Abstract
The aim of the present study was to formulate and evaluate buccoadhesive gels of model drug, glibenclamide indented as an alternate route of drug delivery to have enhanced bioavailability. The buccoadhesive gels were prepared by solution polymerization technique containing Hydroxy Propyl Methyl Cellulose (HPMC, E15 LV) and Carbopol (CP, 934P). An apparatus, simulating the in-vivo conditions of the mouth was designed in order to assess in-vitro release kinetics of the prepared gels. The gels were also evaluated for buccoadhesive strength, hydration, and swelling index, and viscosity. In-vivo evaluation for pharmacodynamic and pharmacokinetic properties of the optimized gel was done in rabbits. The drug release of buccoadhesive gels through rabbit buccal mucosa increased significantly (p < 0.05) by the use of penetration enhancers. The release kinetics followed Higuchi model with release mechanism being Fickian diffusion. In-vivo results of the optimized gel (2% CP: 2% HPMC) revealed that the gel successfully prevented severe hypoglycemia, showed sustained action, and enhanced relative bioavailability with and without penetration enhancers as compared to the marketed formulation. The buccoadhesive gels could possibly be a means for alternative dosage form in avoiding first pass metabolism and ensuring enhanced bioavailability for glibenclamide.
Introduction
Amongst the various routes of drug delivery, the oral route is one of the most preferred routes by the patients. However, within the oral mucosal cavity, the buccal region offers an attractive route of administration for systemic delivery of drugs (CitationSangeetha et al., 2007). Absorption of therapeutic agents from the oral mucosa overcomes premature drug degradation within the GIT, as well as active drug loss due to first pass metabolism (CitationAlur et al., 1999; CitationKataria et al., 2007). As the blood draining from the oral cavity enters the general circulation via the internal jugular vein, oral administration by the buccal or sublingual routes provide a useful strategy for improving bioavailability of the drugs that are susceptible to extensive first-pass metabolism (CitationMcElnay & Hughes, 2002). It has been suggested that drugs with biological half-lives in the range of 2–8 h are good candidates for sustained release formulations (CitationVarshosaz & Dehghan, 2002). Mucoadhesive drug delivery systems are those which utilize the property of bioadhesion of certain polymers, which become adhesive on hydration (CitationDoijad et al., 2006). Mucoadhesive polymers may be used to produce controlled release dosage forms that are able to adhere to the buccal membrane for an extended period of time (CitationPark & Munday, 2004). Buccal mucosa presents a relatively smooth and immobile surface for the placement of a bioadhesive dosage form. The amount of drug that can be incorporated is limited because of the size limitation of the buccal dosage form. In general, a drug with a daily requirement of 25 mg or less is suitable for buccal delivery. Drugs with a short half-life, requiring sustained/controlled delivery, or exhibiting poor aqueous solubility and drugs that are sensitive to enzymatic degradation may be delivered successfully across the buccal mucosa. The dosage forms developed for this purpose include tablets adhesive patches, adhesive gels, and adhesive ointment (CitationAlur et al., 2001).
Diabetes mellitus is a chronic metabolic disorder characterized by high blood glucose concentration, hyperglycemia caused by insulin deficiency, often combined with insulin resistance (CitationDavis & Granner, 1996). Glibenclamide (5-chloro-N-[2-[4- cyclohexyl- carbamoylsulfamoyl] phenyl] ethyl)-2 methoxy- benzamide is a potent oral sulfonylurea hypoglycemic agent. It is sold in doses of 1.25 mg, 2.5 mg, and 5 mg, under the trade names Diabeta, Glynase, and Micronase in the US and Daonil, Semi-Daonil, and Euglucon in the UK. It is also sold in combination with metformin under the trade name Glucovance. It is one of only two oral anti-diabetics in the World Health Organization Model List of Essential Medicines (the other being metformin) (World Health Organization, Citation2007). Because of its poor aqueous solubility (pKa, 5.3) and low half life (3–5 h) (CitationFlorey, 2005), incomplete absorption occurs; thereby necessitating a need for multiple dosing, which leads to gastric disturbances associated with the drug. Glibenclamide undergoes oxidative hepatic metabolism, with the major metabolites having no hypoglycemic activity (Rang et al., Citation2005). Therefore, there is a need and potential market for a drug delivery system which addresses both the issues of route of drug delivery and low bioavailability for the drug.
Therefore, the aim of the present study was to formulate buccoadhesive gels with combinations of polymers, and evaluate them in-vitro to optimize a formulation, which possibly (due to increased bioavailability) would reduce the blood glucose level, in-vivo, in diabetic rabbits.
Materials and methods
Materials
Glibenclamide and glipizide were gift samples from Bestochem Pharma Pvt. Ltd. (Hariddwar, India). Carbopol 934, sodium saccharine, and heparin were obtained from Central Drug House Pvt. Ltd. (New Delhi, India). Hydroxy propyl methyl cellulose (E15 LV) was procured from Titan Biotech Ltd. (Bhiwadi, India). Methyl paraben, propyl paraben, sodium hydroxide from S Merck India Ltd. (New Delhi, India), potassium dihydrogen ortho phosphate and acetronitrile (HPLC grade), sodium carbonate, agar-agar from Qualigens Fine Chemicals Pvt. Ltd. (Mumbai, India), triethanolamine, sodium taurocholate, sodium lauryl sulfate from Ranbaxy Fine Chemicals Pvt. Ltd. (Mumbai, India), were procured from C.N. Chemicals (Mathura, Uttar Pradesh, India). Streptozotocin was procured from Alexis Biochemicals (Switzerland). All glass double distilled water was used throughout the experiment.
Animals
Healthy New Zealand albino (white strain) rabbits of either sex, aged between 2–3 years, and weighing 4–5 kg, were taken from the animal house of Rajiv Academy for Pharmacy, Mathura, India, and housed in the Pharmacology laboratory of the Institute. The experimental protocol was approved by the Institutional animal ethical committee of Rajiv Academy for Pharmacy, Mathura, India (IAEC/RAP/1562/2007).
Preparation of buccoadhesive gels
Buccoadhesive gels were prepared () by dispersing an appropriate amount of the polymer in the phosphate buffer of required pH, i.e. pH 7.4, and allowing it to swell. The polymer solution was stirred to break the lumps formed during swelling. In another beaker, the drug was dissolved in the ethanol, (95% v/v) by slight heating on a water bath. The sweetening agent, namely saccharin sodium (0.05% w/w), and preservatives, namely methyl paraben and propyl paraben (0.2% w/w and 0.02% w/w, respectively) were dissolved in it. To this drug solution, polymer mixture was added, and thoroughly agitated until a clear gel was formed. The final pH of the gel was adjusted by using triethanolamine (CitationAttia et al., 2004).
Table 1. Composition of all the buccoadhesive gels of glibenclamide.
Viscosity study
A Brookfield viscometer (Brookfield DV-II+ Pro, Brookfield Engineering Laboratories, Inc. USA) attached with T-bar (spindle type, S 96) was used for determination of gel viscosity. Buccoadhesive gels were filled in a 50 ml beaker and the spindle lowered perpendicularly, taking care that the spindle did not touch the bottom of the beaker. The gels were rotated at 10 and 50 rpm (g value of 0.012 and 0.28 g/cm3, respectively). The digital readings of the Brookfield viscometer were recorded. The test was carried out at p < 0.05.
Ex-vivo buccoadhesive strength measurement
Buccoadhesive strength of the prepared gels of different pH on rabbit buccal mucosa was measured using a texture analyzer (TA-XT plus, Stable Micro Systems, Surrey, UK). The rabbits were sacrificed immediately before the start of the experiment. The buccal mucosa was freshly excised, and stored in isotonic phosphate buffer solution (PBS; pH 7.4) at 4°C upon removal. The mucosal membrane was separated by removing the underlying muscle and connective tissue with a fine forcep. The tissue was rinsed, and stored in ice-cold PBS. Slice thickness varied between 2.0–2.3 mm. The experiment was performed at room temperature within 5 h of procurement of the mucosa. For the experiment, 50 mg of the gel was applied on the surface of the rabbit buccal membrane. A pre-load of 25 g was applied on the membrane for 10 s (pre-load time) to establish adhesion bonding between the gel and buccal mucosa. After completion of pre-load time, the pre-load was removed, and force was applied on the probe to detach the membrane. The force required for detachment was noted as mucoadhesive strength (BS) (n = 3). The test was carried out at p < 0.05.
Determination of swelling index and surface pH of the hydrating layer
The swelling index of buccoadhesive gels were evaluated by placing 100 mg of gel in an area of 3.14 cm2 on the nutrient agar gel (medium comprising of peptone, beef extract, and agar) kept in a petri plate, which were weighed (W1), covered with aluminum foil, and incubated at 37 ± 1°C. The gels were allowed to swell for 24 h, after which the petri plates were reweighed (W2) and the swelling index calculated using the formula (CitationAgarwal & Mishra, 1999)
where W1 is the initial weight of the gel, W2 is the final weight of the gel.
For determining the surface pH of the hydrating layer of the buccoadhesive gels (in order to investigate the possibility of any irritation to the buccal mucosa, in- vivo), swelling studies were carried out with and without the inclusion of triethanolamine using 2% agar gel plate. The gels with and without triethanolamine, to account for no change or change in pH, respectively, of the prepared gel were applied on the agar gel plate surface in an area of 3.14 cm2. The gels on petri plates were allowed to hydrate for 8 h after which the pH of gels were measured by use of a pH paper.
In-vitro release studies
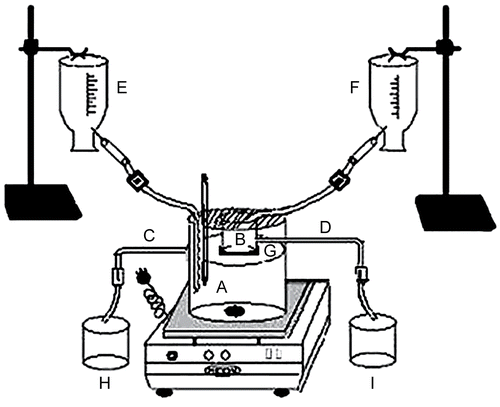
In-vitro drug release studies of the buccoadhesive gels were conducted on a fabricated flow through drug release apparatus (). The fabricated apparatus was validated for the flow through, with an accuracy of 97.99 ± 1.23%, and precision of 98.56 ± 2.10%. The apparatus consisted of a 1 L beaker used as a receptor compartment (A). At a distance of 900 ml from the bottom, a hollow (lower end removed) 50 ml beaker was attached to act as donor compartment (B). The receptor compartment contained 900 ml of phosphate buffer pH 7.4 and a flow through was maintained by providing an outlet (C) in the receptor compartment at 900 ml mark. The donor compartment had an outlet (D) at 10 ml and contained phosphate buffer pH 6.8. Sink condition in the receptor and continuous flow in the donor compartment (to take into account the continuous flow of mucus/saliva which might dissolve partly the drug containing gel) were maintained with the use of two infusion sets separately delivering phosphate buffer pH 7.4 at a flow rate of 4 ml/min in the receptor compartment (E) and phosphate buffer pH 6.8 at a flow rate of 0.6 ml/min in the donor compartment (F). A cellophane membrane (tied to the lower end of the donor compartment (G) to cover its entire circumference) was centrally cut (3.14 cm2) and replaced with similar area of rabbit buccal mucosa. The two membranes were attached to each other with the help of glue and the remaining area of the cellophane membrane was made impermeable with the help of molten wax. The receptor compartment was maintained at 37 ± 0.5°C. Five milligram equivalent of the drug in the gel was applied on the buccal mucosa and from the outlets of the respective compartments, the buffers were collected in two beakers (H and I for receptor and donor compartments, respectively). Five milliliters of the samples were withdrawn at pre-determined time intervals from both the beakers and analyzed at 300.5 nm using a Shimadzu 1700 UV spectrophotometer (Shimadzu, Tokyo, Japan).
Figure 1. Schematic representation of laboratory fabricated in-vitro drug release apparatus, where A is the receptor compartment, B is the donor compartment, C is the outlet for the receptor compartment, D is the outlet for the donor compartment, E is the infusion set maintaining flow through for the receptor compartment, F is the infusion set maintaining flow through for the donor compartment, G is the cellophane membrane tied to the donor compartment, H is for the buffer collection from the receptor compartment, and I is for the buffer collection for the donor compartment.
Effect of permeation enhancer
To investigate the effect of permeation enhancer on glibenclamide release, 1% w/w sodium lauryl sulfate (SLS) and sodium taurocholate (ST) were incorporated in the optimized gel separately along with drug, methyl paraben propyl paraben, and sodium saccharine. The rest of the procedure to formulate the gel and conduct in-vitro drug release were similar to the one described above.
Kinetics of drug release
In order to describe the kinetics of drug release from controlled release formulation, various mathematical equations have been proposed. The zero-order rate (equation 2) (CitationNajib & Suleiman, 1985) describes systems where drug release is independent of its concentration and is generally seen for poorly water-soluble drug in matrix, transdermals, etc. The first-order rate (equation 3) (CitationDesai et al., 1966) describes systems in which the release is dependent on its concentration (generally seen for water-soluble drugs in porous matrix). The Higuchi model (equation 4) (CitationHiguchi, 1963) describes the release of the drug from an insoluble matrix to be linearly related to the square root of time and is based on Fickian diffusion. The Hixson-Crowell cube root law (equation 5) (CitationHixson & Crowell, 1931) describes the release of drug from systems where it depends on the change in surface area and diameter of the particles or tablets with time and mainly applies in the case of systems that dissolute or erode over time. In order to authenticate the release model, dissolution data can further be analyzed by Peppas and Korsmeyer or Power Law (equation 6) (CitationKorsmeyer et al., 1983).
where Qt is the amount of drug released at time t; Q0 is the initial amount of the drug in the formulation; k0, k1, kH, and kHC are release rate constants for zero-order, first-order, Higuchi model, and Hixson-Crowell rate equations. In equation (6), Mt is the amount of drug released at time t, and M∞ is the amount released at time ∞; k is the kinetic constant, and n is the diffusional coefficient. The criteria for the best model were based on goodness of fit (R).
In-vivo evaluation of buccoadhesive gels
The rabbits were housed individually in polypropylene cages, maintained under standard conditions (12 h light and 12 h dark cycle; 25 ± 30°C; 35–60% humidity). The experiment was processed following the internationally accepted ethical guidelines for care of laboratory animals. Prior to the experiments, the rabbits were fed with standard food for 1 week, in order to adapt to the laboratory conditions. Sixteen hours before the experiments, they were fasted overnight, but allowed free access to water. The body weights of all the rabbits were determined before the start of the experiment.
Route of administration and withdrawal of blood samples
The teeth of the rabbit were fixed on a plate, while the tongue was tied to prevent swallowing of the gel. The rabbits were lightly anesthetized (for entire duration of study) by intra-muscular injection of a mixture of xylamine (1.9 mg/kg) and ketamine (9.3 mg/kg). Following induction of anesthesia, a fine cannula hypodermic needle (0.8 mm) with syringe attached to it was inserted in the marginal ear vein for blood sample collection. The syringe was detached from the needle and the cannula closed with the cap to prevent clotting of the blood. To further ensure that clotting of blood did not take place, the cannula before closing was flushed with 10% v/v of heparin/normal saline solution. The needle was kept in its place by means of leucoplast tape (CitationPhilip & Pathak, 2008). Buccoadhesive gel with an equivalent amount of 5 mg of drug was applied on the buccal mucosa of rabbits with the help of a specially designed gel applicator having a flat base of 3.14 cm2 surface area. The gel was placed onto the buccal region of the rabbit and pressed for ∼ 10 ss and the applicator removed. At prerequisite time, 2 ml of blood sample(s) was withdrawn through the cannula into heparinized glass vial at 2, 4, 8, 10, and 24 h for pharmacodynamic studies, and 0, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h following the administration of 5 mg tablet (Glybrol®, U.S Vitamin, India), and the optimized buccoadhesive gels of glibenclamide for pharmacokinetic studies. All the blood samples were centrifuged at 3000 rpm (g value of 503.8 cm/s2) for 10 min to separate plasma, and the retrieved plasma was stored at −20°C. To maintain homeostasis in the rabbits, an injection of same volume of physiological saline was given through the other ear vein.
Pharmacodyanamic evaluation
The hypoglycemic activity (n = 3) was observed both in normal and diabetic rabbits. The rabbits were divided into six groups of six animals each (both for normal and diabetic rabbits) with specifications:
Group 1: consisted of control rabbits which were administered 2 ml of 0.2% w/v oral suspension of HPMC;
Group 2: consisted of rabbits which were administered glibenclamide marketed formulation given as oral suspension in 0.2% w/v HPMC;
Group 3: consisted of rabbits which were administered placebo gel (F4 without the drug);
Group 4: consisted of rabbits which were administered optimized gel buccal gels (F4);
Group 5: consisted of rabbits which were administered optimized gel buccal gels with SLS as penetration enhancer (F4a); and
Group 6: consisted of rabbits which were administered optimized gel buccal gels with ST as penetration enhancer (F4b).
The overnight fasted rabbits were made diabetic by a single intraperitoneal injection of streptozotocin, STZ (55 mg/kg) dissolved in citrate buffer (3 mM, pH 4.5). Seven days later, rabbits were measured for blood glucose levels. Each animal with blood glucose concentration level above 300 mg/dl was considered to be diabetic, and used in the experiments. To prevent the hypoglycemia (this might occur during the first 24 h following the STZ administration), 5% w/v glucose solution was orally given to the diabetic rabbits. Blood was collected from marginal ear vein at prerequisite periods. Blood glucose levels were determined using glucose diagnostic kit (SPAN Diagnostic Ltd. Surat, India).
Pharmacokinetic evaluation
For pharmacokinetic evaluation, the overnight fasted rabbits were taken and divided in the groups with specifications:
Group 1: consisted of rabbits which were administered glibenclamide marketed formulation given as oral suspension in 0.2%w/v HPMC;
Group 2: consisted of rabbits which were administered optimized gel buccal gels (F4);
Group 3: consisted of rabbits which were administered optimized gel buccal gels with SLS as penetration enhancer (F4a); and
Group 4: consisted of rabbits which were administered optimized gel buccal gels with ST as penetration enhancer (F4b).
The plasma concentration time data of prepared glibenclamide buccoadhesive gels with and without the penetration enhancer, and the marketed tablet were fitted in Non Compartment data analysis software (Cure DH 2 v 21, Knoxware, Banglore, India), and the pharmacokinetic parameters calculated. Mass balance model-dependent technique (Wagner Nelson) was also used to calculate the absorption parameter. The area under the concentration-time curve (AUC0–t) was determined using the trapezoidal method. AUCt–∞ was calculated by dividing the last recordable plasma concentration over elimination rate constant. Relative bioavailability was determined by using equation (7).
To compare the main parameters of the different dosages forms, a two-sided unpaired t-test was conducted with Microsoft Excel 2003. Statistical significance was tested at p < 0.01.
Analysis of glibenclamide
Glibenclamide was estimated by reverse phase HPLC method. A Shimadzu Class VP series HPLC system (Shimadzu, Tokyo, Japan) with two LC-10AT pumps, a SPD-10A variable wavelength programmable UV/Vis detector, a SCL-10A system controller, and a RP C-18 column (Luna, Phenomenex, USA; 250 mm × 4.6 mm; particle size 5 μ) was used. The system was equipped with Class VP series version 6.12 software.
The mobile phase consisted of 20 mM monobasic potassium dihydrogen orthophosphate in water, which was adjusted to pH 3.5 with phosphoric acid, and acetonitrile in the proportion of 60:40 v/v. The mobile phase was filtered through a 0.22 μ membrane filter (Sartorius® AG, Goettingen, Germany). The flow rate was 1 ml/min, and the column effluent was monitored at 225 nm. The total run time of the method was set at 20 min. The peaks were well resolved and the retention time for glibenclamide and glipizide (internal standard) were 10.02 and 6.45 min, respectively.
A standard stock solution of glibenclamide (100 mg/ml) was prepared in acetonitrile. The calibration curve standard solutions were prepared by adding a known amount of glibenclamide and glipizide, an internal standard, to the blank plasma.
A volume of 0.1 ml of blank rabbit plasma and 0.1 ml of 0.1 N HCl were mixed thoroughly. The plasma was spiked with standard glibenclamide and glipizide solutions to yield concentrations of 1– 20 mg/ml of glibenclamide and 1 mg/ml of glipizide, respectively. The mixture was gently shaken for 3 min, and to it was added with 5 ml of benzene in a 20 ml glass tube. The tube was gently shaken using a cyclomixer (Remi cyclomixer, Mumbai, India) for 5 min and centrifuged (Remi Centrifuge, Mumbai, India) for 10 min at 3000 rpm (g value of 503.8 cm/s2). After centrifugation, the organic phase was transferred into a conical tube for evaporation to dryness under nitrogen. The residue was dissolved in 0.1 ml of equilibrated mobile phase by vortexing. An aliquot of 20 ml was injected into the chromatograph.
Validation parameters
The method of specificity was assessed by comparing the chromatograms obtained from the drug to its respective internal standard with those obtained from the blank. Linearity, range, limit of quantification, and limit of detection (LOD) were obtained from the standard concentrations (1, 3, 5, 10, 15, and 20 μg/ml), which in turn were obtained from the stock solution. Each concentration was prepared six times (n = 6). The limit of quantitation (LOQ) was the lowest concentration assayed where the signal/noise ratio was at the least 10:1, and the LOD was defined as a signal/noise ratio of 3:1. The accuracy, precision, and recovery in plasma assay validation involved quality control (QC) concentrations prepared from newly prepared spiked stock solution of flurbiprofen (1, 10, and 20 μg/ml). The QC samples were divided into 0.1 ml aliquots in centrifuge tubes and stored at −70°C before use. Intra-day and inter-day variability were tested with 12 replicates of each QC control concentration. Means, standard deviations, and coefficient of variation were calculated by standard methods. Recovery test was performed by adding known amounts of respective stock solution of glibenclamide to the sample with known content and preparing solutions with the respective mobile phase. The percentage of recovery was calculated by comparing the determined amount of these standards with the added amount.
Results and discussion
Viscosity and buccoadhesive strength
The viscosity of the gels were measured at 10 and 50 rpm (). At 50 rpm, the viscosity of all the formulated gels were found to be less than at 10 rpm. Buccoadhesive gels (pH 7.4) containing 2% w/w CP934 showed the best viscosity amongst the other combinations prepared at the same pH. This may be due to the more water absorbing capacity of the polymer and its tendency to swell.
All the formulations prepared with different blends in % w/w of HPMC and CP934 were found to have maximum buccoadhesion strength (), irrespective of pH, probably due to the fact that CP934 absorbed more water and got entrapped in the HPMC network. In addition, the intermolecular complexation between two polymers could also be effective in increasing the bioadhesive strength (CitationSatoh et al., 1989; CitationMohammadi-Samani et al., 2005). The gels of pH 7.4 containing 2% w/w CP934 alone showed minimum buccoadhesion than when used in different combinations with HPMC, although it was having higher viscosity. This could be due to high swellability of CP934, that produced a non-adhesive mucilage layer near the interfacial surface, which was reduced when it was formulated in combination with HPMC.
Table 2. Ex-vivo bioadhesion study of buccoadhesive gels on rabbit buccal mucosa.
Determination of swelling index and surface pH of the hydrating layer
Agar plates were chosen as a simple model of the mucosa, as it can keep an amount of water that resembles the secreting fluid in and around the buccal mucosa required for buccoadhesion and subsequent swelling of the formulations to provide adequate release of drug (CitationMutalik & Udupa, 2006). The results of the swelling index of the buccoadhesive gels of pH 7.4 showed inverse relation to the buccoadhesive strength and had swelling index in the order, F1 (6.543 ± 0.22) > F2 (6.477 ± 0.19) > F3 (6.303 ± 0.27) > F4 (6.106 ± 0.18). Formulation F1 exhibited a higher swelling index, which could be attributed to the property of carbopol to retain water and form a thick swollen mass, which seemed to decrease as the proportion of HPMC increased. However, F2 seemed to be an exception, which could be due to less entry of carbopol into the HPMC network and the swelling ability of HPMC itself, which added to the swelling of this particular combination gel. The inverse relation of buccoadhesive strength to swelling/viscosity observed has also been previously reported (CitationMohammadi-Samani et al., 2005). The report (CitationMohammed & Khedr, 2003) of the increasing combinations of carbopol to HPMC in increasing the swelling and viscosity seems to be justified, however a contradictory result in our case may be attributed to the different grades of polymers used in different ratios. The gels without adjustment for pH of the hydrating layer showed a decrease in the pH of the hydrating layer (pH 6.2) as compared to the gels in which the change in pH of the hydrating layer was accounted for by addition of triethanolamine. Adjustment of the pH of the hydrating layer seems to be justified for two reasons; (1) since the carbopol is said to be acidic in nature it could lend its nature to the preparation and therefore to neutralize the preparation a base (triethanolamine) has to be added (CitationRowe et al., 2006), and (2) a change from higher pH of the core to the lower pH of the hydrating layer will definitely produce complexity in drug transfer between the two different pH layer as compared to a gel of the same pH environment both in the core as well as the hydrating layer.
In-vitro release studies
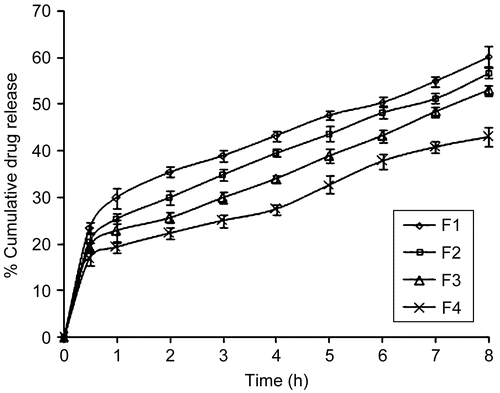
The results of in-vitro drug release profiles () of glibenclamide from buccoadhesive gels showed that the buccoadhesive gel formulation F1 provided maximum release, respectively followed by F3, F2, and F4. The maximum release of glibenclamide from formulation F1 could be attributed to ionization of CP934 at a pH environment higher than its pKa value of 5.3. Ionization of CP934 leads to the development of negative charges along the backbone of the polymer. Repulsion of like charges uncoils the polymer into an extended structure. The counter ion diffusion inside the gel creates an additional osmotic pressure difference across the gel, leading to high water uptake. This water uptake leads to the considerable swelling of the polymer. The continued swelling of the polymer matrix causes the drug to diffuse from the formulation at a faster rate. The release from F4 formulation was least because of the low swelling which caused less amount of drug to diffuse through the gel. The release behavior of glibenclamide from formulation containing CP934 and HPMC at the ratio of 1:1 (F4) was impressive, since these formulations showed effective controlled release pattern with t50% assumed to reach in 10 h, which was more than other formulations in the study.
Figure 2. Viscosities of buccoadhesive gel formulations of pH 7.4 at 10 and 50 rpm.
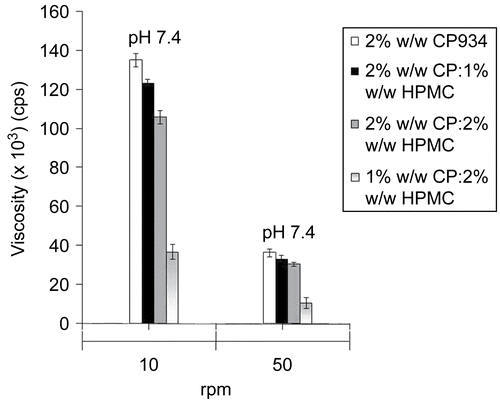
Figure 3. Comparative drug release profiles of buccoadhesive gel formulations of pH 7.4, where ![]()
Effect of permeation enhancer
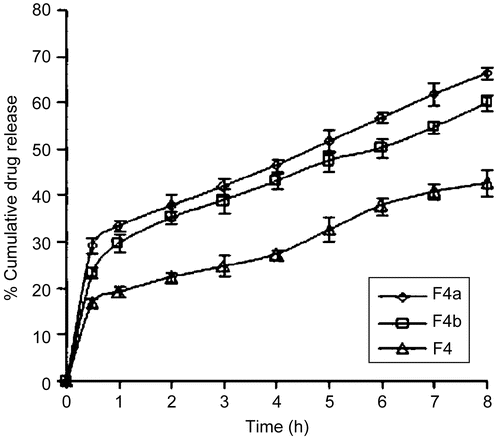
The formulation F4 was further selected based on its good buccoadhesive strength and release pattern to observe the effect of permeation enhancers on the formulation. It was observed that the formulation containing 1% w/w SLS (F4a) showed a higher drug release () than that containing 1% w/w ST (F4b). However, the release through F4a was very high, in that ∼ 41.98% of drug was released in the receptor compartment within 3 h with a loss in the donor compartment of 3.09% at the same time, which would defeat the objective for which the glibenclamide gel was being formulated, while the release through F4b was ∼ 50.42% within 6 h with a negligible loss of 0.47% at the same time. An ideal controlled release system should be able to release the drug immediately to attain the therapeutic level at a faster rate and then maintain this drug level for a prolonged period of time.
Figure 4. Comparative drug release profiles of optimized formulation with permeation enhancers, where ![]()
Kinetics of release
The release mechanism of the drugs was analyzed by the commonly used exponential equation. The best fit model in case of gels of pH 7.4 (F1–F4, F4a, and F4b) could have followed zero-order, Power law, and Higuchi model. However, zero order models are not suited to analyze swellable matrix systems, and were therefore rejected. Among the Power law and Higuchi models, while considering the higher correlation coefficient value (R), the release data seem to fit the Higuchi model better. The drug release data was further analyzed () for curve fitting based on Power Law, and the n-values ranging from 0.2819–0.3982 confirmed that all the formulations followed Fickian diffusion (n < 0.5).
Table 3. Different kinetic models applied on buccoadhesive gels of pH 7.4 (F1-F4, F4a, and F4b).
Validation parameters
The analytical performance parameters, namely specificity, linearity, range, precision, accuracy, LOD, and limit of quantification, were validated according to International Conference on Harmonization ICH Q2B guidelines. Specificity was assessed by comparing the chromatograms obtained from the drug to their respective internal standards and with those obtained from the blank that verified the absence of any interference. The linearity of the method used for the drug was evaluated on a standard curve of the peak area vs the concentration of the analyte. A six-point calibration curve was constructed with working standards and was found linear (R2 = 0.9999) in a range 0.2–20 μg/mL (Rt was found to be 10.02 ± 0.03 and 6.45 ± 0.09 min for glibenclamide and glipizide, respectively). The LOD and LOQ were found to be 0.12 and 0.20 μg/mL, respectively. The results of determination of accuracy using QC concentrations were 98.88 ± 0.12%. Precision assay showed that the averages of the relative standard deviations within 1 day (intra-day) were 0.21–0.78% and among every other day (inter-day) were 0.97–2.82%. The results showed that the method was accurate. Recovery test was performed again on QC samples and the results, 98.99 ± 2.08 μg/mL for 1 μg/mL, 100.05 ± 0.19 for 10 μg/mL, and 99.97 ± 0.67 for 20 μg/mL, validated the method.
Hypoglycemic activity
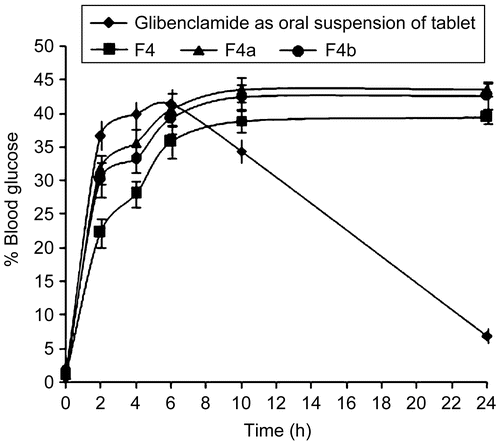
The reduction in blood glucose levels of buccal gels in comparison with oral suspension of the tablet of glibenclamide (5 mg) in rabbits, both normal and diabetic, are shown in and , respectively. The hypoglycemic effect was significant (p < 0.05) between the glibenclamide marketed tablet and the formulated buccal gels upto the 10 h and (p < 0.01) in the 24 h. Glibenclamide (oral tablet in suspension form) produced a decrease in blood glucose levels up to 161.5 ± 4.87 mg/dl (19.14 ± 2.41%) in the case of normal rabbits, and 213.1 ± 3.52 mg/dl (35.45 ± 0.97%) in the case of diabetic rabbits; at 2 h, and the effect lasted ∼ 10 h. In the case of buccal gels, the hypoglycemic response was gradual. A maximum hypoglycemic response was observed at 10 h, and thereafter remained stable up to 24 h. The untreated group did not show any noticeable hypoglycemia.
Figure 5. Blood glucose level profiles for the marketed glibenclamide tablet administered as oral suspension along with the formulated buccoadhesive gels in normal rabbits, where ![]()
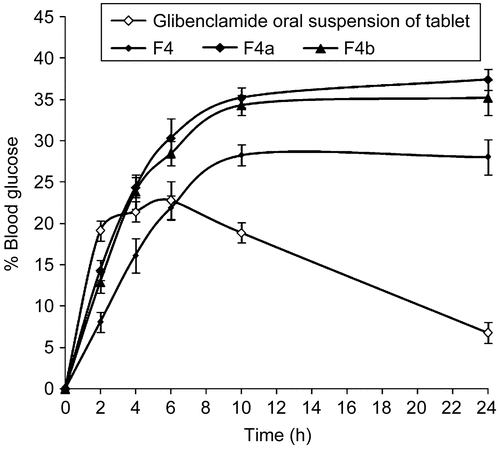
Figure 6. Blood glucose level profiles for the marketed glibenclamide tablet administered as oral suspension along with the formulated buccoadhesive gels in diabetic rabbits, where ![]()
Pharmacokinetic studies
The pharmacokinetic parameters () obtained with buccoadhesive gels were significantly (p < 0.05) different from those obtained with tablet administration, which could be due to the rapid absorption of drugs via the oral route; whereas drug in the buccal route were slowly but continuously absorbed. Although the rise in drug concentration was slower than the tablet formulation, the drug concentration in plasma remained high for a longer period, and thus was able to exert a sustained action when administered as buccoadhesive gel (). This was supported by increased t1/2 values, significantly lower elimination rate constant Ke, and high mean residential time (MRT) values obtained with buccoadhesive gel of glibenclamide. Although the Cmax was less for buccal gel, the AUC values were significantly high (p < 0.05) compared to tablet formulation. The significantly high AUC values with buccal gel indicated increased relative bioavailability (219.2 ± 1.23%) of drug from F8 systems compared to oral tablet formulation. The prepared buccoadhesive gels with penetration enhancers also showed enhanced bioavailability, 272.15 ± 1.45% for F8a and 258.40 ± 1.29% for F8b. On the whole, buccal gel of glibenclamide showed better in-vivo effectiveness in rabbits compared to the tablet formulation. This could be due to a slow and continuous supply of drug at a desirable rate to the systemic circulation.
Figure 7. Plasma concentration-time profiles for the marketed tablet administered as oral suspension along with the formulated buccoadhesive gels, where ![]()
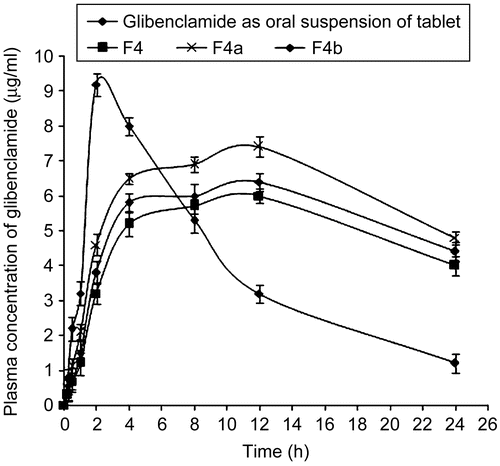
Table 4. Pharmacokinetic parameters of glibenclamide after administration of marketed glibenclamide tablet as oral suspension and buccoadhesive gels.
Conclusion
The buccoadhesive gels of glibenclamide were successfully prepared and optimized in-vitro, and evaluated in-vivo. The prepared buccoadhesive gels of glibenclamide exhibited better in-vivo performance than convention marketed formulation in rabbits, as well as reversing the diabetic complications. The buccoadhesive gels could possibly be a means for alternative dosage form in avoiding first pass metabolism and ensuring enhanced bioavailability for glibenclamide.
Acknowledgements
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Agarwal, V., Mishra, B. (1999). Design development and pharmaceutical properties of buccoadhesive compacts of pentazocine. Drug Dev Ind Pharm. 25, 701–9.
- Alur, H.H., Johnston, T.P., Mitra, A.K. (2001). Peptides and proteins: buccal absorption. In: Swarbrick, J., Boylan, J.C., eds. Encyclopedia of pharmaceutical technology. New York: Dekker, 206.
- Alur, H.H., Pather, I.S., Mitra, K.A., Johnston, P.T. (1999). Transmucosal sustained-delivery of chlorpheniramine maleate in rabbits using a novel, natural mucoadhesive gum as an excipients in buccal tablets. Int J Pharm. 188, 1–10.
- Attia, M.A., El-Gibaly, I., Shaltout, S.E., Fetih, G.N. (2004). Transbuccal permeation, anti-inflammatory activity and clinical efficacy of piroxicam formulated in different gels. Int J Pharm. 276, 11–28.
- Davis, S.N., Granner, D.K. (1996). Insulin, oral hypoglycemic agents, and the pharmacotherapy of the endocrine pancreas. In: Hardman, J.G., Limbird, L.E., eds. The pharmacological basis of therapeutics. New York: McGraw-Hill, 1487–517.
- Desai, S.J., Singh, P., Simonelli, A.P., Higuchi, W.I. (1966). Investigation of factors influencing release of solid drug dispersed in wax matrices. III. Quantitative studies involving polyethylene plastic matrix. J Pharm Sci. 55, 1230–4.
- Doijad, R.C., Manvi, F.V., Rao, V.S.N.M., Patel, P.S. (2006). Buccoadhesive drug delivery system of isosorbide dinitrate: formulation and evaluation. Ind J Pharm Sci., 68, 744–8.
- Florey, K. (2005). Analytical profile of drug substances. 1st ed. New Delhi: Elsevier India, 338.
- Higuchi, T. (1963). Mechanism of sustained action medication, theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 52, 1145–9.
- Hixson, A.W., Crowell, J.H. (1931). Dependence of reaction velocity upon surface and agitation: I—theoretical consideration. Ind Eng Chem. 23, 923–31.
- Kataria, U., Dwivedi, J., Kar, M. (2007). Buccal route for systemic drug delivery. Ind Pharm. 6, 13–20.
- Korsmeyer, R.W., Gurny, R., Doelker, E.M., Buri, P., Peppas, N.A. (1983). Mechanism of solute release from porous hydrophilic polymers. Int J Pharm. 15, 25–35.
- McElnay, J.C., Hughes, C.M. (2002). Drug delivery- buccal route. In: Swarbrick, J., Boylan, J.C., eds. Encyclopedia of pharmaceutical technology. New York: Dekker, 800–9.
- Mohammadi-Samani, S., Bahri-Najafi, R., Yousefi, G. (2005). Formulation and in-vitro evaluation of prednisolone buccoadhesive tablets. Il Farm. 60, 339–44.
- Mohammed, F.A., Khedr, H. (2003). Preparation and in vitro/in vivo evaluation of buccal bioadhesive properties of slow release tablets containing miconazole nitrate. Drug Dev Ind Pharm. 29, 321–37.
- Mutalik, S., Udupa, N. (2006). Glipizide matrix transdermal systems for diabetes mellitus: preparation, in vitro and preclinical studies. Life Sci. 79, 1568–77.
- Najib, N., Suleiman, M. (1985). The kinetics of drug release from ethyl cellulose solid dispersions. Drug Dev Ind Pharm. 11, 2169–81.
- Park, C.R., Munday, D.L. (2004). Evaluation of selected polysaccharide excipient in buccoadheaive tablets for sustained release of nicotine. Drug Dev In. Pharm. 30, 609–17.
- Philip, A.K., Pathak, K. (2008). Wet process-induced phase-transited drug delivery system: a means for achieving osmotic, controlled, and level a ivivc for poorly water-soluble drug. Drug Dev Ind Pharm. 34, 735–43.
- Rang, H.P., Dale, M.M., Ritter, J.M., Moore, P.K. 2005. Pharmacology, 5th ed. New Delhi: Elsevier India, 389.
- Rowe, R.C., Sheskey, P.J., Owen, S.C. (2006). Handbook of pharmaceutical excipients. 5th ed. London: Pharmaceutical Press,112.
- Sangeetha, S., Santhi, K., Ponnusankar, S., Nagasamy, D., Dhanraj, S.A., Suresh, B. (2007). Formulation and evaluation of buccal disks containing fluconazole. Ind Pharm. 6, 62–4.
- Satoh, K., Takayama, K., Machida, Y., Suzuki, Y., Nakagaki, M., Nagai, T. (1989). Factors affecting the bioadhesive property of tablets consisting of hydroxypropyl cellulose and caboxyvinyl polymer. Chem Pharm Bull. 37, 1366–8.
- Varshosaz, J., Dehghan, Z. (2002). Development and characterization of buccoadhesive nifedipine tablets. Eur J Pharm Biopharm. 54, 35–141.
- World Health Organization. 2007. WHO model list of essential medicines. 15th ed. WHO, 21. Available online at: http://www.who.int/medicines/publications/08_English_index FINAL_EMLIS.pdf. Accessed 12 January 2009.