
Abstract
Self-emulsifying drug delivery system (SEDDS) is an isotropic mixture of lipid, surfactant and co-surfactant, which forms a fine emulsion when comes in contact of an aqueous medium with mild agitation. SEDDS is considered as a potential platform for oral delivery of hydrophobic drug in order to overcome their poor and irregular bioavailability challenges. In spite of fewer advantages like improved solubility of drug, bypassing lymphatic transport etc., SEDDS faces different controversial issues such as the use of appropriate terminology (self-microemulsifying drug delivery system; SMEDDS or self-nanoemulsifying drug delivery system; SNEDDS), presence of high amount of surfactant, correlation of in vitro model to in vivo studies, lack of human volunteer study and effect of conversion of SEDDS to final administrable dosage form on pharmacokinetic behavior of the drug. In this review, potential issues or questions on SEDDS are identified and summarized from the pharmacokinetic point of view. Primarily this review includes the conflict between the influences of droplet size, variation in correlation between in vitro lipolysis or ex-vivo intestinal permeation and pharmacokinetic parameters, variation in in vivo results of solid and liquid SEDDS, and potential challenges or limitation of pharmacokinetic studies on human volunteers with orally administered SEDDS. In the past decades, hundreds of in vivo studies on SEDDS have been published. In the present study, only the relevant article on in vivo pharmacokinetic studies with orally administered SEDDS published in past 5–6 years are analyzed for an up to date compilation.
Introduction
Poor bioavailability is a major challenge to formulate an oral dosage form. Poor aqueous solubility is one of the important underlying factors of bioavailability because a drug cannot be absorbed through the gastrointestinal tract unless it is in solution state. Many chemical entities with significant and promising pharmacological effect suffer from poor aqueous solubility. It is stated earlier that over 30% of the most commonly marketed drug and nearly half of the new drug entities reaching to the formulation scientists are hydrophobic in nature or lacking required aqueous solubility (Hauss, Citation2007; Tang et al., Citation2008). As per the BCS (biopharmaceutical classification system), class II drugs bear poor solubility but high permeability and class IV drugs bear poor solubility and poor permeability through the gastrointestinal membrane (Pouton, Citation2006). Both of these types of drugs show erratic absorption behavior with limited oral bioavailability. Several approaches have been adopted to improve the drug solubility and absorption such as modification of crystal habit, reduction of particle size, solid dispersions, solid solutions, salt formation, and miscellaneous methods, including supercritical fluid process and use of surfactant, solubilizers, cosolvency, hydrotrophy, and novel excipients as adjuvant to increase solubility (Savjani et al., Citation2012; Kyaw et al., Citation2015). Lipid-based drug delivery system has gained major attention in the past few decades with the target of bioavailability enhancement of poor water soluble or lipophilic drug administered orally (Gursoy & Benita, Citation2004). The main idea of this type of formulation is to keep the lipophilic drugs in dissolved state as a colloidal dispersion throughout the gastrointestinal tract. There are several forms of lipid-based delivery system such as solutions, suspensions, emulsions, microemulsions, self-emulsifying drug delivery systems (SEDDS) or dry emulsions (Pouton, Citation2000; Araya et al., Citation2005).
The potential of SEDDS for lipophilic compound was well understood long ago with the introduction of cyclosporine A by the pharmaceutical company, Sandoz (Germany) in the microemulsion form (Pouton, Citation1997). For the lipophilic compounds which exhibit dissolution rate limited absorption, SEDDS is gaining popularity to formulation scientists in order to render a reproducible drug–plasma concentration profile after oral administration since early 90s (Shah et al., Citation1994).
In the year 2000, Pouton introduced one useful lipid formulation classification system (LFCS), which was again updated by the same author in the year of 2006 (Pouton, Citation2000,Citation2006). As per the LFCS, SEDDS (type II) is an isotropic mixture of oil or mixture of oils and surfactant/s. When it comes in contact with the aqueous gastric medium, upon slight agitation spontaneously forms oil in water emulsion. Hence, the term comes as self-emulsifying. This SEDDS system was further modified to self-microemulsifying (SMEDDS) or self-nanoemulsifying (SNEDDS) drug delivery system classified as type IIIa or IIIb, which additionally contain one or more co-surfactant or hydrophilic co-solvents. SEDDS can be prepared by empirical way of “trial–error” method altering the components’ ratio or by the application of ternary phase diagram (Balakrishnan et al., Citation2009; Do et al., Citation2011). But, formulation development by the two methods is subjected to a large number of experiments. Recently a HLB coupled to response surface methodology (RSM) approach is developed by Bahloul et al. (Citation2014), which may reduce the number of trials to develop and optimize the SEDDS with desired drug release and satisfactory pharmacokinetic parameters (Bahloul et al., Citation2014,Citation2015).
In a review Kohli et al. (Citation2010) summarized the published reports on various types of SEDDS emphasizing the formulation aspects. They have also discussed the regulatory aspects of oil components highlighting the lipid formulation classification model that was developed by Pouton (Citation2000) (Kohli et al., Citation2010). Since the last decade with the advancement of SEDDS, in vivo studies to evaluate pharmacokinetics parameters are going on in order to establish the applicability of this delivery system.
However, outcomes of these pharmacokinetic studies have been rarely summarized and addressed in recent times. In a nice scientific description, Cherniakov et al. (Citation2015) discussed the biopharmaceutical points of SEDDS after its oral administration. The review was primarily focused on the absorption, bioavailability and disposition of drugs delivered by oral SMEDDS/SNEDDS (Cherniakov et al., Citation2015). Apart from this there is no compilation of reported pharmacokinetic results of this delivery system in recent times. Surmounting the gap between in vitro study and pharmacokinetic studies in different models is a kind of absolute requirement for the development of a drug delivery system. Therefore, this review deals with an extensive account and analysis of published in vivo pharmacokinetic studies on SMEDDS/SNEDDS in last 5–6 years, together with the inherent controversies or questions on the delivery system arising from those studies. From the countless researches published on SEDDS, we have identified the following controversial issues or confusions:
The confusion between the terminologies; SMEDDS or SNEDDS (Anton & Vandamme, Citation2011).
Importance of droplet size difference from the pharmacokinetic aspect (Pouton, Citation2006).
Correlation of in vitro studies by different models with in vivo drug absorption.
Variation in pharmacokinetic parameters between different final dosage forms of SEDDS after oral administration.
Few of these issues have been discussed in different article, but these have never been addressed together (Pouton, Citation2006; Anton & VanDamme, Citation2011). The controversy with pharmacokinetic parameters obtained after orally administered “final dosage form; solid SEDDS or liquid SEDDS” is pointed out for the first time in this review. Instances of lack of correlation between the in vitro and in vivo results after oral administration of SEDDS have never been compiled from a pharmacokinetic point of view. From these aspects, objective of this review is to identify and analyze the potential controversies reported by pharmacokinetic studies of orally administered SEDDS.
General components of SMEDDS/SNEDDS and their role in absorption
Before we delve into the depth of the in vivo pharmacokinetic studies, a general idea of the components of SEDDS/SMEDDS/SNEDDS and their role on drug absorption is provided for better understanding of the main topic.
Main component or active pharmaceutical ingredient (API) for SMEDDS or SNEDDS must have sufficient lipophilicity and should have low dose. The drug must be sufficiently soluble in pharmaceutically acceptable lipids, surfactants and co-surfactants (Morozowich & Gao, Citation2009; Gao, Citation2012). Apart from the API, the SMEDDS or SNEDDS contains three major components; lipids or oil, surfactants and co-surfactants (). There are different well-cited descriptions of the components (Müllertz et al., Citation2010).
Figure 1. Different components of self-emulsifying drug delivery system.
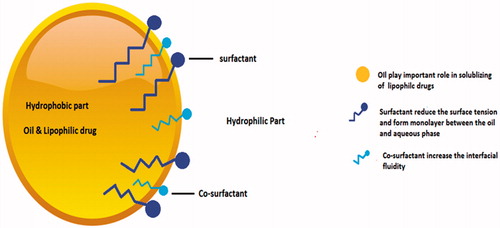
Lipids
Oil (sometimes a combination of different oils) with maximum ability to solubilize a specific drug usually is chosen as a lipid component to form SEDDS (Kang et al., Citation2004; Heshmati et al., Citation2014), but this is not always the case. Larsen et al (Citation2013a) showed that the SEDDS with a lowest oil solubility resulted in highest bioavailability, which indicates that high solubility of drug may not always be a good optimization parameter for better in vivo performance and should not be used alone.
Oil usually has two major positive effects on bioavailability enhancement which includes drug solubility enhancement and favorable effect on lymphatic transport. To explain the in vivo solubility enhancement of a hydrophobic drug in the intestine, the lipid digestion behavior of the small intestine must be considered. The secretion of cholesterol and bile salts in the duodenum is stimulated by the presence of lipid and its digested product, which again forms micelle (Porter et al., Citation2007). The polar groups of micelle remain at the aqueous side while the hydrophobic groups remain at the core. When a poor soluble drug along with some lipid component (oil) is delivered into the duodenum, the miceller solubilization process takes place. This results in the entrapment of drug into colloidal micelle and enhancement of effective solubility ().
Figure 2. Miceller solubilization of hydrophobic drug in the duodenum.
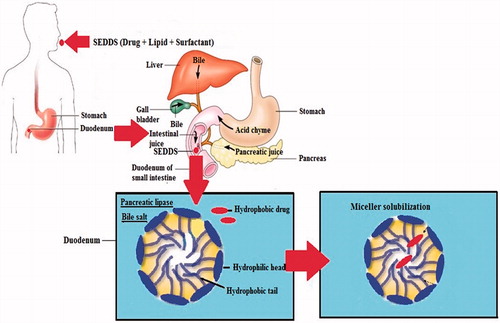
Another important aspect of SEDDS is its potential to be absorbed via the lymphatic system, bypassing the hepatic first pass metabolism (). The formation of lipoprotein induces lymphatic transport for the highly lipophilic drug. The compounds processed by intestinal lymph are transported to the systemic circulation in association with the lipid core of lipoprotein (Kollipara & Gandhi, Citation2014). Among the lipids used in SEDDS, long-chain fatty acids are converted to triglyceride by re-esterification in the small intestine and incorporated into chylomicron, a large lipoprotein, followed by secretion into the lymph vessel by exocytosis.
Figure 3. Mechanism of lymphatic transport from intestine.
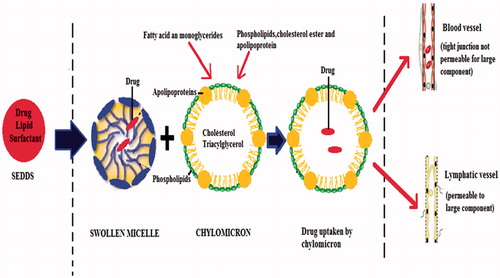
Large chylomicron cannot cross tight junction of blood capillaries, whereas it can easily penetrate to lymphatic junction. By this way drugs delivered by SEDDS may bypass first pass metabolism. All these abovementioned mechanisms are well-depicted in the review published by Cherniakov et al. (Citation2015).
Oils with long hydrocarbon chains or long-chain triglycerides (LCT) have a great ability to increase drug transport by the lymphatic system in GIT, however sometimes they are difficult to nanoemulsify (Chen, Citation2008). On the other hand, medium and short chain oils or triglycerides (MCT or SCT) can easily form nanoemulsion but they do not have a great ability to increase the lymphatic transport of drugs. Thus, a mixture of oil phase can be considered to optimize the SEDDS formulation and result in better pharmacokinetics.
The natural digestible oils with modified LCT and MCT, for example, Olive oil, Castor oil, Peanut oil, Coconut oil, Palm oil and the semisynthetic derivatives, for example, Labrasol, Marlower R40, Caprol ET, Capmul MCM, Labrafac CC, etc., are among the most commonly used lipid component (Chambin & Jannin, Citation2005; Porter et al., Citation2007; Tang et al., Citation2007).
Surfactant
The second obligatory component in SEDDS is surfactants which are amphiphilic molecules with a hydrophilic head and a hydrophobic tail, as illustrated in . These surfactants are added to SEDDS formulation due to their ability to reduce the surface tension and form monolayer between the oil and aqueous phase. By this method, the SEDDS become more stable and stay in GIT for longer time in soluble form, which is useful for the absorption of the drug.
The maximum solubility of the drug in surfactant and the hydrophilic–lipophilic balance (HLB) are two major considerations that must be taken when choosing surfactant in SEDDS. Both HLB values and the concentration of surfactant have the major impact on the emulsion droplet size. The droplet size of the emulsions decreased and then increased with increasing surfactant concentration because at the beginning the amount of adsorbed surfactant around the oil–water interface of a droplet increases, resulting in decreasing the interfacial tension of the system and form fine droplets. But, when the concentration of surfactant increases, excess water penetrates into the bulk oil causing massive interfacial disruption and ejection of droplets into the bulk aqueous phase (An et al., Citation2014).
The nonionic surfactants with relatively high HLB values are the most widely recommended surfactant to be used in the SEDDS, such as Sorbitan ester and Ethoxyl esters of fatty acids and Lecithin with HLB values ranging between 4 and 15, for example, Brij-30, Labrafac CM 10, Tween 20, Tween-80, Cremophor RH40, Pluronic L-64 and Emulphor El-620 (Fernandez-Tarrio et al., Citation2008; Sweetman, Citation2009). Other than fine globule formation, inhibition of the efflux transporter by surfactants is considered as a contributing factor to enhance bioavailability by minimizing the chance of efflux back of the compound to the intestinal lumen. Some nonionic surfactants like Cremophore EL, Cremophore RH40, Span, Tween are reported to inhibit P-gp (P-glycoprotein), which is one of the commonly known efflux transporters (Zhang et al., Citation2003; Katneni et al., Citation2007), but the theory of stimulation of uptake transporter has also been demonstrated by Engel et al. (Citation2012), where the author described the stimulation of OATP family of uptake transporter by some surfactants. The mechanism may be more beneficial for BCS IV-type drugs delivered via oral SEDDS, which are lacking of permeability than BCS II drugs.
Co-surfactant
Co-surfactant mainly helps increase the drug load capacity, resulting in the reduction of the surfactant concentration when it is added to the formulation. The addition of co-surfactant reduces chances of variability and local irritancy caused by the surfactants by increasing the interfacial fluidity (Pouton, Citation2006). It also facilitates the dispersion process in the medium. Commonly used co-surfactants include ethanol, propylene glycol and other newer co-surfactants such as transcutol P and glycofurol, etc (Kale & Patravale, Citation2008, Borhade et al., Citation2009).
Terminology of the delivery system: SMEDDS or SNEDDS?
With the advancement of pharmaceutical researches since the last decade, countless formulation development works have been reported based on the SEDDS system. Some researchers clearly defined their system as either SMEDDS or SNEDDS, whereas some researchers used both SEDDS and SMEDDS as broad terminology. The dilemma begins at this stage. As per some researchers, SEDDS comprises of two systems as SMEDDS (oil droplet size 100–250 nm) and SNEDDS (oil droplet size <100 nm) that can be differentiated based on the emulsion droplet size only (Kohli et al., Citation2010). Each definition of SMEDDS differs from SEDDS in the size of emulsion droplets produced by dilution in an aqueous medium, which are considered lower in the former resulting in a transparent or translucent emulsion (Cannon, Citation2011; Oh et al., Citation2011). Also, the composition of SMEDDS may contain co-solvent (s) which may not be present in SEDDS. Microemulsion by definition is a thermodynamically stable system that forms spontaneously with the involvement of physicochemical variables, mainly temperature and pH. However, the microemulsion formed in in-vivo from SMEDDS is thermodynamically unstable and may require some energy that might be obtained from gastric mobility for emulsification. According to some researchers, type IIIb formulations (as per the LFCS) are termed as SNEDDS as these are thermodynamically unstable, but kinetically stable system (Suresh & Sharma, Citation2011). The characteristic difference between SEDDS, SMEDDS and SNEDDS are well-described by Čerpnjak et al. (Citation2013). For authors, it is better to use SNEDDS terminology instead of SMEDDS as the emulsion generated spontaneously after aqueous dilution is not thermodynamically stable and resembles more to nanoemulsion. The same argument has been earlier expressed by Anton & Vandamme (Citation2011), where the authors concluded that self-emulsifying systems used for drug delivery are mostly SNEDDS, but they should not be classified based on the mean emulsion droplet diameter only. It is to note that the order of mixing the ingredients during preparation of the emulsion is the first basis of differentiation between nanoemulsion and microemulsion. Nanoemulsion forms only when the surfactant is first mixed with oil before adding water. On the contrary, if surfactant is mixed with water first, then it forms “macroscopic” emulsion. This order of addition is not so important for microemulsion (Anton & VanDamme, Citation2011). Taking the droplet size into consideration for “micro” or “nano” SEDDS also creates confusion because the very low droplet size of a SEDDS was sometimes referred as “micro” emulsifying by the researchers and on the other hand comparatively higher droplet size of a SEDDS was reported as SNEDDS. Few examples of terminology used and respective emulsion droplet size are summarized in .
Table 1. Terminology used for SEDDS with respect to droplet size.
This confusion of terminology still remains as both SMEDDS and SNEDDS systems are reported continuously by different researchers. We assume mainly two reasons behind such contradiction: one is structural and visual similarity along with similar modes of characterization and the other is the view of pharmaceutical researchers are different from the view of a physicist with respect to the exact physicochemical classification of a system. But, more attention is required to overcome this ambiguity. In this review, we have referred SMEDDS or SNEDDS specifically as used by different researchers in their respective reports. When the term “SEDDS” is used alone, in this review that refers both SMEDDS and SNEDDS in a broad sense.
Importance of emulsion droplet size on drug absorption
Most of the articles on SMEDDS or SNEDDS highlighted the nano-scale dispersion as one of the major contributing factors behind improved drug absorption after oral administration compared to either normal solution or suspension (Zhu et al., Citation2013; Li et al., Citation2015). However, comparative in vivo studies with variable droplet size with the same SEDDS have been rarely reported. Tarr & Yalkowsky (Citation1989) reported the nano-scale formation of Cyclosporine globule in emulsion produced better absorption after oral administration in rats compared to larger size globules. The point to be noted is there was no difference in formulation other than the preparation method. So, the influencing factor for different levels of bioavailability in different microemulsions was the globule size (Tarr & Yalkowsky, Citation1989). The same observation was supported by de Smidt et al. (Citation2004) by proving better oral bioavailability for Penclomedine in rat from an emulsion with a droplet size about ∼160 nm compared to another emulsion with droplet size of about ∼720 nm. Although there was difference in the oil ratio in two formulations, the difference in bioavailability with respect to the droplet size was significant. Hence, it is commonly considered that the formation of nano-scale dispersion significantly affects drug absorption, but it will not be very wise to generalize that the lower droplet size of emulsion will lead to higher bioavailability. As depicted in a study by Yap & Yuen (Citation2004) with two Tocotrienol SEDDS, one is very susceptible to lipolysis (SES-A) and another with smaller emulsion droplet size, but less susceptible to lipolysis (SES-B), both achieved a faster onset of action as a normal tocotrienol oil solution. Both SES-A and SES-B resulted in the same relative bioavailability despite the fact that SES-B could form an emulsion with lower droplet size. It is worth mentioning that both of these formulations resulted faster onset of action and higher relative oral bioavailability than the normal drug in an oil solution (Yap & Yuen, Citation2004).
In another study, difference in initial emulsion droplet size did not affect the absorption of Halofantrine significantly when formulated with a medium chain triglyceride (MCT) as SEDDS or SMEDDS (Khoo et al., Citation1998). Similarly, droplet size served as a poor indicator of in vivo absorption rate of Atorvaquone from emulsions containing different surfactants where all formulations showed non-significant difference in in vivo results in spite of the different droplet sizes (Sek et al., Citation2006).
As soon as the dispersed formulation leaves the stomach, it encounters the digestive power of the enzymes present in the small intestine. At that point, fate of drug is more important rather than initial droplet size (Pouton, Citation2006) because it not only had the esters that are rapidly hydrolyzed by pancreatic lipase, but also some commonly used surfactants (Ethoxylated esters). Therefore, the influence of droplet size of emulsion in an aqueous medium needs more in-depth study in accordance with pharmacokinetic parameters. To evaluate this, sufficient numbers of in vivo pharmacokinetic studies with the same formulation but, different droplet size are to be carried out.
Correlation between in vitro model and in vivo studies
Apart from the characterization of developed SEDDS such as measurement of particle size and zeta potential, emulsification time, in vitro release rate, etc. two other in vitro models are quite popular to the researchers in order to predict the in vivo behavior of the system delivered orally: in vitro lipolysis model (IVLM) and single pass intestinal permeation model (SPIP) (Kohli et al., Citation2010; Singh & Pai, Citation2015). The two models are diagrammatically explained by and . IVLM can be utilized to assess the lipid digestion rate by the intestinal enzyme and the drug precipitation in simulated intestinal medium, whereas SPIP is used to determine the drug permeation rate through the intestinal mucosa. The correlation of IVLM and SPIP results with in vivo solubilization or bioavailability from a SEDDS is well-established in many reports (Dahan & Hoffman, Citation2008; Gupta et al., Citation2011; Eedara et al., Citation2014). At the same time, contradiction between in vitro and in vivo model for SEDDS has also been reported in another study where the precipitation during in vitro lipolysis of a hydrophobic drug named E804 (Indirubin-3′oxime-2,3dihydroxypropyl ether) was not correlated with the results were obtained by in vivo studies (Heshmati et al., Citation2014). On the other hand, there are a number of studies using precipitation parameters to predict in vivo performance by measuring the maximum supersaturation ratio (SRM) as theoretical drug concentration without precipitation and drug solubility in the aqueous phase (Williams et al., Citation2013). The value of SRM > 2.5 indicates the precipitation is likely to occur, but SRM can reflect the in vivo performance in case of moderate drug load while in high drug load it failed to predict it in a dog model. In the subsequent discussion, we have explained few recent studies indicating the correlation and variation of these models when compared with in vivo pharmacokinetic studies.
Figure 4. In vitro lipolysis model for SEDDS.
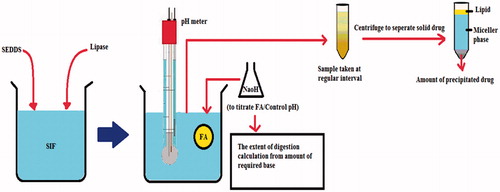
Figure 5. In situ single pass intestinal permeation (SPIP) model.
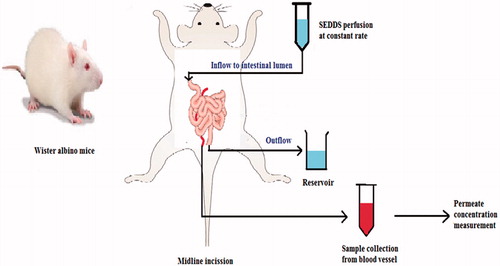
Trans-resveratrol (t-RVT) was formulated as SNEDDS using Lauroglycol FCC, a long-chain triglyceride and Transcutol P, a hydrophilic surfactant and was evaluated for in vivo parameters in rats as well as in situ intestinal permeability evaluation by the SPIP model (Singh & Pai, Citation2015). A “level A” correlation based on point-to-point analysis was possible for t-RVT SNEDDS formulation. According to the author’s view, an increase in Cmax (2.34 folds) and AUC (4.31 folds) by SNEDDS compared to the pure drug suspension was a result of enhanced intestinal permeation and bypassing hepatic first pass metabolism via lymphatic transport. Enhanced intestinal permeation was correlated with the enhanced absorption number from t-RVT SNEDDS in SPIP studies. An increase of absorption number over 3 folds by SNEDDS might be well-correlated with 4.31 folds AUC improvement. Presence of high amount of surfactant and smaller oil globule size (48–100 nm) was considered as the contributing factor for enhanced intestinal permeation. The higher value of in situ absorption parameters ascribes to higher chances of lymphatic transport of drug (Sun et al., Citation2011).
In ex vivo permeation studies, the effective permeability provides direct measures of absorption through the intestinal membrane, as explained by Bandyopadhyay et al. (Citation2012). These researchers formulated and optimized Ezetimibe (EZ), a BCS II drug in two different SNEDDS (globule size: 54 – 66 nm) using Labrasol (surfactant) – Maisine (LCT) and Tween 80 (surfactant) – Capryol (MCT), respectively. A comparative ex vivo permeation study correlated with a pharmacodynamic study established that EZ-LCT provided better absorption in terms of absorption number and permeability efficiency, resulting in better bioavailability than EZ-MCT formulation. The presence of Labrasol enhances paracellular transport of drug (Sha et al., Citation2005) and simultaneously Maisine surmounts hepatic metabolism by lymphatic transport (Khoo et al., Citation1998). Both EZ-LCT and EZ-MCT SNEDDS produced better bioavailability and pharmacodynamic effect compared to marketed formulation.
Another comparison was done in the same study by using an EZ-hydroxy propyl β cyclodextrin (HP-β CD) complex, which is used as a solubilizing approach for poor soluble drug (Bandyopadhyay et al., Citation2012). But, in terms of pharmacodynamic effect, the EZ- HP-β CD complex did not produce significant improvement, which means the reason of bioavailability enhancement in SNEDDS is not only due to the enhancement of drug solubility. Rather, it includes a combined mechanism of presentation of the drug in solubilized form, increase interfacial area of drug absorption, presence of emulgent resulting in increased dissolution, enhanced intestinal permeation, etc. A BCS II drug E804 (Indirubin-3′oxime-2,3 dihydroxypropyl ether) was shown to be absorbed faster (tmax < 60 min) from oral MC-SNEDDS (medium chain) (droplet size < 16.8 ± 5.3 nm) administered in rats compared to LC-SEDDS (long chain) or MC-SEDDS (droplet size < 248 ± 20 nm) (Heshmati et al., Citation2014). As per the author, the MCTs are hydrolyzed easily in intestine resulting in faster drug absorption. But, the LC-SEDDS showed the lowest degree of precipitation in in vitro lipolysis than others. A contributing factor behind such behavior might be the presence of long-chain mono and di glycerides (Peceol) in the LC-SEDDS, which enable the micelle to keep the drug in the solution state longer than the MC-SNEDDS containing medium chain glycerides (Capmul MCM). The result of this study proves that in vitro lipolysis may serve as a tool for understanding lipid digestion behavior, but it may not correlate with the in vivo performance of the drug.
The IVLM does not consider the fact that prior to entering the intestine the formulation needs to cross a lower acidic pH region at stomach. The fate of the drug and behavior of the SNEDDS at the stomach could not be encountered in this model. This has been evidenced and well-explained by Larsen et al. (Citation2013b) by their extensive in vitro as well as in vivo study on dogs for Cinnarizine after administering SNEDDS with three levels of drug loading. Better pharmacokinetic parameters like AUC0–48 h (355–488 ng/ml h−1), Cmax (88–115 ng/ml) and tmax (1.8–3 h) were obtained from all SNEDDS compared to Cinnarizine suspension. The interesting thing is: there were no significant differences among different drug-loaded SNEDDS (the researchers prepared SNEDDS with 50, 25 and 12.5 mg/g of Cinnarizine) despite the fact that in vitro lipolysis resulted in a substantial amount of Cinnarizine precipitation from the SNEDDS with highest drug loading. This report again satisfies the consideration that in vitro lipolysis should not be a surrogate of the in vivo study. Cinnarizine is not a substrate for p-glycoprotein transporter. Therefore, low proportion of vehicle in higher drug loading is not ascribed to same bioavailability with low drug loading. Similar type of observation was reported by Holm et al. (Citation2012), where same dose of Halofantrine were given to rats by high (56.7%) and low (5.67%) drug loaded SNEDDS. They reported no significant differences in bioavailability between two different drug loading in spite of bearing substantial difference (10 times) in excipients or vehicle. Further report also suggests, it is not always possible to get better in vivo performance after oral administration from that SNEDDS which gives less precipitation by in vitro lipolysis. A study by Thomas et al. (Citation2012) depicted two different aspects of comparison regarding SNEDDS: difference in oil component (MCT and LCT) and the presence of a polymer resulting in super saturated SNEDDS. Taking Halofantrine as an API, the researchers developed the MC-SNEDDS (medium chain lipids derived from Coconut oil) and LC-SNEDDS (Soyabean oil, Maisine) and super saturated form of each SNEDDS termed as MC-super-SNEDDS and LC-super-SNEDDS. All formulations have an average droplet size of 33–60 nm. In vivo studies of MC and LC-SNEDDS in dog resulted higher absolute bioavailability in LC-SNEDDS (73.3%) and LC-super-SNEDDS (90.9%) compared to those in MC-SNEDDS (41.0%) and MC-super-SNEDDS (53.2%). This result is expected as per the results of their IVLM study and can be explained by better solubility of Halofantrine by LC-lipids and their digestion product, possible lymphatic uptake of Halofantrine and secretion of bile acid stimulation promoting drug solubilization. Interesting fact is that during in vitro digestion, Halofantrine from MC-super-SNEDDS and LC-super-SNEDDS produced rapid precipitation leading to similar drug concentration in the lipolysis medium like MC-SNEDDS and LC-SNEDDS, which similarities were not reflected in the in vivo study. LC-super-SNEDDS and MC-super-SNEDDS resulted in 10–20% higher absolute bioavailability than LC-SNEDDS and MC-SNEDDS. Their data suggested that the increased dissolution rate for the precipitation of the drug in amorphous form is the driving force for absorption. This report is also an account of dissimilarity between in vitro lipolysis model and in vivo results.
Significant variation in in vivo study from in vitro release was reported by Sangsen et al. (Citation2016). The author described that the type and quantity of the surfactant system did not produce significant alteration in droplet size and in vitro drug release. They formulated Oxyresveratrol as SMEDDS containing Capryol 90 (oil), Cremophore RH40 (surfactant) and Tween 80 (high amount: HT; low amount: LT) or Labrasol (high amount: HL; low amount: LL) (co-surfactant) with varying ratio and amount (Sangsen et al. Citation2016). But, the in vivo pharmacokinetic studies showed that oral SMEDDS containing LT and HT resulted in better (1.8–7.9 folds) relative bioavailability considering unformulated Oxyresveratrol as reference compared to the same proportion of LL and HL (1.4–2.2 folds), respectively. On the other hand, SMEDDS containing higher amount of surfactant:co surfactant (HT: 7.9 times; HL: 2.2 times) resulted in better relative bioavailability than a lower amount (LT: 1.8 times; LL: 1.4 times), which could be correlated to the enhanced in vitro caco-2 cell permeability. However, Cmax of LL (0.64 μg/ml) and HL (0.86 μg/ml) were not significantly different from unformulated drug (0.66 μg/ml). On the contrary, Cmax of LT (0.96 μg/ml) and HT (2.31 μg/ml) were significantly higher, but this difference was not correlated with the caco-2 cell permeability study. Better in vivo absorption in the presence of Tween 80 ascribed to the possible inhibition of efflux transport and increased lymphatic transport. In another research, the presence of higher concentration of Tween 80 (56.4%) and lower concentration of oil (Caprylic acid; 20%) in a SEDDS of Nevirapine resulted in better ex vivo intestinal permeability as well as better Cmax and AUC0–t compared to the SEDDS containing lower Tween 80 (46.8%) and higher oil (20%), although both formulations resulted better oral bioavailability in rats than marketed suspension (Chudasama et al., Citation2015). From all these discussed studies, it can be concluded that the in vitro models, undoubtedly can give idea about drug absorption through the intestine, but in vivo studies are essential to find out the exact outcome of the delivery system.
Effect of final dosage forms (liquid and solid) of SEDDS/SMEDDS/SNEDDS on pharmacokinetic parameters
The conventional form of SEDDS is liquid and administered in soft gelatin capsule that results in several drawbacks, such as high production cost, low portability, low drug loading and low stability that may lead to precipitation of the drug due to the interaction between volatile co-solvents and soft gelatin capsule shells (Prajapati & Patel, Citation2007). To overcome this limitation, solid SEDDS are developed by converting the liquid or semisolid formulation into powders by using different techniques like spray drying (Yi et al., Citation2008) or using porous carriers such as cross-linked porous silicon dioxide, magnesium aluminum silicate and microporous calcium silicate that adsorb the liquid and transfer it to solid SEDDS (Uchino et al., Citation2007; Kang et al., Citation2011). Then, the adsorbed SNEDDS can be formulated into free-flowing powders, granules, pellets, tablets, solid dispersions, microspheres and nanoparticles. Solid SNEDDS disintegrate first and then form fine oil in water emulsion inside the GIT when the formulation is exposed to gastrointestinal fluid with slight agitation due to normal mobility. That means it combines the advantages of liquid emulsions and solid dosage forms (Tang et al., Citation2008). Once disintegrated, the fate of the solid SNEDDS will be same as liquid SNEDDS. Emulsion droplet size should also be the same for both types of delivery system. The in vitro release might be a little slower in case of solid SNEDDS than liquid due to the presence of some extra carrier, which in turn may cause longer tmax in the in vivo study. Regarding the in vivo physicochemical changes of the SMEDDS or SNEDDS delivered orally, it could be assumed that the change of liquid form (L-SNEDDS or L-SMEDDS) to solid (S-SNEDDS or S-SMEDDS) does not possess any significant differences in terms of pharmacokinetic parameters, especially the Cmax or bioavialability. Same degree of in vivo absorption of Lopinavir after oral administration of liquid and solid SNEDDS were reported in terms of equivalent Cmax, tmax and AUC0–48 h values (Garg et al., Citation2016). In that study there was no difference in in vitro characterization parameters like droplet size after emulsification and in vitro dissolution rate between the liquid and solid dosage forms.
However, based on the findings of our analysis of different published articles, we argue that conversion of liquid-to-solid final dosage form of SEDDS may generate different results in pharmacokinetic studies. A few reports have been published describing comparative evaluation of in vivo parameters between liquid and solid SMEDDS or SNEDDS showing variation in pharmacokinetics parameters. Unfortunately, most of these articles lack explanation for such kind of variation.
Darunavir, a poorly soluble drug was formulated as liquid SNEDDS (L-SNEDDS) containing Capmul MCM (a medium chain triglyceride) followed by conversion to a solid powder (S-SNEDDS) (Inugala et al., Citation2015). The in vivo studies showed ∼2–2.5 times higher Cmax and 2–3 times higher AUC0–α values obtained after oral administration of L-SNEDDS and S-SNEDDS to Wistar albino rats compared to normal drug suspension. The initial rate of Darunavir absorption was higher in the case of L-SNEDDS, but the AUC0–α value was higher in the case of S-SNEDDS (32.28 ± 2.5 μg.h/ml) than L-SNEDDS (22.9 ± 2.1 μg.h/ml). The initial higher rate of absorption could be explained by the presence of formulation in liquid state at the absorption site. But, the reason behind higher extent of absorption and bioavailability from S-SNEDDS compared to L-SNEDDS is not clear from the authors view other than general hypothesized mechanisms of improved pharmacokinetics from SNEDDS that have been already explained in this review earlier.
In another study, a different dosage form of the same SNEDDS containing Olmesartan medoxomil as a drug, Oleic acid (oil) and Tween 20 (surfactant) showed almost same pharmacokinetics parameters by in vivo study carried out in Wistar albino rats (Beg et al., Citation2016). The liquid form (L-SNEDDS) with mean droplet size <100 nm and solid form (S-SNEDDS) both have shown same release kinetics in in vitro release study, which was reflected in the in vivo study. It is to be noted that although the authors claimed no significant differences in pharmacokinetic parameters between L-SNEDDS and S-SNEDDS, the L-SNEDDS resulted in better Cmax (646.2 μg/ml) and AUClast (4063.3 μg.h/ml) than S-SNEDDS (Cmax: 586.2 μg/ml; AUClast: 3283.2 μg.h/ml). It is worth mentioning that both dosage forms possessed 2.5–4 folds of increase in Cmax and AUC0–last than pure drug in solution, and a good IVIVC was established between in vitro drug release percentage and in vivo drug absorbed as per linear model fitting for both types of dosage forms. Conversion of liquid SMEDDS (L-SMEDDS) consisting capryol 90 (oil), Cremophore RH40 (surfactant) and Labrasol (co-surfactant) to solid pelletized (P-SMEDDS) dosage form was done by Sermkaew et al. (Citation2013) to deliver andrographolide orally. Both L-SMEDDS and P-SMEDDS showed higher rates of in vivo absorption in rabbit after oral administration compared to the solution form. Apart from the enhanced solubilization capacity, the role of Labrasol in weakening the tight junction of paracellular membrane might be the contributing factor of improved pharmacokinetic parameters from SMEDDS. However, in this research no significant differences in Cmax and AUC0–12 were reported between L-SMEDDS and P-SMEDDS. But, the P-SMEDDS showed a lower degree of increment in Cmax and AUC0–12 (5 folds and 13 folds) than the L-SMEDDS (6 folds and 15 folds) compared to the Andrographolide liquid extract. No specific assumption can be ascribed from this difference. In general, liquid formulation results in better absorption than solid formulation, but in SEDDS once the delivery system disintegrates and the components are released, there should not be theoretically any differences in the fate of the drug.
The solid form of SMEDDS was shown to result in longer tmax and half-life when administered orally compared to liquid SMEDDS and suspension (Cheng et al., Citation2015). The same was evidenced in a study by Christiansen et al. (Citation2014), where Cinnarizine showed lesser oral bioavailability in beagle dogs from solid SNEDDS than liquid SNEDDS filled into capsules. The in vivo results of that study were in good agreement with the in vitro dissolution rate. In order to overcome poor bioavailability of puerarin, a natural alkaloid, Cheng et al. (Citation2015) developed a L-SMEDDS containing Castor oil (oil), Cremophore RH 40(surfactant) and Propanediol (co-surfactant) followed by conversion to S-SMEDDS. The in vivo study resulted in 20–30 folds and 1.9–2.5 folds increase of Cmax and AUC compared to Purerain suspension administered orally in rats. Absorption through lymphatic pathway from SMEDDS is the responsible factor behind such increase as Purearin faces irregular absorption and fast hepatic metabolism. Although the AUC0–t was same for Puerarin from L-SMEDDS and S-SMEDDS, the t1/2 and tmax of the drug was 17.9 h and 2 h from the S-SMEDDS compared to 9.4 h and 0.5 h from L-SMEDDS, respectively. The solid form of SMEDDS had a significant effect on lengthening the duration of action and reducing the elimination rate. At this point, the difference in the in vivo rate of absorption can be correlated to the in vitro drug release study where L-SMEDDS showed drug >80% within 10 min and S-SMEDDS showed 10% drug release within 30 min. This dissolution rate limited absorption is the factor behind longer duration of action from S-SMEDDS. However, a pharmacodynamic study is required to establish the onset and duration of action from such kind of formulation. Most reported studies on pharmacokinetic analysis with orally delivered solid SMEDDS/SNEDDS are summarized in .
Table 2. Comparative pharmacokinetic parameters of liquid and solid SMEDDS/SNEDDS.
In vivo studies on human volunteers
There are hundreds of published research on in vivo studies with SEDDS or SMEDDS or SNEDDS in animals like rat, dog or rabbits, but pharmacokinetic studies on human volunteers are a few reported in recent times. In one of such few studies, Fenofibrate was administered in fasted human volunteer as liquid SMEDDS containing Tween80 (L-SMEDDS) and liquid SMEDDS filled capsule that differs in surfactant type; Tween 80 (SMEDDS80) and Tween20 (SMEDDS20) and compared against the marketed capsule (Fei et al., Citation2013). All SMEDDS formulations resulted in higher AUC0–α (1.7–2.1 times) and Cmax (1.5–2.5 times) compared to marketed capsule. Time taken by SMEDDS20 to reach the maximum plasma concentration (tmax) was the longest among all formulations. This was due to slow absorption rate. Otherwise, all pharmacokinetic parameters were well correlated with the in vitro results which indicate that the drug dissolution, the solubilization capacity and the precipitation property are the contributing factors behind the high AUC0–α and Cmax and the short tmax values derived from L-SMEDDS. But, if a drug is highly susceptible to efflux mechanism of intestinal efflux transporter, then the higher rate of dissolution may not contribute to the betterment of bioavailability.
A recent comparative pharmacokinetic study between spray-dried solid SMEDDS containing Finasteride and marketed tablet after oral administration in human volunteer has revealed the lower bioavailability of Finasteride from marketed tablet despite its excellent in vitro dissolution profile (Fagir et al., Citation2015). The result showed a relative bioavailability of 129.35% with longer tmax, mean residence time (MRT) and longer elimination half-life indicating longer duration of action by the SMEDDS formulation. The lower bioavailability of Finasteride from marketed tablet may be attributed to the efflux action by P-gp transporter. It can be explained that the enhancement of bioavailability by SMEDDS was due to the combined mechanism of good solubilization, possible lymphatic transport and inhibition of the efflux transporter by the components, Tween 80 and Labrasol. Christiansen et al. (Citation2016) have evaluated the effect of placebo SNEDDS in Cinnarizine bioavailability and gastric emptying time following administration of a Cinnarizine tablet with placebo SNEDDS (C-SNEDDS) in human volunteers in the fasted and fed state compared to a commercial Cinnarizine tablet without SNEDDS (C-TAB). From the pharmacokinetic studies, it was observed that C-SMEDDS, which resulted in lower Cmax than C-TAB in the fed state. The components of SMEDDS potentially incorporated the drug in in vivo medium and exhibited different digestion and absorption profile from C-TAB. The total absorption of Cinnarizine from C-SNEDDS in the fasted and fed state was not significantly different as evidenced by the study. Also, as per Cmax value, it is not beneficial to administer a placebo SNEDDS with Cinnarizine tablet (Christiansen et al., Citation2016). But, to draw any firm conclusion, more studies with higher number of subjects are necessary. The possible reasons of lesser number of pharmacokinetic studies of SMEDDS or SNEDDS in human volunteers are discussed in the subsequent section.
Limitations of SEDDS with respect to pharmacokinetic studies
SEDDS is associated with many advantages, but it has fewer limitations nevertheless. Formulation- and characterization-related issues include lack of robust in vitro model, instability of the system, the oxidation potential of lipid component, problem with encapsulation, etc. (Wasylaschuk et al., Citation2007; Patil et al., Citation2007). Apart from these, there are few other issues which limit the scope and outcome of in vivo studies or pharmacokinetics studies, especially in human volunteers.
Absorption through lymphatic system
Absorption through lymphatic system is beneficial, especially to those molecules susceptible to high first pass metabolism. SEDDS is one of the potential candidates to the formulation scientist for lymphatic absorption. Simple mechanism illustrated earlier () is that the drug in association with lipid moiety forms chylomicron, a large lipoprotein that is absorbed through lymphatic duct but not through blood vessel due to the large size. The association of drug into chylomicron is critically the rate-limiting step of ensuring intestinal lymphatic uptake (O’Driscoll, Citation2002). The presence of intracellular drug and the amount of chylomicron in the enterocyte can affect the lymphatic uptake, but lymphatic uptake suffers from high variability from drug to drug as well as different physiological nature of the individual. Occurrence of lymphatic transport even to a small extent of less than 1% of the orally administered dose can significantly affect the plasma concentration of the drug. Therefore, lipophilicity and triglyceride solubility of the drug with respect to lymphatic transport needs in-depth study which requires a robust predictive in vitro or in vivo model (Trevaskis et al., Citation2008).
Safety issues with components
The use of lipid and surfactant as components of a SEDDS needs major attention with respect to safety after the administration. First, the ratio of these two components in a SEDDS usually is very high and second, due to the complexity of properties, these molecules are able to create a complex reaction or interaction with physiological environment that is tough to monitor and control in in vivo studies. Surfactants, especially ionic surfactant, at a very high concentration may lead to serious toxicity by altering the protein structure and malfunctioning the enzyme and phospholipid membrane (Cserháti et al., Citation2002).
Many oil components or lipid compounds are also susceptible to change into toxic material when dispersed in nano-scale. The United States Food and Drug Administration (US FDA) published a listing of materials generally recommended as safe (GRAS) in the Code of Federal Regulation (CFR) (http://www.fda.gov/Food/IngredientsPackagingLabeling/GRAS/). This list may serve as a guide to select the components. In spite of that, in vivo complexity of the reaction or interaction and the subsequent effect create constraint against pharmacokinetics studies, especially in human volunteers.
Possibility of in vivo precipitation of drugs
When a SEDDS is administered orally, the gastric and pancreatic lipase starts digesting the lipid components like glycerides and other fatty acid or alcohol. Not only that the lipase may negatively affect the emulsification and dispersion efficiency of the lipid components, but also the solubilization capacity of the system becomes hampered and the loaded drug tends to precipitate in vivo (Dai, Citation2010). The sharp change of pH and dilution of the delivery platform in the body fluids are also contributing factors of in vivo drug precipitation. While the formulation and in vitro testing factors can be controlled, physiological factors and variation cannot be controlled. Such undesired phenomena decrease available drug concentration for absorption, resulting in delaying and reducing intended pharmacological action. This drug precipitation often leads to inconsistent results and high inter-subject variability in pharmacokinetic studies (Narang et al., Citation2007). In vitro precipitation test and dissolution test in the bio-relevant medium along with suitably designed lipid digestion study may predict the chances of in vivo precipitation from SEDDS.
Effect of food on absorption
Food’s effect on drug absorption was studied widely. The presence of food is attributed to the slower gastric emptying, change of intestinal pH, change of intestinal fluid viscosity and increased secretion of bile in the intestine that facilitates emulsification and digestion (Kostewicz et al., Citation2002). All these physiological processes enhance solubilization of the drug following higher degree of absorption, especially BCS II drugs. SEDDS being a lipid-based drug delivery system can mimic this role of food after oral administration. Unfortunately, there are very few published reports that show the drug absorption from SEDDS with simultaneous administration of food unlike itraconazole SMEDDS study by Woo et al. (Citation2008). Although the reported study revealed higher itraconazole absorption from SMEDDS than commercial capsule in the fasted state and less variation of absorption between fasted and fed state, this requires more studies to establish the food effect on drug absorption from SEDDS. The most applicable model for food–drug interaction study is a beagle dog model, even though the fasted gastric pH of the canine is highly variable and higher than of human (Akimoto et al., Citation2000). But, from a researcher’s point of view, we can explain that availability and handling of dog as animal model is not as easy like a rat or rabbit as animal model. This constraint might ascribe to lesser researches on food effect on SEDDS with respect to pharmacokinetic parameters evaluation. Simulation of feeding state in an in vitro medium is also very challenging. Using a nutritional drink (Ensure + 0.15% pectin) as a surrogate for high-fat, FDA breakfast was well described in an in vitro study by Klein et al. (Citation2004). However, the feasibility of using that drink to correlate in vivo fed state was not established.
Conclusion
The utilization of lipid-based self-emulsifying drug-delivery system is recognized as a potential solution for delivering poorly soluble drug by oral route. With specific advantages of enhanced availability of drug in solution state in the gut and bypassing hepatic first pass metabolism, the SMEDDS/SNEDDS helps overcome poor bioavailability challenges of BCS II and BCS IV drugs. Commercialization of this drug-delivery system has also been done, but not in very large number.
Successful development of SMEDDS/SNEDDS formulation requires the in vitro characterization, including in vitro lipolysis test and in situ intestinal permeation test or ex vivo intestinal permeation test to predict the in vivo behavior of the system. This review summarizes the recent published reports on in vivo pharmacokinetic studies with SMEDDS/SNEDDS focusing on different controversies raised from the reported pharmacokinetic studies. In short, we can conclude that in spite of being advantageous, the SMEDDS/SNEDDS demands more studies, especially suitably designed pharmacokinetic studies to bridge the gap between formulation development and in vivo effect. At this point, we have come up with some specific findings in this review as mentioned below which will definitely help the formulation scientist to identify and overcome the constraint of this dosage form.
The terminology used by the pharmaceutical scientists implies to SEDDS, varies widely from “micro-emulsifying” (SMEDDS) to “nano-emulsifying” (SNEDDS) and use of the terminology does not only belong to the initial droplet size. Rationality of using either “micro” or “nano” terminology in such kind of SEDDS should be explained by the researchers in the future or only SNEDSS term should be used provided the emulsion droplet size is in the nanometer range.
Most of the published articles refer the micro- or nano-scale size distribution from SEDDS as one of the important contributing factors behind improved bioavailability. Although this can be considered well-established fact, there is a lack of comparative in vivo studies between same formulations with different droplet sizes. This lack restricts to draw firm conclusion on the effect of different droplet size on in vivo pharmacokinetic parameters. A combined approach of in vitro lipolysis or ex vivo penetration model and pharmacokinetic studies are advantageous to understand the behavior of a specific drug delivered orally by SEDDS.
Solid dosage form of SEDDS is established as advantageous from formulation aspects and stability. But, in terms of pharmacokinetics benefits, it cannot be concluded that solid SEDDS is much better than liquid SEDDS filled capsule. Overcoming the challenges of pharmacokinetic studies of SEDDS system on human subjects need more attention of researchers to make this system commercially attractive.
Declaration of interest
The authors declare co conflict of interest in this article.
References
- Akimoto M, Nagahata N, Furuya A, et al. (2000). Gastric pH profiles of beagle dogs and their use as an alternative to human testing. Eur J Pharm Biopharm 49:99–102
- An Y, Yan X, Li B, Li Y. (2014). Microencapsulation of capsanthin by self-emulsifying nanoemulsions and stability evaluation. Eur Food Res Technol 239:1077–85
- Anton N, Vandamme TF. (2011). Nano-emulsions and micro-emulsions: clarifications of the critical differences. Pharm Res 28:978–85
- Araya H, Tomita M, Hayashi M. (2005). The novel formulation design of O/W microemulsion for improving the gastrointestinal absorption of poorly water soluble compounds. Int J Pharm 305:61–74
- Bahloul B, Lassoued MA, Seguin J, et al. (2015). Self-emulsifying drug delivery system developed by the HLB-RSM approach: characterization by transmission electron microscopy and pharmacokinetic study. Int J Pharm. http://dx.doi.org/10.1016/j.ijpharm.2015.04.018
- Bahloul B, Lassoued MA, Sfar S. (2014). A novel approach for the development and optimization of self-emulsifying drug delivery system using HLB and response surface methodology: application to fenofibrate encapsulation. Int J Pharm 466:341–8
- Balakrishnan P, Lee BJ, Oh DH, et al. (2009). Enhanced oral bioavailability of Coenzyme Q10 by self-emulsifying drug delivery systems. Int J Pharm 374:66–72
- Bandyopadhyay S, Katare O, Singh B. (2012). Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf B Biointerfaces 100:50–61
- Beg S, Katare O, Saini S, et al. (2016). Solid self-nanoemulsifying systems of olmesartan medoxomil: formulation development, micromeritic characterization, in vitro and in vivo evaluation. Powder Technol 294:93–104
- Borhade VB, Nair HA, Hegde DD. (2009). Development and characterization of self-microemulsifying drug delivery system of tacrolimus for intravenous administration. Drug Dev Ind Pharm 35:619–30
- Cannon JB. (2011). LIPIDS-strategies to formulate lipid-based drug delivery systems. Am Pharm Rev 14:84
- Čerpnjak K, Zvonar A, Gašperlin M, Vrečer F. (2013). Lipid-based systems as a promising approach for enhancing the bioavailability of poorly water-soluble drugs. Acta Pharm 63:427–45
- Chambin O, Jannin V. (2005). Interest of multifunctional lipid excipients: case of Gelucire 44/14. Drug Dev Ind Pharm 31:527–34
- Chen ML. (2008). Lipid excipients and delivery systems for pharmaceutical development: a regulatory perspective. Adv Drug Deliv Rev 60:768–77
- Chen Y, Li G, Wu X, et al. (2008). Self-microemulsifying drug delivery system (SMEDDS) of vinpocetine: formulation development and in vivo assessment. Biol Pharm Bull 31:118–25
- Cheng G, Hu R, Ye L, et al. (2015). Preparation and in vitro/in vivo evaluation of puerarin solid self-microemulsifying drug delivery system by spherical crystallization technique. AAPS PharmSciTech 1–11. doi:10.1208/s12249-015-0469-8
- Cherniakov I, Domb AJ, Hoffman A. (2015). Self-nano-emulsifying drug delivery systems: an update of the biopharmaceutical aspects. Expert Opin Drug Deliv 12:1121–33
- Christiansen M, Holm R, Abrahamsson B, et al. (2016). Effect of food intake and co-administration of placebo self-nanoemulsifying drug delivery systems on the absorption of cinnarizine in healthy human volunteers. Eur J Pharm Sci 84:77–82
- Christiansen ML, Holm R, Kristensen J, et al. (2014). Cinnarizine food-effects in beagle dogs can be avoided by administration in a self nanoemulsifying drug delivery system (SNEDDS). Eur J Pharm Sci 57:164–72
- Chudasama A, Patel V, Nivsarkar M, et al. (2015). Role of lipid-based excipients and their composition on the bioavailability of antiretroviral self-emulsifying formulations. Drug Deliv 22:531–40
- Cserháti T, Forgács E, Oros G. (2002). Biological activity and environmental impact of anionic surfactants. Environ Int 28:337–48
- Dahan A, Hoffman A. (2008). Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J Control Release 129:1–10
- Dai WG. (2010). In vitro methods to assess drug precipitation. Int J Pharm 393:1–16
- de Smidt PC, Campanero MA, Trocóniz IF. (2004). Intestinal absorption of penclomedine from lipid vehicles in the conscious rat: contribution of emulsification versus digestibility. Int J Pharm 270:109–18
- Do TT, Van Speybroeck M, Mols R, et al. (2011). The conflict between in vitro release studies in human biorelevant media and the in vivo exposure in rats of the lipophilic compound fenofibrate. Int J Pharm 414:118–24
- Eedara BB, Veerareddy PR, Jukanti R, Bandari S. (2014). Improved oral bioavailability of fexofenadine hydrochloride using lipid surfactants: ex vivo, in situ and in vivo studies. Drug Dev Ind Pharm 40:1030–43
- Engel A, Oswald S, Siegmund W, Keiser M. (2012). Pharmaceutical excipients influence the function of human uptake transporting proteins. Mol Pharm 9:2577–81
- Fagir W, Hathout RM, Sammour OA, ElShafeey AH. (2015). Self-microemulsifying systems of Finasteride with enhanced oral bioavailability: multivariate statistical evaluation, characterization, spray-drying and in vivo studies in human volunteers. Nanomedicine 10:3373–89
- Fei Y, Kostewicz ES, Sheu MT, Dressman JB. (2013). Analysis of the enhanced oral bioavailability of fenofibrate lipid formulations in fasted humans using an in vitro–in silico–in vivo approach. Eur J Pharm Biopharm 85:1274–84
- Fernandez-Tarrio M, Yañez F, Immesoete K, et al. (2008). Pluronic and tetronic copolymers with polyglycolyzed oils as self-emulsifying drug delivery systems. AAPS PharmSciTech 9:471–9
- Gao P. (2012). Design and development of self-emulsifying lipid formulations for improving oral bioavailability of poorly water-soluble and lipophilic drugs. In: Williams III RO, Watts AB, Miller DA, eds. Formulating poorly water soluble drugs. New York: Springer, 243–66
- Garg B, Katare O, Beg S, et al. (2016). Systematic development of solid self-nanoemulsifying oily formulations (S-SNEOFs) for enhancing the oral bioavailability and intestinal lymphatic uptake of lopinavir. Colloids Surf B Biointerfaces 141:611–22
- Generally Recognized as Safe (GRAS). (2016). Fda.gov. Available at: http://www.fda.gov/Food/IngredientsPackagingLabeling/GRAS/ [last accessed 3 May 2016]
- Gupta S, Chavhan S, Sawant KK. (2011). Self-nanoemulsifying drug delivery system for adefovir dipivoxil: design, characterization, in vitro and ex vivo evaluation. Colloids Surf A Physicochem Eng Asp 392:145–55
- Gursoy RN, Benita S. (2004). Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother 58:173–82
- Hauss DJ. (2007). Oral lipid-based formulations: enhancing the bioavailability of poorly water-soluble drugs. (Vol. 170). New York: Informa Healthcare
- Heshmati N, Cheng X, Dapat E, et al. (2014). In vitro and in vivo evaluations of the performance of an indirubin derivative, formulated in four different self‐emulsifying drug delivery systems. J Pharm Pharmacol 66:1567–75
- Hintzen F, Perera G, Hauptstein S, et al. (2014). In vivo evaluation of an oral self-microemulsifying drug delivery system (SMEDDS) for leuprorelin. Int J Pharm 472:20–6
- Holm R, Tønsberg H, Jørgensen EB, et al. (2012). Influence of bile on the absorption of halofantrine from lipid-based formulations. Eur J Pharm Biopharm 81:281–7
- Inugala S, Eedara BB, Sunkavalli S, et al. (2015). Solid self-nanoemulsifying drug delivery system (S-SNEDDS) of darunavir for improved dissolution and oral bioavailability: in vitro and in vivo evaluation. Eur J Pharm Sci 74:1–10
- Kale AA, Patravale VB. (2008). Design and evaluation of self-emulsifying drug delivery systems (SEDDS) of nimodipine. AAPS PharmSciTech 9:191–6
- Kang MJ, Jung SY, Song WH, et al. (2011). Immediate release of ibuprofen from Fujicalin®-based fast-dissolving self-emulsifying tablets. Drug Dev Ind Pharm 37:1298–305
- Kang BK, Lee JS, Chon SK, et al. (2004). Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm 274:65–73
- Kang JH, Oh DH, Oh YK, et al. (2012). Effects of solid carriers on the crystalline properties, dissolution and bioavailability of flurbiprofen in solid self-nanoemulsifying drug delivery system (solid SNEDDS). Eur J Pharm Biopharm 80:289–97
- Katneni K, Charman SA, Porter CJ. (2007). Impact of cremophor‐EL and polysorbate‐80 on digoxin permeability across rat jejunum: Delineation of thermodynamic and transporter related events using the reciprocal permeability approach. J Pharm Sci 96:280–93
- Khoo SM, Humberstone AJ, Porter CJ, et al. (1998). Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int J Pharm 167:155–64
- Kim MS, Ha ES, Choo GH, Baek IH. (2015). Preparation and in vivo evaluation of a dutasteride-loaded solid-supersaturatable self-microemulsifying drug delivery system. Int J Mol Sci 16:10821–33
- Kim DW, Kang JH, Oh DH, et al. (2012). Development of novel flurbiprofen-loaded solid self-microemulsifying drug delivery system using gelatin as solid carrier. J Microencapsul 29:323–30
- Kim DW, Kwon MS, Yousaf AM, et al. (2014). Comparison of a solid SMEDDS and solid dispersion for enhanced stability and bioavailability of clopidogrel napadisilate. Carbohydr Polym 114:365–74
- Kim GG, Poudel BK, Marasini N, et al. (2013). Enhancement of oral bioavailability of fenofibrate by solid self-microemulsifying drug delivery systems. Drug Dev Ind Pharm 39:1431–8
- Klein S, Dressman JB, Butler J, et al. (2004). Media to simulate the postprandial stomach I. Matching the physicochemical characteristics of standard breakfasts. J Pharm Pharmacol 56:605–10
- Kohli K, Chopra S, Dhar D, et al. (2010). Self-emulsifying drug delivery systems: an approach to enhance oral bioavailability. Drug Discov Today 15:958–65
- Kollipara S, Gandhi RK. (2014). Pharmacokinetic aspects and in vitro-in vivo correlation potential for lipid-based formulations. Acta Pharm Sin B 4:333–49
- Kostewicz ES, Brauns U, Becker R, Dressman JB. (2002). Forecasting the oral absorption behavior of poorly soluble weak bases using solubility and dissolution studies in biorelevant media. Pharm Res 19:345–9
- Kumar GP, Rambhau D, Apte SS. (2015). Improved oral pharmacokinetics of diclofenac sodium from SNEDDS in human volunteers. J Nanotechnol Diagn Treat 3:5–11
- Kyaw OM, Mandal UK, Chatterjee B. (2015). Polymeric behavior evaluation of PVP K30-poloxamer binary carrier for solid dispersed nisoldipine by experimental design. Pharm Dev Technol 1–11. doi:10.3109/10837450.2015.1116568
- Larsen AT, Åkesson P, Juréus A, et al. (2013b). Bioavailability of cinnarizine in dogs: effect of SNEDDS loading level and correlation with cinnarizine solubilization during in vitro lipolysis. Pharm Res 30:3101–13
- Larsen AT, Ohlsson AG, Polentarutti B, et al. (2013a). Oral bioavailability of cinnarizine in dogs: relation to SNEDDS droplet size, drug solubility and in vitro precipitation. Eur J Pharm Sci 48:339–50
- Li F, Song S, Guo Y, et al. (2015). Preparation and pharmacokinetics evaluation of oral self-emulsifying system for poorly water-soluble drug Lornoxicam. Drug Deliv 22:487–98
- Li H, Tan Y, Yang L, et al. (2015). Dissolution evaluation in vitro and bioavailability in vivo of self-microemulsifying drug delivery systems for pH-sensitive drug loratadine. J Microencapsul 32:175–80
- Morozowich W, Gao P. (2009). Improving the oral absorption of poorly soluble drugs using SEDDS and S-SEDDS formulations. In: Qiu Y, Chen Y, Zand GZ, Liu L, Porter WL, eds. Developing solid oral dosage forms: pharmaceutical theory and practice. Cambridge: Academic, 443–68
- Müllertz A, Ogbonna A, Ren S, Rades T. (2010). New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J Pharm Pharmacol 62:1622–36
- Narang AS, Delmarre D, Gao D. (2007). Stable drug encapsulation in micelles and microemulsions. Int J Pharm 345:9–25
- O’Driscoll CM. (2002). Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci 15:405–15
- Oh DH, Kang JH, Kim DW, et al. (2011). Comparison of solid self-microemulsifying drug delivery system (solid SMEDDS) prepared with hydrophilic and hydrophobic solid carrier. Int J Pharm 420:412–18
- Patil P, Patil V, Paradkar A. (2007). Formulation of a self-emulsifying system for oral delivery of simvastatin: in vitro and in vivo evaluation. Acta Pharm 57:111–22
- Porter CJ, Trevaskis NL, Charman WN. (2007). Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov 6:231–48
- Pouton CW. (1997). Formulation of self-emulsifying drug delivery systems. Adv Drug Deliv Rev 25:47–58
- Pouton CW. (2000). Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’drug delivery systems. Eur J Pharm Sci 11:S93–8
- Pouton CW. (2006). Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci 29:278–87
- Prajapati BG, Patel MM. (2007). Conventional and alternative pharmaceutical methods to improve oral bioavailability of lipophilic drugs. Asian J Pharm 1:1–8
- Quan Q, Kim DW, Marasini N, et al. (2013). Physicochemical characterization and in vivo evaluation of solid self-nanoemulsifying drug delivery system for oral administration of docetaxel. J Microencapsul 30:307–14
- Ramasahayam B, Eedara BB, Kandadi P, et al. (2015). Development of isradipine loaded self-nano emulsifying powders for improved oral delivery: in vitro and in vivo evaluation. Drug Dev Ind Pharm 41:753–63
- Sangsen Y, Wiwattanawongsa K, Likhitwitayawuid K, et al. (2016). Influence of surfactants in self-microemulsifying formulations on enhancing oral bioavailability of oxyresveratrol: studies in Caco-2 cells and in vivo. Int J Pharm 498:294–303
- Savjani KT, Gajjar AK, Savjani JK. (2012). Drug solubility: importance and enhancement techniques. ISRN Pharm 2012:1–10
- Sek L, Boyd BJ, Charman WN, Porter CJ. (2006). Examination of the impact of a range of Pluronic surfactants on the in‐vitro solubilisation behaviour and oral bioavailability of lipidic formulations of atovaquone. J Pharm Pharmacol 58:809–20
- Sermkaew N, Ketjinda W, Boonme P, et al. (2013). Liquid and solid self-microemulsifying drug delivery systems for improving the oral bioavailability of andrographolide from a crude extract of Andrographis paniculata. Eur J Pharm Sci 50:459–66
- Sha X, Yan G, Wu Y, et al. (2005). Effect of self-microemulsifying drug delivery systems containing Labrasol on tight junctions in Caco-2 cells. Eur J Pharm Sci 24:477–86
- Shah NH, Carvagal MT, Patel CI, et al. (1994). Self emulsifying drug delivery system (SEDDS) with polyglycolized glycerides for improving in-vitro dissolution and oral absorption of lipophilic drugs. Int J Pharm 106:15–23
- Shanmugam S, Baskaran R, Balakrishnan P, et al. (2011). Solid self-nanoemulsifying drug delivery system (S-SNEDDS) containing phosphatidylcholine for enhanced bioavailability of highly lipophilic bioactive carotenoid lutein. Eur J Pharm Biopharm 79:250–7
- Singh G, Pai RS. (2015). Trans-resveratrol self-nano-emulsifying drug delivery system (SNEDDS) with enhanced bioavailability potential: optimization, pharmacokinetics and in situ single pass intestinal perfusion (SPIP) studies. Drug Deliv 22:522–30
- Sun M, Zhai X, Xue K, et al. (2011). Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the single-pass intestinal perfusion (SPIP) technique and a chylomicron flow blocking approach: linear correlation with oral bioavailabilities in rats. Eur J Pharm Sci 43:132–40
- Suresh PK, Sharma S. (2011). Formulation and in-vitro characterization of self-nanoemulsifying drug delivery system of cinnarizine. Drugs 11:12
- Sweetman SC. (2009). Martindale: the complete drug reference. London: Pharmaceutical press
- Tang B, Cheng G, Gu JC, Xu CH. (2008). Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov Today 13:606–12
- Tang Jl, Sun J, He ZG. (2007). Self-emulsifying drug delivery systems: strategy for improving oral delivery of poorly soluble drugs. Curr Drug Ther 2:85–93
- Tarr BD, Yalkowsky SH. (1989). Enhanced intestinal absorption of cyclosporine in rats through the reduction of emulsion droplet size. Pharm Res 6:40–3
- Thomas N, Holm R, Müllertz A, Rades T. (2012). In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J Control Release 160:25–32
- Trevaskis NL, Charman WN, Porter CJ. (2008). Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev 60:702–16
- Uchino T, Yasuno N, Yanagihara Y, Suzuki H. (2007). Solid dispersion of spironolactone with porous silica prepared by the solvent method. Pharmazie 62:599–603
- Wasylaschuk WR, Harmon PA, Wagner G, et al. (2007). Evaluation of hydroperoxides in common pharmaceutical excipients. J Pharm Sci 96:106–16
- Wei Y, Ye X, Shang X, et al. (2012). Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloids Surf A Physicochem Eng Asp 396:22–8
- Williams HD, Sassene P, Kleberg K, et al. (2013). Toward the establishment of standardized in vitro tests for lipid-based formulations, part 3: understanding supersaturation versus precipitation potential during the in vitro digestion of type I, II, IIIA, IIIB and IV lipid-based formulations. Pharm Res 30:3059–76
- Woo JS, Song YK, Hong JY, et al. (2008). Reduced food-effect and enhanced bioavailability of a self-microemulsifying formulation of itraconazole in healthy volunteers. Eur J Pharm Sci 33:159–65
- Yap SP, Yuen KH. (2004). Influence of lipolysis and droplet size on tocotrienol absorption from self-emulsifying formulations. Int J Pharm 281:67–78
- Yi T, Wan J, Xu H, Yang X. (2008). A new solid self-microemulsifying formulation prepared by spray-drying to improve the oral bioavailability of poorly water soluble drugs. Eur J Pharm Biopharm 70:439–44
- Zhang H, Yao M, Morrison RA, Chong S. (2003). Commonly used surfactant, Tween 80, improves absorption of P-glycoprotein substrate, digoxin, in rats. Arch Pharm Res 26:768–72
- Zhu JX, Tang D, Feng L, et al. (2013). Development of self-microemulsifying drug delivery system for oral bioavailability enhancement of berberine hydrochloride. Drug Dev Ind Pharm 39:499–506