tat peptide (GRKKRRQRRRQPPG) was synthesized in-house using a 433A Applied Biosystems synthesizer (ABI, Sciex, Canada) according to a standard Fmoc (N-(9-fluorenyl) methoxycarbonyl) protocol. The synthetic peptide-polymer hybrid (PEI600-Tat conjugate) was obtained from mixture according to the described protocol (Citation14) () and analyzed by analytical HPLC and MALDI-TOF mass spectrometry.
Figure 1. Chemical structure of PEI600 conjugated to a Tat peptide through the formation of an amide bond. PEI 600 was conjugated to the last glycine residue of Tat peptide to synthesize a peptide–polymer hybrid [14].
![Figure 1. Chemical structure of PEI600 conjugated to a Tat peptide through the formation of an amide bond. PEI 600 was conjugated to the last glycine residue of Tat peptide to synthesize a peptide–polymer hybrid [14].](/cms/asset/92a02f54-4e06-4324-b6a1-5345923d8a00/idrd_a_375942_f0001_b.gif)
The lyophilized material was dissolved in 50% acetic acid/H2O (v/v) at a concentration of 1 mg/ml and 50 μl of this peptide solution was subjected to analytical HPLC on a C18 nucleosil column (250 × 4 mm, 5 μ). The column was eluted at 1 ml/min by a linear gradient of acetonitrile on 0.1% TFA/H2O; rising within 30 min from 0 to 100% [HPLC-grade trifluoroacetic acid was purchased from Fluka (Buchs, Switzerland), HPLC-grade H2O from Romil Ltd (Amman Technik SA, Kölliken, Switzerland) and acetonitrile was purchased from Biosolve Ltd (Chemie Brunschwig, Basel)]. The detection was performed at 220 and 280 nm using a Waters 991 photodiode array detector.
Mass spectrometry (MS) analyses were performed using a Perceptive Biosystems MALDI-TOF Voyager DE-RP Mass Spectrometer (Framingham, MA) operated in the delayed extraction and linear mode. The lyophilized material was dissolved in 50% acetic acid/H2O (v/v) at a concentration of 1 mg/ml, mixed with the matrix α-cyano-4-hydroxycinnamic acid [α-cyano-4-hydroxycinnamic acid was purchased from Sigma (Sigma Chemical Co., St. Louis, MO)] at 1:20 (v/v) and was then subjected to analysis by MALDI-TOF.
Preparation of condensed cationic PEI600-Tat/DNA complexes and gel retardation assay
The synthetic peptide-polymer hybrid (PEI600-Tat conjugate) was dissolved in water and DMSO at 50 μg/ml as a stock solution. To prepare DNA complexes, conjugate work solutions were added dropwise to the DNA encoding E7 gene, while vortexing. The plasmid DNA was mixed with PEI600-Tat at ratios of 50:2, 50:5, 50:10, and 50:25 (w/w: E7 DNA/ PEI-Tat) in PBS (pH = 7.4), and incubated for 60 min at room temperature. To characterize the complexes in agarose gel retardation assay, 25 μl of each complex solution was mixed with 5 μl of a 6 x loading buffer and loaded onto ethidium bromide containing 0.8% agarose gel.
Transfection of pEGFP and pEGFP-E7 plasmids using PEI600-Tat in COS-7 cells
The enhanced green fluorescent protein encoding plasmid (pEGFP-N1, Clonetech, Palo Alto, CA) and the E7-expressing plasmid (pEGFP-E7) as previously described (Citation24) were prepared in large scale with high purity (Midi-kit, Qiagen). COS-7 cells were seeded at 37°C in the presence of 5% CO2 into the 4-well plates (Greiner) in complete RPMI 10%; 10 μg of each pEGFP and pEGFP-E7 was incubated for 1 h at room temperature with different amounts of PEI600-Tat (2, 5, 80, and 150 μg) as transfection reagent in PBS in a total volume of 100 μl. PEI600-Tat/DNA complexes were added to 0.5 × 105 COS-7 cells in serum-free media. The medium was replaced after 8 h incubation at 37°C with complete RPMI 10%. Transfection efficacy (the level of GFP expression) was determined by fluorescence microscopy at 24 and 48 h after transfection.
Mice immunization
Inbred C57BL/6 female mice, 6–8 weeks old, were obtained from the breeding stocks maintained at the Pasteur Institute of Iran. Nine groups of 10 mice were selected. For priming, groups 1–5 were immunized subcutaneously at the footpad with 50 μg of plasmid DNA encoding E7 (pcDNA-E7) and different ratios of PEI600-Tat conjugate/pcDNA-E7 complexes (2:50, 5:50, 10:50, and 25:50; w/w), respectively. The control groups (groups 6–9) were injected empty pcDNA3.1 (-) plasmid, PEI600-Tat conjugate/pcDNA3.1 (-) at a ratio of 25:50 (w/w), PEI600-Tat conjugate (25 μg), and PBS, respectively. For boosting (3 weeks after priming), all groups received the same regimen as priming. shows the immunization regimens of each group.
Table 1. DNA immunization in C57BL/6 mice using E7 in the different formulations.
Antibody assay
Mice were bled from retro-orbital and sera were prepared from the whole blood samples of each mouse at different time intervals before, 3, and 6 weeks after the booster injection. Pooled sera of each group were stored at −20°C. ELISA was performed with the recombinant E7 protein for the identification of antigen-specific total IgG, IgG1, and IgG2a. Briefly, 96-microwell plates (NUNC) were coated overnight at 4°C with 100 μl of rE7 (5 μg/ml) diluted in PBS (pH 7.2). Plates were washed with PBS containing 0.05% (v/v) Tween 20 (PBS-Tween) and blocked with 1% BSA in PBS for 2 h at 37°C. Then, 100 μl of mice sera (diluted 1:50 in 1% BSA/PBS-Tween) was added and the plates were incubated at 37°C for 2 h. After washing, 100 μl of either biotinylated goat anti-mouse IgG (diluted 1:10,000 in 1% BSA/PBS-Tween, Cedarlane Laboratories, Hornby, Ontario, Canada), or rat anti-mouse IgG1 or IgG2a conjugated with Biotin (diluted 1:1000 in 1% BSA/PBS-Tween, Cedarlane Laboratories, Hornby, Ontario, Canada) was added, and the plates were incubated at 37°C for 2 h. After that, the plates were washed and incubated with 1:600 dilution of streptavidin-horseradish peroxidase (Cedarlane Laboratories, Hornby, Ontario, Canada) at 37°C for 1 h. Detection was done with 100 μl of O-Phenylenediamine (OPD, Sigma) as the substrate in citrate phosphate buffer (citric acid 0.1 M, Na2HPO4 0.2 M, pH 4.5). After 30 min, the enzyme reaction was stopped by 1 M sulfuric acid and the absorbance was measured at 492 nm.
Cytokine analysis
Three weeks after the booster injection, two mice from each group were sacrificed and the spleens were removed. A single cell suspension of splenocytes was seeded in complete RPMI medium 1640 supplemented with 5% FCS, 2 mM glutamine, 5 × 10−5 mM 2- mercaptoethanol (2-ME), 10 mM HEPES, and 40 μg/ml gentamycin. Cells were incubated in U-bottomed, 96-well microtiter plates (Costar, Cambridge, MA) at a density of 2 × 106 cells/ml in the presence of 10 μg/ml of the recombinant E7 protein and 5 μg/ml of concanavalinA (ConA, as positive control) and incubated for 5 days at 37°C in an atmosphere of 5% CO2. After supernatant extraction, 1 μCi of [3H]-thymidine (Amersham, UK) was added to each well and incubated overnight. Cells were harvested on filters and [3H]-thymidine incorporation was determined by liquid scintillation counting. All tests were performed in triplicate. Proliferative responses were expressed as their stimulation indices (SI), which represents the ratio of mean proliferation after stimulation to the mean proliferation of medium. The collected supernatants were analysed for IFN-γ production by a commercially available, sandwich-based ELISA kit (R&D, Minneapolis, MN), as described in the kit manual. The lower detection limit of IFN-γ was 2 pg/ml. All tests were performed in duplicate for each mouse and the median was recorded for each set of samples.
Statistical analysis of the data
The differences in the level of cellular and antibody responses were determined by one-way ANOVA. They were considered statistically significant when the p-value was < 0.05.
Results
Production of E7 DNA construct and its recombinant protein
DNA construct encoding HPV16E7 (pcDNA-E7) was prepared in large scale with high purity. The recombinant E7 protein was expressed in E.coli and purified by affinity chromatography. The recombinant E7 protein migrated as a 23 kDa protein in SDS-PAGE, as reported previously (Citation24).
PEI600-Tat/DNA complex formation
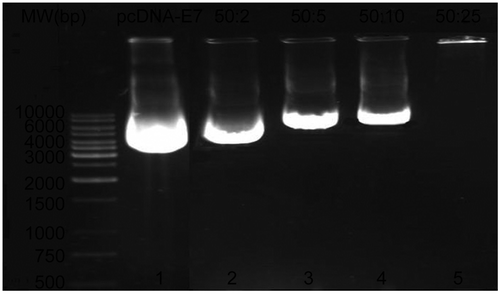
Fifty micrograms of pcDNA-E7 was mixed with increasing amounts of PEI600-Tat conjugate (2, 5, 10, and 25 μg) for 60 min at room temperature. Samples were subjected to agarose gel electrophoresis and stained by ethidium bromide. Addition of increasing amounts of conjugate showed a dose-dependent change in the electrophoretic mobility of the DNA. Indeed, negatively charged plasmid DNA interacts with positively charged cationic peptide-polymer conjugate. At quantities greater than 25 μg of conjugate, the polynucleotide molecule did not migrate into the gel. These analyses revealed quantitative complex formation of the conjugate and pcDNA-E7. In fact, PEI600-Tat conjugate bound to and condensed E7-encoding plasmid DNA into small nanoparticles, as demonstrated in a DNA retardation assay. shows the formation of PEI600-Tat/DNA complexes.
Figure 2. Characterization of plasmid DNA/PEI-Tat complexes by agarose gel electrophoresis. pcDNA-E7 (50 μg) was incubated for 60 min at room temperature with increasing amounts of PEI-Tat conjugate (0, 2, 5, 10, and 25 μg), as shown in lines 1–5. The DNA complexed with PEI-Tat that was not able to migrate into the gels, was observed at a ratio of 50:25 (w/w: E7DNA/PEI-Tat) (line 5).
Binding of PEI-Tat to plasmid DNA promotes cell transfection in vitro
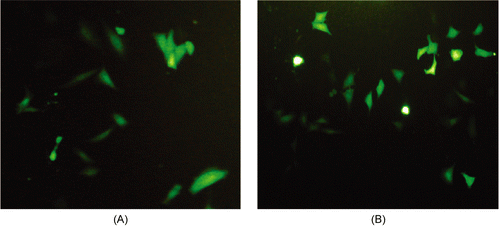
GFP expression was evaluated by incubating COS-7 cells for 8 h with plasmid DNA encoding either GFP or E7-GFP as naked plasmid DNA or complexed with increasing amounts of PEI600-Tat (2, 5, 80, and 150 μg) in vitro. As a control, incubation with naked plasmid DNA did not show any fluorescence at 24 and 48 h after transfecting COS-7 cells. The cells expressing GFP alone started to show fluorescence at 80 μg of PEI600-Tat. In contrast, the fluorescence of pEGFP-E7 acted differently and elevated at 5 μg of PEI600-Tat with maximum fluorescence at 150 μg of PEI600-Tat. Therefore, PEI600-Tat/DNA complexes in certain amounts facilitate uptake of DNA by cells that leads to expression of the respective protein. Our results indicated that PEI600-Tat/DNA complexes were efficient to transfect cells in vitro. The transfection efficiency of E7 gene using PEI600-Tat (5 and 150 μg) has been shown in .
Figure 3. Transfection efficiency of pEGFP-E7 using PEI600-Tat in COS-7 cells. GFP expression was assessed by fluorescence microscopy at 24 h after transfecting of COS-7 cells with 10 μg of pEGFP-E7 plasmid combined by 5 (A) and 150 μg (B) of PEI600-Tat in serum-free medium.
PEI600-Tat/DNA complexes increase the humoral response of HPV16 E7 in DNA vaccination
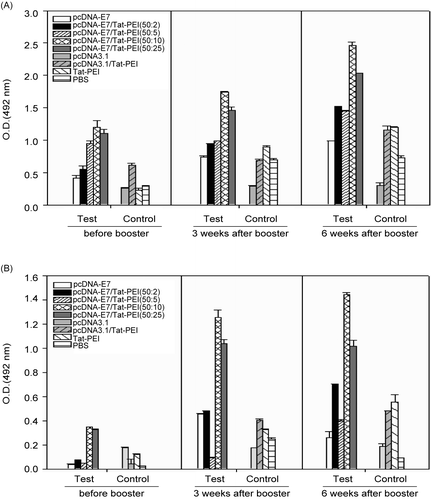
Nine groups of 10 mice were considered for immunization, as described in . Four different ratios of PEI600-Tat/E7DNA (2:50, 5:50, 10:50, and 25:50; w/w) were applied to determine the most efficient DNA delivery in vivo. The E7-specific humoral response was evaluated from the sera of immunized and control animals by ELISA, before, 3, and 6 weeks after booster immunization. The levels of total IgG and IgG1 antibodies with respect to rE7 have been depicted in , respectively. The mice immunized with the E7DNA/PEI-Tat constructs at ratios of 50:10 and 50:25 (w/w, groups 4 and 5) displayed a higher humoral response as compared with E7 injected mice (p < 0.05). Similar distribution of the IgG1 isotype was observed for the mice immunized with the E7-containing vaccine formulations. The results showed that PEI-Tat conjugate at above ratios can stimulate the production of IgG1, even after first DNA immunization. The evaluation of IgG isotypes indicated that IgG1 was the predominant isotype in groups 4 and 5 in comparison with control groups, especially 6 weeks after booster (p < 0.05). There was no significant difference for IgG2a production in different groups as compared with that in control groups 6–9 (p > 0.05, data not shown). However, an increased level of the specific IgG response in E7DNA/PEI-Tat-vaccinated animals indirectly demonstrates that PEI-Tat conjugate enhances the E7-specific T helper-cell response.
Figure 4. Analysis of total IgG (A) and IgG1 isotype (B) antibody levels with respect to rE7 by using ELISA, respectively. Total IgG and IgG1 responses were similar in groups 4 and 5. Each vaccine formulation has been shown in . Data shows antibody responses of pooled mice sera with independent experiments, and error bars indicate SD.
E7DNA/PEI600-Tat complex at ratio of 50:10 (w/w) efficiently enhances IFN-γ production by E7-specific T-cells
To characterize E7-specific, cell-mediated responses, splenic mononuclear cells were obtained at 3 weeks after booster and stimulated in vitro with the recombinant E7 protein (rE7) and ConA as positive control. The stimulation indices against rE7 and ConA are shown in . PEI-Tat/E7DNA immunization at group 4 resulted in E7-specific Th-cell proliferative responses as compared with control mice (p < 0.05). There are no significant differences between test groups. For further evaluation, the supernatants were examined for specific IFN-γ production in different groups. The level of IFN-γ with respect to rE7 was significantly higher in group 4 (E7DNA/PEI600-Tat immunized mice at ratio of 50:10 w/w) as compared with the other groups (p < 0.05). The results confirmed a significantly higher level of IFN-γ production in group 4 in comparison with E7-injected mice (group 1, p < 0.05), as indicated in .
Figure 5. Lymphocyte Transformation Test [LTT] (A) and analysis of cytokine secretion (B) in mice immunized by E7 in different formulations at 3 weeks after booster. Splenocytes of two mice from each group were isolated and stimulated by either E7 (10 μg/ml) or ConA (5 μg/ml) for 5 days and finally IFN-γ levels were measured. All assays were performed in triplicate and at least for two mice. Error bars indicate mean values and SD are expressed in pg/ml.
![Figure 5. Lymphocyte Transformation Test [LTT] (A) and analysis of cytokine secretion (B) in mice immunized by E7 in different formulations at 3 weeks after booster. Splenocytes of two mice from each group were isolated and stimulated by either E7 (10 μg/ml) or ConA (5 μg/ml) for 5 days and finally IFN-γ levels were measured. All assays were performed in triplicate and at least for two mice. Error bars indicate mean values and SD are expressed in pg/ml.](/cms/asset/bf93c157-959f-4864-9701-9fa6103bf79c/idrd_a_375942_f0005_b.gif)
Discussion
Viral proteins expressed in tumors represent good targets for the immunotherapy of virally-induced cancer. Vaccines or immunotherapies targeting E7 or E6 proteins may provide an opportunity to prevent and treat HPV-associated cervical malignancies. We chose the HPV16 E7 oncoprotein as a vaccine target. E7 is the main target of current therapeutic vaccination studies, since it is continuously expressed during the oncogenic process in HPV infected cells (Citation25–Citation28).
E7 protein is a very poor inducer of immune responses, when being used as an antigen in DNA vaccination. Therefore, different approaches have been undertaken by others to improve the immunogenicity of E7 in DNA vaccination, such as mutating the pRb binding pocket, fusion with heat shock protein, and directing the protein into lysosomal compartments (Citation25,Citation29–Citation34).
Recently, non-viral delivery systems have been served as an effective approach in DNA vaccination. Non-viral gene delivery systems have been determined to be more efficient than viral gene vectors for their advantages in inducing relatively low toxicity and nearly no immune responses. Among various non-viral gene delivery methods, the utilization of natural or artificial cell-penetrating peptides (CPPs) with certain biological functions is considered as one promising approach. The current studies regarding non-viral gene delivery are now focused on understanding the mechanisms involved in cell entry, endosomal escape, and nuclear import of the transgene (Citation1–Citation3,Citation6,Citation8).
Cationic domains are abundant in many viral proteins, for example the full length Tat protein of HIV or Tat-derived peptides as short as Tat49–57 (RKKRRQRRR). The HIV-1 Tat protein is capable of delivering biologically-active proteins to the cytoplasmic compartment via the plasma membrane. Soluble Tat-antigen conjugates have been shown to deliver the antigen directly to the MHC class I processing pathway and thereby increase the generation of antigen-specific CD8+ T cells in vitro (Citation35–Citation37). A fusion protein containing the carboxy-terminal end of Tat (amino acids: 49–86) linked to the HPV16 E7 oncoprotein could enhance tumor- specific immune responses in vivo. Also, a similar strategy including the linkage of E7 to herpes simplex virus type1 VP22 has been shown to improve the potency of DNA vaccination (Citation30,Citation31).
The aim of this study was to evaluate whether a novel delivery system including PEI600-Tat conjugate could enhance E7-specific immune responses in vivo or not. According to the published data by others (Citation14,Citation15), this delivery system as a peptide–polymer hybrid could be a transfection enhancer. Also, this newly developed conjugate maintains the desirable property of low cytotoxicity displayed by low-Mw PEIs and Tat peptides compared to PEI 25 kDa (Citation14,Citation15).
Some reports suggested that Tat-PEG-PEI could be an interesting pulmonary delivery system for DNA, offering new treatment modalities for different lung diseases. The enhanced transfection efficiency under in vivo conditions is most likely due to Tat-derived oligopeptide mediated cell uptake of DNA, increased DNA condensation and polyplex stability. The PEG spacer seems to be essential for both the enhanced gene expression and the reduced toxicity (Citation38).
In the current study, we applied PEI600-Tat with low toxicity instead of Tat-PEG-PEI, and studied its potency in eliciting immune responses. One of our first goals was to determine the degree of plasmid/conjugate complex formation. To achieve this, E7 DNA was complexed with various amounts of PEI600-Tat conjugate to create different ratios. In a DNA retardation assay, the DNA mobility through an agarose gel during electrophoresis was significantly reduced when the ratio of the PEI-Tat to pcDNA-E7 reached 25:50 (w/w). The driving forces for DNA migration in electrophoresis are the number of negative charges and the size of complexes. However, more positively charged peptide–polymer hybrid bound to the plasmid would cause more retardation in the gel. When the complex is too big, it is expected to remain stationary in the well and not enter the gel matrix.
The E7DNA delivery was confirmed using the GFP reporter construct and PEI600-Tat conjugate in vitro. In this experiment, the COS-7 eukaryotic cells transfected with pEGFP-E7 exhibited increasing levels of GFP fluorescence at higher ratios of PEI600-Tat conjugate (w/w). Therefore, the transfection efficacy of PEI600-Tat/DNA improves with increasing the amount of conjugate and the best concentration is 150 μg of PEI600-Tat.
Furthermore, we evaluated the footpad DNA immunization using E7 and PEI-Tat/E7 complex at four different ratios in C57BL/6 mice (groups 1–5). To understand the immunological mechanisms, we analysed the humoral immune responses at three different periods. The results indicated that there is strong IgG1 response similar to total IgG against rE7 in groups 4 and 5 as compared with the control groups. This result confirmed that certain ratios of PEI600-Tat conjugate to E7 DNA could enhance humoral immune responses in vivo. Also, E7-specific, cell-mediated response, were determined at 3 weeks after booster. IFN-γ level in the supernatant of splenocytes from mice injected with E7DNA/PEI-Tat complex at a ratio of 50:10 (w/w, group 4) was significantly higher than the other groups (p < 0.05). In fact, PEI-Tat/E7 complex induced significantly higher production of IFN-γ in comparison with E7 in DNA immunization. This illustrates that E7 in the presence of PEI-Tat drives T-cell responses towards a Th1-type fashion.
Collectively, our observations illustrate the ability of PEI-Tat conjugate to augment immune responses in vivo. Therefore, PEI600-Tat could effectively improve E7 protein expression. Furthermore, the ratio of PEI600-Tat/E7DNA complex formation has significant influence on the level of protein expression and consequently immune responses in the mice model.
In our previous published data for improving DNA vaccine potency using heat shock protein (Gp96), it was indicated that co-delivery of naked E7 + Gp96 DNA plasmid induced Th1 response (Citation24). In this study, a similar result was obtained by using E7DNA/PEI600-Tat complex at ratio of 50:10 (w/w), significantly enhanced at the level of IFN-γ (149 pg/ml vs 110 pg/ml related to E7 + Gp96 DNA, p < 0.05). Also, IgG1 was the predominant isotype produced in both DNA immunizations. Therefore, further investigation is needed to demonstrate the co-delivering effect of E7 and Gp96 by using PEI600-Tat as new delivery system in order to show the synergistic effects.
In summary, the mixture of a DNA vaccine (pcDNA-E7) with short, synthetic cationic peptide (Tat49–57) linked to low Mw PEI (600 Da) is immunologically more potent than E7 alone. Binding of cationic peptide– polymer to plasmid DNA encoding an antigen (E7) enhances the uptake of the plasmid DNA and consequently induces both humoral and cellular immune responses in vaccinated mice. However, this new delivery system (peptide–polymer hybrid) would be worth exploring further as an in vivo delivery system for gene therapy of cancer or other disorders.
Acknowledgments
The financial support by the Research Council of the Republic Presidentship is gratefully acknowledged. We would like to thank A. Javadi (Pasteur Institute of Iran, Department of Immunology), N. Moradin, F. Zahedifard, and S. Alizadeh (Pasteur Institute of Iran, Molecular Immunology and Vaccine Research Laboratory) for their technical assistance.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Dubensky TW, Liu MA, Ulmer JB. (2000). Delivery systems for gene-based vaccines. Molec Med, 6, 723–32.
- Hellgren I, Gorman J, Sylven C. (2004). Factors controlling the efficiency of Tat-mediated plasmid DNA transfer. J Drug Target, 12, 39–47.
- Jarver P, Langel U. (2004). The use of cell-penetrating peptides as a tool for gene regulation. DDT, 9, 395–402.
- Zorko M, Langel U. (2005). Cell-penetrating peptides: mechanism and kinetics cargo delivery. Adv Drug Deliv Rev, 57, 529–45.
- Gupta B, Levchenko TS, Torchilin VP. (2005). Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev, 57, 637–51.
- Noguchi H, Matsumoto S. (2006). Protein transduction technology: a novel therapeutic perspective. Acta Med, 60, 1–11.
- Wagstaff KM, Jans DA. (2006). Protein transduction: cell penetrating peptides and their therapeutic applications. Curr Med Chem, 13, 1371–87.
- Martin ME, Rice KG. (2007). Peptide-guided gene delivery. The AAPS J, 9, E18–29.
- Brooks H, Lebleu B, Vives E. (2005). Tat peptide-mediated cellular delivery: back to basics. Adv Drug Deliv Rev, 57, 559–77.
- Tung CH, Mueller S, Weissleder R. (2002). Novel branching membrane translocational peptide as gene delivery vector. Bioorg Med Chem, 10, 3609–14.
- Ignatovich IA, Dizhe EB, Pavlotskaya AV, Akifiev BN, Burov SV, Orlov SV, Perevozchikov AP. (2003). Complexes of plasmid DNA with basic domain 47–57 of the HIV-1 Tat protein are transferred to mammalian cells by endocytosis-mediated pathways. J Biol Chem, 278, 42625–36.
- Riedl P, Reimann J, Schirmbeck R. (2004). Peptides containing antigenic and cationic domains have enhanced, multivalent immunogenicity when bound to DNA vaccines. J Mol Med, 82, 144–52.
- Dizhe EB, Ignatovich IA, Burov SV, Pohvoscheva AV, Akifiev BN, Efremov AM, Perevozchikov AP, Orlov SV. (2006). Complexes of DNA with cationic peptides: conditions of formation and factors effecting internalization by mammalian cells. Biochemistry, 71, 1350–6.
- Alexis F, Lo SL, Wang S. (2006). Covalent attachment of low molecular weight poly (ethyleneimine) improves Tat peptide mediated gene delivery. Adv Mater, 18, 2174–8.
- Wang S. (2006). Tat peptide conjugates of low molecular weight polyethylenimine as effective non-viral gene delivery vectors. Molec Ther, 13, S76.
- Boussif O, Lezoualch F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. (1995). A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci, 92, 7297–301.
- Godbey WT, Wu KK, Mikos AG. (1999). Poly (ethylenimine) and its role in gene delivery. J Contr Rel, 60, 149–60.
- Putnam D, Gentry CA, Pack DW, Langer R. (2001). Polymer-based gene delivery with low cytotoxicity by a unique balance of side-chain termini. Proc Natl Acad Sci, 98, 1200–5.
- Tang GP, Guo HY, Alexis F, Wang X, Zeng S, Lim TM, Ding J, Yang YY, Wang S. (2006). Low molecular weight polyethylenimines linked by β-cyclodextrin for gene transfer into the nervous system. J Gene Med, 8, 736–44.
- Hausen HZ, Villiers EMD. (1994). Human papillomaviruses. Ann Rev Microbiol, 48, 427–47.
- Schiller J, Davies P. (2004). Delivering on the promise: HPV vaccines and cervical cancer. Nature Rev, 2, 343–7.
- Damania B. (2006). DNA tumor viruses and human cancer. Trends Microbiol, 15, 38–44.
- Yan Q, Cheung YK, Cheng SC, Xian-Hua W, Yong X. (2007). A DNA vaccine constructed with human papillomavirus type 16 (HPV16) E7 and E6 genes induced specific immune responses. Gynecol Oncol, 104, 199–206.
- Bolhassani A, Zahedifard F, Taghikhani M, Rafati S. (2008). Enhanced immunogenicity of HPV16E7 accompanied by Gp96 as an adjuvant in two vaccination strategies. Vaccine, 26, 3362–70.
- Chen CH, Wang TL, Hung CF, Yang Y, Young R, Pardoll D, Wu TC. (2000). Enhancement of DNA vaccine potency by linkage of antigen gene to an HSP70 gene. Cancer Res, 60, 1035–42.
- Leitner WW, Hammerl P, Thalhamer J. (2001). Nucleic acid for the treatment of cancer: Genetic vaccines and DNA adjuvants. Curr Pharm Des, 7, 1641–67.
- Doria-Rose NA, Haigwood NL. (2003). DNA vaccine strategies: candidates for immune modulation and immunization regimens. Methods, 31, 207–16.
- Lewis J. (2004). Therapeutic cancer vaccines: using unique antigens. PNAS, 101, 14653–6.
- Kim TY, Myoung HJ, Kim JH, Moon IS, Kim TG, Ahn WS, Sin JI. (2002). Both E7 and CpG oligodeoxynuclotide are required for protective immunity against challenge with human papillomavirus 16 (E6/E7) immortalized tumor cells: involvement of CD4+ and CD8+ T cells in protection. Cancer Res, 62, 7234–40.
- Michel N, Osen W, Gissmann L, Schumacher TNM, Zentgraf H, Muller M. (2002). Enhanced immunogenicity of HPV16 E7 fusion proteins in DNA vaccination. Virology, 294, 47–59.
- Giannouli C, Brulet JM, Gesche F, Rappaport J, Burny A, Leo O, Hallez S. (2003). Fusion of a tumor-associated antigen to HIV-1 Tat improves protein-based immunotherapy of cancer. Anticancer Res, 23, 3523–32.
- Li H, Ou X, Xiong J. (2007). Modified HPV16 E7/HSP70 DNA vaccine with high safety and enhanced cellular immunity represses murine lung metastatic tumors with downregulated expression of MHC class I molecules. Gynecol Oncol, 104, 564–71.
- Huang CY, Chen CA, Lee CN, Chang MC, Cheng WF. (2007). DNA vaccine encoding heat shock protein 60 co-linked to HPV16 E6 and E7 tumor antigens generates more potent immunotherapeutic effects than respective E6 or E7 tumor antigens. Gynecol Oncol, 107, 404–12.
- Kim D, Gambhira R, Karanam B, Monie A, Hung CF, Roden R, Wu TC. (2008). Generation and characterization of a preventive and therapeutic HPV DNA vaccine. Vaccine, 26, 351–60.
- Kim DT, Mitchell DJ, Brockstedt DG, Fong L, Nolan GP, Fathman CG, Engleman EG, Rothbard JB. (1997). Introduction of soluble proteins into the MHC class I pathway by conjugation to an HIV tat peptide. J Immunol, 159, 1666–8.
- Riedl P, Buschle M, Reimann J, Schirmbeck R. (2002). Binding immune-stimulating oligonucleotides to cationic peptides from viral core antigen enhances their potency as adjuvants. Eur J Immunol, 32, 1709–16.
- Riedl P, Reimann J, Schirmbeck R. (2004). Complexes of DNA vaccines with cationic, antigenic peptides are potent, polyvalent CD8+ T cell-stimulating immunogens. Methods Molec Med, 127, 159–69.
- Kleemann E, Neu M, Jekel N, Fink L, Schmehl T, Gessler T, Seeger W, Kissel T. (2005). Nano-carriers for DNA delivery to the lung based upon a Tat-derived peptide covalently coupled to PEG-PEI. J Contr Rel, 109, 299–316.