Abstract
Nasal drug delivery is an interesting route of administration for metoclopramide hydrochloride (MTC) in preventing different kind of emesis. Currently, the routes of administration of antiemetics are oral or intravenous, although patient compliance is often impaired by the difficulties associated with acute emesis or invasiveness of parenteral administration. In this perspective, nasal dosage forms (solution, gel, and lyophilized powder) of MTC were prepared by using a mucoadhesive polymer sodium carboxymethylcellulose (NaCMC). In vitro and ex vivo drug release studies were performed in a modified horizontal diffusion chamber with cellulose membrane and excised cattle nasal mucosa as diffusion barriers. The tolerance of nasal mucosa to the formulation and its components were investigated using light microscopy. In vivo studies were carried out for the optimized formulations in sheep and the pharmacokinetics parameters were compared with oral solution and IV dosage form. The release of MTC from solution and powder formulations was found to be higher than gel formulation (p < 0.05). Histopathological examination did not detect any severe damage. Hydroxypropyl-β-cyclodextrin (HPβCD) used in powder formulations was found to be effective for enhancing the release and absorption of MTC. In contrast to in vitro and ex vivo experiments nasal bioavailability of gel is higher than those of solution and powder (p < 0.05). In conclusion, the NaCMC gel formulation of MTC with mucoadhesive properties with increased permeation rate is promising for prolonging nasal residence time and thereby nasal absorption.
Introduction
Metoclopramide hydrochloride (MTC) is a potent antiemetic, effective for preventing different kinds of emesis. This drug is characterized by a short half-life (3–4 h), and for this reason it is usually administered orally three or four times daily (Citation1). Antiemetics play an important role in the quality of life of patients in different pathological situations, and in particlar during and after chemothrapy, since they determine adequate control of the adverse effects of the medical treatment. Currently, the routes of administration of antiemetics are oral or intravenous, although patient compliance is often impaired by the difficulties associated with acute emesis or invasiveness of parenteral administration (Citation2). In this perspective, nasal administration of metoclopramide represents an interesting alternative administration route to others. The intranasal route for drug administration probably is one of the most attractive routes for distributing drugs to the systemic circulation. This is because of serious drawbacks of parenteral route in chronic therapy, rich vascularization of nasal route, and rapid absorption of drugs, highly permeable tissue, fast onset of action, ease of administration, and familiarity to the population at large and circumvention of first-pass metabolism (Citation3). In acute situations the rapid-onset of action can be of vital importance. In the case of emesis, the use of a nasal spray may be preferred to intravenous or oral administration for practical reasons, it is amenable to self-medication, it is essentially painless, and does not require sterile technique (Citation4). There are, however, limitations; for example, low permeability across the mucosa, degradation of drug by enzymes in the nasal cavity, and drug loss by mucociliary clearance (Citation5). Strategies to overcome these limitations include: the use of bioadhesive polymers that increase residence time of the formulation in the nasal cavity, thereby improving absorption; the use of nontoxic enhancers to improve the permeability of the nasal membrane to high molecular weight compounds; and the use of enzyme stabilizers that prevent degradation of peptide drugs in the nasal cavity (Citation6).
Cellulose derivatives have been investigated for use in nasal drug delivery due to their ability to slow down mucociliary clearance, and cyclodextrins, especially methylated β-cyclodextrins, have proven to be excellent solubilizers and absorption enhancers in nasal drug delivery (Citation7,Citation8). So in this study we developed nasal dosage forms (solution, gel, and lyophilized powder) of MTC by using carboxymethylcellulose sodium (NaCMC) as mucoadhesive polymer and hydroxypropyl-β-cyclodextrin (HPβCD) as penetration enhancer. In vitro and ex vivo release characteristics of the formulations and histological examinations of nasal mucosa were evaluated. In vivo studies were carried out in a sheep model.
Materials and methods
Materials
MTC and NaCMC (high viscosity) (Sigma, USA), HPβCD (Capital Limited, UK). All other chemicals were commercial products of reagent grade and were used without further purification.
Preperation of formulations
The contents of all prepared formulations are given in .
Table 1. Compositions of the formulations.
Solution formulations
NaCMC was added to the 0.9% NaCI solution. Benzalconium chloride was used as preservative at the concentration of 0.02%. After standing overnight, MTC was dissolved in the rest of the isotonic solution and was added slowly to the polymeric solution. The pH of the solution formulation was adjusted to 6.5 with 0.1 N HCI.
Gel formulations
Gel formulations were prepared as described before (Citation9). The required amount of NaCMC was wetted in some part of water overnight. MTC was dissolved in the rest of the water and added to gel base by mixing gently. Benzalconium chloride was used as a preservative at the concentration of 0.02%. The pH of the gel formulation was adjusted to 6.5 with 0.1 N HCI.
Powder formulations
MTC was added to purified water in lyophilization vials and sonicated until complete dissolution. NaCMC was dispersed into the same solution. The mixture was frozen for 1 h (−20°C) and than lyophilized at 0°C for 24 h. The powders were sieved (150 μm mesh) and stored at room temperature until used.
In vitro release studies of MTC with cellulose membrane
MTC release from formulations was tested with a modified horizontal diffusion chamber with a receiver compartment volume of 32 mL, and the effective diffusion area was 1 cm2. Formulation containing 2 mg of MTC was placed at the donor compartment and complemented to 2 mL with Krebs Henseleit Solution (KHS). The receptor phase (KHS) was continuously stirred and kept at a temperature of 37 ± 0.5°C. A sample of 500 μL was taken from the receiver solution at predetermined time intervals and replaced with an equal amount of KHS.
Ex vivo release studies of MTC through nasal mucosa
Tissue preparation
The use of natural membranes is very important to predict the real drug release characteristic. So in this experimental section of the study cattle nasal mucosa was chosen for the following reasons: (Citation1) It is easy to obtain cattle nasal mucosa at slaughterhouses in Turkey; and (Citation2) The area of the respiratory mucosa in the snout is relatively large, and this makes it possible to obtain more material from each animal. It also makes it easier to handle the tissue and increases the chance of getting intact, unstrained pieces of mucosa. On the other hand, an ethical advantage is that material is obtained from the slaughterhouse, and the sacrifice of animals for only a small piece of the animal thus avoided.
Cattle nasal cavity mucosa was obtained from the local slaughterhouse (çubuk, Ankara, Turkey). Within 10 min of the killing of the animal, the cavity mucosa was carefully removed, then immediately immersed in KHS (approx 900 mL), placed on ice and O2/CO2 (95%/5%) was applied.
Permeation studies
Within 80 min of removal, the cavity mucosa were carefully cut with a scalpel and mounted in the diffusion chambers with the mucosal and serosal sides facing the donor and receiver phases, respectively. Both sides of the nasal mucosa were filled with the KHS solution and bubbled with 95% O2/5% CO2. Following a preincubation period of 45–60 min, used in order to reach 37°C and electrophysiological equlibrium, permeability experiments were started by replacing the buffer on both sides of the tissue with prewarmed (37°C) gassed buffer. The other experimental procedures and samples collection were performed in a same fashion as explained earlier.
Histological evaluation
Each piece of mucosa was carefully removed from the diffusion chamber, and rinsed with KHS after the transport experiments. Mucosa samples were fixed in Bouien’s fixative (picric acid:saturated formaldehyde:acetic acid, 15:5:1, v/v) for 6 h. The specimens were dehydrated gradually by incubating for 1 h in each of 70, 80, 96%, and finally absolute alcohol. The samples were then incubated in methyl benzoate for 24 h in order to soften the materials. The samples were immersed first into benzene/paraplast (1:1) mixture, and then into pure Paraplast® for 6 h in a vacuum oven and embedded in paraplast. The blocks were cut in sections 5–6 μm in thickness with a rotary microtom (Leica RM 2025, Germany). The sections were stained with Crossmon Modified Triple stain for light microscopic examinations (Karl-Zeiss, Germany). The mucosa, fixated directly after isolation at the slaughterhouse, was used as a control. The examined mucosa were then scored according to coarse ranking system based on observation, where 0 = no damage, equal to control, 1 = less than 25% of epithelial cells lost, 2 = less than 50% of epithelial cells lost, 3 = less than 75% of epithelial cells lost, 4 = only basal cells left, and 5 = all epithelial and basal cells lost. The results of this scoring are presented as the mean value of individual scorings from all mucosa exposed to the same formulation (Citation10).
Nasal absorption of MTC in sheep model, sample collection, and preparation
In vivo studies were carried out for the optimized formulations in sheep for the following reasons. Sheep make an ideal model for evaluation of nasal delivery of drugs because of their large nares, the anatomical similarity to the human nasal cavity in terms of nasal cavity surface area per kg body weight, easily accessibility of veins for blood sampling, and their mild temperament under experimental conditions. Furthermore, results obtained in the sheep model have been found to correlate well to data obtained in humans (Citation11). Fifteen female sheep (Akkaraman, ∼50 kg) were divided into three groups. Formulations (0.4 mg/kg of MTC) were divided equally between both nostrils and administered to unanesthetized, awake sheep with polyethlene tubes. The formulations were released from the tubes using a syringe containing compressed air. We also administered orally (simple solution) and IV dosage forms at the same dose. A wash-out period of 7 days was left between two administrations. After application, blood samples, approximately 5 mL, were taken from the two external jugular veins in an alternating fashion at predetermined time intervals. Sera was seperated by centrifugation and stored frozen at −20°C until required for analysis.
The sample preparation: 300 μL of sodium hydroxide and 30 μL propylparaben at a concentration of 1000 μg/mL were added to 600 μL of the sera sample in a conical test tube and vortexed for a few seconds. Diethylether (2.5 mL) was then added. The solution was vortexed for 15 s and centrifuged for 5 min, at 4400 rpm. Organic layer was taken and evaporated to dryness under a stream of nitrogen, then reconstituted with 300 μL of mobile phase.
In vivo and ex vivo experiments have been carried out under approval of Gulhane Military Medical Academy Animal Experiment Ethics Committee with the decision number of 07/64.
Analytical procedure, data analysis, and statistics
The amount of MTC in the samples was determined using a HPLC system consisting of the following components: a Hewlett-Packard (Avondale, USA) Model 1100 series with a Model Agilent series G-13159 fluorescence detector and Model Agilent 1100 series G-1329 ALS auto sampler and HP chemstation. The chromatographic separation was performed using an isocratic elution. The mobile phase components were acetonitrile:0.05 M KH2PO4 (20:80, v/v) adjusted to pH 6.2 with 0.1 N o-phosphoric acid and delivered at a flow rate of 1 ml/min. The seperation was carried out at 20°C, on a reversed phase Waters® Bondopak C18 column (300 × 3. 9 mm, 5 μm particle size). A fluorescence detector was set at an excitation wavelength of 272 nm and emission wavelength of 355 nm. An injection volume of 20 μL was used for in vitro and ex vivo experiments and 100 μL for in vivo studies. Propyl paraben was used as an internal standard.
In in vitro and ex vivo experiments, the cumulative of MTC permeated per unit area was plotted against time, and the slope of the linear portion of the plot was used as steady state flux (JSS). The permeability coefficient (Kp) was calculated with equation (1), in which CV is the total donor concentration of the formulation
In in vivo studies, a non-compartmental model analysis was performed using pharmacokinetic software Pharmacologic Calculation System (v 4.1) to estimate the pharmacokinetic parameters of MTC in sera. The area under the curves (AUC) from time zero to the last experimental data point of 4 h were used in calculating the bioavailability of each formulation. The Cmax and Tmax were also obtained from Pharmacologic Calculation System. The bioavailability of each formulation was calculated with equation 2 (Citation12):
Statistical analysis of the data obtained was performed using SPSS (v 11). For comparisons of data, one-way analysis of variance using Duncan’s multiple comparison test and independent samples t-test were used. The level of statistical significance was chosen as less than 0.05 (i.e. p < 0.05).
Results and discussion
In vitro release studies of MTC with cellulose membrane
Drug release parameters of different formulations are shown in . A lower percentage of drug release was observed from gel and powder formulations compared to solution ones (p < 0.05). The steady state flux (Jss) and permeability coefficient (Kp) values were also found to be lower (p < 0.05). It is obvious that the drug release rates decreased due to high concentration of NaCMC in gel and powder formulations. The effect of high polymer content on low drug release can be interpreted in terms of (a) increased number of micelles results in a more entangled system and more rigid structure; (b) high degree of entanglement reduces the effective molecular diffusion area; (c) increased diffusional path length make drug molecules traverse, (d) active substance’s trapping in smaller polymer cells results in close proximity to the polymer molecules, leading to increased diffusional resistance; and (e) high density of chain structure limits the active substance’s movement area (Citation13–16).
Table 2. Polymer content, viscosity, and drug release parameters (by using cellulose membrane at the end of a 4-h period) of the formulations.
Another explanation of low drug release from gel formulations is their high viscosity values compared to solution ones (p < 0.05) (). The ability of a hydrogel system to serve as a reservoir for drug delivery is influenced by the macro- and micro-rheological properties of the matrix. Viscosity is the most widely utilized reference for the characterization of polymer structure, although it is not sufficiently comprehensive for the full determination of hydrogel strength. If the viscosity of the formulation is very high, the release rate of the drug may be slowed due to a decrease in the diffusion of the drug through the base to reach the membrane for penetration. This demonstrates that the viscosity of the polymer matrices showed an inverse relationship with the drug flux. The permeation process of MTC may be consistent with a diffusion barrier controlled mechanism, since the viscosity of formulations plays an important role in controlling the release of the drug if the diffusion of drug through the polymer matrix is a rate-determining step (Citation17,Citation18).
In order to describe the kinetics of the release process of drug in all formulations, various equations were used, such as the zero-order rate equation, first-order equation, Higuchi square root equation, and Hixson-Crowell cube root law. The results, as shown in , revealed that the dug release from the formulations followed first order kinetic r > 0.99, which means an excellent model fit. The release rate constants (K1) were different for each kind of formulation. It appears that diffusion is the main mechanism of drug release from the formulations.
Table 3. The kinetic parameters of the release of MTC from the formulations by using cellulose membrane.
Following the studies with cellulose membrane, one optimal formulation in each dosage form group should have been chosen, according to maximum drug release profile. However, some critical points must be paid attention, especially in nasal drug delivery studies.
The viscosity of a formulation is of relevance with respect to its administration and clearance from the nasal cavity. A formulation should have sufficiently low viscosity to permit delivery through the narrow orifice of a spray device or dosing canula, but be sufficiently viscous to retain the dose in the nasal cavity (Citation19).
It was preferred the maximum polymer content for solution formulations (S3) to prevent drainage and result in an increase in the retention time of the drug in contact with the mucus membranes (). Among the gel formulations, the medium viscosity one (G2) was chosen to prevent drainage and to be administered easily compared to low and high viscosity ones ().
For powder formulations we paid attention to the ratio of active substance/polymer ratio and chose the 2/1 (P2) ratio to apply maximum drug amount at minumum powder mass ().
Permeation studies with excised cattle concha mucosa
As it was observed with cellulose membrane, the gel formulation gave the minumum drug release profile (, p < 0.05). The release of the MTC from solution formulation was found to be higher when compared with the powder and gel formulation (, p < 0.05).
Figure 1. Comparison of ex vivo drug release from different dosage forms (S3 solution, G2 gel, P2 and EP2 powder) prepared with NaCMC by using nasal mucosa (S3-G2, S3-P2, G2-P2, P2-EP2: p < 0.05).
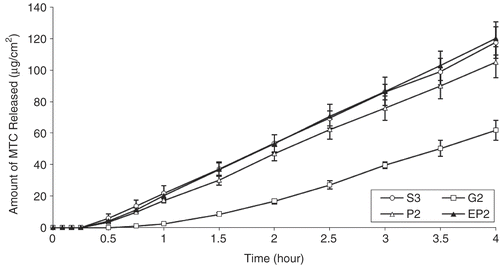
Table 4. Drug release parameters of the formulations in ex vivo experiments by using nasal mucosa and the mean scoringa values from the light microscopy study subsequent to the transport study of formulations.
In our first part of the study, we used Carbopol 981 (CRB 981) as mucoadhesive polymer for powder formulations. These formulations were acidic due to chemical structure of CRB 981 (Citation13). When we compared powder formulations prepared with these two polymers, NaCMC and CRB 981, for drug release, NaCMC exhibited more drug release (5.25 ± 0.19%) compared to CRB 981 (4.81 ± 0.21%) (p > 0.05). This can be explained with the pH-partition theory (Citation20). This theory states that drugs are absorbed from the biological membranes by passive diffusion, depending on the fraction of un-ionized form of the drug at the pH of the fluids close to that biological membrane. The degree of ionization of the drug depends on both the pKa and the pH of drug solution. The nasal mucosa acts as a lipophilic barrier and thus ionized drugs, compared to un-ionized ones, are more hydrophilic and have minimal membrane transport. The donor solution pH affects the overall partition coefficient of an ionizable substance. The pKa of the molecule is the pH at which there is a 50:50 mixture of conjugate acid-base forms. The conjugate base form is present at a pH higher than that of the pKa. MTC, as a weak basic drug, will be in un-ionized form at high pH value in NaCMC powder formulation compared to CRB 981 one, and this makes it more permeable to nasal mucosa.
The complex and lipophilic nature of nasal mucosa exhibited more resistance to the penetration of active substance compared to cellulose membrane (, p < 0.05). The low release of MTC through nasal mucosa can also be explained by the active substance’s hydrophilic nature (Citation1,Citation9).
Figure 2. Comparison of in vitro and ex vivo release of MTC from different dosage forms (S3 solution, G2 gel, P2 and EP2 powder) by using cellulose membrane and nasal mucosa at the end of 4 h period.
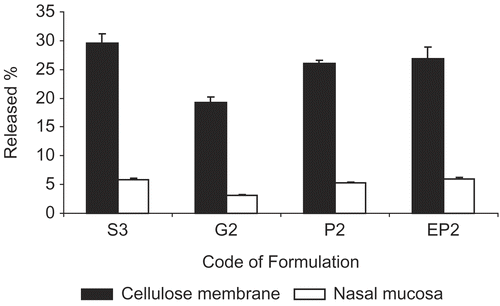
HPβCD, used in powder formulation, was effective in enhancing the nasal absorption of MTC (). The release enhancing effect of HPβCD can be explained via one of the following mechanisms: increasing membrane fluidity, disaggregation of protein aggregates and inhibiting of enzyme activity, reducing mucus viscosity or elasticity, direct effect on the paracellular pathway by a transient effect on tight junctions, and increasing solubility of the drug (Citation5,Citation21).
Histological study
The microscopy pictures of nasal mucosa after permeation studies, including washing period, are shown in . The microscopic observations indicate that solution and especially gel formulation have no severe damage on the structure of mucosa. Neither cell necrosis nor removal of all parts of epithelium from the nasal mucosa was observed after permeation studies. Signs of irritation such as vascular congestion and subepithelial edema were not observed. Moreover, no severe signs such as appearance of epithelial necrosis, sloughing of epithelial cells, and hemorrhage were detected in any of the nasal mucosa. There were no alterations in basal membrane and superficial part of submucosa as compared with non-treated mucosa (Citation22). Thus, solution and gel formulation seem to be safe with respect to nasal administration.
Figure 3. Images from the histological evaluation of nasal mucosa; control (a), solution (b; S3), gel (c; G2), powder (d; P2) and powder + HPβCD (e; EP2).
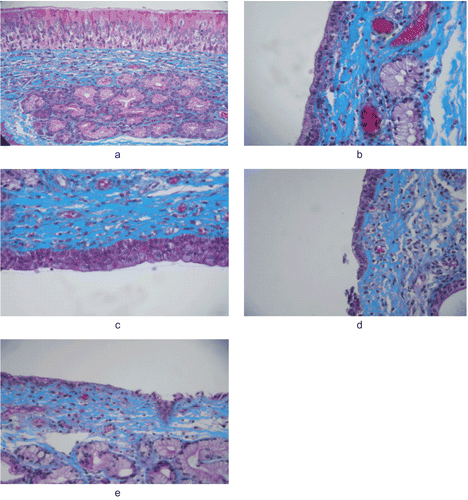
On the other hand, the effect powder formulation (P2) on nasal mucosa can be summarized as epithelium disruption and complete loss of some parts of the epithelium. This untoward effect of powder formulations may be explained with their lack of soothing/emolient excipients that reduce irritation (Citation23,Citation24). The findings of the epithelia exposed to EP2 coded formulation was similar to that of the formulation P2. So, HPBCD did not cause any irritation to the nasal mucosa (Citation25,Citation26).
Nasal absorption of MTC in sheep model
The mean sera concentration-time curve of MTC after nasal, intravenous, and oral adminstrations to five sheep is shown in . The pharmacokinetic parameters and the values of absolute bioavailabilities of three different nasal preparations of MTC are summarized in .
Figure 4. Mean sera concentration-time profiles in sheep after oral, IV, and intranasal administations of different formulations at a dose of 0.4 mg/kg applied to five sheep (G2-S3, P2, EP2: p < 0.05; P2-EP2: p < 0.05).
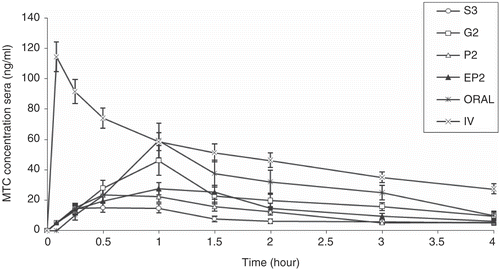
Table 5. Pharmacokinetic parameters after intravenous, oral and nasal administration of MTC in sheep (n = 5).
In contrast to in-vitro and ex-vivo experiments, intranasal administration of the gel formulation resulted in increased sera levels of MTC as compared to those for the powder and liquid formulation (, p < 0.05).
The absolute bioavailabilty of gel formulation using equation (Citation2) was 42.08 ± 5.53%, whereas the bioavailability of nasal solution and powder was 16.71 ± 3.72% and 24.57 ± 3.86%, respectively. Thus, approximately 2-fold increase in bioavailability was obtained using gel formulation in comparison to solution and powder one. Gels might enhance the absorption of MTC in several ways; (a) they adhere more strongly to the mucosal surface and increase the contact time of the drug, thereby providing a better chance for the drug to be absorbed; (b) the hypo-osmotic environment on the cell surface, because of the hypo-osmotic character of administered gel formulation, could create a condition that will promote the cell swelling and thereby promote the absorption active material; (c) gels reduce post-nasal drip into the back of the throat and therefore minimize loss of drug formulation from the nasal cavity; and (d) reduce anterior leakage of the drug out of the nasal cavity, and help localize formulation on the mucosa (Citation23,Citation27,Citation28).
This result is consistent with the findings that gel formulations are suitable carriers for nasal applications of drugs (Citation13,Citation29–33).
Powder formulation was found more numerically effective than solution formulation (). This result is in good agreement with reports in the literature; nasal particulate dosage forms have been shown to give an improved bioavailability of the delivered drugs compared with solutions, mainly due to their ability to reside longer in the nasal cavity before being cleared by the mucociliary clearance system and an increased topical concentration of the active substance by attaching to the powder particle (Citation34,Citation35). Low bioavailability from the solution formulations may be explained with faster drainage of intranasally applied solution. This result is agreement with the other studies (Citation36–38).
HPβCD enhanced the absorption of drug 1.23 ± 0.19-times via the mechanisms explained above. In our previous report, DMβCD was used as a penetration enhancer, and its enhancement ratio was found to be 1.34 ± 0.13 (Citation13). As observed, the enhancing activity of HPβCD was found to be lower than DMβCD (p > 0.05). This finding is aggreement with previous reports (Citation39–41).
Conclusion
The advantages of the nasal gel are its enhancing nasal residence time, owing to increased viscosity, and mucoadhesive characteristic, providing a higher absorption rate of the incorporated drug. Histopathological study of the nasal mucosa after permeation study suggested that gel formulation was safe for nasal administration. The NaCMC-based gel delivery system described within the study therfore represents a good starting point in order to make nasal MTC delivery feasible in the near future.
Acknowledgement
This work was supported by Gülhane Military Medical Academy Research and Development Center with the project number AR2003-38.
Presented at the 12th International Pharmaceutical Technology Symposium (IPTS-2004), 12–15 September 2004, Istanbul, Turkey.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Martindale (2002). The complete drug reference. In: Sweetman SC, ed. (2002). Martindale: The complete drug reference, thirty-fifth edition, London: Pharmaceutical Press, 1235–7.
- Gavini E, Rassu G, Sanna V, Cossu M, Giunchedi P. (2004). Mucoadhesive microspheres for nasal administration of an antiemetic drug, metoclopramide: in-vitro/ex-vivo studies. J Pharm Pharmacol, 57, 287–94.
- Chien YW. (1992). Nasal drug delivery systems. In: Swacbrick, J, ed. Novel drug delivery systems. New York: Marcel Dekker, 139–96.
- Zaki NM, Award GA, Mortada ND, Elhady SS. (2006). Rapid onset intranasal delivery of metoclopramide hydrochloride. Part II: safety of various absorption enhancers and pharmacokinetic evaluation. Int J Pharm, 327, 97–103.
- Chavanpatil MD, Vavia PR. (2004). The influence of absorption enhancers on nasal absorption of acyclovir. Eur J Pharm Biopharm, 57, 483–7.
- Dondeti P, Zia H, Needham TE. (1996). Bioadhesive and formulation parameters affecting nasal absorption. Int J Pharm, 127, 115–33.
- Merkus FWHM, Verhoef JC, Marttin E, Romeijn SG, Van der Kuy PHM, Hermens WAJJ, Schipper NGM. (1999). Cyclodextrins in nasal drug delivery. Adv Drug Del Rev, 36, 41–57.
- Ugwoke MI, Agu RU, Vanbilloen H, Baetens J, Augustijns P, Verbeke N, Mortelmans L, Verbruggen A, Kinget R, Bormans G. (2000). Scintigraphic evaluation in rabbits of nasal drug delivery systems based on carbopol 971p® and carboxymethylcellulose. J Contr Rel, 6, 207–14.
- Tas C, Ozkan Y, Savas¸er A, Baykara T. (2003). In vitro release studies of chlorpheniramine maleate gels prepared by different cellulose derivatives. II Farmaco, 58, 605–11.
- Osth K, Paulsson M, Björk E, Edsman K. (2002). Evaluation of drug release from gels on pig nasal mucosa in a horizontal Ussing chamber. J Contr Rel, 83, 377–88.
- Cheng Y-H, Watts P, Hinchcliffe M, Hotchkiss R, Nankervis R, Faraj NF, Smith A, Davis SS, Illum L. (2002). Development of a nasal nicotine formulation comprising an optimal pulsatile and sustained plasma nicotine profile for smoking cessation. J Contr Rel, 79, 243–54.
- Shargel L, Yu ABC. (1985). Applied biopharmaceutics and pharmacokinetics, 2nd ed. Norwalk: Appleton-Century-Crofts, 231–43.
- Tas C, Ozkan CK, Savaser A, Ozkan Y, Tasdemir U, Altunay H. (2006). Nasal absorption of metoclopramide from different carbopol® 981 based formulations: in vitro, ex vivo and in vivo evaluation. Eur J Pharm Biopharm, 64, 246–54.
- Escudero JJ, Ferrero C, Jimenez-Castellanos MR. (2008). Compaction properties, drug release kinetics and fronts movement studies from matrices combining mixtures of swellable and inert polymers: effect of HPMC of different viscosity grades. Int J Pharm, 351, 61–73.
- Ma WD, Xu H, Wang C, Nie S-F, Pan W-S. (2008). Pluronic F127-g-poly(acrylic acid) copolymers as in situ gelling vehicle for opthalmic drug delivery system. Int J Pharm, 350, 247–56.
- Rajinikanth PS, Mishra B. (2008). Floating in situ gelling system for stomach site-spesific delivery of claritromycin to eradicate H.pylori. J Contr Rel, 125, 33–41.
- Fang J-Y, Hwang T-L, Leu Y-L. (2003). Effect of enhancers and retarders on percutaneous absorption of flurbiprofen from hydrogels. Int J Pharm, 250, 313–25.
- D’Souza R, Mutalik S, Venkatesh M, Vidyasagar S, Udupa N. (2005). Nasal insulin gel as an alternate to parenteral insulin: formulation, preclinical, and clinical studies. AAPS Pharm Sci Tech, 6, 184–9.
- Charlton ST, Davis SS, Illum L. (2007). Evaluation of bioadhesive polymers as delivery systems for nose to brain delivery: in vitro characterisation studies. J Contr Rel, 118, 225–34.
- Mahato RI. (2004). Pharmaceutical delivery systems and dosage forms. New York: Castle Connoly, 37–64.
- Zhang Y, Zhang Q, Sun Y, Sun J, Wang X, Chen M. (2005). Nasal recombinant hirudin-2 delivery: absorption and its mechanism in vivo and in vitro studies. Bio Pharm Bull, 28, 2263–7.
- Zaki NM, Awad GA, Mortada ND, Elhady SSA. (2007). Enhanced bioavailability of metoclopramide HCI by intranasal administration of a mucoadhesive in situ gel with modulated rheological and mucociliary transport properties. Eur J Pharm Sci, 32, 296–307.
- Behl CR, Pimplaskar HK, Sileno AP, deMeireles J, Romeo VD. (1998). Effects of physicochemical and other factors on systemic nasal drug delivery. Adv Drug Del Rev, 29, 89–116.
- Aurora J. (2002). Development of nasal delivery systems. A Review. Drug Deliv Technol, 2(7), 70–3.
- Martin E, Romeijn SG, Verhoef JC, Merkus FWHM. (1997). Nasal absorption of dihydroergotamine from liquid and powder forrmulations in rabbits. J Pharm Sci, 86, 802–7.
- Asai K, Morishita M, Katsuta H, Hosoda S, Shinomiya K, Noro M, Nagai T, Takayama K. (2002). The effects of water-soluble cyclodextrins on the histological integrity of the rat nasal mucosa. Int J Pharm, 246, 25–35.
- Arora P, Sharma S, Garg S. (2002). Permeability issues in nasal drug delivery. Drug Discov Today, 7, 967–75.
- Najafabadi AR, Moslemi P, Tajerzadeh H. (2004). Intranasal bioavailability of insulin from carbopol-based gel spray in rabbits. Drug Del, 11, 295–300.
- Morimoto K, Tabata H, Morisaka K. (1987). Nasal absorption of nifedipine from gel preparations in rats. Chem Pharm Bull, 35, 3041–4.
- Tuncel T, Otuk G, Kuscu I, Ates S. (1994). Nasal absorption of roxitromycin from gel preparations in rabbits. Eur J Pharm Biopharm, 40, 24–6.
- Yetkin G, çelebi N, Agabeyoglu I, Gokcora N. (1999). The effect of dimethyl-β-cyclodextrin and sodium taurocholate on the nasal bioavailability of salmon calcitonin in rabbits. STP Pharm Sci, 9, 249–52.
- Ozsoy Y, Tuncel T, Can A, Akev N, Birteksoz S, Gerceker A. (2000). In vivo nasal studies on nasal preparations of ciprofloxacin hydrochloride. Pharmazie, 55, 607–9.
- Leitner VM, Guggi D, Bernkop-Schnurch A. (2004). Thiomers in noninvasive poypeptide delivery: in vitro and in vivo characterization of a polycarbophil-cysteine/glutathione gel formulation for human growth hormone. J Pharm Sci, 93, 1682–91.
- Ishikawa F, Katsura M, Tamai I, Tsuji A. (2001). Improved nasal bioavailability of elcatonin by insoluble powder formulation. Int J Pharm, 224, 105–14.
- Leitner VM, Guggi D, Krauland AH, Bernkop-Schnürch A. (2004). Nasal delivery of human growth hormone: in vitro and in vivo evaluation of a thiomer/glutathione microparticulate delivery system. J Contr Rel, 100, 87–95.
- Ugwoke MI, Exaud S, Mooter GVD, Verbeke N, Kinget R. (1999). Bioavailability of apomorphine following intranasal administration of mucoadhesive drug delivery systems in rabbits. Eur J Pharm Sci, 9, 213–9.
- Nakamura K, Maitani Y, Takayama K. (2002). The enhancing effect of nasal absorption of FITC-Dextran 4,400 by β-Sitosterol β-D-Glucoside in rabbits. J Contr Rel, 79, 147–55.
- Gu F, Cui F, Gao Y. (2005). Preparation of prostaglandin E1-hydroxypropyl-β-cyclodextrin complex and its nasal delivery in rats. Int J Pharm, 290, 101–8.
- Irie T, Wakamatsu K, Arima H, Aritomi H, Uekama K. (1992). Enhancing effects of cyclodextrins on nasal absorption of insulin in rats. Int J Pharm, 84, 129–39.
- Sinswat P, Temgamnuay P. (2003). Enhancing effect of chitosan on nasal absorption of salmon calcitonin in rats: comparison with hydroxypropyl- and dimethyl-β-cyclodextrins. Int J Pharm, 257, 15–22.
- Yang T, Hussain A, Paulson J, Abbruscato TJ, Ahsan F. (2004). Cyclodextrins in nasal delivery of low-molecular-weight heparins: in vivo and in vitro studies. Pharm Res, 21, 1127–36.