
Abstract
In the present study, pluronic lecithin based organogels (PLO gels) were formulated as topical carrier for controlled delivery of mefenamic acid. Ten organogel formulations were prepared by a method employing lecithin as lipophilic phase and pluronic F-127 as hydrophilic phase in varying concentrations to study various parameters using in vitro diffusion study and in vivo studies. All formulations were found to be off-white, homogenous, and reluctant to be washed easily and have pH value within the range of 5.56–5.80 which is nonirritant. Polymer concentration increased in formulations of F1 to F5 (lecithin) and F6 to F10 (pluronic) resulted in decrease of the gelation temperature, increase of viscosity and reduction of spreadability of gels having polymer tendency to form rigid 3D network. Organogels with higher viscosity were found to be more stable and retard the drug release from the gel. The formulations of F2 and F3 were selected for kinetic studies and stability studies, as they found to have all physical parameters within acceptable limits, highest percent drug content and exhibited highest drug release in eight hours. The order of drug release from various formulations was found to be F2 > F3 > F10 > F4 > F1 > F9 > F8 > F5 > F7 > F6. The optimized formulation F2 was found to follow zero order rate kinetics showing controlled release of the drug from the formulations. In vivo anti-inflammatory activity of optimized mefenamic acid organogel (F2) against a standard marketed preparation (Volini gel) was found satisfactory and significant.
Introduction
Mefenamic acid is a drug of NSAIDs category having anti-inflammatory and analgesic activity. It is used in management of pain and inflammation, swelling and uterine contractions by inhibition of prostaglandin synthesis. Conventional oral dosage form of mefenamic acid is available in the form of capsules but like other NSAIDs, mefenamic acid is also prone to cause gastrointestinal ulceration, rashes and intestinal bleeding when administered by oral route. The aqueous solubility of mefenamic acid is very low (20 mg/l) and major metabolism occurs in liver and excretion by kidney, which further reduces its oral bioavailability. Therefore an attempt is made to deliver mefenamic acid through skin for better bioavailability along with local action and to avoid gastric side effects. It is 90% protein bound drug with apparent volume of distribution 1.06 l/kg having half-life of two hours. Log p value of mefenamic acid is 4.1 and molecular weight is 241.285 g/mol, which makes it a suitable candidate for transdermal route. Therefore, transdermal administration of mefenamic acid will be promising, as transdermal route has many advantages over other conventional oral routes such as avoid drug degradation in stomach due to internal body conditions and various metabolizing enzymes, first pass effect, irritation of GI mucosa, ease of application, controlled rate drug release, constant drug blood level, patient compliance and decreased side effects with minimum discomfort (Kumar et al., Citation2010). But, it is not easy to deliver a drug through skin such as stratum corneum; the outermost and toughest layer of the skin is the major barrier which is composed of corneocytes containing dead keratin (Arunachalam et al., Citation2010). To enhance the permeation of drug through the skin and to increase the bioavailability of the drug, various approaches have been utilized such as application of permeation enhancers, novel drug delivery systems such as vesicular systems (Schmid & Korting, Citation1994), nanoparticles, microspheres (Abrol et al., Citation2004) and polymeric lipid gels. Pluronic lecithin based organogels (PLO gels) have the potential to permeate through skin and deliver sufficient quantity of drug for local as well as systemic action. Therefore, we have chosen PLO gels for the topical delivery of mefenamic acid, which is expected to deliver the drug across the skin for better results. PLO gels are nonirritant and biocompatible composed of phospholipid (lecithin) as surfactant, an organic solvent as outer continuous phase and an aqueous polar phase/solvent. The entangled reverse micelles forms a 3D network which entraps the outer continuous nonpolar phase and immobilizes it turning into a viscous gel by self-association of individual gelator molecules (Scartazzini & Luisi, Citation1988; Schurtenberger et al., Citation1990). Several categories of drugs such as NSAIDS, hormones, opiods, local anesthetics and anti-emetic drugs have successfully been incorporated in PLO gels (Dumortier, Citation2006).
Both hydrophilic and lipophilic types of drugs having molecular weight less than 400 Da can be easily incorporated into PLO gel formulation. The composition of PLO gel includes oil phase containing phospholipid lecithin as permeation enhancer, isopropyl myristate (IPM)/palmitate as a solvent for lecithin and emollient for skin having a good spreading rate and sorbic acid as preservative and aqueous phase containing pluronic F-127 as surfactant, purified water as solvent and potassium sorbate as preservative (Kumar & Katare, Citation2005; Vintiloiu & Leroux, Citation2007; Belgamwar, Citation2009). Various studies show that PLO gel formulations are much better than oral tablets in the treatment of inflammation. So, the present study was undertaken to develop PLO gel formulation of mefenamic acid for anti-inflammatory activity using in vivo method in rat model.
Materials and methods
Materials
Mefenamic acid was obtained as a gift sample from K Pharma (Ambala, India). Pluronic F-127 was purchased from Sigma Aldrich (Delhi, India), Soya lecithin and disodium hydrogen phosphate were purchased from Himedia laboratories Pvt. Ltd. (Mumbai, India), IPM, sorbic acid potassium sorbate, potassium dihydrogen orthophosphate, and sodium chloride and sodium hydroxide were purchased from Qualikems Fine Chem. Pvt. Ltd. (New Delhi, India). Menthol was purchased from Avon Flavors (Mumbai, India). All other chemicals were also of analytical grade.
Methods
Preparation of organogel
PLO gel is a microemulsion prepared by mixing of aqueous and oil phase at a high shear. Ten formulations of PLO gel () with varying concentrations of lecithin and pluronic F-127 were prepared by the below method comprising of the two phases, i.e. oil phase and aqueous phase (Umamaheswari et al., Citation2002; Belgamwar, Citation2009).
Table 1. Formulation of PLO gels from F1 to F10.
Oil phase: The oil phase is prepared by taking a measured amount of lecithin and sorbic acid in IPM as a solvent. The above mixture was kept at room temperature for 12 h to ensure complete dissolution of lecithin and sorbic acid in IPM.
Aqueous phase: Aqueous phase was prepared by dissolving weighed quantity of pluronic F-127 and potassium sorbate and menthol in cold water. The mixture was kept below 4 °C in refrigerator for 12 h for complete dissolution of pluronic F-127.
Next day, gel was prepared by adding slowly oil phase to aqueous phase at a high shear using mechanical stirrer so that a uniformly dispersed microemulsion is formed. Drug was incorporated in oil phase by making a paste with polyethylene glycol 400 before mixing with pluronic phase (Garg et al., Citation2011). Menthol was added as soothing agent and permeation enhancer.
Visual characters of organogel system
The knowledge of molecular packing and formation of cross linking bridges within the organogel network and entrapment of aqueous phase in the lipid polymer phase was observed using light microscope and scanning electron microscope (Kasliwal et al., Citation2008).
Organoleptic characteristics
Each formulation was tested for various organoleptic characteristics such as odor, color, texture, phase separation and greasiness (Kasliwal et al., Citation2008).
Homogeneity test
Gel (100 mg) was pressed between the thumb and the index finger in order to notice the consistency of gel that any coarse particles being attached or detached on finger (Agrawal et al., Citation2010).
Washability
PLO gel (100 mg) was rubbed on backside skin of the hand. After drying the layer was washed with tap water and observed whether it is washable or not (Agrawal et al., Citation2010).
pH determination
The pH of all the gel formulations was determined using digital pH meter which was calibrated before measurements using standard buffer solutions of pH 7 and pH 4. After calibration, the electrode was dipped in an aqueous solution of gel (1 g in 20 ml water) at 25 °C which directly gives the pH of the gel formulation (Murthy & Kishore, Citation2008; Singh, Citation2009).
Viscosity (rheological characters)
Viscosity (in terms of centipoises: cps) of all the formulations was determined using Brookfield digital viscometer (DV-I Prime, Brookfield Engineering Laboratory, Inc., Middleboro, MA). Tests were performed at 25 °C and temperature was controlled using a water bath. Viscosity was determined using the spindle (LV 61) rotating at 10 rpm (Singh, Citation2009).
Spreadability
Spreadability of each formulation was determined by an apparatus defined by Mutimer et al. (Citation1956) containing two glass slides. Gel (1 g) was placed between the slides and a weight of 1000 g was placed on upper slide to allow the gel to spread uniformly. Now weights were added to the weight pan and the time traveled by the movable slide for a particular distance was noted and spreadability of different gels was calculated by the below formula (Mutimer et al., Citation1956):
where S is the spreadability of gel in g cm/s, M is the weight placed in the weight pan, L is the distance traveled by the movable slide in cm, and T is the total time taken by the slide to travel the distance L (Kumar & Katare, Citation2005).
Gel transition temperature
The gel transition temperature was determined in a 20 ml transparent vial that contains a magnetic bead. Each formulation (10 g) was placed in a water bath maintained at 4 °C and temperature was increased gradually at a rate 1 °C/min while stirring the magnetic bead at 60 rpm. The temperature at which the magnetic bead stopped moving was noted as gelation temperature of gels (Baloglu et al., Citation2011).
Drug content
Drug content of different formulations was calculated using UV spectrophotometer. Gel (100 mg) was dissolved in 100 ml buffer and filtered through 0.45 μm syringe filter. After filtration the drug content was found out by taking absorbance at λmax 285 nm (Nemutlu, Citation2005).
In vitro diffusion study
In vitro diffusion study was performed to calculate the percent cumulative drug release with respect to time. Franz diffusion cell having two compartments, i.e. upper donor compartment and lower receiver compartment separated by a semi permeable cellophane membrane was used. PLO gel (1 g) was placed in donor compartment and receiver compartment was filled with phosphate buffer pH 7.4. Whole assembly was maintained at 37 °C with constant stirring. Samples were taken at time intervals of 0, 1, 2, 3, 4, 5, 6, 7 and 8 h. One milliliter sample was withdrawn and an equal quantity of phosphate buffer pH 7.4 was replaced after each sampling. Samples were analyzed after making proper dilutions using UV spectrophotometer at λmax 285 nm to determine the percent cumulative drug release (Nemutlu, Citation2005).
Stability study
The stability testing of PLO gel formulations was carried out in humidity chamber (I-Therm Inc.) according to the ICH guidelines. The chamber was operated at 4 °C, 25 °C/65% of relative humidity and 60 °C/75% of relative humidity for three months and evaluated for percent drug content, pH and physical stability to check phase separation (Agrawal et al., Citation2010).
Animals
Albino rats (Wistar strain) of either male or female of about 200–250 g were used after obtaining the approval of the Institute Animal Ethics Committee (MMCP/IAEC/12/25). The animals were housed under standard conditions of temperature (24 ± 28 °C) and relative humidity (60–70%) with a 12:12 light–dark cycle. The animals were fed a standard pellet diet (Lipton India, Ltd., Kolkata, India) and water ad libitum.
Skin irritation study
Skin irritation study was performed in each group of six rats. The hairs were removed from the back of the rat with the help of hair removing cream. An area of 4 cm2 was marked on both the sides. After 24 h of depilation, the formulations were applied (100 mg/rat) once a day for seven days and sight was covered with cotton bandage (Kulkarni & Jain, Citation2001). The rat was observed for sign and symptoms of any sensitivity and reaction that may occur and were classified as: 0 No reaction, 0.5 Slight, patchy erythema, 1 Slight but confluent or moderate but patchy erythema, 2 Moderate erythema and 3 Severe erythema with or without edema.
Experimental design for in vivo studies (analgesic and inflammation activity)
Rats were divided into three different groups (six rats each) and treated as follows. Group 1: Normal treated control group, Group 2: Positive control group, Group 3: Standard marketed formulation treated group (Volini gel; 100 mg), Group 4: Test formulation treated group (PLO gel; 100 mg).
Eddy’s hot plate method
In this method, heat was the source of pain to check the analgesic activity of drug. Individually animals were placed on Eddy’s hot plate (Techno Instrument India) maintained at constant temperature of 55 ± 1 °C. Time taken by animal for any of the reaction either paw licking or jumping or raising the limbs, which ever will be observed first will be taken as end point. Reaction time was noted before and 30 min after administration of the drug (Rohit et al., Citation2006).
Carrageenan induced rat paw edema model
The drug was applied 30 min before carrageenan administration. Each 0.1 ml of 1% w/v carrageenan was injected into the rat paw of all the groups except normal control group after 30 min of the drug administration. The paw edema volume was measured with digital plethysmometer (Model 7140, UGO Basile, Monvalle, Italy) after a gap of 0 h, 1 h, 2 h, 3 h and 4 h (). The percentage inhibition of edema compared with that of the control was taken as anti-inflammatory activity. The percentage inhibition of edema was calculated by the formula:
Figure 1. Carrageenan induced paw edema test.
Percentage inhibition of edema = (A−B)/A × 100 where A represents the paw volume of the control group and B represents the paw volume of the test drug treated group (Crunkhorn & Mencock, Citation1971).
Statistical analysis
The data were expressed as mean ± standard error of mean (SEM). The statistical significance between mean was analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. A p value of <0.05 was considered as statistically significant.
Results
Ten formulations were prepared with varying concentrations of lecithin (F1–F5) and pluronic F 127 (F6–F10) and evaluated for various formulation parameters.
Organoleptic characteristics
The gel formulations were observed for physical appearance and other organoleptic characteristics. All formulations were found homogenous, odorless, off-white in color, greasy and reluctant to wash and stable without any sign of phase separation.
Surface morphology of organogel
Light microscopic examination of organogel (F2) at 10 × 40 resolutions has shown organogel as bi-continuous systems containing water molecules entrapped within the self-assembled three-dimensional network of the gelator ().
Figure 2. Light microscope structure at 10 × 40 resolution (1) and SEM image at 10 × 1000 resolution of organogel formulation (F2).
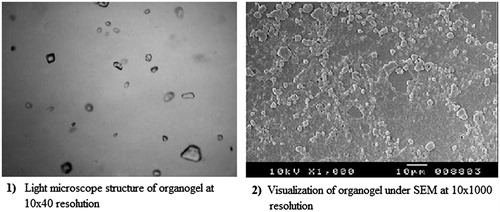
SEM photographs of the gel (F2) at 10 × 1000 resolutions clearly showed the gel forming the highly viscous three-dimensional network structure. The three-dimensional networked structure, so formed, prevents the flow of external apolar phase ().
pH
The pH of all PLO gel formulations were found in the range 5.57–5.80 which is in the normal range of skin pH. Therefore all PLO gel formulations are nonirritant to the skin ().
Table 2. Evaluation parameters of organogel formulations (F1–F10).
Spreadability
The values of spreadability indicate that the gel is easily spreadable with small shear but increase in percentage of lecithin resulted in decrease in the spreadability of organogels from F1 to F5 (30.18–10.42 g.cm/s). Likewise same trend was observed when percentage of pluronic was increased in formulations from F6 to F10 (40.50–8.26 g.cm/s) (). This decrease in spreadability of gels with increase in polymer concentration is because of increase in the cross linking between polymers which resist it to spread easily.
Viscosity
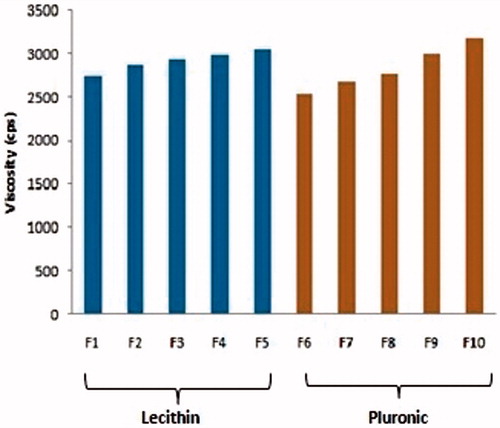
The viscosity of gel formulations was found in the range from 2738 cps to 3052 cps (F1–F5) and from 2542 cps to 3176 cps (F6–F10). The proportional increase in the viscosity with polymer concentration can be attributed to the incidence of more cross linking in polymer with increase in polymer concentration, as viscosity curve given in . PLO gels show shear thinning, non-Newtonian behavior with pseudo plastic flow ().
Figure 3. Viscosity values of formulations from F1 to F10.
Skin irritation study
Absence of skin irritation in gel formulation is acceptable. There was no erythema, edema or reddening of skin. All gel formulations found were free from any sign of irritation ().
Gel transition temperature
The gel transition temperature of 10 organogel formulations were evaluated between the range of 35.4 °C and 29.1 °C (F1–F5) and 33.9–29.8 °C (F6–F10). As the polymer concentration increases the gel strength also increases and the gelation occurred at lower temperature ().
Percentage drug content
The percentage drug content of organogels was found in the range from 96.25% to 98.36%. It was calculated by using the standard curve of MA in PBS 7.4 ().
Table 3. Percentage drug content of organogels.
Percent cumulative drug release
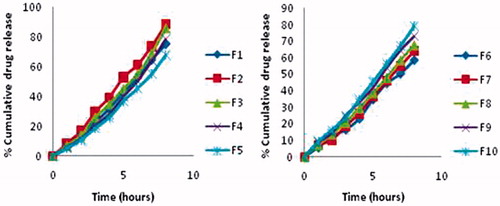
Formulations with higher percentage of lecithin retarded the drug release and have higher viscosity. The percent cumulative drug release in eight hours was found in the order F2 > F3 > F10 > F4 > F1 > F9 > F8 > F5 > F7 > F6 ().
Table 4. Percentage cumulative drug release in 8 h .
It may be noted that increase in lecithin concentration showed retardation in release of MA in F1–F5 formulations but in F6–F10 formulations, the increase in pluronic concentration showed less variation in drug release profile. Since the number and dimension of aqueous region available for diffusion of the drug are reduced with the possible increase in microviscosity in the three-dimensional structure of the gel. shows percentage cumulative drug release profile from F1 to F5 and from F6 to F10.
Figure 4. Percentage cumulative drug release profile of F1–F5 and F6–F10 formulations.
Based on above studies, we found that formulations F2 and F3 are suitable for further kinetic data fitting stability study, as these have shown all physical parameters within acceptable limits and showed the highest percent cumulative drug release in eight hours. Therefore F2 and F3 formulations were further tested for different rate equations. F2 formulation was found to follow zero order rate kinetics () with r2 value 0.996 when compared to F3 which indicates it as a controlled release formulation.
Table 5. Kinetic data of release studies for F2 and F3 formulations.
Stability study
Formulations F2 and F3 were found to be sufficiently stable during study period at all temperature ranges. Pluronic gels were in semisolid to liquid state at temperature 60 °C depending on polymer concentration but no phase separation occurs up to 60 days in F2 formulation and up to 30 days in F3 formulation. However on prolonged study at 60 °C PLO gels started showing phase separation (). Therefore finally F2 formulation was then selected for in vivo anti-inflammatory activity.
Table 6. Stability study data of F2 and F3 formulations.
In vivo study
Hot plate method
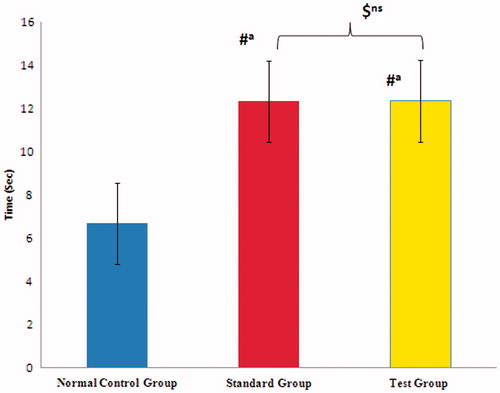
The analgesic effect of PLO gel of mefenamic acid was compared with control group and standard group (Volini gel). Normal reaction time in control group animals was 4–8 s. Reaction time was significantly increased in test group animals but it was almost within the range of standard group, i.e. 10–13 s. The statistical analysis also showed non-significant effect when compared between standard and test formulation (F2), which also confirmed that both the formulation showed same type of effect ().
Figure 5. Analgesic activity of mefenamic acid organogel formulation using Eddy’s hot plate.
Carrageenan induced paw edema
The results () showed that both the formulations (F2 and marketed) produced statistically significant inhibition (p < 0.0001) of edema when compared with positive control group after second hour onwards but at third and fourth hour, our F2 formulation showed non-significant effect when compared with marketed formulation indicating similar type of inhibitory activity. Test formulation was found to be 28.55% and 33% and marketed formulation was produced 30.16% and 35.52% at third and fourth hour ().
Figure 6. Anti-inflammatory mefenamic acid organogel using digital plethysmograph.
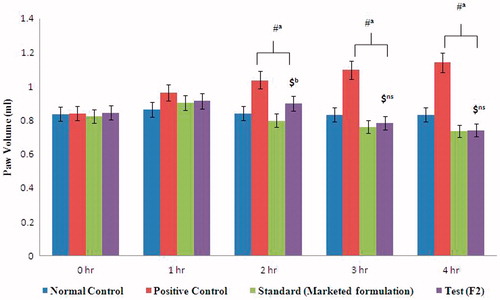
Table 7. Mean values of paw volume at different time intervals.
Discussion
Searching of new drug molecule and development of pharmaceutical product is a tedious method in the laboratory now days. On the other side, marketed products are well known for its therapeutic activity and with serious adverse effects. Best drug delivery system is our target for the drug to achieve its goal without any serious side effects. The objective of the present study was to formulate and evaluate different organogel tropical formulations of mefenamic acid with varying concentrations of pluronic and lecithin. In our study, all gel formulations were found to have all physical parameters within acceptable limits such as homogenous, off-white in color, odorless, greasy and stable without any sign of phase separation. The organogel formulations were somewhat reluctant to wash. The pH of our organogel formulations was found to be near skin pH range and showed good compatibility with skin. Polymer concentration is one of the parameter to measure the viscosity, spreadability and percent cumulative drug release. Our study showed that, when the polymer concentration increases then the viscosity was also elevated and it is directly proportional to the polymer concentration. Therefore increase in lecithin concentration (F1–F5) or pluronic concentration (F6–F10) increases the viscosity of gel formulations and reduces the spreadability due to increase in resistance between layers. Thus, the percent cumulative amount of drug release was reduced, as the viscosity of the formulations increased and these results were in accordance with earlier study findings (Pandey et al., Citation2010). The gelation temperature range of our formulations lies below normal body temperature because our formulation exists in gel state and similar findings were also reported by Ba et al. (Citation2016). The skin irritation study was also performed on rats and all formulations showed no detectable sign of skin irritation, indicating excellent compatibility with skin. Based on different evaluation parameters (), the optimized formulations F2 and F3 were selected for further rate kinetics study and stability study. In stability study, data of F2 and F3 formulations for three months are within range in accordance with the ICH guidelines and same result was found in the literature reported by Pandey et al. (Citation2010). Our F2 and F3 formulations showed no sign of phase separation during storage and no significant macroscopic changes in physical parameters, percentage drug content and pH. In the rate kinetic data, formulation F2 follows zero order release kinetic as compared to F3 which will deliver the drug at controlled rate, therefore, F2 formulation was then selected for in vivo analgesic and anti-inflammatory activity. In vivo analgesic activity studies on animals ( and and ) indicated that, our F2 formulation and marketed diclofenac gel (Volini gel) formulation showed similar type of activity indicating non-significant effect when compared with each other, whereas in inflammation induced rat model, at 2 h, test formulation showed slightly significant effect (p < 0.01) when compared with marketed formulation but at third hour and fourth hour, F2 formulation showed non-significant effect when compared with marketed formulation which indicated that, our F2 formulation is having same therapeutic activity than that of marketed formulations (Volini gel). Although, the effectiveness of F2 formulation and marketed gel formulation was almost same for analgesic and anti-inflammatory activities but our formulation (Topical organogel) could be better effective alternative to diclofenac marketed gel as diclofenac gel have short duration and low intensity activity. Being organic in nature and presence of lecithin (a component of skin phospholipids) these have a very high potential to permeate the drug through the skin so these can easily deliver the drug across the skin. Therefore, more studies are needed to optimize the organogel formulations to get the superior results.
Conclusion
Therefore, from the above study we conclude that organogels are novel base for drugs through topical route and could be an alternative to the marketed diclofenac gel formulations. Our F2 formulation may come as a new drug in the market for the treatment of inflammation after following various phases of clinical trials and ethical guidelines.
Declaration of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Acknowledgements
The authors are grateful to the management for offering the requisite technical help to accomplish this study.
References
- Abrol S, Trehan S, Katare OP. (2004). Formulation, characterization, and in vitro evaluation of silymarin loaded lipid microspheres. Drug Deliv 11:185–91.
- Agrawal V, Gupta AV, Ramteke S, Trivedi P. (2010). Preparation and evaluation of tubular micelles of pluronic lecithin organogel for transdermal delivery of sumatriptan. AAPS PharmSciTech 11:1718–25.
- Arunachalam A, Karthikeyan M, Kumar VD, et al. (2010). Transdermal drug delivery system: a review. Curr Phar Res 1:70–81.
- Ba W, Li Z, Wang L, et al. (2016). Optimization and evaluation of pluronic lecithin organogels as a transdermal delivery vehicle for sinomenine. Pharm Dev Technol 21:535–45.
- Baloglu E, Karavana SY, Senyigit ZA, et al. (2011). Rheological and mechanical properties of poloxamer mixtures as a mucoadhesive gel base. Pharm Dev Technol 16:627–36.
- Belgamwar VS. (2009). Topical delivery of flurbiprofen from pluronic lecithin organogel. Indian J Pharm Sci 71:87–90.
- Crunkhorn P, Mencock SC. (1971). Mediators of inflammation induced in rat paw carrageenan. Br J Pharmacol 42:371–402.
- Dumortier G. (2006). A review of poloxamer 407 pharmaceutical and pharmacological characteristics. Pharm Res 12:2709–28.
- Garg T, Bilandi A, Kapoor B, et al. (2011). Organogels: advanced and novel drug delivery system. Int Res J Pharm 2:15–1.
- Kasliwal N, Derle D, Negi J, Gohil J. (2008). Effect of permeation enhancers on the release and permeation kinetics of meloxicam gel formulations through rat skin. Asian J Pharm Sci 3:193–9.
- Kulkarni SK, Jain NK. (2001). Pharmacological and pharmacokinetic studies on marketed gel formulations of nimesulide. Ind Drugs 38:63–6.
- Kumar R, Katare OP. (2005). Lecithin organogels as a potential phospholipid-structured system for topical drug delivery: a review. AAPS PharmSciTech 6:E298–10.
- Kumar A, Pullakandam N, Prabu SL, Gopal V. (2010). Transdermal drug delivery system: an overview. Int J Pharm Sci Rev Res 3:49–54.
- Murthy TEGK, Kishore VS. (2008). Formulation and evaluation of transdermal gels of diltiazem hydrochloride. Ind J Pharm Educ Res 42:272–6.
- Mutimer MN, Riffikin C, Hill JA, et al. (1956). Synthesis of methylsilyl derivates of procaine and their diffusion. J Am Pharm Assoc Sci 45:212.
- Nemutlu E. (2005). Determination of lornoxicam in pharmaceutical preparations by zero and first order derivative UV spectrophotometric methods. Pharmazie 60:421–5.
- Pandey M, Belgamwar V, Gattani S, et al. (2010). Pluronic lecithin organogel as a topical drug delivery system. Drug Deliv 17:38–47.
- Rohit Rao C, Krishna G. (2006). Effect of captopril and losartan on thermal and chemical induced pain in mice. Ind J Physiol Pharmacol 50:169–74.
- Scartazzini R, Luisi PL. (1988). Organogels from lecithins. J Phys Chem 92:829–33.
- Schmid MH, Korting HC. (1994). Liposomes: a drug carrier system for topical treatment in dermatology. Crit Rev Ther Drug Carrier Syst 11:97–18.
- Schurtenberger P, Scartazzini R, Magid LJ, et al. (1990). Structural and dynamic properties of polymer-like reverse micelles. J Phys Chem 94:3695–701.
- Singh S. (2009). Enhanced transdermal delivery of ketoprofen from bioadhesive gels. Pak J Pharm Sci 22:193–8.
- Umamaheswari RB, Jain P, Jain NK. (2002). Hydrogel – a novel drug delivery systems. Ind Drugs 39:243–56.
- Vintiloiu A, Leroux JC. (2007). Organogels and their use in drug delivery – a review. J Control Release 125:179–92.