
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Being one of the highly effective drugs in treatment of Alzheimer’s disease, Rivastigmine brain targeting is highly demandable, therefore liposomal dispersion of Rivastigmine was prepared containing 2 mol% PEG-DSPE added to Lecithin, Didecyldimethyl ammonium bromide (DDAB), Tween 80 in 1:0.02:0.25 molar ratio. A major challenge during the preparation of liposomes is maintaining a stable formulation, therefore the aim of our study was to increase liposomal stability by addition of DDAB to give an electrostatic stability and PEG-DSPE to increase stability by steric hindrance, yielding what we called an electrosteric stealth (ESS) liposomes. A medium nano-sized liposome (478 ± 4.94 nm) with a nearly neutral zeta potential (ZP, −8 ± 0.2 mV) and an entrapment efficiency percentage of 48 ± 6.22 was prepared. Stability studies showed no major alteration after three months storage period concerning particle size, polydispersity index, ZP, entrapment efficiency and in vitro release study confirming the successful formation of a stable liposomes. No histopathological alteration was recorded for ESS liposomes of the sheep nasal mucosa. While ESS liposomes showed higher % of drug permeating through the sheep nasal mucosa (48.6%) than the drug solution (28.7%). On completing the in vivo pharmacokinetic studies of 36 rabbits showed 424.2% relative bioavailability of the mean plasma levels of the formula ESS compared to that of RHT intranasal solution and 486% relative bioavailability of the mean brain levels.
Introduction
Alzheimer’s disease (AD) is the most widespread dementia, where about 60–80% of the patients with dementia were diagnosed as Alzheimer patients (Rodgers, Citation2011; Association, Citation2012). AD is characterized by diffuse atrophy of the cerebral cortex, reflecting loss and shrinkage of neurons (Braak & Braak, Citation1991; Squire & Zola-Morgan, Citation1991). Patients with AD have an abundance of two abnormal structures in their brain; the amyloid plaques and the neurofibrillary tangles that are made of misfolded proteins (Francis et al., Citation1999; Capone et al., Citation2009; Rodgers, Citation2011). Acetylcholine (Ach), a neurotransmitter, is particularly important for memory, and loss of cholinergic neurons that may cause memory loss in AD (Francis et al., Citation1999). During its early stages, short-term memory loss is the most common presenting symptom of AD, that it might be designated as amnesic mild cognitive impairment. Rivastigmine hydrogen tartarate (RHT) is an acetylcholinesterase (AChE) inhibitor that facilitates cholinergic neurotransmission by declining the degradation of released Ach by intact cholinergic neurons. RHT inhibits both AChE and butyrylcholinesterase (BChE), as a result it can optimize cholinergic function, resulting in a clinically realistic benefit (Moretti et al., Citation2007).
Intranasal (IN) delivery is noninvasive, painless, does not require sterile preparation and is readily and easily administered by the patient (Johnson & Quay, Citation2005). The large surface area of the nasal mucosa affords a rapid onset of therapeutic effect, direct central nervous system delivery (Costantino et al., Citation2007) and bypassing first-pass metabolism. Liposomes play an important role in nasal delivery into the systemic circulation by overcoming barriers of the nasal route such as ciliary clearance and nasal peptidase enzyme (Alsarra et al., Citation2010).
One of the challenges during liposomal preparation is stability. The stability of the liposomes suspensions requires a repulsive interaction that is comparable to the range and magnitude of the Van der Waals force. Increasing the surface charge by the addition of charged molecules in the bilayer (i.e. a positive charge inducer, such as didecyldimethyl ammonium bromide (DDAB)) leads to electrostatic repulsion. Another method is steric stabilization achieved by covering the surface with an adsorbed layer of long, bulky molecules to prevent the close approach of the liposomes (i.e. ligands, such as polyethylene glycol, PEG). The combination of both, termed electrosteric stabilization, leads to a highly stable dispersions (Yadav et al., Citation2011). Polyethylene glycol, the most widely used stabilizer in pharmaceutical applications, is non-biodegradable, highly soluble in aqueous solution, bind to a large number of water molecules, with high flexibility of its polymer chain, and limited accumulation in cells of the reticular endothelial system (RES) (Woodle, Citation1995). Moreover, PEGylated liposomes (Stealth® liposomes) showed enhanced pharmacokinetic parameters such as increased half-life, decreased plasma clearance, and decreased distribution volume (Vd) along with a favored accumulation on diseased tissues, and affords the ability to adjust the liposomal drug loading and leakage (Yadav et al., Citation2011; Milla et al., Citation2012).
In earlier studies, Kreuter et al. (Citation1995) concluded that after peripheral administration of Dalargin and loperamide nanoparticles, no therapeutic effect was observed because they did not diffuse through the blood brain barrier (BBB). But, when the same nanoparticles were coated with Tween 80, a nonionic surfactant, a distinct analgesic effect was attained (Kreuter et al., Citation1995; Schroeder et al., Citation1998). It was suggested that tween 80 acted as an anchor for apolipoproteins (APO-B and APO-E), at the surface of the nanoparticles which interact with LDL receptor, thus being taken up via receptor-mediated endocytosis (Borchard et al., Citation1994; Kreuter et al., Citation1995; Alyaudtin et al., Citation2001).
So, the aim of this study is to prepare electrosterically stabilized (using DDAB and PEG) IN liposomal dispersions that is bounded to Tween 80 for optimum RHT brain targeting.
Materials and methods
Materials
Rivastigmine hydrogen tartarate was kindly provided by Mepaco – Arab Co. For Pharmaceuticals & Medicinal Plant, Heliopolis, Cairo, Egypt. l-α-Lecithin from soybean, type II-S 14-23% choline basis (L-II-S) was purchased from MP Biomedicals, LLC, Paris, France. Didecyldimethyl ammonium bromide were purchased from Sigma Chemical Co. (St. Louis, MO). Tween 80 (polyoxyethylene sorbitan monooleate), disodium hydrogen phosphate, potassium dihydrogen phosphate, sodium chloride, potassium chloride, methanol and chloroform were purchased from Adwic, El-Nasr Pharmaceutical Chemicals Co., Cairo, Egypt. 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethyleneglycol)-2000] (PEG-DSPE) was a kind gift as a medical sample from Lipoid GMBH, Ludwigshafen, Germany. Spectra/Pore® dialysis membrane (12 000–14 000 molecular weight cutoff) was purchased from Spectrum Laboratories Inc. (Los Angeles, CA). Ethyl acetate, chloral hydrate, formalin, methanol, ethanol, absolute ethyl alcohol, xylene, Paraffin bees wax, hematoxylin and eosin stain were purchased from El-Nasr Pharmaceutical Chemicals Co., Cairo, Egypt. Sodium tetraphenyl borate (NaTPB) was purchased from Sigma Aldrich (Steinheim, Germany). Poly vinyl chloride (PVC) of high molecular weight, and 2-nitrophenyl octyl ether (NPOE) were purchased from Fluka Chemie GmbH, St. Louis, MO. Tetrahydrofuran (THF) was purchased from BDH, Poole, England. Quitiapine was purchased from Astrazeneca, Cairo, Egypt. Acetonitrile and ammonium formate were purchased from Sigma Chemical Co. (St. Louis, MO). All other reagents were of analytical grade or HPLC grade.
Preparation of electrosteric stealth (ESS) liposomes
As a result of a previous 32 full factorial design (El-Helaly et al., Citation2014) that was conducted to study the joint influence of DDAB molar ratio, as a positive charge inducer, and Tween 80 molar ratio, numerical optimization was performed using the statistical program Design-Expert software (V. 7.0.0, Stat-Ease Inc., Minneapolis, MN) and accordingly the following molar ratios were used. Thin film hydration method was conducted as stated by Burgess & Wright (Citation2012). Briefly, 50 mg Rivastigmine together with 2 mol% PEG-DSPE was added to 200 mg Lecithin, DDAB, Tween 80 (1:0.02:0.25 molar ratio) in a round-bottom flask and dissolved in 10 ml chloroform at 40 °C. The organic solvent was evaporated at the same temperature under vacuum, using a rotary evaporator (Rotavapor, Heidolph VV 2000, Burladingen, Germany) at 90 rpm such that a thin film was formed inside the flask. The deposited thin film was then hydrated with 5 mL of phosphate buffer saline (PBS), pH = 7.4, by rotating the flask in a water bath at 40 °C using rotary evaporator under normal pressure to ensure complete hydration of the film. The resulting vesicles were sonicated for 10 minutes in an ultrasonic water bath to reduce their particle size (PS) (Woodbury et al., Citation2006; Jesorka & Orwar, Citation2008). The obtained dispersion was left to mature overnight at 4 °C. Liposomal dispersion was then subjected to three freeze–thaw cycles, freezing at −4 °C and then thawing at room temperature (Arumugam et al., Citation2008). The liposomal dispersion was stored at 4 °C until analysis.
Characterization of ESS liposomes
Morphological examination
The liposomes were examined by optical microscope (Lecia Image, Solms, Germany), by means of a fitted camera (JVC, Yokohama, Japan) and photographed at a magnification power of 40×, and by transmission electron microscope (TEM) operated at 80 kV (model JEM-1230, Jeol, Tokyo, Japan).
Particle size, polydispersity index (PDI), zeta potential (ZP) and entrapment efficiency (EE%)
The mean PS, PDI and ZP were determined by Zetasizer at 25 °C (Malvern Instrument Ltd., Worcestershire, UK) after being diluted with PBS (Yang et al., Citation2007a). Triplicates were taken for each sample. Measuring the EE% of Rivastigmine was conducted by ultracentrifugation at 15 000 rpm for two hour using a cooling centrifuge (Beckman, Fullerton, Canada) at 4 °C. The precipitated liposomes were ruptured using methanol, filtered, then the concentration of the entrapped drug was measured spectrophotometrically, by first derivative (Shimadzu, model UV-1601 PC, Kyoto, Japan) at λmax=274.8 nm.
In vitro release studies
In vitro release of Rivastigmine from rivastigmine solution and ESS liposomes were performed using the dialysis bag diffusion technique (Avgoustakis et al., Citation2002). Phosphate buffer (PBS, pH 7.4) was used as the dissolution medium. The dialysis bags (molecular weight cutoff 12 000–14 000 Da, Sigma, St. Louis, MO) were soaked in PBS for 12 h before use. Two milliliters of ESS liposomal dispersion were ultracentrifuged (to remove the free drug) and the precipitate was reconstituted in 2 ml PBS. These 2 ml of ESS Rivastigmine loaded liposomes and 2 ml of Rivastigmine solution (both equivalent to ∼8–9 mg) were placed in dialysis bags with the two ends fixed by thread. Each bag was put into a glass stoppered vials containing 20 ml of phosphate buffer (PBS, pH 7.4) as the dissolution medium. The vials were placed in a water bath shaker at 37 ± 0.5 °C and 100 rpm. Aliquots of the dissolution medium (500 μl) were withdrawn at each time interval and the same volume of fresh PBS was added to the vials to maintain constant volume. Drug concentrations in the dissolution medium were finally analyzed spectrophotometrically, by first derivative (Shimadzu, model UV-1601 PC, Kyoto, Japan) at λmax = 273.2 nm. The release experiments were carried out in duplicates. The results are expressed as means ± standard deviation.
Stability studies
To investigate their physical stability, the ESS liposomes were stored in sealed glass vials (20 ml capacity) in refrigerator (4–8 °C) for 3 months. After one month, and at the end of the storage period, they were evaluated with respect to their PS, ZP and EE%. Statistical analysis of the obtained results was performed by t-test with unequal variance using SPSS 19.0® software (SPSS Inc., Chicago, IL). Difference at p ≤ 0.05 was considered significant. The release profile of the stored formulae was compared to that of the freshly prepared ones according to the model independent mathematical approach of Moore & Flanner (Citation1996). The similarity factor (f2) was calculated according to the following equation:
where n is the number of sampling points, Rt and Tt are the mean percent released from reference (fresh) and from test (stored) at time t. An f2 value ≥50 indicates that the release profiles are similar, whereas smaller values may imply dissimilar release profiles.
Ex vivo studies
Ex vivo estimation of nasal toxicity
Safety, both as a one-off dose, as well as after long term repeated application is one of the most important considerations in the administration to the nose. A nasal formulation that promotes drug absorption will be considered useless if it causes damage to the nasal mucosa.
The nasal-cavity mucosa of a one-year old sheep, weighing 30 kg, was obtained from the local slaughter house (Cairo, Egypt). Within one hour of the sacrifice of the animal, the nasal cavity was fully exposed by a longitudinal incision through the lateral wall of the nose while avoiding the damage of the septum. Subsequently, the mucosa was carefully removed and immediately immersed in normal saline (Du et al., Citation2006). Two segments were carefully separated from the anterior and the posterior regions of the mucosa in the nasal cavity. Each segment was sectioned into three pieces. The pieces were treated with normal saline (negative control), isopropyl alcohol (positive control) and ESS liposomal dispersion. After 2 h of treatment, the pieces were washed with distilled water and preserved in 10% formalin solution (Seju et al., Citation2011).
The histopathological studies were conducted according to the protocol described by Bancroft & Gamble (Citation2008). Briefly, the samples were dehydrated by treatment with serial dilutions of methyl alcohol, ethyl alcohol, and absolute ethyl alcohol, respectively. Specimens were cleared in xylene embedded in paraffin in a hot air oven at 56 °C and the samples were kept for 24 h. Paraffin-beeswax tissue blocks were sectioned by a sledge microtome. The obtained sections (3–4 μm thick) were collected, de-paraffinized, stained by hematoxylin and eosin, and examined under a light microscope.
Ex vivo permeation through sheep nasal mucosa
The permeation studies were started immediately after the mucosa samples were excised. It was conducted by attaching the nasal mucosa to open ended 10 mm column by a thread, then suspended in a beaker filled with 20 ml PBS pH 7.4 at 37 °C, 100 rpm. Two formulations were applied: 1.5 mg Rivastigmine solution (PBS pH 7.4) and ESS liposomal dispersion. Percentage RHT permeated at 0.5, 1, 2, 4 and 6 h was determined (Seju et al., Citation2011) by Ion Selective Electrodes method (El-Kosasy et al., Citation2008).
The transport parameters, i.e. steady-state flux and permeability coefficient of RHT across the membrane, were calculated (Xiang et al., Citation2002) according to the steady-state solution of Fick’s equation:
where Pe is the apparent permeability coefficient of diffusion (cm s−1), C is the initial concentration and JSS is the flux at steady-state (mg s−1 cm−2); dM is the amount of drug (mg) transported across the membrane during the time dt and A is the diffusion area (cm2). The flux at steady-state across the mucosa was calculated from the slope of the linear part of the line obtained by plotting mass transported per unit area against time (Tas et al., Citation2006).
After 6 h, the mucosa is detached, washed with distilled water, dissolved in 10 ml chloroform, then solvent is filtered, and evaporated to dryness. The residue is reconstituted in 10 ml methanol. The % retained was determined by first derivative spectra.
In vivo pharmacokinetic studies
Assay of RHT in rabbit plasma and brain
The method involved a liquid–liquid extraction procedure with ethyl acetate. RHT and internal standard "Quitiapine 50 ng/ml" (IS) were measured by reversed phase high performance liquid chromatography and detected by LC–MS/MS. Chromatography was carried out at ambient temperature, on a Waters SunFire™ C18 analytical column, 5 μm (50 mm × 4.6 mm). Mobile phase consisted of 80% acetonitrile, 20% 0.01 M ammonium formate, at a flow rate of 1 mL/min. The injection volume was 25 μl and the total run time was set for 0.8 min.
Study design
Thirty six male albino rabbits (each ≈1.5 kg) administered the formulae under study by IN route. The protocol of the study was reviewed and approved (PI 1197) by the institutional review board; Research Ethics Committee-Faculty of Pharmacy, Cairo University (REC-FOPCU), Egypt. The rabbits were housed three per cage at room temperature with free access to food and water with a 12-h light–dark cycle. The rabbits were divided into two groups, Group 1: RHT nasal solution, Group 2: ESS liposomal dispersion. Three rabbits for each formulation per time point were used in the study, 18 rabbits total were used for each formulation. The groups received a dose of 1 mg/kg RHT (Arumugam et al., Citation2008). The rabbits were anesthetized by diethyl ether. A sufficient dose was given by inhalational route, to keep the rabbits sedated for about three minutes during instillation of formulations to prevent sneezing (Fazil et al., Citation2012).
The rabbits were sacrificed by decapitation at different time intervals and the blood samples were collected into heparinized tubes at the following set points: 0.5, 1, 2, 4, 8 and 12 hours after administration of each treatment. Plasma was obtained by centrifugation at 4000 rpm for 15 min at 25 °C. The plasma was transferred directly into 5 ml plastic tubes and stored frozen at –20 °C until drug analysis. Plasma samples were extracted using same technique mentioned earlier in the assay. Subsequently, the brain was dissected, washed twice using normal saline, made free from adhering tissue/fluid and weighed. An aliquot (1:1) of normal saline solution was added. The brain was then homogenized on ice, transferred directly into 5 ml plastic tubes and stored frozen at –20 °C until drug analysis. Aliquots of brain homogenates were extracted using same technique mentioned earlier in the assay.
Statistical analysis of the pharmacokinetic parameters
Results were expressed as mean values ± S.D. One-way analysis of variance (ANOVA) was performed for the log transformed data derived from the pharmacokinetic parameters; Cmax, AUC0–t, AUC0–∞, K and t1/2, and the nonparametric Kruskal–Wallis test for Tmax and MRT using SPSS 19.0 software (SPSS Inc., Chicago, IL), in order to investigate the statistical significance among groups. A p value < 0.05 was considered statistically significant.
Results
Morphological examination
Both photomicrograph and transmission electron micrograph () demonstrated that the prepared ESS liposomes were well identified, with nearly perfect spherical shape, clearly showing multilayered structure of the formed liposomes.
Figure 1. (A) Photomicrographs (40×) of ESS liposomes; (B) transmission-electron micrograph (TEM) of ESS liposomes.

Particle size, polydispersity index, zeta potential and entrapment efficiency
ESS liposomes showed medium nano-ranged PS of about 478 ± 4.94 nm with a PDI of 0.557 ± 0.04. The ZP of the prepared liposomes was found to be nearly neutral –8 ± 0.2 mV which favors their transport through the negatively charged BBB. Hence the prepared liposomes would neither be repelled away from the BBB (if negatively charged) nor be retained on its surface (if positively charged). ESS liposomes entrapped 48 ± 6.22% of rivastigmine inside it.
In vitro release studies
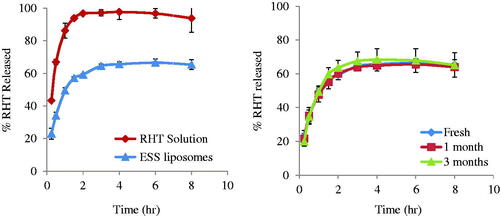
The in vitro release of RHT from ESS liposomes compared with that of RHT solution is represented in . The release of the drug from the solution is faster than that from ESS liposomes which showed delayed release pattern.
Figure 2. (a) In vitro release profile of RHT from ESS liposomes in comparison with RHT solution. (b) In vitro release profile of RHT from ESS liposomes; fresh, after storage for one, and three months.
Stability studies
After one month of storage, the ESS liposomes showed a decrease in PS to 408.5 ± 3 with PDI of 0.5195 ± 0.05 and –7.385 ± 0 mV ZP. While the EE% remained almost the same (48.89 ± 4.96%). After three months, the PS was 427.55 ± 3.6 with PDI of 0.591 ± 0.01 and 6.64 ± 0.4 mV ZP. The results of the t-test with unequal variance of PS and ZP of ESS liposomes after one and three months were significantly different at a significance level of α = 0.05 than the fresh formulation in case of PS while the ZP was non-significantly different after one month but significantly different after three months. While the EE% showed non-significant difference after one month (48.89 ± 4.96) and after three months (37.03 ± 0.41).
The release profiles; fresh, after one, and three months of storage are shown in . The similarity factor was 85 after one month and 74 after 3 months. These values are above the critical value (50) indicating similarity in the release profiles.
Ex vivo estimation of nasal toxicity
The anterior segment of the negative control and ESS liposomes showed no histopathological alteration and the normal histological structure of the covering stratified keratinized epithelium with the underlying connective tissue as a dermal lamina propria were observed. As for the posterior segments, it has shown normal histological structure of the glandular structure with the interstitial connective tissue stroma as well as the cartilaginous structure. On the other hand, the anterior segment of the positive control showed destruction in the focal area of the dermal connective tissue in lamina propria as inflammatory reaction, in addition to destruction in the cartilaginous structure in the posterior segment.
Ex vivo permeation through sheep nasal mucosa
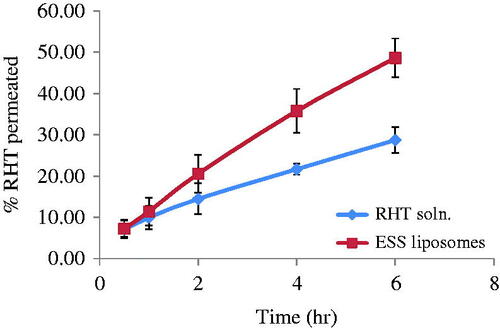
The percentage of drug permeated in six hours from RHT solution, and ESS liposomes is represented in . The percentage of drug permeated after six hours from RHT solution, and ESS liposomes was 28.7 ± 3.1% and 48.6 ± 4.7%, respectively. While about 45.6 ± 0.026% of RHT were retained in the sheep nasal mucosa in case of RHT solution against 51 ± 0.9% in case of rivastigmine ESS loaded liposomes. The steady state flux and permeability coefficient of drug solution through the nasal mucosa were 0.0263 ± 0.0069 g cm−2 h−1 and 0.0175 ± 0.0046 cm−2 h−1 respectively whereas steady-state flux and permeability coefficient of ESS liposomes were higher 0.0515 ± 0.0030 g cm−2 h−1 and 0.0343 ± 0.0020 cm−2 h−1, respectively.
Figure 3. Permeation profile of Rivastigmine through sheep nasal mucosa in six hours from RHT solution, and ESS liposomes.
In vivo pharmacokinetic studies
When the drug is administered via IN route, it can enter into the brain via three different paths (Vyas et al., Citation2005). The first one is the systemic pathway by which the drug is absorbed into the systemic circulation and subsequently reaches the brain by crossing BBB (especially lipophilic drug). The others are the olfactory region and the trigeminal neural pathway by which the drug is transported directly from the nasal cavity to CNS (cerebrospinal fluid and brain tissue). The trigeminal nerve receptors which are present in the nasal cavity are responsible for most chemoperception and are suggested to transport the drug directly to CNS (Thorne et al., Citation2004).
Mean plasma and brain concentration time data for RHT following IN administration of the two groups are shown in . It is evident that drug concentration in rabbit plasma and brain reached its maximum 676.37 ± 139.8 ng/ml and 484.7 ± 225.3 ng/g, respectively, after 30 minutes following administration of the IN drug solution, while in case of ESS two peaks were observed 196.93 ± 10.9 ng/ml and 382.5 ± 63.5 ng/g, respectively, after 30 minutes and 489.24 ± 64.7 ng/ml and 875.1 ± 110.3 ng/g, respectively, after four hours. The increase in plasma and brain drug concentration is followed, in all cases, by a gradual decrease with increasing time. The mean pharmacokinetic parameters of RHT in blood and brain determined for both groups are summarized in .
Figure 4. (a) Mean plasma concentration time curve; (b) mean brain concentration time curve of RHT in plasma after administration of groups 1 and 2.
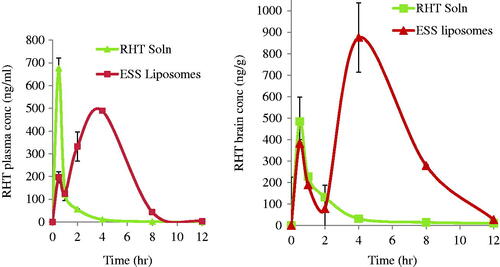
Table 1. Mean pharmacokinetic parameters of Rivastigmine (rabbits plasma and brain) after receiving groups 1 and 2 intranasally.
Concerning the plasma data, post hoc's LSD test showed a non-significant difference between RHT solution and ESS, in case of mean peak plasma concentration (Cmax), elimination half-life (t1/2) and elimination rate constant (K). On the other hand, the area under the plasma concentration time curve (AUC0–∞ and AUC0–t) showed a significant difference between RHT solution and ESS. Also, the statistical non-parametric Kruskal–Wallis test showed a significant difference between RHT solution and F-ESS of the median time to peak plasma concentration (tmax), and mean residence time (MRT) in plasma.
As for the brain data, the post hoc LSD test showed a non-significant difference between RHT solution and ESS, in case of mean peak brain concentration (Cmax), and a significant difference between the area under the brain concentration time curve (AUC0–∞ and AUC0–t) of RHT solution and ESS. Also, the statistical non-parametric Kruskal–Wallis test showed a significant difference between RHT solution and ESS of the median time to peak brain concentration (tmax), and did not differ significantly in MRT in brain.
According to the mean plasma levels of 36 rabbits completing the study, the relative bioavailability was found to be 424.2% based on the mean AUC0–∞ of the formula ESS compared to that of RVT IN solution. The post hoc LSD test and the non-parametric Kruskal–Wallis test showed significant differences in AUC0–t, AUC0–∞, MRT, tmax. However, a non-significant difference was observed in Cmax, K and t1/2. Moreover, the mean brain levels of 36 rabbits completing the study presented 486% relative bioavailability based on the mean AUC0–∞ of the formula ESS compared to that of RVT IN solution. The post hoc LSD test and the non-parametric Kruskal–Wallis test showed significant difference in AUC0–t, AUC0–∞, and tmax. However, a non-significant difference is observed in Cmax, and MRT.
Discussions
The thin film hydration method is considered method of preparation of multilayered vesicles (MLVs) (Burgess & Wright, Citation2012). The photomicrographs images and TEM, confirmed the formation of MLVs that was also confirmed by the increased PS, 478 nm, as described by Siepmann et al. (Citation2011). The increase observed in PS could be due to presence of PEG-DSPE, as it appears as a mushroom like structure which gives a slight increase in PS (Rovira-Bru et al., Citation2002). These lipids typically have PEG chains of molecular weights between 1000 and 2000, extending approximately 5–6 nm from the liposome surface (Burgess & Wright, Citation2012). It must be highlighted that the significant decrease in ZP occurred due to the same reason in addition to the presence of the positive charge inducer DDAB .The presence of Tween 80 also contributed to such decrease in the ZP. This might be attributed to the penetration of its hydrocarbon tail in the lecithin lipid bilayer thus leaving its polyethylene oxide groups on the surface of the liposomes thereby introducing a steric barrier on the surface of the liposomes, which decreases liposomes fusion and consequently decreases ZP. This steric barrier on the surface masks the negative charge of the phospholipid (Yang et al., Citation2007b). As for the decreased EE%, it must be taken into consideration that RHT is a water-soluble, weakly basic drug with the large molecular weight of 400.43. Hence, the high encapsulation of Rivastigmine in liposomes was a challenging process. As indicated by Schnyder & Huwyler (Citation2005), the choice, the optimization, and the validation of a specific loading technique may be a complex problem depending on the physicochemical properties of a given drug. Szoka and Papahadjopoulos studied the effect of several types of lipids, cholesterol and long chain alcohols, used in liposomal preparation, on the EE%. They calculated the captured volume in the internal aqueous space and correlated it to the change in the entrapment that was reported. Szoka and Papahadjopoulos attributed the increase in entrapment to the increase in the internally captured volume which was synchronized with the increase in liposomal PS (Szoka & Papahadjopoulos, Citation1978). Similarly Arumugam et al. (Citation2008) prepared multilamellar liposomes by thin film hydration method containing soya lecithin and cholesterol in a 4:1 molar ratio. EE% of the developed formulation was found to be 80.0 ± 5.0% with a large PS of 10.0 ± 2.8 μm. Attempts to decrease the PS to the nano range, yielded decreased EE% as described by Degim et al. (Citation2010) where EE% of liposomes was 35.4%, 25.2% and 29.9% for RHT, RHT-sodium taurocholate, RHT-dimethyl-beta-CD liposomes, respectively. Similar findings were stated by Yang et al. (Citation2011). Even when RHT was loaded into liposome via ammonium sulfate gradient method, the EE% of RHT liposome was (33.41 ± 6.58)%, with the mean diameter of 154–236 nm. And again in 2013, Yang et al. (Citation2013) formulated RHT liposomes and cell penetrating peptide modified liposomes to improve RHT distribution in brain and proceed to enhance pharmacodynamics by IN administration. Liposomes diameter (nm) was 166.3 ± 17.4 and 178.9 ± 11.7, with EE (%) equals to 33.4 ± 6.6, and 30.5 ± 8.0. A similar trend was observed by Were et al. Liposomes sizes varied from 85 to 239 nm depending on lipid composition, encapsulated matter, and incubation time of liposomes (Were et al., Citation2003). Similar finding was reported by Brgles et al. who showed how different compositions of lipid and aqueous phases can be used to vary the EE%, liposome size and ZP (Brgles et al., Citation2008). In our study, electrosteric stealth liposomal dispersion showed 48 ± 6.22 EE% with medium nano-sized droplets of 478 ± 4.94 nm. RHT has a phenethylamine moiety, which consists of a phenyl group substituted at the second position by an ethan-1-amine and a carbamide moiety from the other side, whereas Lecithin (L-II-S) is a combination of phosphatidylcholine (14–23%), phosphatidylethanolamine (traces), and inositol phosphatides. Consequently, phosphatidyl-choline is a quaternary ammonium compound that hinders the entrapment of RHT between hydrophilic parts of the MLVs due to chemical repulsion with RHT. However, these results showed higher encapsulation percentage than the previously mentioned researches meanwhile the PS was still kept in the nano range in order to bypass the BBB and achieve the main aim of our study that is brain targeting of RHT.
The non-significant difference in the EE% shown after one month (48.89 ± 4.96) and after three months (37.03 ± 0.41) is attributed to the presence of PEG-DSPE. As it is generally believed that the greater the bilayer stability (in terms of drug retention by the vesicles), the longer the vesicle half-life in the circulation. PEG-DSPE leads to the development of long circulating liposomes, i.e. ESS liposomes. ESS liposomes can circumvent recognition by the body as foreign and hence avoid opsonization and phagocytic uptake. Hence, they can circulate in the blood stream for a prolonged time. This effect is mostly attributed to the steric stabilization induced by the incorporated PEGylated lipids, PEG-DSPE. A class of synthesized lipids composed of polyethylene glycol-phosphatidylethanolamine (PEG-PE) (Burgess & Wright, Citation2012).
RHT showed a fast release profile where 43.3 ± 0.5% of drug were released in the first 15 minutes and 96.8 ± 0.4 is reached after two hours, followed a plateau release profile. On the other hand, 23.1 ± 3.3%, 59.3 ± 0.4%, 65.4 ± 3% of RHT were released from ESS liposomes after 15 min, 2 and 8 hours, respectively. The release of the drug from the solution is faster than that from ESS liposomes. This might be explained by the fact that the delayed release was attributed to the lipophilic character of the drug (Kurz et al., Citation2009) that was held by the small fragment of the liposomal membrane and the drug encapsulated in lipid membrane that released mainly through dissolution and diffusion from the lipid bilayer. Also, liposomes serve as a rate-limiting membrane barrier thus providing a controlled system. This result suggests that it takes time for RHT to be released once encapsulated in the liposomes because the lipid bilayers are stabilized by PEG-DSPE and DDAB. Thus, a sustained effect could be achieved using our ESS liposomal formulation. These results came in agreement with Yang et al. who studied the in vitro release profile of the conventional and PEGylated liposomes. The conventional and PEGylated liposomes released 55% and 33% of paclitaxel within 24 h of dialysis, respectively (Yang et al., Citation2007b).
The release of the drug after a storage period of three months (stability study) showed similar in vitro release profile to that freshly prepared confirmed by similarity factor results. Such a similarity is attributed to the presence of PEG-DSPE and DDAB that causes the formation of electrosterically stable liposomes. This similarity contributed to the success in maintaining the stability of the prepared formula during the storage period.
ESS liposomes showed higher percentage of drug permeating through the sheep nasal mucosa (48.6%) than the drug solution (28.7%). This may be attributed to the permeation enhancing capabilities of the phospholipids used in ESS liposomal dispersion (Romeo et al., Citation1998). Moreover, vesicular drug delivery systems (i.e. liposomes) overcame limitations of the nasal route such as ciliary clearance and breakdown by nasal peptidase enzyme, providing a promising alternatives with many advantages over the conventional systems (Alsarra et al., Citation2010). The percentage of drug retained in the nasal mucosa at the end of the study showed higher results in case of ESS liposomes (51%±0.9) than RHT solution (45.6%±0.026) due to interaction of the phospholipids with intercellular lipids in the deeper layers of the nasal mucosa. These results come in agreement with Law et al. who studied the permeability of positively charged liposomes through the nasal mucosa. Law et al. stated that a high local desmopressin concentration, entrapped within the liposomes, on the penetration site promoted an effective penetration of desmopressin through the nasal mucosa (Law et al., Citation2001).
The plasma concentration time profile shown in suggests that the percentage of ESS liposomes, permeated through the nasal mucosa, and reached the blood circulation maintained long circulation time keeping the drug stable in the blood circulation with an MRT of 3.5 ± 0.3 h in comparison to 1.1 ± 0.1 h in case of RHT solution.
The presence of a doubled-peak in the mean plasma concentration time data for RHT following IN administration in case of ESS at 30 minutes and four hours may be due to free drug reaching the blood first. Then the second peak was delayed due to time taken by the ESS liposomes to permeate through the nasal mucosa, by disrupting intercellular lipid domain, reaching the blood circulation and releasing RHT into the blood stream. Similar findings were reported by Chen et al. who found a double-peak in plasma of rats after administering intragastric harmine (HM) liposomes to rats. Chen et al. speculated that the first peak may be caused by part of HM was rapidly split from HM-liposomes in the GIT and rapidly absorbed, then another part of HM-lip transferred into the small intestine and was absorbed later, resulting in the second peak (Chen et al., Citation2016).
For the doubled-peak in the mean brain concentration time data for RHT, may be due to free drug reaching the brain through two routes. The first is through the blood circulation while the second is through the olfactory extraneuronal pathway (epithelial pathway) which is a faster route for direct nose-to-brain transfer as compounds pass paracellularly across the olfactory epithelium into the perineural space, which is continuous with the subarachnoid space before transport to basolateral side of the olfactory epithelium which delivers drugs directly to the brain parenchymal tissue and/or CSF. Then the second peak may be due to encapsulated RHT reaching the brain through two routes. The first one was from RHT released from the ESS liposomes in the blood stream bypassing the BBB. While the second route maybe the olfactory intraneuronal pathway which involves axonal transport and is considered a slow route where substance enters the olfactory neuron via endocytotic or pinocytotic mechanisms and diffuses to the olfactory bulb by utilizing the same mechanisms the cell uses to transport endogenous substances to the rest of the brain (Yamada, Citation2004).
The results of the in vitro release profile which showed sustained release profile of RHT from ESS liposomes (∼65% of RHT after 8 h) together with the results of ex vivo permeability supported evidence on the reported delayed release of RHT in vivo which appeared in the form of second peak after 4 h of ESS RHT loaded liposomes IN administration. Both the plasma and brain concentration time curves made it obvious that even though the concentration of RHT after IN administration of RHT solution in plasma was high, yet the drug failed to bypass the BBB in equal amounts at the same time or even later. On the other hand, ESS RHT loaded liposomes succeeded in maintaining close brain levels of RHT as the solution in addition to sustaining profound increase in brain levels after 4 h. This suggests that our formulation showed higher stability levels in the blood stream and that it succeeded to circulate for a longer time in the blood circulation to bypass the BBB and at the same time paving its way through direct nose to brain delivery, meeting the requirement it was designed for.
Conclusions
ESS liposomes showed outstanding results concerning the in vitro, ex vivo and in vivo studies that showed a four-fold increase in both plasma and brain drug levels. Consequently, Rivastigmine IN liposomal dispersion containing 0.02 molar ratio DDAB, 0.25 molar ratio Tween 80, and 2 mol% PEG-DSPE is a promising drug delivery system in treatment of Alzheimer disease that needs further investigation concerning new dosage adjustment, toxicological studies, etc.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
supplementary_pictures_drug_delivery.docx
Download MS Word (1.1 MB)Acknowledgements
We would like to acknowledge Mepaco – Arab Co. For Pharmaceuticals & Medicinal Plant, and Lipoid GMBH, for gifting us free medical samples that aided in this work.
Related Research Data
References
- Alsarra IA, Hamed AY, Alanazi FK, El Maghraby GM. (2010). Vesicular systems for intranasal drug delivery. New York: Springer-Verlag
- Alyaudtin RN, Reichel A, Lobenberg R, et al. (2001). Interaction of poly(butylcyanoacrylate) nanoparticles with the blood–brain barrier in vivo and in vitro. J Drug Target 9:209–21
- Arumugam K, Subramanian GS, Mallayasamy SR, et al. (2008). A study of rivastigmine liposomes for delivery into the brain through intranasal route. Acta Pharm 58:287–97
- Association AS. (2012). 2012 Alzheimer’s disease facts and figures. Alzheimer's Dement 8:131–68
- Avgoustakis K, Beletsi A, Panagi Z, et al. (2002). PLGA-mPEG nanoparticles of cisplatin: in vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J Control Release 79:123–35
- Bancroft JD, Gamble M. (2008). Theory and practice of histological techniques. Philadelphia: Elsevier Health Sciences
- Borchard G, Audus KL, Shi F, Kreuter J. (1994). Uptake of surfactant-coated poly (methyl methacrylate)-nanoparticles by bovine brain microvessel endothelial cell monolayers. Int J Pharm 110:29–35
- Braak H, Braak E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–59
- Brgles M, Jurasin D, Sikiric MD, et al. (2008). Entrapment of ovalbumin into liposomes-factors affecting entrapment efficiency, liposome size, and zeta potential. J Liposome Res 18:235–48
- Burgess D, Wright J. (2012). Long acting injections and implants, advances in delivery science and technology. Berlin, Germany: Springer
- Capone R, Quiroz FG, Prangkio P, et al. (2009). Amyloid-beta-induced ion flux in artificial lipid bilayers and neuronal cells: resolving a controversy. Neurotox Res 16:1–13
- Chen WL, Yuan ZQ, Liu Y, et al. (2016). Liposomes coated with N-trimethyl chitosan to improve the absorption of harmine in vivo and in vitro. Int J Nanomed 11:325–36
- Costantino HR, Illum L, Brandt G, et al. (2007). Intranasal delivery: physicochemical and therapeutic aspects. Int J Pharm 337:1–24
- Degim Z, Mutlu NB, Yilmaz S, et al. (2010). Investigation of liposome formulation effects on rivastigmine transport through human colonic adenocarcinoma cell line (CACO-2). Pharmazie 65:32–40
- Du G, Gao Y, Nie S, Pan W. (2006). The permeation of nalmefene hydrochloride across different regions of ovine nasal mucosa. Chem Pharm Bull (Tokyo) 54:1722–4
- El-Helaly SN, Elbary AA, Kassem MA, El-Nabarawi MA. (2014). Design, development and statistical evaluation of positively charged rivastigmine liposomes. Inventi Rapid: Pharm Tech 4:1–8
- El-Kosasy AM, Salem MY, El-Bardicy MG, El-Rahman MK. (2008). Miniaturized membrane sensors for the determination of rivastigmine hydrogen tartrate. Chem Pharm Bull (Tokyo) 56:753–7
- Fazil M, Md S, Haque S, et al. (2012). Development and evaluation of rivastigmine loaded chitosan nanoparticles for brain targeting. Eur J Pharm Sci 47:6–15
- Francis PT, Palmer AM, Snape M, Wilcock GK. (1999). The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry 66:137–47
- Jesorka A, Orwar O. (2008). Liposomes: technologies and analytical applications. Annu Rev Anal Chem (Palo Alto, CA) 1:801–32
- Johnson PH, Quay SC. (2005). Advances in nasal drug delivery through tight junction technology. Expert Opin Drug Deliv 2:281–98
- Kreuter J, Alyautdin RN, Kharkevich DA, Ivanov AA. (1995). Passage of peptides through the blood–brain barrier with colloidal polymer particles (nanoparticles). Brain Res 674:171–4
- Kurz A, Farlow M, Lefevre G. (2009). Pharmacokinetics of a novel transdermal rivastigmine patch for the treatment of Alzheimer's disease: a review. Int J Clin Pract 63:799–805
- Law SL, Huang KJ, Chou HY. (2001). Preparation of desmopressin-containing liposomes for intranasal delivery. J Control Release 70:375–82
- Milla P, Dosio F, Cattel L. (2012). PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr Drug Metab 13:105–19
- Moore J, Flanner H. (1996). Mathematical comparison of curves with an emphasis on in-vitro dissolution profiles. Pharm Technol 20:64–74
- Moretti R, Torre P, Vilotti C, et al. (2007). Rivastigmine and Parkinson dementia complex. Expert Opin Pharmacother 8:817–29
- Rodgers AB. (2011). Alzheimer's disease unraveling mystery. Available at: http://www.nia.nih.gov/alzheimers/publication/alzheimers-disease-unraveling-mystery/preface
- Romeo VD, de Meireles JC, Gries WJ, et al. (1998). Optimization of systemic nasal drug delivery with pharmaceutical excipients. Adv Drug Deliv Rev 29:117–33
- Rovira-Bru M, Thompson DH, Szleifer I. (2002). Size and structure of spontaneously forming liposomes in lipid/PEG-lipid mixtures. Biophys J 83:2419–39
- Schnyder A, Huwyler J. (2005). Drug transport to brain with targeted liposomes. NeuroRx 2:99–107
- Schroeder U, Sommerfeld PSabel BA. (1998). Efficacy of oral dalargin-loaded nanoparticle delivery across the blood–brain barrier. Peptides 19:777–80
- Seju U, Kumar A, Sawant KK. (2011). Development and evaluation of olanzapine-loaded PLGA nanoparticles for nose-to-brain delivery: in vitro and in vivo studies. Acta Biomater 7:4169–76
- Siepmann J, Siegel RA, Rathbone MJ. (2011). Fundamentals and applications of controlled release drug delivery. Boston, MA: Springer Science & Business Media
- Squire LR, Zola-Morgan S. (1991). The medial temporal lobe memory system. Science 253:1380–6
- Szoka F, Papahadjopoulos D. (1978). Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc Natl Acad Sci 75:4194–8
- Tas C, Ozkan CK, Savaser A, et al. (2006). Nasal absorption of metoclopramide from different Carbopol 981 based formulations: in vitro, ex vivo and in vivo evaluation. Eur J Pharm Biopharm 64:246–54
- Thorne RG, Pronk GJ, Padmanabhan V, Frey WH 2nd. (2004). Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 127:481–96
- Vyas TK, Shahiwala A, Marathe S, Misra A. (2005). Intranasal drug delivery for brain targeting. Curr Drug Deliv 2:165–75
- Were LM, Bruce BD, Davidson PM, Weiss J. (2003). Size, stability, and entrapment efficiency of phospholipid nanocapsules containing polypeptide antimicrobials. J Agric Food Chem 51:8073–9
- Woodbury DJ, Richardson ES, Grigg AW, et al. (2006). Reducing liposome size with ultrasound: bimodal size distributions. J Liposome Res 16:57–80
- Woodle MC. (1995). Sterically stabilized liposome therapeutics. Adv Drug Deliv Rev 16:249–65
- Xiang J, Fang X, Li X. (2002). Transbuccal delivery of 2′,3′-dideoxycytidine: in vitro permeation study and histological investigation. Int J Pharm 231:57–66
- Yadav A, Murthy M, Shete AS, Sakhare S. (2011). Stability aspects of liposomes. Indian J Pharm Educ Res 45:402–13
- Yamada T. (2004). The potential of the nasal mucosa route for emergency drug administration via a high-pressure needleless injection system. Anesth Prog 51:56–61
- Yang T, Choi MK, Cui FD, et al. (2007a). Preparation and evaluation of paclitaxel-loaded PEGylated immunoliposome. J Control Release 120:169–77
- Yang T, Cui FD, Choi MK, et al. (2007b). Enhanced solubility and stability of PEGylated liposomal paclitaxel: in vitro and in vivo evaluation. Int J Pharm 338:317–26
- Yang ZZ, Wang ZZ, Wu K, Qi XR. (2011). Preparation of rivastigmine liposome and its pharmacokinetics in rats after intranasal administration. Yao Xue Xue Bao 46:859–63
- Yang ZZ, Zhang YQ, Wang ZZ, et al. (2013). Enhanced brain distribution and pharmacodynamics of rivastigmine by liposomes following intranasal administration. Int J Pharm 452:344–54