Abstract
A chitosan derivative micelle system was developed as the delivery system for a novel anti-tumor drug, gambogic acid (GA). The physicochemical and pharmaceutical properties of GA-loaded micelles (GA-M) were evaluated compared with the formulation GA-L-arginine (GA-L) injection, which entered phase I clinical trials. The results showed that GA-M had high GA-loading rate (29.8 ± 0.17%), high entrapment efficiency (63.8 ± 0.52%), and small particle size (108.2 ± 0.8 nm). After i.v. administration at the dose of 4 mg/kg, the area under concentration-time curve (AUC) and elimination half-life (T1/2β) of GA-M were all increased by 1.7-fold compared with GA-L in rat. Biodistribution study indicated that ∼ 67% of GA in the GA-M group was distributed in the liver, while the value of the GA-L group was ∼ 55%. Additionally, GA amount in the kidney was greatly reduced in the GA-M group. Also, GA-M was shown to reduce the acute toxicity after i.v. administration in mice compared with GA-L. The present study indicated that GA was rapidly eliminated from the blood and transferred to the tissues, especially the liver. Moreover, GA acute toxicity and irritation to vein were decreased.
Introduction
Gambogic acid (GA), a polyprenylated xanthone, is the major active ingredient of gamboges (CitationAuterhoff et al., 1962) used as a coloring material in the printing industry and as antidote in Chinese traditional medicine (CitationLin et al., 1993; CitationAsano et al., 1996). Recent studies from several groups had demonstrated that GA possessed potent anti-cancer activity both in vitro and in vivo in animal models (CitationZhang et al., 2004; CitationZhao et al., 2004; CitationWu et al., 2004; CitationLiu et al., 2005; CitationGuo et al., 2006a; CitationYu et al., 2007). The potent anticancer activity of GA was mainly attributed to its activation effect of GA by binding to the transferrin receptor (CitationKasibhatla et al., 2005) and suppression of nuclear factor-kappa B (NF-kappa B) signaling pathway (CitationPandey et al., 2007). The poor water-solubility and toxic side-effect of GA once retarded the preclinical study and further application. In some patents of GA injectable formulations, L-arginine was added to form the complex (CitationDai, 2003; CitationJin et al., 2003; CitationYou et al., 2003) or polyoxylated castor oil (Cremophor EL) was used as solubilization agent (CitationZhao, 2006). A parenteral formulation, in which GA was complexed with L-arginine, had entered phase I clinical trials in China for tolerance testing (CitationZhou and Wang, 2007).
The data of preclinical tests were reported recently, which were obtained in rodent and dog models (CitationGuo et al., 2006b;CitationQi et al., 2008). In the chronic toxicity study, GA was administered to rats by parenteral administration, and ankylenteron with ascites was observed in the repeated intraperitoneal injection. A universal problem that cannot be neglected was lethal allergic reaction, which appeared in the animal model at a broad dosage range. In our preliminary experiment, ulceration and necrosis were also observed in the control group (GA-L), which was intravenously administered via caudal vein. In addition, a large amount of L-arginine had to be added to keep the solubility equilibrium in the formulation, and the potential risk was unknown. In the formulation solubilized with Cremophor EL, a series of side-effects associated with solubilizing agent such as life-threatening hypersensitivity reaction, nephrotoxicity, neurotoxicity, and cardiotoxicity would probably occur (CitationHennenfent and Govindan, 2005; CitationWang et al., 2005; CitationMarupudi et al., 2007). An alternative drug delivery system had been designed and studied to enhance the water-solubility and reduce toxicity. Among these efforts, the polymeric micelles system was recognized as one of the most promising formulations for anti-tumor drug with poor water-solubility (CitationKwon and Okano, 1996; CitationKwon, 2003; CitationAliabadi and Lavasanifar, 2006; CitationNishiyama and Kataoka, 2006; CitationMahmud et al., 2007; CitationYasuhiro, 2008). Polymeric micelles possess a core-shell structure formed by self-assembly of hydrophobic segments as internal core and hydrophilic segments as surrounding corona in aqueous environment. The hydrophobic core acts as a micro-reservoir for the encapsulation of hydrophobic drugs, and the hydrophilic shell interfaces the biological media (CitationTorchilin, 2001). As a result, polymeric micelles could substantially improve the solubility and bioavailability of various hydrophobic drugs. Additionally, the polymeric micelles had been proven as efficient drug delivery systems for intravenous administration. The propensity to locate tumor position by enhanced permeability and retention (EPR) effect because of nano-scaled size was an additional attractive characteristic of polymeric micelles (CitationMaeda et al., 2009). Among a number of natural or synthetic polymers used to form polymeric micelles, chitosan, a polysaccharide derived from chitin by complete or incomplete alkaline deacetylation, is the most attractive candidate due to its biochemical activity, biocompatibility, biodegradability, and low toxicity (CitationChandy and Sharma, 1990; CitationMuzzarelli and Muzzarelli, 2005; CitationVarshosaz, 2007; CitationKim et al., 2008).
In our previous study, amphipathic chitosan derivatives, N-octyl-O-sulfate chitosan (NOSC) and N-mPEG-N-octyl-O-sulfate chitosan (mPEGOSC), were synthesized and characterized (CitationZhang et al., 2003; CitationYao et al., 2007), and the biological properties of NOSC were evaluated as a drug carrier for intravenous injection (CitationZhang et al., 2008b). Furthermore, NOSC and mPEGOSC were successfully used to significantly improve the solubility of paclitaxel with high drug-loading content and biodistribution (CitationZhang et al., 2008a; CitationQu et al., 2009).
GA was also successfully solubilized in the micelles based on N-octyl-O-sulfate chitosan (NOSC), and the physical properties and stability of micelles loading with GA were characterized (CitationZhu et al., 2008). Thus, in this paper, we prepared and characterized the polymeric micelles loading with GA. The pharmacokinetics, biodistribution, and acute toxicity of GA-M were compared with the control formulation GA-L.
Materials and methods
Materials
Chitosan was purchased from the Suanglin Biochemical Co. Ltd. (Nantong, China), with deacetylation degree of 97% and viscosity average molecular weight of 65 kDa. GA was extracted and isolated from the resin of the gamboges by our group with the purity of 99.7%. N-octyl-O-sulfate chitosan (NOSC) was synthesized using the chitosan mentioned above, and the substitution of octyl degree and sulfonic degree were 0.38 and 2.56, respectively, and viscosity average molecular weight was 65–70 kDa. HPLC/spectra-grade reagents were used as the mobile phase in HPLC analysis. All other reagents were analytical grade and used without further purification. Distilled and deionized water was used in corresponding experiments.
Animals
Sprague-Dawley (SD) rats and Kunming mice were obtained from the Laboratory Animal Center of Nantong University. All the animals were pathogen free and allowed access to food and water freely. The experiments were carried out in compliance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Preparation and characterization of GA-M
GA-loaded micelles based on NOSC (GA-M) were prepared using the method reported in our previous article. Briefly, according to the orthogonal experimental design, GA (8 mg) and NOSC (12 mg) were dissolved in ethanol (0.3 ml) and water (2 ml), respectively, then the two solutions were mixed before dialysis using cellulose membrane dialysis tubing (Molecule Weight Cut-Off (MWCO) 10,000, Sigma-Aldrich Co., USA) at 25°C. Then the dialyzed solution was centrifuged at 3000 rpm (TGL-16 Centrifuge, China) for 40 min, and the GA level in micelle solution was analyzed after filtration through 0.22 μm pore-sized millipore films. GA-free polymeric micelles were produced in a similar manner without adding the drug. The dried powder of GA-M was obtained via lyophilization using a freeze-dryer system (Yuhua, China). GA-loading rate and entrapment efficiency of the micelles were calculated using the following equations:
where C, V, Wm, and WGA represented the GA concentration of micelle solution, the volume of micelle solution, the weight of micelles after freeze-drying and the weight of GA feed, respectively.
The hydrodynamic diameter and Zeta potential of the micelle solution were measured by dynamic light scattering (Zetasizer 3000 HAS, Malvern, UK) with 633 nm He-Ne lasers at 25°C. Physicochemical properties were studied by WAXD spectra which were obtained using an XD-3A powder diffraction meter with Cu Kα radiation in the diffraction range of 5–40° (40 kV and 30 mA) and TG spectra which were obtained with a NETZSCH TG 209 thermogravimetric analyzer, with a temperature range from 30–350°C and a heating rate of 10°C/min. The morphology of GA-M was studied using transmission electron microscopy JEM-200CX (TEM, JEOL, Japan) at 200 kV, sample solution was dropped on the copper grid with a film and dried before observation. 1H NMR spectra (BRUKER AVANCE 500 AV system, Bruker Biospin GmbH, Rheinstetten, Germany) and FTIR spectra (Nicolet 2000 FT-IR spectrophotometer, Thermo Electron Corp., WI) were used to identify the structure of the micelle.
HPLC analysis of GA
The amount of GA was measured using a reverse-phase HPLC method. The standard curve was set up and satisfactory linearity was obtained. Agilent 1200 series (Agilent Technologies, USA) was used and chromatographic separation was achieved using a Lichrospher C18 column (4.6 × 250 mm, Hanbon, China) at 30°C. Mobile phase consisted of water and HPLC grade methanol (6:94 (V/V)) which was adjusted to pH 3.5 using phosphoric acid. The samples were delivered at a flow rate of 1.0 ml/min and detected at 360 nm using UV detection. The GA amount in GA-M solution was analyzed after dilution with 50-times volume of methanol to disrupt the micelle structure, and the injected volume of sample was 20 μl. Otherwise, the GA levels in blood or tissue samples were determined with pretreatment.
Pharmacokinetic study
The pharmacokinetic behavior of GA-M was evaluated by the determination of the GA level in rat plasma. As control, GA-L-arginine injection (GA-L) was prepared following the method in the patent specification. The lyophilized powder of GA-M was reconstituted with 5% glucose injection and all the drug solutions were sterilized through 0.22 μm pore-sized micropore films before intravenous injection. Fifteen Sprague-Dawley rats (180–230 g) were used and randomly divided into three groups (n = 5). Then the solutions were injected into the tail vein of rats at a dose of 4 mg/kg. At 2, 5, 10, 15, 20, 30, 40, 50, and 60 min after injection, blood samples (0.5 ml) were collected from the plexus venous in eyeground; 0.15 ml plasma was obtained by centrifugation at 3000 rpm for 10 min, 50 μl hydrochloric acid solution (1 mol/l) and 0.3 ml acetonitrile were added before vortexing for 3 min. After centrifugation at 10,000 rpm for 10 min, 20 μl of clear supernatant was injected into the HPLC system directly and the GA concentration in rat plasma was calculated by standard curve. The GA plasma concentration over time was analyzed by compartmental model analysis using the software package program 3P97 (the Committee of Mathematic Pharmacology of the Chinese Society of Pharmacology) and the following pharmacokinetic parameters were obtained: area under the plasma concentration time curve (AUC), plasma half life (T1/2), total plasma clearance (Cl), and apparent distribution volume (Vd).
Tissue distribution determination
The tissue distributions of two preparations (GA-M and GA-L) were investigated in mice. Seventy-two mice were randomly divided into two groups (n = 36), each group comprised half-female and half-male. GA-M and GA-L solutions were intravenously administrated via tail vein in a dose of 6 mg/kg after sterilization. At the time points of 5, 10, 15, 20, 40 and 60 min after injection, six mice in one group were sacrificed by cervical dislocation after drawing blood from the eyeball. The blood was immediately treated as described above. The organs (heart, liver, spleen, lung, kidney, and brain) were excised and thoroughly washed with normal saline, then blotted dry and weighed. Subsequently, the weighted tissues were homogenized (Tearork, BioSpec Products Inc., Bartlesville, OK) with 2-fold weight of normal saline. Seventy-five microliters of homogenate, 25 μl of hydrochloric acid solution (1 mol/l) and 300 μl of acetonitrile were added into a glass tube. After vortexing for 3 min, the mixture was centrifugalized at 10,000 rpm for 10 min, and 50 μl of the supernatant was injected into the HPLC system. The concentrations of GA in samples were analyzed under the condition described above. The areas under the plasma or organ concentration-time curves from time 0 to time t (AUC (t)) were calculated by the trapezoidal method, and T1/2β was calculated from elimination rate constant according to the statistical moment principle. Relative exposure (re) to tissues and drug distribution to tissue j (%) were calculated as follows (CitationGupta and Hung, 1989):
Acute toxicity test
Acute toxicity, as a critical part of intravenous injection safety assessment, was tested in healthy mice. The LD50 of both GA-M and GA-L were calculated and the toxic effects on major organs were examined. Fifty Kunming mice (half male and half female, 18–22 g) were randomly divided into five groups (n = 10) and the freeze-dried power of GA-M was re-dissolved in 5% glucose solution and injected via tail vein at the dose of 56, 47.6, 40.5, 34.4, and 29.2 mg/kg, respectively. Mice were observed for 2 weeks in all groups, and the number of mice surviving was recorded. The LD50 was calculated using the Bliss method. The LD50 of GA-L was calculated in the same way as described above for the control group, while the doses were 27.4, 21.9, 17.5, 14, and 11.2 mg/kg, respectively.
Statistical analysis
Statistical analysis was performed by Student’s t-test for two groups. All results were expressed as the mean ± SD unless noted exceptionally, a probability (p) of less than 0.05 is considered statistically significant.
Results and discussion
Preparation and characterization of GA-M
Dialysis method was used to screen and prepare drug-loading micelles based on chitosan derivate NOSC. After the optimization with orthogonal design and lyophilization, the stable and homogeneous powder of GA-M was obtained. GA-loading rate and entrapment efficiency in the GA-M system were calculated to be 29.8 ± 0.17 and 63.8 ± 0.52%, while the particle size of the drug-loaded micelles was 108.2 ± 0.8 nm, determined using dynamic light scattering technology. The morphology observation of GA-M was showed in , which demonstrated a spherical structure with particle diameter ∼ 100 nm.
Figure 1. GA loaded micelle in water (×59,000) visualized by TEM.

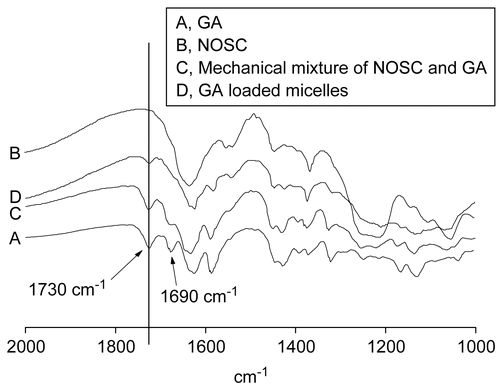
IR analysis was used to identify the micellar structure. It was obvious that a series of GA characteristic peaks weakened in the drug loaded micelle system and some peaks disappeared (), which was different from the spectrogram of GA in the physical mixture of drug and NOSC. The result suggested that GA was entrapped into the micelles.
Figure 2. IR spectrograms of GA (A), NOSC (B), Mechanical mixture of NOSC and GA (C), and GA loaded micelle (D).
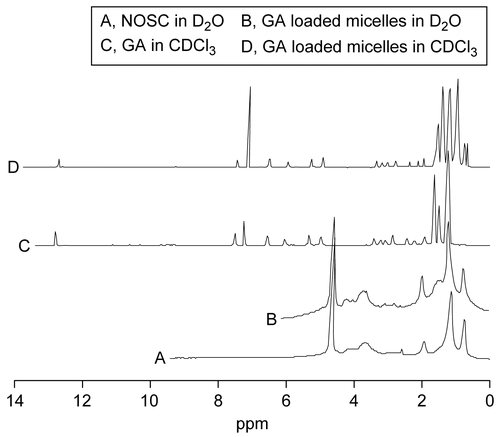
The 1H NMR analysis () showed the identical result with IR analysis. The 1H NMR spectrum of drug loaded micelle in D2O and the 1H NMR spectrum of the blank micelle made of NOSC in D2O resembled each other. However, the spectrum of drug loaded micelle in CDCl3 resembled that of GA in CDCl3. Due to dissociation of the micelle structure and precipitation of NOSC caused by CDCl3, only GA solved in the organic solvent and provided the characteristic signal response in the 1H NMR spectrum. This analysis in also indicated that GA was encapsulated by the aggregated structure of NOSC.
Figure 3. 1H NMR spectrum of NOSC in D2O (A), GA loaded micelles in D2O (B) and in CDCl3 (D), and GA in CDCl3 (C).
X-ray powder diffraction () and DSC thermograms () were used to further characterize the structure and physicochemical properties of drug-loaded micelles. As shown in , there were three characteristic crystal peaks of GA, at 10°, 18.5°, and 20°, while the crystal peak of NOSC was at 20.5°. Obviously, characteristic diffraction peaks of GA, which were visible in the pattern obtained for the physical mixture of GA and NOSC, disappeared in the pattern of GA-loaded micelles, especially around 10°. A new broad and blunt peak between 17–23° arose in the pattern of GA-loaded micelle, which indicated that GA was encapsulated in the polymeric micelles in molecular or amorphous state and there was no free drug in the surface of micelles.
Figure 4. WAXD spectra of GA (A), NOSC (B), Mechanical mixture of NOSC and GA (C), and GA loaded micelles (D).
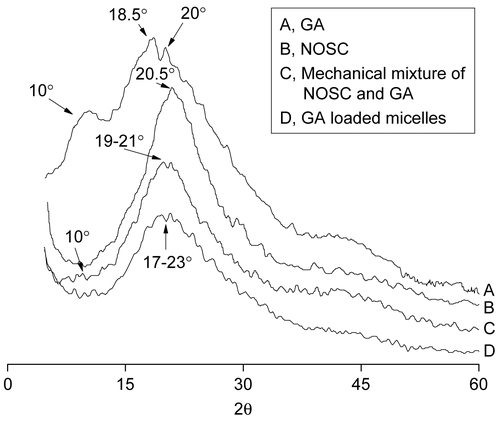
Figure 5. DSC thermograms of GA (A), NOSC (B), Mechanical mixture of NOSC and GA (C), and GA loaded micelles (D).
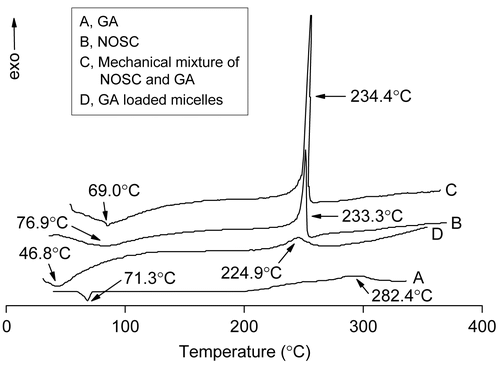
DSC thermograms () revealed exothermic peaks of GA and NOSC at 224.9°C and 233.3°C, respectively. For physical mixture, peaks were observed with small shifts at 234.4°C. The lyophilized GA-loaded micelles showed a characteristic exothermic peak at 282.4°C, which suggested that a new solid dispersion of GA and the NOSC formed during the procedure of self-assembly and freeze-drying.
Pharmacokinetics of GA-M and GA-L
The concentration-time curves () showed that the GA levels of the GA-M and GA-L groups decreased rapidly after injection in the two groups, which indicated a rapid elimination of GA from blood circulation.
Figure 6. The concentration-time curves of GA preparations in rat plasma after i.v. administration (M ± SD, n = 6).
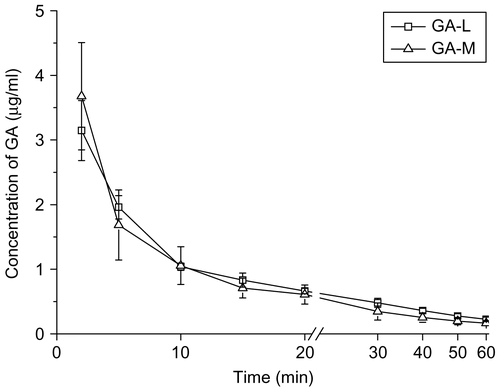
The GA plasma concentrations over times fitted to two-compartment model analyzed by compartmental model, and the pharmacokinetic parameters are listed in . The GA was distributed widely in the body (Vd >110 ml) and the mean elimination half-life time was less than 30 min. Compared with the GA-L group, the T1/2β of GA in GA-M was increased by 1.7-times (p < 0.001) and the Vd of GA in GA-M was increased by 1.6-fold (p < 0.05). However, the Cl of GA-L was 1.4-fold higher than that of the GA-M (p < 0.05), and AUC of GA-M was 1.7-fold higher than that of the GA-L, but there was no significant difference (p > 0.05).
Table 1. Pharmacokinetic parameters of two GA formulations in rats after i.v. administration at dosage of 4 mg/kg (M ± SD, n = 5).
The prolonged half-life, decreased clearance, and comparative AUC contributed to the stability of GA-M in the systemic circulation. The nano-system of GA-M based on graft polymeric micelle could keep more stable in blood under severe dilution effects, which might also result in the widespread distribution of GA and larger Vd beyond the blood system (CitationNarang et al., 2007).
Tissue distribution of GA-M and GA-L
All the profiles were listed in and shown in . The targeting property of drug delivery system could be evaluated according to the value of AUC and Re in , more amounts of GA, of the two groups were distributed to liver, lung, and kidney, compared to other tissues and plasma. According to , ∼ 67% of the in vivo distribution of GA in formulation GA-M was found in the liver, demonstrating a liver-passive targeting property of the nano-particle system, while the value of the GA-L group was ∼ 55%. The more selective localization of PTX in the liver was consistent with the uptake by the RES. Particulates with an average size below 7 μm were generally taken up by the Kupffer cells of liver (CitationPatel, 1992). In the GA-M group, GA amounts in kidney had been greatly reduced with a Re of 0.44, which demonstrated the reduced risk of potential toxicity on kidney.
Table 2. AUC (t)a, relative exposure (re)b, and AUCi (t) (μg/min) of GA in mouse plasma and organs after i.v. administration of GA-M or GA-L at a dose of equivalent GA 4 mg/kg (n = 6).
Figure 7. GA distribution in: liver, heart, spleen, brain (A), and plasma, lung, and kidney (B) of mice receiving GA-M and GA-L at a dose of 6 mg/kg by intravenous administration (M ± SD, n = 6).
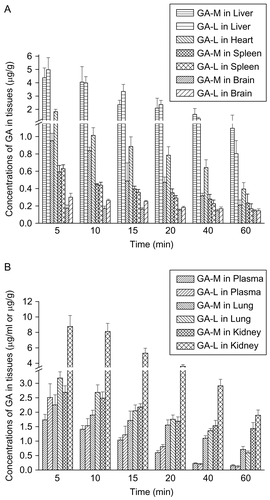
As shown in , GA was widely distributed in the main tissues following i.v. administration. The tissue distribution profiles of GA-L in this study agreed with the data obtained by CitationHao et al. (2007). GA reached its maximal concentration in all tissues at 5 min after administration. At 5 min, the GA content in kidney of GA-L group was three times that of the GA-M group (p < 0.05). A marked difference also could be found in the heart, in which the GA content of the GA-L group was 1.5-times higher than the GA-M group (p < 0.05). The extra high concentration of GA in kidney and heart might bring out toxicity in corresponding tissues. GA content in spleen and brain were low, while it still had significant deviation in brain. The concentrations of GA in liver and lung were higher than that in plasma and other tissues, but the distribution of GA in GA-M and GA-L has no significant difference in liver and lung (p > 0.05). This might be the basis that GA could specially inhibit cancer cells in liver and lung.
Acute toxicity of GA-M and GA-L
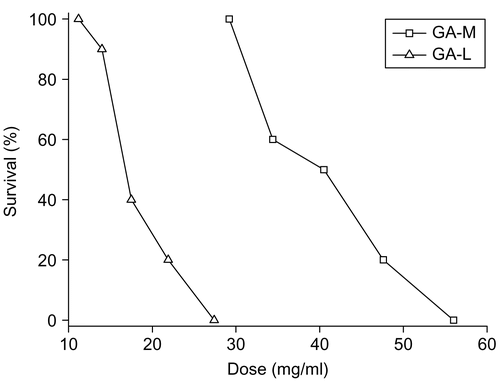
The median lethal dose (LD50) was used to determine the acute toxicity as mice were injected with various doses of GA-M or GA-L. In the GA-L group, less activity, piloerection, less food and drink consumption were observed, while insignificant toxic response was observed in the GA-M group. The main organs of mice, such as heart, liver, spleen, lung, and kidney, were subject to macroscopic examination; no obvious changes were observed in both groups. However, the ulceration and suppuration occurred at the injection site of tail in the GA-L group, rats in the GA-M group showed better tolerance. The dose-toxicity relationship was shown in . The LD50 of GA-M and GA-L administered by intravenous injection were 39.17 mg/kg and 19.6 mg/kg, respectively, and the 95% confidence limits were found to fall in the range of 34.58–44.37 mg/kg and 15.97–19.4 mg/kg, respectively. The value of LD50 of GA-M was 2-fold higher than that of GA-L, which showed its advantage of lower toxicity.
Figure 8. Acute toxicity of GA-M and GA-L injected intravenously in mice. The percentage survival at day 7 was shown.
Conclusions
A micelle formulation based on N-octyl-O sulfate chitosan was prepared and characterized as an effective delivery system of GA. Pharmacokinetic studies demonstrated that GA-M had longer elimination half-life time and 1.7-fold larger AUC compared with the GA-L group. After i.v. administration, GA-M was widely distributed in most tissues of mice, but the higher concentrations were observed in liver and lung. The LD50 of the GA-M was 2-fold lower than GA-L, which showed enhancive safety for GA delivery with no obvious hypersensitivity associated with injection. This fact has been well documented in our previous study (CitationZhang et al., 2008a), paclitaxel-loaded micelle formulation based on NOSC had lower acute toxicity and comparable anti-tumor activity compared with the commercial formulation TAXOL®. Taken together, in contrast to GA-L, these results indicated that the polymeric micelle formulation GA-M might be the better possible approach which would bypass the limitations of poor water-solubility and high toxicity of natural anti-cancer agent GA, and great expectations were centered on this formulation for higher targeting and less peripheral toxicity.
Acknowledgements
This study is financially supported by the National Natural Science Foundation of China (30772662), Program for New Century Excellent Talents in University (NCET- 06-0499), Ministry of Education key project (107062).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Aliabadi, H. M., and Lavasanifar, A. (2006). Polymeric micelles for drug delivery. Expert Opin Drug Deliv, 3, 139–62.
- Asano, J., Chiba, K., Tada, M., and Yoshii, T. (1996). Cytotoxic xanthones from Garcinia hanburyi. Phytochemistry, 41, 815–20.
- Auterhoff, H., Frauendorf, H., Liesenklas, W., and Schwandt, C. (1962). The chief constituents of gamboge resins. 1. Chemistry of gamboge. Arch Pharm, 295, 833–46.
- Chandy, T., and Sharma, C. P. (1990). Chitosan-as a biomaterial. Biomat Art Cells Art Org, 18, 1–24.
- Dai, J. G. (2003). The preparation of a kind of gambogic acid injection. CHN Patent CN 03131511.9.
- Guo, Q. L., Lin, S. S., You, Q. D., Gu, H. Y., Yu, J., Zhao, L., Qi, Q., Liang, F., Tan, Z., and Wang, X. T. (2006a). Inhibition of human telomerase reverse transcriptase gene expression ganbogic acid in human hepatoma SMMC-7721 cells. Life Sci, 78, 1238–45.
- Guo, Q. L., Qi, Q., You, Q. D., Gu, H. Y., Zhao, L., and Wu, Z. Q. (2006b). Toxicological studies of gambogic acid and its potential targets in experimental animals. Basic Clin Pharmacol Toxicol, 99, 178–84.
- Gupta, P. K., and Hung, C. T. (1989). Quantitative evaluation of targeted drug delivery systems. Int J Pharm, 56, 217–26.
- Hao, K., Liu, X. Q., Wang, G. J., and Zhao, X. P. (2007). Pharmacokinetics, tissue distribution and excretion of gambogic acid in rats. Eur J Drug Metab Pharmacokinet, 32, 63–68.
- Hennenfent, K. L., and Govindan, R. (2005). Novel formulations of taxanes: a review. Old wine in a new bottle? Ann Oncol, 17, 735–49.
- Jin, B., Dong, H., and Qiao, L. (2003). The preparation of a kind of gambogic acid injection. CHN Patent CN 02124510.X.
- Kasibhatla, S., Jessen, K. A., Maliartchouk, S., Wang, J. Y., English, N. M., Drewe, J., Qiu, L., Archer, S. P., Ponce, A. E., Sirisoma, N., Jiang, S., Zhang, H. Z., Gehlsen, K. R., Cai, S. X., Green, D. R., and Tseng, B. (2005). A role for transferrin receptor in triggering apoptosis when targeted with gambogic acid. Proc Natl Acad Sci USA, 102, 12095–100.
- Kim, I. Y., Seo, S. J., Moon, H. S., Yoo, M. K., Park, I. Y., Kim, B. C., and Cho, C. S. (2008). Chitosan and its derivatives for tissue engineering applications. Biotechnol Adv, 26, 1–21.
- Kwon, G. S. (2003). Polymeric micelles for delivery of poorly water- soluble compounds. Crit Rev Ther Drug Carrier Syst, 20, 357–403.
- Kwon, G. S., and Okano, T. (1996). Polymeric micelles as new drug carriers. Adv Drug Deliv Rev, 21, 107–16.
- Lin, L. J., Lin, L. Z., Pezzuto, J. M., Cordell, G. A., and Ruangrungsi, N. (1993). Isogambogic acid and isomorellinol from Garcinia hanburyi. Magn Reson Chem, 31, 340–7.
- Liu, W., Guo, Q. L., You, Q. D., Zhao, L., Gu, H. Y., and Yuan, S. T. (2005). Anticancer effect and apoptosis induction of gambogic acid in human gastric cancer line BGC-823. World J Gastroenterol, 11, 3655–9.
- Maeda, H., Bharate, G. Y., and Daruwalla, J. (2009). Polymeric drugs for efficient tumor targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm, 71, 409–19.
- Mahmud, A., Xiong, X. B., Aliabadi, H. M., and Lavasanifar, A. (2007). Polymeric micelles for drug targeting. J Drug Target, 15, 553–84.
- Marupudi, N. I., Han, J. E., Li, K. W., Renard, V. M., Tyler, B. M., and Brem, H. (2007). Paclitaxel: a review of adverse toxicities and novel delivery strategies. Expert Opin Drug Saf, 6, 609–21.
- Muzzarelli, R. A. A., and Muzzarelli, C. (2005). Chitosan chemistry: relevance to the biomedical sciences. Adv Polym Sci, 186, 151–209.
- Narang, A. S., Delmarre, D., and Gao, D. (2007). Stable drug encapsulation in micelles and microemulsions. Int J Pharm, 345, 9–25.
- Nishiyama, N., and Kataoka, K. (2006). Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery, Pharmacol Ther, 112, 630–48.
- Pandey, M. K., Sung, B., Ahn, K. S., Kunnumakkara, A. B., Chaturvedi, M. M., and Aggarwal, B. B. (2007). Gambogic acid, a novel ligand for transferrin receptor, potentiates TNF-induced apoptosis through modulation of the nuclear factor-kappaB signaling pathway. Blood, 110, 3517–25.
- Patel, H. M. (1992). Serum opsonin and liposomes: their interaction and opsonophagocytosis. Crit Rev Ther Drug Carrier Syst, 9, 39–90.
- Qi, Q., You, Q. D., Gu, H. Y., Zhao, L., Liu, W., Lu, N., and Guo, Q. L. (2008). Studies on the toxicity of gambogic acid in rats. J Ethnopharmacol, 117, 433–8.
- Qu, G. W., Yao, Z., Zhang, C., Wu, X. L., and Ping, Q. N. (2009). PEG conjugated N-octyl-O-sulfate chitosan micelles for delivery of paclitaxel: in vitro characterization and in vivo evaluation. Eur J Pharm Sci, 37, 98–105.
- Torchilin, V. P. (2001). Structure and design of polymeric surfactant-based drug delivery systems. J Contr Rel, 73, 137–72.
- Varshosaz, J. (2007). The promise of chitosan microspheres in drug delivery systems. Expert Opin Drug Deliv, 4, 263–73.
- Wang, J., Mongayt, D., and Torchilin, V. P. (2005). Polymeric micelles for delivery of poorly soluble drugs: preparation and anticancer activity in vitro of paclitaxel incorporated into mixed micelles based on poly(ethylene glycol)-lipid conjugate and positively charged lipids. J Drug Target, 13, 73–80.
- Wu, Z. Q., Guo, Q. L., You, Q. D., Zhao, L., and Gu, H. Y. (2004). Gambogic acid inhibits proliferation of human lung carcinoma SPC-A1 cells in vivo and in vitro and represses telomerase activity and telomerase reverse transcriptase mRNA expression in the cells. Biol Pharm Bull, 27, 1769–74.
- Yao, Z., Zhang, C., Ping, Q. N., and Yu, L. (2007). A series of novel chitosan derivatives: synthesis, characterization and micellar solubilization of paclitaxel. Carbohyd Polym, 68, 781–92.
- Yasuhiro, M. (2008). Polymeric micellar delivery systems in oncology, Jpn J Clin Oncol, 38, 793–802.
- You, Q. D., Guo, Q. L., Ke, X., Xiao, W., Dai, L. L., and Lin, Y. (2003). The preparations of gambogic acid and its compound. CHN Patent CN03132386.3.
- Yu, J., Guo, Q. L., You, Q. D., Zhao, L., Gu, H. Y., Yang, Y., Zhang, H. W., Tan, Z., and Wang, X. (2007). Gambogic acid-induced G2/M phase cell-cycle arrest via disturbing CDK7-mediated phosphorylation of CDC2/p34 in human gastric carcinoma BGC-823 cells. Carcinogenesis, 28, 632–8.
- Zhang, C., Ping, Q. N., Zhang, H. G., and Shen, J. (2003). Preparation of N-alkyl-O-sulfate chitosan derivatives and micellar solubilization of taxol. Carbohyd Polym, 54, 137–41.
- Zhang, C., Qu, G. W., Sun, Y. J., Wu, X. L., Yao, Z., Guo, Q. L., Ding, Q. L., Yuan, S., Shen, Z. L., Ping, Q. N., and Zhou, H. P. (2008a). Pharmacokinetics, biodistribution, efficacy and safety of N-octyl-O-sulfate chitosan micelles loaded with paclitaxel. Biomaterials,29, 1233–41.
- Zhang, C., Qu, G. W., Sun, Y. J., Yang, T., Yao, Z., Shen, W. B., Shen, Z. L., Ding, Q. L., Zhou, H. P., and Ping, Q. N. (2008b). Biological evaluation of N-octyl-O-sulfate chitosan as a new nano-carrier of intravenous drugs. Eur J Pharm Sci, 33, 415–23.
- Zhang, H. Z., Kasibhatla, S., Wang, Y., Herich, J., Guastella, J., Tseng, B., Drewe, J., and Cai, S. X. (2004). Discovery, characterization and SAR of gambogic acid as a potent apoptosis inducer by a HTS assay. Bioorg Med Chem Lett, 12, 309–17.
- Zhao, A. G. (2006). The preparation of a new kind of gambogic acid injection. CHN Patent CN 1857529.A.
- Zhao, L., Guo, Q. L., You, Q. D., Wu, Z. Q., and Gu, H. Y. (2004). Gambogic acid induces apoptosis and regulates expressions of Bax and Bcl-2 protein in human gastric carcinoma MGC-803 cells. Biol Pharm Bull, 27, 998–1003.
- Zhou, Z. T., and Wang, J. W. (2007). Phase I human tolerability trail of gambogic acid. Chinese J New Drugs, 16, 79–83.
- Zhu, X., Zhang, C., Wu, X. L., Tang, X. Y., and Ping, Q. N. (2008). Preparation, physical properties, and stability of gambogic acid-loaded micelles based on chitosan derivatives. Drug Dev Ind Pharm, 34, 2–9.