Abstract
An ethylene-vinyl acetate (EVA) matrix containing glimepiride was prepared as a potential transdermal drug delivery system. Permeation studies of quinupramine through the EVA copolymer membrane were carried out using a two-chamber diffusion cell. The rate of drug permeation through the EVA membrane was proportional to the PEG 400 volume fraction. The release of glimepiride from the EVA matrix was examined using a modified Franz diffusion cell. A plasticizer was added to prepare the pore structure of the EVA matrix in order to increase the rate of drug release. The effects of PEG 400, drug concentration, temperature, and plasticizer on the drug release rate were investigated. Various types of enhancers were added to an EVA matrix containing 2% glimepiride in an attempt to increase the level of skin permeation of quinupramine through an EVA matrix. The effects of the enhancers on the level of glimepiride permeation through the skin were evaluated using Franz diffusion cells fitted with intact excised rat skin. The rate of drug release from the EVA matrix increased with increasing PEG 400 volume fraction, temperature, and drug loading. The estimated activation energy of drug release was 7.274 kcal/mol for 2% drug loading dose. The release of glimepiride from the EVA matrix followed a diffusion-controlled model, where the quantity released per unit area was proportional to the square root of time. The controlled release of glimepiride was achieved using the EVA polymer including the plasticizer. Among the plasticizers used, such as the alkyl citrates and phthalates groups, diethyl phthalate slightly increased the rate of glimepiride release. Among the various enhancers used, such as the non-ionic surfactants, the glycerides, the propylene glycol derivatives, fatty acids (saturated or unsaturated), and pyrrolidones, linoleic acid showed the highest permeation rate; 3.17-times higher than the control. In conclusion, an EVA matrix containing a permeation enhancer can be used for the transdermal controlled delivery of glimepiride.
Introduction
Glimepiride is a new oral anti-diabetic drug in a third generation sulfonylurea class that is used to treat type 2 diabetes (CitationDraeger, 1995). Despite its wide acceptance, glimepiride can cause nausea and vomiting as well as hemolytic anemia due to the transient high blood concentration that occurs after oral administration. In order to avoid these problems and obtain a sustained release rate, glimepiride may be administered using a transdermal drug delivery system. Compared to their corresponding classical oral, injectable, and inhaler systems, transdermal drug delivery systems offer many advantages, such as improved systemic bioavailability of drugs due to avoidance of the first-pass metabolism by the liver and digestive system and a controlled constant drug delivery profile (i.e. controlled zero-order absorption). Other benefits include a longer duration of therapeutic action from a single application, and reversible action, i.e. ease of use and withdrawal. For example, patches can be removed to reverse any adverse effects caused by overdosing. However, skin is resistant to the permeation of most chemicals and drugs. A variety of physical and chemical methods have been used to increase the absorption of drugs through the skin (CitationShah et al., 1993).
Skin is a complex, dynamic layer organ with many functions that extend far beyond its role as a barrier to the environment. The skin is an attractive potential route for drug administration because the first-pass hepatic metabolism of drugs intended for systemic action can be avoided, thereby offering potentially lower drug dosages and reduced side effects (CitationBarry, 1983; CitationGuy & Hadgraft, 1985; CitationSmith & Maibach, 1995). One long-standing approach to increase the range of drugs that can be delivered effectively by this route has been to use penetration enhancers, which are chemicals that interact with the skin constituents to promote drug permeation. To date, large numbers of chemicals have been evaluated as penetration enhancers. The chemical methods involve the incorporation of specific chemicals in topical drug formulations to increase the level of drug penetration. Penetration enhancers facilitate the absorption of penetrants through the skin by temporarily increasing the permeability of the skin.
Basic components of transdermal devices are polymer matrix, penetration enhancer, and excipient (CitationGuy & Hadgraft, 1987). The use of a release controlling membrane is one method to regulate the drug release. The use of drugs dispersed in inert polymer matrices to achieve controlled release by diffusion has considerable attention. Among the many polymers, ethylene-vinyl acetate (EVA) copolymer is a heat processible, flexible and inexpensive material. An EVA copolymer membrane has been used successfully in membrane-controlled transdermal drug delivery systems (CitationKrishnaiah, Satyananrayana, & Bhaskar, 2002). In our laboratory, EVA has been used in studies of transdermal controlled drug delivery using a polymer membrane (CitationShin & Lee, 2002). However, pure polymer coatings are often brittle and relatively impermeable to many drugs. External plasticizers are frequently added to the polymeric networks to overcome these restrictions. They increase the flexibility of the coatings, increase the permeability of the drug, and promote film formation (CitationSiepmann, Brun, & Siepmann, 2006).
This study evaluated the possibility of using a polymer EVA membrane as a controlling membrane, and examined whether the EVA matrix system is suitable as a transdermal delivery of glimepiride. Several tests were carried out to formulate and develop the transdermal delivery system. In addition, release studies according to the loading dose were performed to confirm the controlled release of the drug. The effect of temperature, adding plasticizers on the release studies was evaluated. A glimepiride-EVA matrix containing the best enhancer and plasticizer was formulated to develop new formulations with enhanced transdermal delivery.
Materials and methods
Materials
Glimepiride was supplied by Sudo Pharm. Co., Ltd. (Korea). Ethylene vinyl acetate (EVA, 40%) was purchased from Sigma-Aldrich Chemical Co., Inc. (USA). Acetyl tributyl citrate (ATBC), tributyl citrate (TBC), acetyl triethyl citrate (ATEC), and triethyl citrate (TEC) were obtained from Morflex, Inc. (USA). Diethyl phthalate (DEP) and di-n-butyl phthalate (DBP) were acquired from Junsei Chemical Co., Ltd. (Japan). Lauric acid, oleic acid, and caprylic acid were purchased from Tokyo Kasei Kogyo Co., Ltd. 1-methyl-2-pyrrolidone and 2-pyrrolidone were supplied by Acros organics (USA). Myristic acid, linoleic acid, polyoxyethylene- 23-lauryl ether (Brij 35), polyoxyethylene-2-oleyl ether (Brij 92), and polyoxyethylene-23-lauryl ether (Brij 72), tetraethylene glycol (TEG), and diethylene glycol (DEG) were obtained from Sigma-Aldrich Co. (USA). Oleyl macrogol-6 glycerides, caprylocaproyl mcarogol-8 glycerides, propylene glycol mono caprylate, propylene glycol laurate, and propylene glycol monolaurate were a kind gift from Gattefose (France). Acetonitrile was of HPLC grade and acquired from J.T.Baker Inc. (USA). All other chemicals were of regent grade and used without further purification.
Determination of drug solubility
Excess amounts of glimepiride were equilibrated with saline containing various concentrations of PEG 400. Each solution was shaken at 37°C for 8 h, and the solutions were filtered through a 0.45 μm filter membrane. The concentration of glimepiride was determined using by UV spectrophotometer at a wavelength of 230 nm after proper dilution.
Preparation of the EVA copolymer membrane
The ethylene vinyl acetate membrane was prepared using a casting method. Approximately 2 g of EVA copolymer beads was dissolved in 20 ml of methylene chloride in a glass beaker. The polymer solution was poured onto a glass plate and the solvent was allowed to evaporate off overnight at room temperature. The membrane was then removed from the plate.
Drug permeation through the EVA membrane
Two chamber-diffusion cells were used to determine the steady state permeation of glimepiride through the EVA membrane. Each half-cell has a volume of ~ 7 ml and an effective diffusion area of 0.79 cm2. A piece of the EVA membrane was clamped between the two halves of the assembled cell at 37°C. The donor compartment was filled with a suspension containing the drug at concentrations above its solubility in a solvent containing various concentrations of a PEG 400 solution. The same concentration of a PEG 400 solution (without the drug) was added to the receptor compartment to prevent solvent permeation from the donor to the receptor side on the glimepiride permeation through the membrane. The cell was shaken horizontally at 150 rpm to minimize the boundary effect. The total volume of the receptor solution was removed at predetermined intervals and replaced with 7 ml of a fresh solution. The amount of drug permeated was determined at 230 nm using a UV spectrophotometer.
HPLC determination of glimepiride
Glimepiride was assayed by high-performance liquid chromatography (HPLC). The HPLC system consisted of pump (Knauer, DE/K-120, USA), ultraviolet detector (Waters 484, USA), C18 column (250 × 4.6mm, 5 μm), degaser, and an integrator (D520A, Youngin scientific Co., Ltd., Korea). The mobile phase was composed of a mixture (50:50, v/v) of acetonitrile and 1 g/L formic acid. A flow rate of 1. 0 ml/min yielded an operating pressure of ~ 1000 psi. The UV detector was operated at a wavelength of 230 nm. Under these conditions, the glimepiride peak appeared at a retention time of 13.1 min.
Drug-containing EVA matrix preparation
The drug-containing EVA matrix was prepared using a casting process. A weighed amount of EVA copolymer beads was dissolved in 20 ml of methylene chloride in a beaker, which was followed by the addition of the drug solution. This mixture was poured onto a glass plate and the solvent was allowed to evaporate off overnight at room temperature. The membrane was removed from the plate, and a piece of matrix was cut properly before the experiment. The drug content was calculated from the weight ratio of the drug and copolymer used.
Enhancer-containing EVA matrix preparation
An enhancer-containing EVA matrix was prepared by the casting process. A weighed amount of EVA copolymer beads was dissolved in 20 ml of chloroform in a beaker, which was followed by the addition of the drug solution and enhancer with constant stirring. This mixture was poured onto a glass plate and the solvent was allowed to evaporate off overnight at room temperature. The membrane was removed from the plate, and a piece of matrix was cut properly before the experiment. The drug content was calculated from the weight ratio of the drug and copolymer used.
In vitro release studies
The in vitro release of glimepiride from the EVA matrix was examined using a modified Keshary-Chien cell. One unit of the EVA matrix was clamped between the cell cap and receptor cell. The diameter of the cell was 2 cm, which provide a 3.14 cm2 effective constant area between the matrix and bulk solution of 20 ml. The receptor, 40% PEG 400 solution was maintained at 37°C and stirred at 350 rpm. Before the experiment, the system was tested to remove the remaining air bubbles in the receptor site. At a predetermined time, the entire solution was taken from the receptor cell and replaced with fresh solution. The cumulative amount of glimepiride released from the matrix was determined using a UV spectrophotometer at 230 nm. The effects of the drug concentration on its release from the EVA matrix were examined at drug concentrations of 0.5%, 1%, 1.5%, 2%, and 2.5% (w/w). The effect of temperature on drug release was examined at 27°C, 32°C, 37°C, and 42°C. Each data point represents the average of three determinations.
In vitro release studies from the plasticizer-containing EVA matrix
The plasticizer reduces the brittleness, improves flow, imparts flexibility, and increases toughness, strength, tear resistance, and impact resistance of the polymer. Increasing the amount of plasticizer can lead to an increase in free film elongation as well as a decrease in tensile strength and Young’s modulus. The plasticizer was dropped into a drug-containing EVA solution and mixed for 1 h. This method was chosen to produce large undamaged pieces of membrane with no particular molecular orientation. This mixture was poured onto a glass plate and the solvent was allowed to evaporate off overnight at room temperature. Plasticizers were added at a ratio of 5% (w/w) of the EVA matrix. The plasticizers used were alkyl citrates, such as acetyl tributyl citrate (ATBC), tributyl citrate (TBC), acetyl triethyl citrate (ATEC), triethyl citrate (TEC), and phthalates, such as diethyl phthalate (DEP) and di-n-butyl phthalate (DBP).
Skin preparation
A male rat (Sprague Dawley rat strain) was sacrificed by ether anesthesia. The hair of the abdominal area was removed carefully with electric clippers. A square section of the abdominal skin was excised. After the incision, the adhering fat and other visceral debris were removed carefully from the undersurface of the skin with tweezers (CitationDurrhein et al., 1980). The excised skin was used immediately. The experiments were carried out in accordance with the ‘Guiding Principles in the Use of Animals in Toxicology’ adopted by the Society of Toxicology (USA) in July 1989 and revised in March 1999. The Animal Care Committee of Chonnam National University (Gwangju, Republic of Korea) approved the design and conduct of this study.
Effects of enhancer on the permeation of glimepiride from the EVA matrix through rat skin
The matrix containing 5% enhancer was prepared using a casting process. The following enhancers were used: saturated fatty acid groups such as capric acid, myristic acid, and lauric acid; unsaturated fatty acid groups, such as oleic acid and linoleic acid; pyrrolidones such as 2-pyrrolidone, N-methyl-2-pyrrolidone, and poly vinyl pyrrolidone; propylene glycol groups such as propylene glycol monolaurate, propylene glycol laurate, and propylene glycol monocaprylate; non-ionic surfactants such as polyoxyethylene-2-oleyl ether, polyethylene-2-stearyl ether, and polyoxyethylene-23-lauryl ether; and glycerides such as oleyl macrogol-6 glycerides and caprylocaproyl macrogol-8 glycerides. The freshly excised full-thickness skin sample was mounted on the receptor site of the diffusion cell with the stratum corneum side facing upwards to the donor compartment and the dermal side facing downwards to the receptor compartment. The appropriate size of the matrix was placed on the stratum corneum side and covered with a round glass plate and clamped. The receptor medium was a 40% PEG 400 solution to achieve a sink condition and was maintained at 37°C by a circulating water bath. The total samples were withdrawn at pre-determined times and replaced immediately with an equal volume of fresh medium. The permeation quantities of glimepiride were analyzed by HPLC at 230 nm. Each data point represents the average of three determinations.
The permeation rate was calculated from the slope of the linear region of the permeation profile. The flux was calculated from the slope of the linear region of the Q vs time (t) permeation profile. The cumulative amount of glimepiride through the rat skin was plotted as a function of time (t). A linear profile was observed over an 8 h period and the slope of the linear portion of the curve was determined by linear regression. The effectiveness of the penetration enhancers was defined as the enhancement factor (EF), which was calculated using the following equation:
Scanning electron microscopy (SEM)
The membrane structure was examined by scanning electron microscopy (SEM). The membrane samples were gold coated to a thickness of ~ 500 × 10−8 cm using a Hitachi E-1030 coater under high vacuum (0.1 Torr), high voltage (1.2 kV), and 50 mA. The coated samples were examined using a Hitachi S-4700 scanning electron microscope.
Results and discussion
Solubility of glimepiride
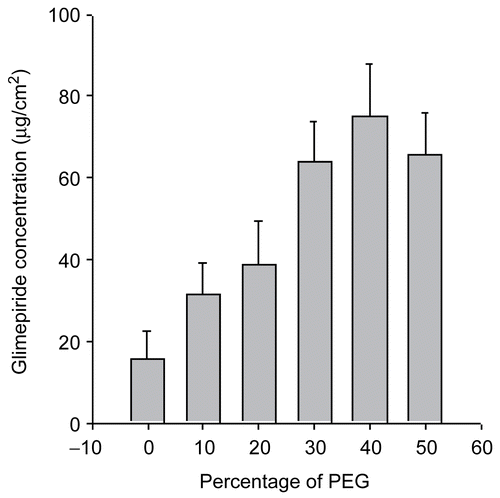
The aqueous solubility of glimepiride is extremely low and can be improved by adding a water-miscible hydrophilic polymer, such as PEG 400, into the aqueous solution as a solubilizer for glimepiride. PEG 400 was reported to be an excellent solubilizer for many steroids (CitationChien & Lambert, 1975). The solubility of glimepiride increased with increasing volume fraction of PEG 400 and was highest at 40% PEG 400 ().
Figure 1. Solubility of glimepiride according to the PEG 400 concentration in solution.
Permeation studies through the EVA membranes
The cumulative amount of the drug (Q) permeating through a unit surface area under constant stirring can be expressed mathematically by the following relationship:
where P is the permeability coefficient and CD and CR are the drug concentration in the donor (D) and the receptor (R) solution, respectively.
When the drug concentration in the donor solution (CD) was maintained at a level greater than the equilibrium solubility and the drug concentration in the receptor solution (CR) was maintained under sink conditions (i.e. CR << Ce), equation (1) can be simplified to the following:
and a constant permeation profile should be yielded. The rate of permeation can then be defined as follows:
As expected from equation (2), a constant permeation profile would be achieved when the glimepiride concentration in the donor solution was at a level greater than its equilibrium solubility (data not shown). The rate of permeation (Q/t), which was measured from the slope of the Q vs t plots (equation 2), increased with increasing PEG 400 concentration in solution until 40% (v/v), but was slightly lower in the 50% (v/v) PEG 400 solution. As expected from equation (3), an increase in the permeation rate (Q/t) was found to be dependent on the equilibrium solubility (Ce) of furosemide in the PEG 400 solutions.
The effect of PEG 400 on the permeability coefficient (P) of glimepiride across the EVA membrane was determined using equation (4):
The permeability coefficient (P) decreased with increasing volume fraction of PEG 400 in the saline solution (data not shown).
Release of glimepiride from the EVA matrix
A characteristic drug release profile of matrix-type drug delivery systems can be represented by Higuchi’s equation (Higuchi, 1961). The release from system with a dispersed drug in a homogeneous matrix should follow the following relationship:
where Q is the amount of drug released after time t per unit exposed area, D is the diffusivity of the drug in the matrix, A is the initial drug loading dose dispersed in the polymer matrix, and Cs is the drug solubility in the matrix. He later derived a similar relationship for release from a granular matrix system, in which diffusion occurs through channels (CitationHiguchi, 1963):
where D and Cs refer to the diffusivity and solubility in the permeability field, respectively, τ is the tortuosity of the matrix, and ϵ is the porosity of the matrix. Although the two equations are for different mechanisms, they both describe drug release as being linear with the square root of time (CitationDesai, Simonelli, & Higuchi, 1965; CitationDesai et al., 1966; CitationLapidus & Lordi, 1966; CitationSingh et al., 1967):
where for a homogeneous matrix system:
and for a granular matrix system
The validity of the relationships has been confirmed experimentally by many studies using a variety of systems (CitationLapidus & Lordi, 1966; CitationFarhadieh, Boradkin, & Buddenhagen, 1971).
Effect of drug loading dose
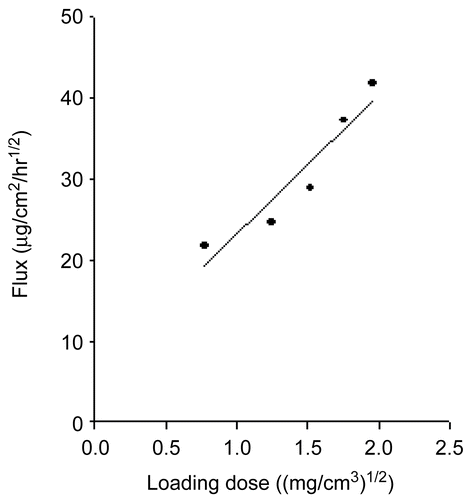
The release profiles of glimepiride from EVA matrices with different drug loadings over an 8-h period were studied. The cumulative amount of glimepiride released (Q) vs the square root of time (t1/2) plot shows good linearity for all five concentrations. As expected from equations (7) and (8), a plot of Q/t1/2 vs the square root of the loading dose (A) yielded a straight line (Figures, not shown). The Q/t1/2 increased in proportion to the increase in the square root of the glimepiride loading dose ().
Figure 2. Relationship between the quinupramine flux and the drug-loading dose in the EVA matrix at 37°C.
Effect of release media temperature
The effects of the temperature of the release medium on the drug release from the EVA matrix were examined at 27, 32, 37, and 42°C. shows the amount of drug released as a function of temperature. The cumulative amount of the drug released (Q) was plotted as a function of the square root of time (t1/2). After an initial period of drug release, the release was approximately linear with respect to t1/2. The steady-state rate of the drug release (Q/t1/2) was estimated from the slope of the linear Q − t1/2 profile from 4 to 24-h.
Figure 3. Effects of temperature on the release of quinupramine from the EVA matrix containing various loading doses.
The permeability coefficient can then be defined by:
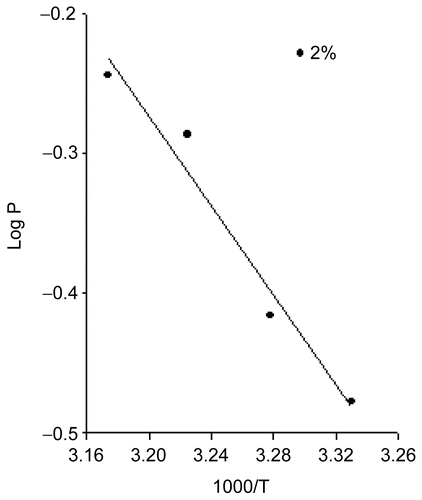
As expected from equation (12), a plot of log P vs 1000/T yielded a straight line (). The Ea (activation energy of release) was measured from the slope of the log P vs 1000/T plots (equation 14).
The drug flux from the EVA matrix containing 1.5% glimepiride at 27, 32, 37, and 42°C was 23.95, 27.57, 37.12, and 41.01 μg/cm2/h1/2, respectively. It should be noted that the rate of drug release increased with increasing temperature. The rate of drug release at 42°C was ~ 2.49-times higher than that at 27°C. The estimated activation energy of release was 7.274 kcal/mol for 2% loading dose (). This clearly shows that the release of glimepiride from the EVA matrix is an energy-linked process (CitationMiyazake et al., 1982). The temperature effects could be due to either the increased solubility of the drug and/or its effects on diffusion.
Effect of plasticizers on the drug release from EVA matrix
Generally plasticizers increase the amount of drug release with the increased chain mobility of polymer. The plasticizer will interpose itself between the polymer chains and interact with the forces held together by extending and softening the polymer matrix (CitationEntwistle & Rowe, 1979). The plasticizer reduces the brittleness, improves flow, imparts flexibility, and increases the toughness, strength, tear resistance, and impact resistance of the polymer. The selection of a suitable plasticizer and its concentration has a profound effect on the mechanical properties and permeability of drugs (CitationCrawford & Esmerian, 1971). Increasing the amount of plasticizer can lead to an increase in free film elongation and a decrease in tensile strength. A strong interaction between the drug and a polymer has been reported to influence significantly the amount of drug release through a polymeric film (CitationBodmeier & Paeratakul, 1989; CitationJenquin et al., 1990). shows the release profiles of glimepiride from an EVA matrix containing a citrate group and phthalate group as plasticizers. In the case of the citrate group, there was a slight increase in the amount of drug released. On the other hand, the phthalate group show a much higher release rate.
Table 1. Effects of the citrates on the release of glimepiride from the EVA matrix with 4% loading dose at 37°C.
Effect of enhancers on the permeation of glimepiride across the rat skin
The highly organized structure of SC forms an effective barrier to the penetration of a wide range of substances. The transdermal route for systemic drug delivery has attracted considerable attention in recent years (CitationShaw, 1984; CitationBanerjee & Ritschal, 1989; CitationKim et al., 1993; CitationFuhrman et al., 1997; CitationMura et al., 2000). However, the major problem encountered using this route of administration is the low permeability of the skin. One way of reducing this problem and improving the bioavailability after the topical application of drugs is to include a penetration enhancer in the formulation. Penetration enhancers, accelerants, or promoters are believed to interact with some components of the skin, causing increased fluidity in the intercellular lipid lamellae, the SC, to swell and/or leach out some of the structural components, which increases the level of drug penetration through the barrier membrane (CitationElfbaum & Laden, 1968; CitationCarelli et al., 1993; CitationMagnusson & Runn, 1999). These substances may also reduce the capacity for drug binding to the skin, thereby improving drug transport. Consequently, the use of penetration enhancers has become more prevalent, and is a growing trend in transdermal drug delivery.
Many penetration enhancers have a molecular structure and charge density suitable for interacting with the polar head groups of lipids. Therefore, they may disturb hydrated regions and head group interactions, as well as fluidize these regions, which would increase the penetration of polar (hydrophilic) compounds. A more aqueous fluid may enter the tissue, increasing the interlamellar volume and hence the volume available for polar diffusion. Disruption of the interfacial structure can lead to increased disorder of the lipid chains, resulting in the increased permeation of non-polar (hydrophobic) compounds. Many penetration enhancers exert some, if not most, of their activity by modifying the organization of the intercellular lipids.
Surface-active molecules, particularly ionic substances organic acids, tend to denature the keratin filaments in corneocytes. Small polar enhancers, such as dimethyl sulfoxide and dimethylformamide, as well as pyrrolidones have effects on the protein and lipid structures. Other polar solutes/solvents, such as urea, natural moisturizing factor, polyols, and water, may solvate or hydrate the proteins both in the corneocytes and intercellular region, causing swelling and enhanced penetration.
The LPP (Lipid Protein Partitioning) theory proposes that one action of a penetration enhancer may be to increase the concentration of a penetrant, or a coenhancer, in the SC. An increased level of the drug in the membrane would then promote permeation by increasing its concentration gradient and increasing the flux. An increased level of coenhancer may further increase the drug concentration in the membrane, or reduce the barrier resistance of the tissue by lipid or protein interactions. Small polar penetration enhancers, such as dimethyl sulfoxide, ethanol, and propylene glycol, may accumulate in the horny layer to such an extent that they alter the solubility of the penetrant in the SC. Synergism is often shown by propylene glycol combined with many accelerants, such as Azone, long chain alcohols and terpenes.
The precise mechanisms for the function of many penetration enhancers are unclear. However, it is almost certain that they will have multiple effects once absorbed into the SC. Williams and Barry (Citation2004) introduced the lipid protein partitioning (LLP) theory to describe the modes of action of penetration enhancers. According to their theory, enhancers would act by one or more of three main mechanisms. This may depend to a large extent on the mechanism by which propylene glycol increases the partitioning of the main promoter into the SC. CitationBarry and Bennett (1987) suggested that penetration enhancers, such as propylene glycol plus 2-pyrrolidone, N-methylpyrrolidone, and N-methylformamide, enhance the permeation of polar and non-polar compounds. Decylmethylsulfoxide plus propylene glycol affects the polar route, and accelerants, propylene glycol plus oleic acid, propylene glycol alone, and to a limited extent, water, mainly modify the non-polar route.
In these experiments, various enhancers, such as fatty acids (saturated, unsaturated), specialized oils, pyrrolidones, and non-ionic surfactants were used to increase the rate of glimepiride permeation through rat skin. Fatty acids are currently attracting considerable attention as penetration enhancers (CitationTanojo et al., 1997; CitationOh et al., 1998). This class of enhancer has the advantage of being an endogenous component in human skin. Fatty acids can differ in several features: chain length, characteristics of the double bonds (position, number, and configuration), branching schema, and substituents. These structural variations can influence their effects as skin penetration enhancers (CitationElyan, Sidhom, & Plakogiannis, 1996; CitationBhatia & Singh, 1998; CitationTakeuchi et al., 1998; CitationTaguchi et al., 1999). Fatty acids are capable of inserting between the hydrophobic tails of the stratum corneum lipid bilayer, thus disturbing their packing, increasing their fluidity, and, subsequently, decreasing the diffusional resistance to permeants (CitationGolden, Mckie, & Potts, 1987). Fatty acids (FAs) have been shown, via interactions with intercellular lipid domains, to promote the skin permeation of drugs with a wide range of polarities (CitationCooper, 1984; CitationBarry & Bennett, 1987). The efficacy of FAs is intrinsically linked to their structure, with significant differences between the saturated and unsaturated forms as well as between those of different hydrocarbon chain lengths (CitationTanojo et al., 1997). Unsaturated FAs, particularly those with a cis conformation and C18 chain lengths, have been shown to be more effective enhancers than their saturated counterparts, promoting the permeation of such penetrants as naloxone (CitationAungst, Rogers, & Shefter, 1986) and flurbiprofen (CitationChi, Park, & Kim, 1995). When introduced into the predominantly saturated, straight-chained lipid environment of the SC, these kinked FAs intercalate and disrupt the ordered lipid array (CitationGreen, Guy, & Hadgraft, 1988). This results in the formation of separate fluid states that disorder the endogenous lipids (CitationOngpipattanakul et al., 1991; CitationChi et al., 1995). Saturated fatty acids with a linear shape and low solubility are less capable of disrupting the lipid packing of the stratum corneum and inserting themselves into the lipid bilayers than kinked unsaturated fatty acids with high solubility.
shows the permeation aspects of glimepiride from the EVA matrix including various enhancers. Compared to the control, the saturated fatty acid group showed a slightly higher permeation rate. Among the saturated fatty acid group, myristic acid produced the largest increase in permeation rate. In the case of the glimepiride EVA matrix, the unsaturated fatty acid group showed a higher permeation rate than the saturated fatty acid group. Surfactants have been reported to enhance the permeability of drugs (CitationSarpotdar & Zatz, 1986; CitationCappel & Kreuter, 1991; CitationIwasa et al., 1991; CitationNaik et al., 1995; CitationLopez et al., 2000; CitationShin, Cho, & Oh, 2001). They have effects on the permeability characteristics of several biological membranes, including skin (CitationIwasa et al., 1991; CitationShokri et al., 2001). For this reason, they can enhance the skin penetration of other compounds present in the formulation. Therefore, in recent years, they have been used to enhance the permeation rates of a variety of drugs (CitationAungst et al., 1986; CitationFlorence, Tuker, & Walters, 1994) examined in the mechanism for the effect of non-ionic surfactants, such as permeation enhancers. The skin pre-treated with a non-ionic surfactant showed that the SC was loosely layered with wide intercellular spaces (CitationLopez et al., 2000).
Table 2. Enhancement factor according to various enhancers.
With other experiments performed at our laboratory, Brij 92 (polyoxyethylene 2-oleyl ether) produced the best enhancing effect. Brij 35 (polyoxyethylene 2-stearyl ether) and Brij 72 (polyoxyethylene 23-lauryl ether) produced a similar increase in permeation rate. A permeation study of glycerides and propylene glycol groups as enhancers was performed. Caprylocaproyl macrogol-glyceride (Labrasol) increased the passive transport of drug molecules, and has high tolerance and low toxicity. It was included as a pharmaceutical excipient in European Pharmacopoeia in 1998. Oleoyl macrogo-6 glyceride (Labrafil) is a PEG derivative that is used as a co-surfactant in pharmaceutical systems, such as microemulsions. This substance is biocompatible and biodegradable (CitationChowan & Pritchard, 1978). Among the glycerides, Oleoyl macrogo-6 glyceride produced a significantly high permeation rate of glimepiride.
Propylene glycol (PG) is widely used as a vehicle for penetration enhancers and permeates well through human stratum corneum. PG readily permeates the skin and in so doing may carry the drug molecules across (CitationGao et al., 1995). The permeation of PG through the tissue can alter the thermodynamic activity of the drug in the vehicle, which would in turn modify the driving force for diffusion. PG may partition into the tissue, facilitating uptake of the drug into the skin, and there may be some minor disturbance to intercellular lipid packing within the stratum corneum bilayers (CitationChowan & Pritchard, 1978). Propylene glycol monolaurate produced a significant increase in the rate of glimepiride permeation. Pyrrolidones have been used as penetration enhancers in human skin for hydrophilic and lipophilic permeants (CitationWilliams & Barry, 2004). In terms of the mechanisms of action, pyrrolidones partition well into the human corneum stratum. Within the tissue, they may act by altering the solvent nature of the membrane, and pyrrolidones have been used to generate ‘reservoirs’ within the skin membranes. Such a reservoir effect has the potential for the sustained release of a permeant from the stratum corneum over extended periods of time (CitationSquillante et al., 1998). Among the enhancers examined, linoleic acid showed the best enhancing effect.
Scanning electron microscopy (surface morphology)
shows SEM micrographs of drug-EVA matrix and drug-EVA matrix containing DEP. The drug was well dispersed in the EVA matrix. The surface morphology determined by SEM indicated that the drug was released from the glimepiride-EVA matrix containing the plasticizer through the micro-channels produced by the plasticizer. The drug-EVA matrix structure () was homogeneously dense and has no visual pores, indicating that the porosity is very small and the release rate of glimepiride from the EVA matrix is low. To increase the porosity of EVA matrix and to prevent the shrinkage of membranes during the formation process, a plasticizer such as DEP was added to the casting solution (). It was found that the addition of plasticizers could have an effect on membrane morphology and could be used to effectively control the porosity of EVA matrix, and the effects of plasticizers on membrane porosity could be well correlated with the release rate of glimepiride from EVA matrix. This characteristic structure of EVA matrix was suitable for transdermal drug delivery because the porous sublayer can function as a drug reservoir and increase the release rate of glimepiride from the EVA matrix, so the drug release rate could be controlled by the addition of plasticizers in casting solution.
Figure 4. Surface morphology of the matrix before (a, b) or after release (c, d) test by scanning electron microscopy. (a) EVA/Drug; (b) EVA/Drug/DEP; (c) EVA/Drug; (d) EVA/Drug/DEP.

Conclusions
The rate of drug release from the EVA matrix increased with increasing PEG 400 volume fraction, temperature, and drug loading dose. The estimated activation energy of drug release was 7.274 kcal/mol for 2% drug loading dose. The release of glimepiride from the EVA matrix followed a diffusion-controlled model, where the quantity released per unit area is proportional to the square root of time. Among the plasticizers used, diethyl phthalate produced a slight increase in the rate of glimepiride release. Among the various enhancers used, linoleic acid showed the highest permeation rate; 3.17-times that of the control. Overall, an EVA matrix containing a permeation enhancer can be used for the transdermal controlled delivery of glimepiride.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Aungst, B.J., Rogers, N.J., Shefter, E. (1986). Enhancement of naloxon penetration through human skin in vitro using fatty acids, fatty alcohols, surfactants, sulfoxides and amides. Int J Pharm, 33, 225–34.
- Banerjee, P., Ritschal, W. (1989). Transdermal permeation of vasopressin. II. Influence of Azone on in vitro and in vivo permeation. Int J Pharm, 49, 199–204.
- Barry, B.W. (1983). Properties that influence percutaneous absorption. In: Barry, B.W., ed. Dermatological formulations; percutaneous absorption. New York: Marcel Dekker, 127–233.
- Barry, B.W., Bennett, S.L. (1987). Effect of penetration enhancers on the permeation of mannitol, hydrocortisone and progesterone through human-skin. J Pharm Pharmacol, 39, 535–46.
- Bhatia, K.S., Singh, J. (1998). Synergistic effect of iontophoresis and a series of fatty acids on LHRH permeability through porcine skin. J Pharm Sci, 87, 462–9.
- Bodmeier, R., Paeratakul, O. (1989). Evaluation of drug containing polymer films prepared from aqueous latexes. Pharm Res, 6, 725–30.
- Cappel, M.J., Kreuter, J. (1991). Effect of nonionic surfactants on transdermal drug delivery: I. Polysorbates. Int J Pharm, 69, 143–53.
- Carelli, V., Di, Colo G, Nanoripieri, E., Serafini, M. (1993). Bile acids as enhancers of steroid penetration through excised hairless mouse skin. Int J Pharm, 89, 81–9.
- Chi, S.C., Park, E.S., Kim, H. (1995). Effect of penetration enhancers on flurbiprofen permeation through rat skin. Int J Pharm, 126, 267–74.
- Chien, Y.W., Lambert, H.J. (1975). Solubilization of steroids by multiple co-solvent systems. Chem Pharm Bull, 23, 1085.
- Chowan, Z.T., Pritchard, R. (1978). Effect of surfactants on percutaneous absorption of naproxen. I: comparisons of rabbit, rat and human excised skin. J Pharm Sci, 67, 1272–4.
- Cooper, E.R. (1984). Increased skin permeability for lipophilic molecules. J Pharm Sci, 73, 1153–6.
- Crawford, R.R., Esmerian, O.K. (1971). Effect of plasticizers on some physical properties of celluose acetate phthalate films. J Pharm Sci, 60, 312–4.
- Desai, S.J., Simonelli, A.P., Higuchi, W.I. (1965). Investigation of factors influencing release of solid drug dispersed in inert matrices. J Pharm Sci, 54, 1459.
- Desai, S.J., Singh, P., Simonelli, A.P., Higuchi, W.I. (1966). Factors influencing release of solid drug dispersed in inert matrices II. Quantitation of procedures. J Pharm Sci, 55, 1224.
- Draeger, E. (1995). Clinical profile of glimepiride. Diabetes Res Clin Pract, 28 (Suppl), 5139–5146.
- Durrhein, H., Flynn, G.L., Higuchi, W.I., Behl, C.R. (1980). Permeation of hairless mouse skin: experimental methods and comparison with human epidermis permeation by alkanols. J Pharm Sci, 69, 781.
- Elfbaum, S., Laden, K., (1968). The effect of dimethyl sulphoxide on percutaneous absorption. J Soc Cosmet Chem, 19, 119–127.
- Elyan, B.M., Sidhom, M.B., Plakogiannis, F.M. (1996). Evaluation of the different fatty acids on the percutaneous absorption of metaproterenol sulfate. J Pharm Sci, 85, 101–5.
- Entwistle, C.A., Rowe, R.E. (1979). Plasticization of cellulose ethers used in the film coating of tablets. J Pharm Pharmacol, 31, 269–72.
- Farhadieh, B., Boradkin, S., Buddenhagen, J. (1971). Drug release from methyl acrylate-methyl methacrylate copolymer matrix: kinetics of release. J Pharm Sci, 60, 209.
- Florence, T., Tuker, I.G., Walters, K.A. (1994). Interaction of non-ionic alkyl and aryl ethers with membranes and other biological systems. In: Rosen MJ, ed. Structure performance relationships in surfactants. ACS symposium Series, 253, 189–207.
- Fuhrman, L.C., Michniak, B.B., Behl, C.R., Malick, A.W. (1997). Effect of novel penetration enhancers on the transdermal delivery of hydrocortisone: an in vitro species comparison. J Contr Rel, 45, 199–206.
- Gao, Z., Crowley, W.R., Shukla, A.J., Johnson, J.R., Reger, J.F. (1995). Controlled release of contraceptive steroids from biodegradable and injectable gel formulations; in vivo evaluation. Pharm Res, 12, 864–8.
- Golden, G.M., Mckie, J.E., Potts, R.O. (1987). Role of stratum corneum lipid fluidity in transdermal drug flux. J Pharm Sci, 76, 25–8.
- Green, P.G., Guy, R.H., Hadgraft, J. (1988). In vitro and in vivo enhancement of skin permeation with oleic-acid and lauric acids. Int J Pharm, 37, 251–5.
- Guy, R.H., Hadgraft, J. (1985). Transdermal drug delivery: the ground rules are emerging. Pharm Int, 6, 112.
- Guy, R.H., Hadgraft, J. (1987). The effect of penetration enhancers on the kinetics of percutaneous absorption. J Contr Rel, 5, 43–51.
- Higuchi, T. (1961). Rate of release of medicaments from ointment bases containing drug in suspension. J Pharm Sci, 50, 874.
- Higuchi, T. (1963). Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci, 52, 1145.
- Iwasa, A., Irimoto, K., Kasai, S., Okuyama, H., Nagai, T. (1991). Effect of nonionic surfactants on percutaneous absorption of diclofenac sodium. Yakuzaigaaku, 51, 16–21.
- Jenquin, M.R., Liebowitx, S.M., Sarabia, R.E., McGinity, J.W. (1990). Physical and chemical factors influencing the release of drugs from acrylic resin films. J Pharm Sci, 79, 811–6.
- Kim, C., Kim, J., Chi, S., Shim, C. (1993). Effect of fatty acids and urea on the penetration of ketoprofen through rat skin. Int J Pharm, 99, 109–18.
- Krishnaiah, YSR, Satyananrayana, V., Bhaskar, P. (2002). Influence of limonene on the bioavailability of nicardipine hydrochloride from membrane-moderated transdermal therapeutic systems in human volunteers. Int J Pharm, 247, 91–102.
- Lapidus, H., Lordi, N.G. (1966). Some factors affecting the release of a water-soluble drug from a compressed hydrophilic procedures. J Pharm Sci, 55, 840.
- Lopez, A., Llinares, F., Cortell, C., Herraez, M. (2000). Comparative enhancer effects of span 20 with Tween 20 and Azone on the in vitro percutaneous penetration of compounds with different lipophilicities. Int J Pharm, 202, 133–40.
- Magnusson, B.M., Runn, P. (1999). Effect of penetration enhancers on the permeation of the thyrotropin releasing hormone analogue pGlu-3-methyl-His-Pro amide through human epidermis. Int J Pharm, 178, 149–59.
- Miyazake, S., Ishii, K., Sugibayashi, K., Morimoto, Y., Takada, M. (1982). Antitumor effect of ethylene-vinyl acetate copolymer matrices containing 5-fluorouracil on Ehrlich Ascites carcinoma in mice. Chem Pharm Bull, 30, 3770.
- Mura, P., Faucci, M.T., Bramanti, G., Corti, P. (2000). Evaluation of transcutol as a clonazepam transdermal permeation enhancer from hydrophilic gel formulations. Eur J Pharm Sci, 9, 365–72.
- Naik, A., Pechtold, L., Potts, R.O., Guy, R.H. (1995). Mechanism of oleic acid-induced skin penetration enhancement in vivo in humans. J Contr Rel, 37, 299–306.
- Oh, S.Y., Jeong, S.Y., Park, T.G., Lee, J.H. (1998). Enhanced transdermal delivery of AZT (Zidovudine) using iontophoresis and penetration enhancer. J Contr Rel, 51, 161–8.
- Ongpipattanakul, B., Burnete, R.R., Potts, R.O., Francoeur, M.L. (1991). Evidence that oleic-acid exists in a separate phase within stratum-corneum lipids. Pharm Res, 8, 350–4.
- Sarpotdar, R., Zatz, J.L. (1986). Percutaneous absorption enhancement by nonionic surfactants. Drug Dev Ind Pharm, 12, 1625–47.
- Shah, V.P., Peck, C.C., Williams, R.L., Walters, K.A., Hadgraft, J. (eds) (1993). Pharmaceutical skin penetration enhancement. New York: Marcel Dekker, 417–27.
- Shaw, J. (1984). Pharmacokinetics of nitroglycerine and clonidine delivered by transdermal route. Am Heart J, 108, 217–22.
- Shin, S.C., Cho, C.W., Oh, I.J. (2001). Effects of non-ionic surfactants as permeation enhancers towards piroxicam from the poloxamer gel through rat skins. Int J Pharm, 222, 199–203.
- Shin, S.C., Lee, H.J. (2002). Enhanced transdermal delivery of triprolidine from the ethylene-vinyl acetate matrix. Eur J Pharm Biopharm, 54, 325–8.
- Shokri, J., Nokhodchi, A., Kashbolaghi, A., Hassan-Zad, K., Ghafourian, T., Barzegar-Jalali, M. (2001). The effect of surfactants on the skin penetration of diazepam. Int J Pharm, 228, 99–107.
- Siepmann, F., Brun, V.L., Siepmann, J. (2006). Drugs acting as plasticizers in polymeric systems: a quantitative treatment. J Contr Rel, 115, 298–306.
- Singh, P., Desai, S.J., Simonelli, A.P., Higuchi, W.I. (1967). Release rates of solid drug mixtures dispersed in inert matrices I. Noninteracting drug mixtures. J Pharm Sci, 56, 1542.
- Smith, E.W., Maibach, H.I. (eds) (1995). Percutaneous penetration enhancers. Boca Raton, FL: CRC Press, 1–50.
- Squillante, E., Needham, T., Maniar, A., Kislaliglu, S., Zia, H. (1998). Codiffusion of propylene glycol and dimethyl isosorbide in hairless mouse skin. Eur J Pharm Biopharm, 46, 265–71.
- Taguchi, K., Fukushima, S., Yamaoka, Y., Takeuchi, Y., Suzuki, M. (1999). Enhancement of propylene glycol distribution in the skin by high purity cis-unsaturated fatty acids with different alkyl chain lengths having different double bond position. Biol Pharm Bull, 22, 407–11.
- Takeuchi, Y., Yamaoka, Y., Jukushima, S., Miyawaki, K., Taguchi, K., Yasuka, H., Kishimoto, S., Suzuki, M. (1998). Skin penetration enhancing action of cis-unsaturated fatty acids with ω-9 and ω-12 chain lengths. Biol Pharm Bull, 21, 462–9.
- Tanojo, H., Bouwstra, J.A., Junginger, H.E., Bodde, H.E. (1997). In vitro human skin barrier modulation by fatty acid: skin permeation and thermal analysis studies. Pharm Res, 14, 42–9.
- Williams, A.C., Barry, B.W. (2004). Penetration enhancers. Adv Drug Deliv News, 6, 603–18.