
Abstract
Oral delivery is the most common mode of systemic drug application. Inhalation is mainly used for local therapy of lung diseases but may also be a promising route for systemic delivery of drugs that have poor oral bioavailability. The thin alveolar barrier enables fast and efficient uptake of many molecules and could deliver small molecules and proteins, which are susceptible to degradation and show poor absorption by oral application. The low rate of biotransformation and proteolytic degradation increases bioavailability of drugs but accumulation of not absorbed material may impair normal lung function. This limitation is more relevant for compounds that should be systematically active because higher doses have to be applied to the lung. The review describes processes that determine absorption of orally inhaled formulations, namely dissolution in the lung lining fluid and uptake and degradation by alveolar epithelial cells and macrophages. Dissolution testing in simulated lung fluid, screening for cytotoxicity and pro-inflammatory action in respiratory cells and study of macrophage morphology, and phagocytosis can help to identify adverse effects of pulmonary formulations.
1. Introduction
Oral and enteral routes are the preferred routes for systemic administration of drugs; 38% of all drugs are delivered by sublingual, buccal, oral, and rectal application (Marketsandmarkets, Citation2017). Pulmonary delivery, by contrast, is used almost exclusively for the treatment of pulmonary diseases. Medication for asthma, chronic obstructive pulmonary disease (COPD), and the combination of both, accounts for 73% of all inhaled medicines (http://www.gilberttechnologies.nl/market-opportunity/). Systemic applications (diabetes, hormone therapy, analgesics, anaphylaxis, influenza, and multiple sclerosis) represent only 18% of the marketed products. Fast absorption by the respiratory epithelium, reduced drug inactivation by first pass metabolization and independence from food uptake make drug delivery by the pulmonary route an attractive option for fast pain relief in migraine, vaccines in pandemic diseases, and emergency applications in labor. Natural peptides are rapidly inactivated by proteases in the gastrointestinal tract but can be applied by the pulmonary route (Patton et al., Citation2004). An important limitation of pulmonary delivery is the maximal amount of material that can be delivered, and the reduced capacity of the lung to clear not absorbed materials. In local therapy, with the exception of antibiotic treatment of cystic fibrosis with tobramycin (2 × 300 mg (Ramsey et al., Citation1999)), the required amounts of active pharmaceutical ingredients (APIs) are low. For systemic therapy delivered doses have to be higher in order to achieve effective blood levels. Furthermore, for the treatment of chronic diseases, such as diabetes, APIs have to be applied repeatedly and for longer time periods. This poses the question how formulations are degraded, removed and metabolized at the respiratory epithelium. This is important because accumulation of insoluble material at the respiratory barrier may impair lung function. Respiratory impairment in pulmonary alveolar proteinosis is an extreme example for accumulation of not degraded material at the alveolar barrier (Huaringa & Francis, Citation2016).
More than 380 medications can induce pulmonary toxicity as side effect and illustrate the particular sensitivity of the lung. The most common manifestation is drug-induced interstitial lung disease (DILD (Schwaiblmair et al., Citation2012)). The large surface of the lung and the challenge by high levels of oxygen are seen as major contributors to the high vulnerability of lung tissue. Drugs with the highest potential for lung damage are the chemostatic drugs bleomycin, busulfan, and cyclophosphamide, the cardiovascular drugs amiodarone and hydroxymethylglutaryl-CoA reductase inhibitors, the anti-inflammatory drugs aspirin, methotrexate, gold, penicillamine, azathioprine and sulfasalazine, and antimicrobials like nitrofurantoin, amphotericin B, and sulfonamides. Furthermore, biological agents, such as tumor necrosis factor-alpha (TNF-α) blockers, anti-CD20 antibodies, recombinant Interferon-alpha (INF-α), and T-cell antiproliferative agents as well as bromocriptine and cabergoline may cause lung damage. For most of the agents the exact mechanism is not known. The higher accumulation of certain drugs in the lung than in other organs plays a role as well as the lung-specific bioactivation and the reaction to the activation. Due to its high incidence and high mortality of 40–50%, amiodarone-induced lung disease is one of the best-studied diseases. The intracellular accumulation of phospholipids in Μφs and alveolar type II cells is regarded as pathognomonic for DILD. The link of lipid-loaded Μφs to pulmonary toxicity has been confirmed for the antiarrhythmic agent amiodarone, various hydroxymethylglutaryl-CoA reductase inhibitors, and the antidepressant fluoxetine hydrochloride (Dean et al., Citation1987; Israel-Biet et al., Citation1987; Lapinsky et al., Citation1993; Gonzalez-Rothi et al., Citation1995; Huang et al., Citation2013). Animal studies suggested the link of the intracellular amiodarone accumulation to decreased pulmonary clearance, while another study reported macrophage activation by the compound (Ferin, Citation1982; Reasor et al., Citation1996). In lung toxicity induced by the serotonin reuptake inhibitor venlafaxine also interaction with the drug metabolizing CYP450 system was involved (Ferreira et al., Citation2014). Furthermore, propellants of pressurized metered dose inhalers and carriers in dry powder inhalers can cause pulmonary irritation (Patil & Sarasija, Citation2012; Myrdal et al., Citation2014).
2. Defense systems of the lungs
The architecture of the airways presents a barrier for the deposition of inhaled particulates in the respiratory system. Deposition is size-dependent and particles for inhalation are designed for optimal deposition in the respiratory tract, which is maximal (around 40%) for particles of 2–4 μm (Miller, Citation2000). Once particles get into the lungs they can be removed by acellular systems (mucociliary escalator) and by cellular defense mechanisms (alveolar macrophages, AMs). If compensation systems are exhausted lung overload occurs with morphological lung changes and respiratory dysfunction. It has been speculated that impairment of AMs is the underlying reason (Morrow, Citation1988). At an amount of 6% phagocytized material decrease in the function of AMs occurs and at 60% cessation of AM function is assumed. The phenomenon of lung overload has been identified in rats and it is still unclear whether it occurs in the same way in humans and how rat effects can be extrapolated to the human situation.
2.1 Lung lining fluid
The lung lining fluid represents a protective barrier for the underlying epithelium. In the proximal parts of the lung (large airways), the thickness of the lung lining fluid ranges from 5 to 10 μm and can completely surround inhaled particles (Olsson et al., Citation2011). The lung lining fluid consists of mucus and particles sticking or being immersed in the mucus are cleared from the lungs by transport via motile elements of bronchial epithelial cells (cilia) to the pharynx. The mechanism is termed mucociliary clearance and most efficient for particles >6 μm (El-Sherbiny et al., Citation2011). Up to 90% of the inhaled particles are removed within 24 h (Evans & Koo, Citation2009). The mucus layer measures 2–8 μm in the bronchi and 1.8–3 μm in bronchioles (National Research Council, Citation1977; Patton & Byron, Citation2007; Wauthoz & Amighi, Citation2015). Focal increases of the layer up to 20 times can occur but some small bronchi may completely lack a mucus layer (Hiemstra, Citation2010). The mucus layer consists of 97% water and 3% solids (mucins, non-mucin proteins, salts, lipids, cellular debris). Mucins are large glycoproteins with charged parts and hydrophobic regions. They form fibers of 3–10 nm in diameter with coverage of typically 20–30 carbohydrates per 100 amino acids. The high content in sialic acid and sulfate creates a strongly negative charge density and renders the polymer rigid by charge repulsion (Lai et al., Citation2009). Entangled mucins and other mucus constituents with reversible linkage to the polymer, such as lipids and associated proteins (e.g. IgA), primarily form the mucus mesh and also determine the viscosity of mucus. The effective mesh spacing is 30–100 nm (Murty et al., Citation1984). Hydrophilic APIs are better soluble in mucus than hydrophobic molecules and permeation of the mucus layer is best for uncharged small (<10 nm) APIs (Fröhlich & Roblegg, Citation2014).
The thickness of the lining fluid in the alveoli (surfactant layer) measures 0.07–0.3 μm and is arranged as a thicker aqueous and a thin lipid layer (National Research Council, Citation1977; Patton & Byron, Citation2007; Wauthoz & Amighi, Citation2015). While respiratory mucus does not show prominent differences to gastrointestinal mucus, the surfactant layer of the alveolus has a unique composition. Surfactant consists of 92% lipids with 41% dipalmitoyl phosphatidylcholine (DPPC) as the main component. Unsaturated phosphatidylcholine contributes with 25%, other lipids with 26% and proteins only with 8% (Parra & Perez-Gil, Citation2015). Out of these proteins surfactant protein (SP) A has the main contribution with 6%, SP-B and SP-C make up 1% each, and SP-D <0.5% (Green et al., Citation2010). Analysis of bronchoalveolar lavage fluids showed a mixture of lamellar bodies with different numbers of layers and tubular myelin (Goerke, Citation1998). Ultrastructural images of the surfactant layer in lung sections indicated various arrangements, partly due to the respiration state in which the lungs were fixed and on the age of the animal. The lining in young animals consists of membranes that show a mesh-like regular structure of up to 200 nm thickness (Walski et al., Citation2009), while the surfactant layer in senescent rats had an irregular appearance with membranous blebs and absence of a regular myelin-tubular mesh (Tomashefski & Farver, Citation2008). Modeling data indicate that pure DPPC is interspersed with regions of mixed lipid and protein, which are arranged as bilayers and multilayers (Harishchandra et al., Citation2010).
The different presentations of surfactant can be described as follows. Surfactant is produced in alveolar type II cells as lamellar inclusion bodies (), which are secreted into the alveolar space as lamellar bodies consisting of different numbers of myelin layers () and form a partly crystallized hypophase of tubular myelin (). The DPPC-enriched layer on top of the aqueous sub- or hypophase has a double- or multi-layered structure ().
Figure 1. Presentations of surfactant. Alveolar type II cells contain surfactant as intracellular lamellar bodies (a). In the alveolar hypophase lamellar bodies with different numbers of myelin layers (b) and membranes arranged as tubular myelin (c) are seen. On top of the hypophase the surfactant is arranged as multilayer (double arrow) or bilayer (one arrow) (d). Examples were taken from (Schurch et al., Citation1995; Walski et al., Citation2009).

2.2 Cellular clearance
While particles in the upper parts of the respiratory tract are removed by mucociliary clearance, AMs are responsible for degradation and elimination of API particles that were deposited in the deep lung. AMs are a member of the diverse group of phagocytic cells. Macrophages (Mφs) can polarize into different classes, which are roughly described as inflammatory M1 cells and immune modulatory M2 cells. M2 cells can further polarize into types that play an important role in tumor development and progression (Allavena & Mantovani, Citation2012). M2a cells induce Th2 response, promote type II inflammation, and help in killing parasites. M2b cells suppress tumor growth, induce Th1 response, and control metastasis. M2c are involved in matrix deposition and tissue remodeling and M2d tumor-associated Mφs promote angiogenesis (Weagel et al., Citation2015).
Mφs in the lung include AMs and interstitial Mφs (IMs). The latter are located between alveolar epithelium and vascular endothelium and can migrate into the alveolus to become AMs (Boorsma et al., Citation2013). IMs have a lower capacity for phagocytosis but a higher rate of IL-10 secretion. In addition to M1 and M2 cells, M2-like cells have been identified in the human lung (Satoh et al., Citation2013). The populations of Mφs undergo specific changes in lung pathologies. M2-like cells decrease, M2 cells increase, and M1 cells first increase and then decrease in asthma. M2-like cells in COPD decrease slightly, while M1 and M2 cells increase. All Mφs in COPD are dysfunctional. In lung fibrosis M2-like, M2, and M1 cells are increased (Boorsma et al., Citation2013).
In the healthy lung AMs are not activated. They are tethered to alveolar epithelial cells and show slow turnover. Proliferation and survival is regulated by macrophage colony-stimulating factor (M-CSF, CSF-1)/granulocyte macrophage colony-stimulating factor (GM-CSF) (Vlahos & Bozinovski, Citation2014). These growth factors have slightly different roles by promoting preferentially M1 polarization (GM-CSF) or M2 polarization (M-CSF) (Mia et al., Citation2014). The attachment to the alveolar epithelium via integrin αvβ6 is important to keep AMs in the quiescent state. Furthermore, tumor growth factor beta (TGF-β), secreted by various cell types in the lung, inhibits AM activation. Stimulation by pathogens induces the switch to the M1 state with secretion of pro-inflammatory cytokines, monocyte recruitment, stimulation of alveolar cells and effector T cells. After removal of the stimulus, reprogramming toward M2 state with abrogation of inflammation, cessation of cell recruitment, apoptosis of inflammatory cells, interaction with regulatory T cells, secretion of lipoxins and resolvins and growth factors for epithelial cell repair, and AT2 to AT1 transition take place (Aggarwal et al., Citation2014). The secreted proteases activate latent TGF-β and reconstitute the resting state of the AMs (Vlahos & Bozinovski, Citation2014).
AMs migrate in the alveolar lining layer of around 200 nm thickness (Bastacky et al., Citation1995). In human lungs obtained from surgery and cadavers 22% of the lung cells were classified as alveolar epithelial cells compared to 3.25% AMs (Crapo et al., Citation1982). This would correspond to a relative ratio of ∼1:7 but ratios of one AM to forty alveolar epithelial cells in human lungs have also been reported (Crabbe et al., Citation2011). Fourteen to fifteen AMs have been determined in one human alveole (Geiser, Citation2010). Although human AMs are more than two times larger than rat and baboons cells, when referred to lung surface, little differences in surface coverage between rodents and humans were detected. On the average, one AM per 18,800 μm2 (rodents) or one AM/17,100 μm2 (humans) was seen (Miller, Citation2000; Geiser, Citation2010). When taking an average size of murine AMs of 121 μm2 into account less than one percent of the alveolar surface area is covered by AMs. Based on an alveole surface in mice of 3620 μm2 (Knust et al., Citation2009) and the AM area of 121 μm2 this leads to coverage of 3.34% of the alveolar surface (Rodero et al., Citation2015). This indicates that AMs have to move to ingest and remove deposited particles. Cell mobility is determined by speed (how fast a cell is moving) and persistence (time a cell spends moving in a given direction) ( based on (Lauffenburger & Linderman, Citation1993)).
Figure 2. Illustration of cell mobility as result of speed and persistence.
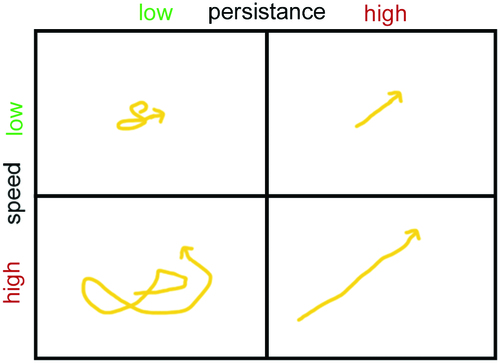
Persistence time can be influenced for instance by the secretion of chemoattractant. Neutrophilic granulocytes, which possess high speeds of 20 μm/min and low persistence times of 4 min in the normal situation can be guided quickly to inflamed areas by increasing persistence time through secretion of cytokines. In rat AMs speeds of 2 μm/min and persistence times of 30 min have been determined, while human Mφs appear to have lower speeds of 1 μm/min (Bzymek et al., Citation2016). Studies on murine lung explants determined the velocities of AMs as 2 ± 1.5 μm/min (Rodero et al., Citation2015).
Calculations by Pollmächer and Figge indicate that a speed of 4 μm/min would be sufficient to find a conidium of Aspergillus fumigatus within 6 h before it starts to germ (CitationPollmacher & Figge, 2015). In the presence of cytokines and with recruitment of additional AMs 2 μm/min would be sufficient. This situation cannot be directly transferred to the situation of inhalation exposure because not one but many particles deposit in one alveole. Other simulations estimate about 5 min as sufficient for AM to patrol across the entire alveolus (Gradon & Podgorski, Citation1995). High efficacy of AMs for particle removal has been found in vivo. Particles of 0.1, 1, and 2 μm size were cleared to 85–90% from the airways (Hofmann & Asgharian, Citation2002). Non-biodegradable particles of 3–6 μm in diameter were phagocytized by Mφs to more than 80% within 24 h (Geiser, Citation2002) and biodegradable particles containing growth hormone were removed by Mφs within 24 h to 70% (Patton et al., Citation1989). In the range of 0.5–4 μm instilled insoluble particles are cleared within 100–200 d from human lungs and lungs of large animal species compared to <50 d in rodent lungs (Kreyling, Citation1990). The lower extent of clearance suggests that the particle material plays a prominent role in the velocity of the clearance.
2.3 Degradation and metabolization
Absorption of substances into the systemic circulation takes place, mainly, at the alveolar epithelium. Alveolar type I (AT1) cells represent only 8.3% of the entire cell population of the lung but cover about 93% of the lung surface (Simon, Citation1992). They are flat thin cells with long processes and a surface of about 5 μm2/cell. Alveolar type II (AT2) cells are about twice as frequent as AT1 cells but the ratio is 1:43 according to surface coverage (Stone et al., Citation1992). Their main functions include synthesis and secretion of surfactant, xenobiotic metabolism, transepithelial movement of water, and regeneration of AT1 cells (Castranova et al., Citation1988). Furthermore, AT2 cells are a rich source of antioxidant enzymes; they contain much higher levels of superoxide dismutase, glutathione peroxidase, and glucose-6-phosphate dehydrogenase than AMs and they are more resistant to oxygen exposure than other lung cells, in particular AT1 cells. AT2 cells secrete surfactant lipid and proteins, complement components, prostaglandins, lysozyme, and glutathione.
Both types of alveolar cells can ingest particles up to 200 nm by active mechanisms (endocytosis) (Lankoff et al., Citation2012). Dissolved material can be absorbed by diffusion and carrier-mediated uptake. Absorption of small hydrophobic molecules is fast and occurs within 1–2 min, for small hydrophilic molecules absorption takes 65 min and the absorption of peptides is still unclear (Liao & Wiedmann, Citation2003; Patton & Byron, Citation2007; Mansour et al., Citation2009).
APIs are metabolized by the same group of enzymes as in the gastrointestinal tract. The cytochromes P450 (CYPs) constitute the major enzyme family capable of catalyzing the oxidative biotransformation. CYP450 enzymes are so named because they are bound to membranes within a cell (cyto) and contain a colored heme pigment (chrome and P). There are more than 50 CYP450 enzymes, but the CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, and CYP3A5 enzymes metabolize 90% of drugs (Lynch & Price, Citation2007). Gene polymorphism of these enzymes is a major reason for the inter-individual variations in drug levels and has lead to the classification of individuals into poor, intermediate, extensive, and ultrarapid metabolizers. Activities of metabolizing enzymes in the human alveolar epithelium are 1–10% of those in hepatocytes (Somers et al., Citation2007) and analysis of 10 human lung samples ranked mRNA expression in the following order: CYP1B1, CYP2B6 > CYP2E1 > CYP2C9 > CYP1A1, CYP3A4 and CYP3A5 (Castell et al., Citation2005). Variations were highest for CYP1A1 followed by CYP2E1 expression, and relatively little variation for the remaining enzymes. Protein expression in human lungs confirmed the gene expression of CYP1A1, CYP1B1, CYP2B6, CYP2E1, and CYP3A5 (Hukkanen et al., Citation2002). AT1 cells mainly express CYP1A1 and CYP2B1 (McElroy & Kasper, Citation2004) and AMs contain predominantly CYP3A5 (Hukkanen et al., Citation2003) (). In contrast to rodent lungs, human non-ciliated bronchiolar epithelial cells (Club or Clara cells) possess only low levels of CYP enzymes and do not play a prominent role in pulmonary metabolization of APIs. The ABC (ATP-binding cassette) transporters contribute toward detoxification of xenobiotics by cellular export. Multidrug resistance protein 1/p-glycoprotein (MDR-1/P-gp) is the main exporter for gastrointestinal absorption, biliary, and urinary excretion and regulation of entry into the central nervous system (Vrbanac & Slauter, Citation2013). MDR-1/P-pg is also highly expressed in the bronchial epithelium and in AMs (van der Deen et al., Citation2005). AT1 cells express the transporter at the luminal site and in intermediate levels (Campbell et al., Citation2003), while AT2 cells express MDR-1/P-pg only under oxidative stress and not in the normal condition (Weidauer et al., Citation2004).
Figure 3. Dissolution and enzymatic degradation of drug-loaded particles by alveolar type I cells (AT1) with indication of the main degrading and metabolizing enzymes. Particle dissolution is slow because particles are only partly immersed in the alveolar lining fluid (hypophase). AT1 cells secrete proteases (EP24.15) in the hypophase. AT1 cells possess various membrane-associated proteases (CPM, APA, APB, APN, EP24.11, and γ-GT), and the lysosomal protease cathepsin D (CatD). The metabolizing enzymes CYP1A1 and CYP2B1 are located in the endoplasmic reticulum. The main transporter MDR-1/P-pg is located at the apical plasma membrane. Abbreviations: CPM: carboxypeptidase M; APA: aminopeptidase A; APB: aminopeptidase B; APN: aminopeptidase N; EP24.11: endopeptidase 24.11; γ-GT: gamma-glutamyltransferase; Cyt: cytoplasm; N: nucleus; P-pg: P-glycoprotein.
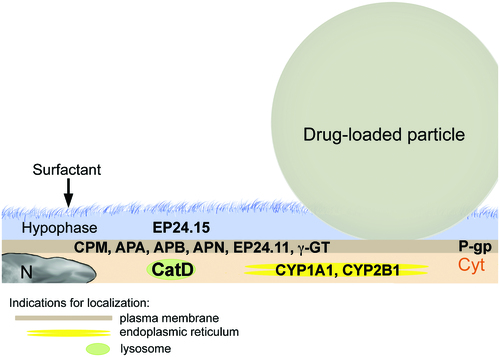
Not only metabolizing enzymes are present at a lower level than in the gastrointestinal tract, also proteolytic activity of the lung is relatively low and partly insufficient to degrade delivered proteins (Okumura et al., Citation1992). Inhaled superoxide dismutase was found as proteinous aggregates in AMs and in the lung lining fluid (Welty-Wolf et al., Citation1997). Respiratory epithelial cells contain aminopeptidase N (APN), and dipeptidyl peptidase IV, neutral endopeptidase, and various cathepsins (Cohen et al., Citation1996; Juillerat-Jeanneret et al., Citation1997). Aminopeptidases A, B, and N, gamma-glutamyltransferase, endopeptidases 24.11 (enkephalinase), and 24.15 (metalloendopeptidase) are localized at the plasma membrane of AT1 cells (Horalkova et al., Citation2009). AT2 cells express lysosomal enzymes cathepsin C (dipeptidyl peptidase I), tripeptidyl peptidase I and cathepsin H in their lamellar bodies (Ishii et al., Citation1991). Out of the lysosomal enzymes, only cathepsin D is expressed by AT1 cells to a greater extent than by AT2 cells (Kasper et al., Citation1996). The lysosomal enzymes cathepsin B, H, and L are present in both AMs and alveolar epithelial cells (Ishii et al., Citation1991; Yayoi et al., Citation2001; Yin et al., Citation2005). Although Mφs of the lungs contain higher concentrations of proteolytic enzymes than the alveolar cells, they did not play a prominent role in the degradation of insulin. It was reported that degradation of insulin occurred mainly in AT2 cells and only to a low extent in AMs (Finch, Citation2006). In general, degradation of insulin is lower in the lung than in subcutaneous tissue. Clearance of particles and APIs can further by impaired by external stressors. Smoke, air-borne particulate matter and carbon nanotubes and slowly biodegradable nanoparticles in general decrease AM function, mainly phagocytosis (Fick et al., Citation1984; Kotani et al., Citation2000; Renwick et al., Citation2001; Moss & Wong, Citation2006; Brown et al., Citation2007; Hodge et al., Citation2007; Boyles et al., Citation2015). The consequence of accumulation of non-biodegradable particles, such as airborne particulate matter, in the deep lung was inflammation leading to fibrous transformation and lung cancer (Bonner, Citation2007; Winterbottom et al., Citation2014). Critical evaluation on the pulmonary effects of inhaled human insulin (rDNA origin in Exubera®) could not exclude adverse effects of the inhaled insulin in combination with additional stressors of the respiratory system, such as smoking (Seymour, Citation2006).
In summary, dissolution, degradation, and metabolization of APIs at the alveolar barrier plays an important role for systemic efficacy and pulmonary effects of orally inhaled formulations ().
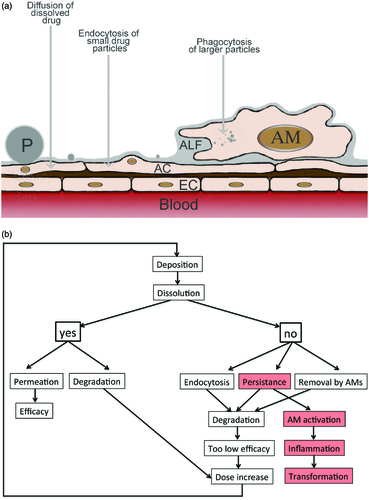
Physiological mechanisms to prevent overload of the lungs with foreign substances and particles comprises the following protective mechanisms. (1) The architecture of the respiratory tract reduces the deposition of particles. (2) Particles deposited on the mucus layer of the larger airways (trachea, bronchi, and bronchioles) are transported to the pharynx to be swallowed. (3) In the smaller parts of the airways (terminal bronchioles, alveoli) non-dissolved API particles are removed by AMs. (4) Particles in the nanosize (<200 nm) can be ingested by alveolar epithelial cells. (5) Degradation and metabolization of APIs occurs at the epithelial membrane, inside epithelial cells and AMs, and by enzymes in the hypophase of the surfactant layer (). (6) Dissolved APIs are removed from the respiratory epithelium by permeation (diffusion, carrier-mediated transport, and paracellular transport) across the respiratory epithelium.
Figure 4. Fate of API formulations in the alveoli as scheme (a) and flow diagram (b). a: Particles can dissolve and diffuse across the alveolar epithelium. Alveolar epithelial cells (ACs) actively ingest small particles while larger particles are phagocytized by AMs. Abbreviations: EC: endothelial cell; P: particle. b: When API particles dissolve fast, either therapeutic levels can be reached in the blood or degradation in alveolar epithelial cells occurs leading to insufficient activity. When dissolution is insufficient small API particles can be taken up by the alveolar epithelial cells and be degraded. AMs can ingest and degrade larger particles that persist at the alveolar barrier. Degradation may result in low systemic drug levels and be counteracted by increase of the applied dose. Persistent particles may also activate AMs and cause inflammation and tissue transformation.
3. In vitro testing
For the preclinical safety studies of formulations for oral inhalation experiments in two species are mandatory and, usually, in vivo experiments are started with rodents. By nebulization in the air only few particles will reach the deep lung because rodents are obligatory nose breathers. Application directly to the lung via intratracheal instillation is invasive and bypasses the defense system of the upper respiratory tract. Particles applied by oropharyngeal aspiration or by intratracheal intubation can partly be cleared by mucociliary clearance because solutions are applied at the beginning of the trachea. Since the latter techniques are atraumatic, they are regarded as more physiologic than intratracheal instillation (Fröhlich & Salar-Behzadi, Citation2014; Ribero et al., Citation2015). The applied particle dose is influenced by variable loss in the application device. Furthermore, the commonly used volumes of 50–100 μl are much higher than the total amount of epithelial lining fluid in rodents (45–55 μl in rats and 5–15 μl in mice). The application by itself, therefore, may alter the normal lung physiology (Fernandes & Vanbever, Citation2009).
There are also species differences between rodents and man that have to be taken into account in the interpretation of experimental data from rodents (). The branching of the airways is dichotomous in humans, while the rat bronchial tree has a monopodial airway branching, which may result in different particle deposition (Hofmann et al., Citation1989). The lower viscosity of mucus produced by serous glands in rodents compared to the mixed submucosal glands and intraepithelial glands may influence lung physiology. Mucus clearance was reported to be faster in human than in rat lungs (Fernandes & Vanbever, Citation2009). Further differences regard the cellular composition of the lower respiratory tract and the higher levels of metabolizing enzymes in rat lungs compared to human lungs. Ethoxyresorufin deethylase (EROD, substrate for CYP1A/B) activity, ethoxycoumarin O-deethylase (ECOD, substrate for several CYPs) activity and aryl hydrocarbon hydroxylase (AHH, substrate for CYP1A1) activity per mg of lung tissue were considerably higher in rats than in humans (Carlson, Citation2008). This can explain the about 10 times higher metabolization of cyclosporine per mg protein in rat compared to human lungs (Vickers et al., Citation1997). The distribution of specific cell types differs in the way that Club cells are present in the terminal bronchioles of rat lungs but not of human lungs (Harkema, Citation2000). Serous cells are present in rat lungs, not in human lungs (Hruban, Citation1984). In order to estimate the availability of APIs in a given formulation at the respiratory barrier in vitro tests can be performed in order to identify the most promising formulations for in vivo testing and support data from animal experiments. Dissolution testing provides an idea how long non-dissolved particles may remain on the alveolar surface and may be ingested by AMs. A second assessment can identify cellular accumulation and the effect of the API particles on morphology and function of AMs.
Table 1. Differences between rodent and human lungs.
3.1 Dissolution
Due to the low amount of lung lining fluid (∼20 ml) dissolution of APIs in the respiratory tract is slower than in the gastrointestinal tract. It is estimated that the particles used for inhalation (1–3 μm), which are deposited in the alveoles are only partly surrounded by fluid because the height of the alveolar lining fluid has been indicated as 70–300 nm (National Research Council, Citation1977; Patton & Byron, Citation2007). The speed of dissolution determines whether particles can be removed by AMs rather than permeate the alveolar barrier. Once the API particles are dissolved log p values indicate by which route and how fast absorption across the epithelial layer occurs. Lipophilic drugs with a log p > 0 are rapidly absorbed via the transcellular route within ∼1 min. Hydrophilic drugs with a log p < 0 are absorbed via the paracellular route with an absorption time of approximately 1 h (Patton et al., Citation2004). The permeation of poorly soluble inhaled corticoids (fluticasone propionate, mometasone furoate, beclomethasone dipropionate) and antimicrobials (ciprofloxacin betaine, amphotericin B) at high doses is limited by dissolution (Riley et al., Citation2012). While fluticasone propionate and beclomethasone dipropionate have dissolution times of >5 h, the mean dissolution time of budesonide is 0.1 h suggesting that the former APIs may accumulate at the respiratory barrier. Slow dissolution of drugs might be advantageous for local therapy with anti-inflammatory drugs and with APIs for the treatment of pulmonary infections and pulmonary arterial hypertension. For these indications sustained release is intended and formulations with modified release are being developed (Tiwari et al., Citation2012; Loira-Pastoriza et al., Citation2014). For systemic therapy, however, the advantage of persistence at the alveolar epithelium is less clear.
Since no standardized setup and protocols are available for dissolution testing of inhaled formulations, various methods have been published ().
Table 2. Overview on dissolution methods used for orally inhaled formulations.
Jaspart et al. described a method where powder was sealed in a filter membrane and immersed within the basket of a Type I dissolution apparatus (Jaspart et al., Citation2007). This method demonstrated some issues related to the contact area with the powder and appears to be suitable mainly for high-solubility drug products. In most protocols, powders are collected onto polycarbonate membranes or directly onto the stainless steel collection base of the impactors (Son & McConville, Citation2009; Son et al., Citation2010). Particle collection on the plates of the impactors may influence the aerodynamic flow profiles of the particles (Riley et al., Citation2012). In order to avoid this modified cups with a removable impaction insert have been developed. Specified powder cut from each stage is collected on a polycarbonate membrane and covered with a membrane of the same material (Son et al., Citation2010). The drug contained within the two membranes is clamped into the holder and immersed into the dissolution vessel, for instance a standard USP Method 2 apparatus containing 300 ml of dissolution fluid. Smaller particles on the impactor stage with a cutoff diameter of 0.94 μm have a large surface area to volume ratio and are expected to exhibit a faster dissolution rate in accordance with the standard Noyes–Whitney model (Son et al., Citation2010). Available studies used filter pore sizes of 0.45 μm and it has been reported that orientation of the filters had an impact on the dissolution rate (Jensen et al., Citation2011). Furthermore, the dissolution rate was dependent on drug loading (Son & McConville, Citation2009; Arora et al., Citation2010). The role of the membrane material has been studied systematically in Franz cells using ibuprofen (MW 206.4, log p 3.5, pKa 4.5) as the model drug. Membrane materials included regenerated cellulose, cellulose esters, cellulose nitrate, polyacrylonitrile, polyamide (nylon), polyethersulfone, polysulfone, polycarbonate, polypropylene, and polydimethylsiloxane. The membranes differed also in other parameters, thickness (10 μm 400 μm), pore size (0.05–0.45), porosity (8–84), and tortuosity (1–1.5). The authors concluded that the ideal high flux membrane for formulation analysis should have high porosity (> 60%), tortuosity of 1, and be relatively thin (∼10 μm) (Ng et al., Citation2010). A systematic study of various dissolution setup systems (paddle apparatus with membrane holder, flow through cell or Franz diffusion cell) identified the paddle apparatus as most appropriate to discriminate between good and poorly soluble substances (May et al., Citation2014). The main concern for the prediction of in vivo dissolution is the fact that the dissolution volume in the paddle apparatus (300 ml) is much higher than the amount of lung lining fluid, which has been indicated in most publications as 12–26 ml (Fröhlich et al., Citation2016). Another approach to determine dissolution is the combined chemical and microscopical evaluation of particle dissolution commercialized as DissolvIT®. The dissolution cell is positioned on top on an inverted microscope and perfused in flow-past configuration. The cell consists of an injection-molded polycarbonate cell with a porous polycarbonate membrane. There are currently too few data obtained with this system to conclude whether this system has advantages over the other setups (Börjel et al., Citation2014).
Several recipes for simulated lung fluid (SLF) are available in the literature ().
Table 3. Composition of simulated lung fluids used for dissolution studies.
Gamble’s solution, at acidic and neutral pH, has been developed for the testing of environmental particles (for instance: (Wragg & Klinck, Citation2007; Colombo et al., Citation2008; Sdraulig et al., Citation2008; Gray et al., Citation2010; Julien et al., Citation2011)). The acidic pH should mimic the situation in lysosomes of AMs and IMs, while the neutral pH should represent the interstitial fluid. Similar recipes were later employed for the testing of pulmonary drug delivery systems (examples: (Taylor et al., Citation2006; Yang et al., Citation2008; Ungaro et al., Citation2009)). Since alveolar lining fluid in vivo contains a high amount of DPPC (see section 2.1), the use of buffer + DPPC has been tested. Addition of 0.02% DPPC increased the dissolution of inhaled corticosteroids (Davies & Feddah, Citation2003). The positive value of adding DPPC to the dissolution solution is not unanimously accepted. It has been postulated that the formation of liposome aggregates may hinder the passage of drug through the membranes. To produce DPPC-liposomes for pulmonary delivery, Cook et al. used hydration of dry films produced by chloroform:methanol evaporation and sequential extrusion through 1, 0.4, and 0.2 μm membranes. The resultant liposomes had sizes of 168 ± 4.2 nm (Cook et al., Citation2005). In another protocol DPPC liposomes in SLF were prepared by sonication instead of extrusion without indication of size distribution (Son et al., Citation2010). The influence of the preparation method on the observed dissolution profiles is unknown. In addition to salts, SLF in some studies contained antioxidants (Wragg & Klinck, Citation2007) or DPPC (Stopford et al., Citation2003; Yang et al., Citation2008; Julien et al., Citation2011) and mucin plus albumin (Boisa et al., Citation2014). Llinas et al. screened dissolution of APIs with different logP, pKa, and intrinsic solubility in buffers containing either 0.5% sodium dodecyl sulfate (SDS), 0.025% DPPC or 0.003% Curosurf® (porcine lung surfactant extract). There was no SLF composition that provided optimal dissolution for all APIs. The main conclusions were that no differences in the dissolution between the SLFs was seen for hydrophilic APIs, that hydrophobic APIs profited from addition of SDS, that the effect of DPPC was similar to the natural surfactant Curosurf®, and that high ionic strength of the SLF decreased API solubility (Llinas et al., Citation2014). The evaluation of the best SLF should also take compatibility with cells into account. Determination of permeability as apparent permeability (Papp) value is a common parameter in formulation development. To create more physiologically relevant conditions, the routine protocol can be modified in the way that compounds are applied in physiological solutions, such as fasted simulated intestinal fluid (FaSSIF) for oral compounds (Mercuri et al., Citation2016). This requires the use of a non-cytotoxic ingredient in the simulated fluids. SDS in this respect is less ideal than DPPC and natural surfactants like Curosurf®.
3.2 Cellular screening
Cytotoxicity screening is the first assessment of drugs for all delivery routes. Generally, this testing uses cell lines relevant for the application and cell number, total protein, total DNA, cellular ATP content, enzyme activity, etc. of exposed cells compared to control cells are common readout parameters. Most often, cellular dehydrogenase activity by metabolization of tetrazolium salts to colored formazan salts is performed (Ekwall et al., Citation1990; Fröhlich et al., Citation2009). More specific assays to elucidate the mode of cellular action by assessment of membrane integrity, apoptosis, proliferation, generation of oxidative stress, and organelle function can follow. For cytotoxicity screening of APIs/formulations for pulmonary application various respiratory cells can be used. In general, A549 cells are deemed the most suitable because their CYP enzyme expression pattern is more typical for respiratory cells than that of other cell lines (Castell et al., Citation2005). A549 cells are derived from an adenocarcinoma of the lung and the phenotype resembles AT2 cells (Shapiro et al., Citation1978). In case difference between bronchial epithelial cells and alveolar cells is of interest, researchers usually use BEAS-2B bronchial cells. These cells are immortalized bronchial epithelial cells, which have been suggested as representative model for bronchial epithelial cells due the similarity of the expression pattern of metabolizing enzymes (Courcot et al., Citation2012). More recent data of CYP enzyme expression in BEAS-2B cells, however, confirm the expression pattern only in part (Garcia-Canton et al., Citation2013). The different cytotoxicity and genotoxicity of multi-walled carbon nanotubes in A549 and BEAS-2B cells may illustrate that bronchial and alveolar epithelial cells react differently (Ursini et al., Citation2014). The main limitation of BEAS-2B cells is the lack of mucus production. If action of mucus, therefore, is of interest, Calu-3 cells are the most appropriate models because they produce mucus when cultured in the physiologically relevant air-liquid interface culture, where cells are cultured on transwell inserts and supplied with medium only from the basolateral side (Meindl et al., Citation2015b). Furthermore, Calu-3 cells express metabolizing enzymes similar to BEAS-2B cells and, in addition, are useful indicators for disruption of the cell monolayer because they form a tight epithelial barrier (Foster et al., Citation2000; Ehrhardt et al., Citation2008). NuLi-1 cells may be also good models for evaluation of pulmonary effects as they also form tight intercellular junction formation (Molina et al., Citation2015). However, they capacity for mucus production and expression of CYP enzymes is not clear.
Routine cytotoxicity testing with cells seeded in plastic wells and exposure to different dilutions of the compound is the established procedure for compound screening. It is suggested to include also an assay for membrane integrity as gold standard for cytotoxic action (Niles et al., Citation2009). In the screening of new formulations physiologically more relevant exposure systems, such as air-liquid interface culture with exposure to the formulation as aerosol or suspended in simulated lung fluid, may improve the predictive value for reaction in vivo. In addition to cytotoxicity, evaluation of cellular oxidative stress may be a useful readout parameter as respiratory cells are subjected to higher concentrations of oxygen than other cells in the body. For poorly biodegradable compounds cellular content (accumulation) after repeated dosing of cells might be of interest. Prolonged culture (28 d) of Calu-3 cells in air-liquid interface culture is a way to identify such effects (Fröhlich & Meindl, Citation2015).
The identification of pro-inflammatory effects triggered either by epithelial cells of the respiratory tract or by AMs is very important. Increased secretion of pro-inflammatory cytokines (for instance IL-6, TNF-α, IL-1β, and IL-8) usually detected by enzyme-linked immunosorbent assay (ELISA), serves as indicator. Additional assays are available to identify adverse effects on Mφ function, for instance on chemotaxis, nitric oxide formation, phagocytosis, and oxidative burst (Prietl et al., Citation2014). Impairment of phagocytosis is induced when Mφs are exposed to poorly soluble particles, for instance air-borne particulate matter and carbon black particles (Lundborg et al., Citation2006). This exposure leads to morphological changes of Mφs in vivo, usually described as ‘foamy macrophages’, which were then proposed as indicators for adverse effects on Mφs. Similar changes are also observed after exposure to cationic amphiphilic drugs, for instance amiodarone, chloroquine, desipramine, and azithromycin (Shayman & Abe, Citation2013). According to one theory these drugs form intracellular complexes with phospholipids, which become then resistant to degradation. The cellular changes induce lysosomal fragility and proteolytic enzyme leakage (Forbes et al., Citation2014). Another theory hypothesizes lysosomal dysfunction as the cause, not the consequence, for the pathological changes (Shayman & Abe, Citation2013). Formation of the phospholipids may be caused either by inhibition of lipases or by increase of intralysosomal pH. The morphological changes have been termed ‘phospholipidosis’ (PLD) and are characterized by membrane-bound inclusions, primarily lysosomal in origin, with a lamellar structure (‘lamellar bodies’) (Nonoyama & Fukuda, Citation2008).
In the screening for adverse effects on Mφs mostly murine and rat cells are used. This is due to the fact that immortalized human Mφs are not available and the cells have to be differentiated from monocyte cell lines (THP-1, U937, etc.). Phorbol 12-myristate 13-acetate (PMA) induces the differentiation to Mφs in THP-1 cells, which can further differentiate into M1 and M2 class (Genin et al., Citation2015). Alternatively, differentiation of monocytes isolated from peripheral blood mononuclear cells (PBMCs) by stimulation with GM-CSF or M-CSF is possible (for instance: (Hassan et al., Citation1986; Jones et al., Citation1989; Daigneault et al., Citation2010)). The differentiation from circulating monocytes with GM-CSF could be an option to study human AMs because monocyte-derived Mφs and AMs isolated from bronchoalveolar fluid of the same individual showed similar rates of phagocytosis while expression of activation surface markers, differed (Forbes et al., Citation2014). It cannot be excluded that both isolations, AMs from bronchoalveolar lavage and Mφs differentiated from PBMCs, change the original phenotype. It is not possible to decide if data obtained with one of the systems is better than the other because human in vivo data for validation are lacking. By comparing Mφ differentiation from PBMCs and from THP-1 cells the authors reported higher cell yield after differentiation from THP-1 cells and higher increase of cell size in Mφs differentiated from PBMCs (Chitra et al., Citation2014). It is not known to which extent the different sources influence Mφ functions. Cell size, granularity, and surface marker expression are the main parameters for the characterization of the differentiated Mφs. It is assumed that the cell population is not homogenous and contains cells with different extent of differentiation and this heterogeneity may influence the assay results. This heterogeneity can be avoided by the use of murine cell lines because murine Mφs phagocytize particles similarly to human macrophages but behave more homogenous in culture (Gantt et al., Citation2001).
Murine RAW264.7 cells show morphological changes of PLD upon addition of serum and it is hypothesized that lipoproteins, cytokines and growth factors in the serum trigger these changes. Yao et al. identified micropinocytosis as main mechanism in the formation of lamellar bodies (Yao et al., Citation2009). In addition to inducing morphological changes, amiodarone impaired phagocytosis of J774A.1 cells, leaving the reaction to endotoxin challenge unchanged (Hoffman et al., Citation2015). Brasey et al., by contrast, reported only induction of morphological changes in RAW264.7 cells by amiodarone without impairment of phagocytosis (Brasey et al., Citation2011). It cannot be excluded that cell lines differ in their sensitivity to drug-induced PLD. Phagocytosis has, in any case, been suggested as very sensitive parameter for macrophage impairment (Renwick et al., Citation2001; Lundborg et al., Citation2006; Hoffman et al., Citation2015).
PLD can be detected using lipophilic dyes, such as Oil Red O, Sudan black, Nile red, osmium tetroxide, LipidTox®, paraphenylenediamine, etc. that accumulate in lipid-rich organelles (Brown et al., Citation1992; Hopkins et al., Citation2010). Vital dyes for lysosome function, namely acridine orange, Lyso-ID®, and Lysotracker® are also suitable to identify PLD-inducing drugs like chloroquine (Fröhlich et al., Citation2012; Meindl et al., Citation2015a). Furthermore, immunoreactivity against lysosome-associated membrane protein 2 (LAMP2) has been identified as an earlier marker for PLD (Mahavadi et al., Citation2015).
Various methods are suitable to quantify phagocytosis in vitro. Commonly used targets for phagocytosis are fluorescently labeled bacteria (mainly Escherichia coli, Staphylococcus aureus), IgG-coated and uncoated latex particles, and zymosan (Gu et al., Citation2014; Kapellos et al., Citation2016). Quantification is performed by spectrofluorometry, confocal microscopy, flow cytometry, imaging flow cytometry, and automated image analysis. Automated image analysis combines sensitivity with flexibility in magnification, real time kinetics, low cell numbers, and parallel assessment of viability (Kapellos et al., Citation2016). This techniques, also termed ‘high-content screening’, is the main technology for high-throughput cytotoxicity screening of drug candidates and can also identify changes in cell function or adaptive responses by the evaluation of organelle damage, changes in intracellular signaling, oxidative stress, etc. (Nichols, Citation2007). One problem in the screening for morphological changes in Mφs is that the clinical relevance of PLD is not entirely clear because >50 drugs, that caused PLD in different tissues, did rarely induce toxicity in patients when taken in prescribed doses (Forbes et al., Citation2014). This may suggest that pulmonary toxicity only occurs in combination with another stressor. One of the typical inducers of ‘foamy macrophages’ in vitro, amiodarone causes pulmonary toxicity in 10–20% of patients (Schwaiblmair et al., Citation2010). Histologic findings in these patients show morphological changes of PLD in AMs and AT2 cells (Nacca et al., Citation2012). These findings appear to indicate that screening for effects of inhaled formulations in Mφs has some predictive value for adverse effects in human lungs. Since there is no official FDA policy, drugs that exhibit PLD have been dealt with on a case-by-case basis by industry and FDA.
4. Conclusion
Accumulation of APIs at the respiratory barrier, cytotoxicity, and overload of Μφs accompanied by decreased function might be a problem for oral inhalation of drugs. The risk for accumulation is expected to be higher for systemic therapy with higher doses than for the low-dose local medication. In addition to a good physicochemical characterization (mass median diameter, geometric standard deviation, hygroscopicity, zeta potential, etc.) formulations should undergo also in vitro biological testing. Given the differences between rodents and human lungs combination of in vitro and in vivo experiments may improve the value of the preclinical studies. Important in vitro screening parameters are particle dissolution, cellular accumulation, cytotoxicity, generation of oxidative stress, and cytokine release in respiratory cells and Mφs, as well as phagocytosis and induction of phospholipidosis in Mφs. Approximate in vivo concentrations can be estimated by using the amount of lung lining fluid as distribution volume. APIs and formulations that cause cytotoxicity in the expected dose range should not further be developed. Non-cytotoxic formulations that also do not induce cytokine release and rapidly dissolve in SLF may not need more detailed investigations. For poorly soluble APIs characterization of effects on Mφs may be indicated. These studies should include cellular accumulation, morphological changes, and phagocytosis. In the evaluation of formulations it should be taken into account that not only the API but also excipients can cause the observed adverse effects.
Disclosure statement
The author reports no conflicts of interest.
Related Research Data
References
- Aggarwal NR , King LS , D’alessio FR. (2014). Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol 306:L709–25.
- Allavena P , Mantovani A. (2012). Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol 167:195–205.
- Arora D , Shah KA , Halquist MS , Sakagami M. (2010). In vitro aqueous fluid-capacity-limited dissolution testing of respirable aerosol drug particles generated from inhaler products. Pharm Res 27:786–95.
- Bastacky J , Lee CY , Goerke J , et al . (1995). Alveolar lining layer is thin and continuous: low-temperature scanning electron microscopy of rat lung. J Appl Physiol 79:1615–28.
- Boisa N , Elom N , Dean JR , et al . (2014). Development and application of an inhalation bioaccessibility method (IBM) for lead in the PM10 size fraction of soil. Environ Int 70:132–42.
- Bonner JC. (2007). Lung fibrotic responses to particle exposure. Toxicol Pathol 35:148–53.
- Boorsma CE , Draijer C , Melgert BN. (2013). Macrophage heterogeneity in respiratory diseases. Mediators Inflamm 2013:769214.
- Börjel M , Sadler R , Gerde P. (2014). The dissolvIt: an in vitro evaluation of the dissolution and absorption of three inhaled dry powder drugs in the lung. Respiratory Drug Delivery to the Lungs Conference, Poster. Stockholm, Swedan: Karolinska Institutet.
- Boyles MS , Young L , Brown DM , et al . (2015). Multi-walled carbon nanotube induced frustrated phagocytosis, cytotoxicity and pro-inflammatory conditions in macrophages are length dependent and greater than that of asbestos. Toxicol In Vitro 29:1513–28.
- Brasey A , Igue R , Djemame L , et al . (2011). The effect of in vitro exposure to antisense oligonucleotides on macrophage morphology and function. J Nucleic Acids Invest 2:77–84.
- Brown D , Kinloch I , Bangert U , et al . (2007). An in vitro study of the potential of carbon nanotubes and nanofibres to induce inflammatory mediators and frustrated phagocytosis. Carbon 45:1743–56.
- Brown WJ , Sullivan TR , Greenspan P. (1992). Nile red staining of lysosomal phospholipid inclusions. Histochemistry 97:349–54.
- Bzymek R , Horsthemke M , Isfort K , et al . (2016). Real-time two- and three-dimensional imaging of monocyte motility and navigation on planar surfaces and in collagen matrices: roles of Rho. Sci Rep 6:25016.
- Campbell L , Abulrob AN , Kandalaft LE , et al . (2003). Constitutive expression of p-glycoprotein in normal lung alveolar epithelium and functionality in primary alveolar epithelial cultures. J Pharmacol Exp Ther 304:441–52.
- Carlson GP. (2008). Critical appraisal of the expression of cytochrome P450 enzymes in human lung and evaluation of the possibility that such expression provides evidence of potential styrene tumorigenicity in humans. Toxicology 254:1–10.
- Castell JV , Donato MT , Gomez-Lechon MJ. (2005). Metabolism and bioactivation of toxicants in the lung. The in vitro cellular approach. Exp Toxicol Pathol 57:189–204.
- Castranova V , Rabovsky J , Tucker JH , Miles PR. (1988). The alveolar type II epithelial cell: a multifunctional pneumocyte. Toxicol Appl Pharmacol 93:472–83.
- Cheng YS , Dahl AR , Jow HN. (1997). Dissolution of metal tritides in a simulated lung fluid. Health Phys 73:633–8.
- Chitra S , Ganesan N , Lokeswari TS. (2014). Comparison of differentiation to macrophages in isolated monocytes from human peripheral blood and THP1 cells. Sri Ramacha J Med 7:1–7.
- Cohen AJ , Bunn PA , Franklin W , et al . (1996). Neutral endopeptidase: variable expression in human lung, inactivation in lung cancer, and modulation of peptide-induced calcium flux. Cancer Res 56:831–9.
- Colombo C , Monhemius AJ , Plant JA. (2008). Platinum, palladium and rhodium release from vehicle exhaust catalysts and road dust exposed to simulated lung fluids. Ecotoxicol Environ Saf 71:722–30.
- Cook RO , Pannu RK , Kellaway IW. (2005). Novel sustained release microspheres for pulmonary drug delivery. J Control Release 104:79–90.
- Courcot E , Leclerc J , Lafitte JJ , et al . (2012). Xenobiotic metabolism and disposition in human lung cell models: comparison with in vivo expression profiles. Drug Metab Dispos 40:1953–65.
- Crabbe A , Sarker SF , Van Houdt R , et al . (2011). Alveolar epithelium protects macrophages from quorum sensing-induced cytotoxicity in a three-dimensional co-culture model. Cell Microbiol 13:469–81.
- Crapo JD , Barry BE , Gehr P , et al . (1982). Cell number and cell characteristics of the normal human lung. Am Rev Respir Dis 126:332–7.
- Daigneault M , Preston JA , Marriott HM , et al . (2010). The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 5:e8668.
- Davies NM , Feddah MR. (2003). A novel method for assessing dissolution of aerosol inhaler products. Int J Pharm 255:175–87.
- Dean PJ , Groshart KD , Porterfield JG , et al . (1987). Amiodarone-associated pulmonary toxicity. A clinical and pathologic study of eleven cases. Am J Clin Pathol 87:7–13.
- Ehrhardt C , Forbes B , Kim K. 2008. In vitro models of the Tracheo-Bronchial epithelium. In: Ehrhardt C & Kim K , eds. Drug absorption studies. New York: Springer; 235–57.
- Ekwall B , Silano V , Paganuzzi-Stammati A , Zucco F. 1990. Toxicity tests with mammalian cell cultures. In: Bourdeau P , Somers E , Richardson G & Hickman J , eds. Short-term toxicity tests for non-genotoxiceEffects. Chichester: John Wiley & Sons Ltd.
- El-Sherbiny I , Villanueva D , Herrera D , Smyth H. 2011. Overcoming lung clearance mechanisms for controlled release drug delivery. In: Smyth H & Hickey AJ , eds. Controlled pulmonary drug delivery. New York: Springer; 101–26.
- Evans CM , Koo JS. (2009). Airway mucus: the good, the bad, the sticky. Pharmacol Ther 121:332–48.
- Ferin J. (1982). Alveolar macrophage mediated pulmonary clearance suppressed by drug-induced phospholipidosis. Exp Lung Res 4:1–10.
- Fernandes CA , Vanbever R. (2009). Preclinical models for pulmonary drug delivery. Expert Opin Drug Deliv 6:1231–45.
- Ferreira PG , Costa S , Dias N , et al . (2014). Simultaneous interstitial pneumonitis and cardiomyopathy induced by venlafaxine. J Bras pneumol 40:313–8.
- Fick RB, Jr ., Paul ES , Merrill WW , et al . (1984). Alterations in the antibacterial properties of rabbit pulmonary macrophages exposed to wood smoke. Am Rev Respir Dis 129:76–81.
- Finch G. 2006. Nonclinical pharmacology and safety studies of insulin administered to the respiratory tract. In : H. Salem & S. Katz , eds. Inhal toxicol. Boca Raton: CRC Press; 609–22.
- Forbes B , O’lone R , Allen PP , et al . (2014). Challenges for inhaled drug discovery and development: induced alveolar macrophage responses. Adv Drug Deliv Rev 71:15–33.
- Foster KA , Avery ML , Yazdanian M , Audus KL. (2000). Characterization of the Calu-3 cell line as a tool to screen pulmonary drug delivery. Int J Pharm 208:1–11.
- Fröhlich E , Meindl C. 2015. In vitro assessment of chronic nanoparticle effects on respiratory cells. In: Soloneski S & Larramendy M , eds. Nanomaterials – toxicity and risk assessment. Rijeka: InTech; 69–91.
- Fröhlich E , Meindl C , Roblegg E , et al . (2012). Action of polystyrene nanoparticles of different sizes on lysosomal function and integrity. Part Fibre Toxicol 9:26.
- Fröhlich E , Mercuri A , Wu S , Salar-Behzadi S. (2016). Measurements of deposition, lung surface area and lung fluid for simulation of inhaled compounds. Front Pharmacol 7:181.
- Fröhlich E , Roblegg E. 2014. Mucus as physiological barrier to intracellular delivery. In: Prokop A , Iwasaki Y & Harada A , eds. Intracellular delivery fundamentals and applications. Dordrecht: Springer Science + Business Media; 139–63.
- Fröhlich E , Salar-Behzadi S. (2014). Toxicological assessment of inhaled nanoparticles: role of in vivo, ex vivo, in vitro, and in silico studies. Int J Mol Sci 15:4795–822.
- Fröhlich E , Samberger C , Kueznik T , et al . (2009). Cytotoxicity of nanoparticles independent from oxidative stress. J Toxicol Sci 34:363–75.
- Gantt KR , Goldman TL , Mccormick ML , et al . (2001). Oxidative responses of human and murine macrophages during phagocytosis of Leishmania chagasi. J Immunol 167:893–901.
- Garcia-Canton C , Minet E , Anadon A , Meredith C. (2013). Metabolic characterization of cell systems used in in vitro toxicology testing: lung cell system BEAS-2B as a working example. Toxicol In Vitro 27:1719–27.
- Geiser M. (2002). Morphological aspects of particle uptake by lung phagocytes. Microsc Res Tech 57:512–22.
- Geiser M. (2010). Update on macrophage clearance of inhaled micro- and nanoparticles. J Aerosol Med Pulm Drug Deliv 23:207–17.
- Genin M , Clement F , Fattaccioli A , et al . (2015). M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 15:577.
- Goerke J. (1998). Pulmonary surfactant: functions and molecular composition. Biochim Biophys Acta 1408:79–89.
- Gonzalez-Rothi RJ , Zander DS , Ros PR. (1995). Fluoxetine hydrochloride (Prozac)-induced pulmonary disease. Chest 107:1763–5.
- Gradon L , Podgorski A. (1995). Displacement of alveolar macrophages in air space of human lung. Med Biol Eng Comput 33:575–81.
- Gray JE , Plumlee GS , Morman SA , et al . (2010). In vitro studies evaluating leaching of mercury from mine waste calcine using simulated human body fluids. Environ Sci Technol 44:4782–8.
- Green FH , Schnürch S , Amrein M , et al . 2010. The role of surfactant in particle exposure. In: Gehr P , Mühlfeld C , Rothen-Rutishauser B & Blank F , eds. Particle-lung interactions. Boca Raton: Taylor & Francis Group; 204–25.
- Gu BJ , Sun C , Fuller S , et al . (2014). A quantitative method for measuring innate phagocytosis by human monocytes using real-time flow cytometry. Cytometry A 85:313–21.
- Harishchandra RK , Saleem M , Galla HJ. (2010). Nanoparticle interaction with model lung surfactant monolayers. J R Soc Interface 7:S15–S26.
- Harkema J. 2000. Comparative structure of the respiratory tract: airway architecture in humans and animals. In: Cohen M , Zelikoff J & Schlesinger R , eds. Pulmonary immunotoxicology. Dordrecht: Kluwer Academic Publishers Group; 1–53.
- Hassan NF , Campbell DE , Douglas SD. (1986). Purification of human monocytes on gelatin-coated surfaces. J Immunol Methods 95:273–6.
- Hiemstra P. 2010. Host defense against infection in the airways. In Sethi S , ed. Respiratory infections. Boca Raton: CRC Press.
- Hodge S , Hodge G , Ahern J , et al . (2007). Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 37:748–55.
- Hoffman E , Kumar A , Kanabar V , et al . (2015). In Vitro Multiparameter Assay Development Strategy toward Differentiating Macrophage Responses to Inhaled Medicines. Mol Pharm 12:2675–87.
- Hofmann W , Asgharian B. (2002). Comparison of mucociliary clearance velocities in human and rat lungs for extrapolation modeling. Ann Occup Hyg 46:323–5.
- Hofmann W , Koblinger L , Martonen TB. (1989). Structural differences between human and rat lungs: implications for Monte Carlo modeling of aerosol deposition. Health Phys 57:41–6. discussion 46-7.
- Hopkins PM , Kermeen F , Duhig E , et al . (2010). Oil red O stain of alveolar macrophages is an effective screening test for gastroesophageal reflux disease in lung transplant recipients. J Heart Lung Transplant 29:859–64.
- Horalkova L , Radziwon A , Endter S , et al . (2009). Characterisation of the R3/1 cell line as an alveolar epithelial cell model for drug disposition studies. Eur J Pharm Sci 36:444–50.
- Hruban Z. (1984). Pulmonary and generalized lysosomal storage induced by amphiphilic drugs. Environ Health Perspect 55:53–76.
- Huang LK , Tsai MJ , Tsai HC , et al . (2013). Statin-induced lung injury: diagnostic clue and outcome. Postgrad Med J 89:14–9.
- Huaringa AJ , Francis WH. (2016). Pulmonary alveolar proteinosis: a case report and world literature review. Respirol Case Rep 4:e00201.
- Hukkanen J , Pelkonen O , Hakkola J , Raunio H. (2002). Expression and regulation of xenobiotic-metabolizing cytochrome P450 (CYP) enzymes in human lung. Crit Rev Toxicol 32:391–411.
- Hukkanen J , Vaisanen T , Lassila A , et al . (2003). Regulation of CYP3A5 by glucocorticoids and cigarette smoke in human lung-derived cells. J Pharmacol Exp Ther 304:745–52.
- Ishii Y , Hashizume Y , Watanabe T , et al . (1991). Cysteine proteinases in bronchoalveolar epithelial cells and lavage fluid of rat lung. J Histochem Cytochem 39:461–8.
- Israel-Biet D , Venet A , Caubarrere I , et al . (1987). Bronchoalveolar lavage in amiodarone pneumonitis. Cellular abnormalities and their relevance to pathogenesis. Chest 91:214–21.
- Jaspart S , Bertholet P , Piel G , et al . (2007). Solid lipid microparticles as a sustained release system for pulmonary drug delivery. Eur J Pharm Biopharm 65:47–56.
- Jensen B , Reiners M , Wolkenhauer M , et al . (2011). Dissolution testing for inhaled products. RDD Europe 2:303–8.
- Jones BM , Nicholson JK , Holman RC , Hubbard M. (1989). Comparison of monocyte separation methods using flow cytometric analysis. J Immunol Methods 125:41–7.
- Juillerat-Jeanneret L , Aubert JD , Leuenberger P. (1997). Peptidases in human bronchoalveolar lining fluid, macrophages, and epithelial cells: dipeptidyl (amino)peptidase IV, aminopeptidase N, and dipeptidyl (carboxy)peptidase (angiotensin-converting enzyme). J Lab Clin Med 130:603–14.
- Julien C , Esperanza P , Bruno M , Alleman LY. (2011). Development of an in vitro method to estimate lung bioaccessibility of metals from atmospheric particles. J Environ Monit 13:621–30.
- Kanapilly GM , Raabe OG , Goh CH , Chimenti RA. (1973). Measurement of in vitro dissolution of aerosol particles for comparison to in vivo dissolution in the lower respiratory tract after inhalation. Health Phys 24:497–507.
- Kapellos TS , Taylor L , Lee H , et al . (2016). A novel real time imaging platform to quantify macrophage phagocytosis. Biochem Pharmacol 116:107–19.
- Kasper M , Lackie P , Haase M , et al . (1996). Immunolocalization of cathepsin D in pneumocytes of normal human lung and in pulmonary fibrosis. Virchows Arch 428:207–15.
- Knust J , Ochs M , Gundersen HJ , Nyengaard JR. (2009). Stereological estimates of alveolar number and size and capillary length and surface area in mice lungs. Anat Rec (Hoboken) 292:113–22.
- Kotani N , Hashimoto H , Sessler DI , et al . (2000). Smoking decreases alveolar macrophage function during anesthesia and surgery. Anesthesiology 92:1268–77.
- Kreyling W. (1990). Interspecies comparison of lung clearance of “Insoluble” particles. J Aerosol Med 3:S93–S110.
- Lai SK , Wang YY , Wirtz D , Hanes J. (2009). Micro- and macrorheology of mucus. Adv Drug Deliv Rev 61:86–100.
- Lankoff A , Sandberg WJ , Wegierek-Ciuk A , et al . (2012). The effect of agglomeration state of silver and titanium dioxide nanoparticles on cellular response of HepG2, A549 and THP-1 cells. Toxicol Lett 208:197–213.
- Lapinsky SE , Mullen JB , Balter MS. (1993). Rapid pulmonary phospholipid accumulation induced by intravenous amiodarone. Can J Cardiol 9:322–4.
- Lauffenburger DA , Linderman JL. 1993. Receptor-mediated cell behavioral responses. In: Lauffenburger DA & Linderman JL , eds. Receptors: models for binding, trafficking, and signaling. New York: Oxford University Press.
- Liao X , Wiedmann TS. (2003). Solubilization of cationic drugs in lung surfactant. Pharm Res 20:1858–63.
- Llinas A , Box K , Comer J , et al . (2014). Study of the impact on physicochemical properties of inhaled products in several simulated lung fluid media. AAPS Annual Meeting Exposition; T3057; San Diego, CA.
- Loira-Pastoriza C , Todoroff J , Vanbever R. (2014). Delivery strategies for sustained drug release in the lungs. Adv Drug Deliv Rev 75:81–91.
- Lundborg M , Dahlen SE , Johard U , et al . (2006). Aggregates of ultrafine particles impair phagocytosis of microorganisms by human alveolar macrophages. Environ Res 100:197–204.
- Lynch T , Price A. (2007). The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician 76:391–6.
- Mahavadi P , Knudsen L , Venkatesan S , et al . (2015). Regulation of macroautophagy in amiodarone-induced pulmonary fibrosis. J Pathol Clin Res 1:252–63.
- Mansour HM , Rhee YS , Wu X. (2009). Nanomedicine in pulmonary delivery. Int J Nanomedicine 4:299–319.
- Marketsandmarkets (2017). North American Drug Delivery Technologies Market 2017 [online]. Available from: http://www.beforeitsnews.com [Accessed 11/02/2017]
- May S , Jensen B , Weiler C , et al . (2014). Dissolution testing of powders for inhalation:iInfluence of particle deposition and modeling of dissolution profiles. Pharm Res 31:3211–24.
- May S , Jensen B , Wolkenhauer M , et al . (2012). Dissolution techniques for in vitro testing of dry powders for inhalation. Pharm Res 29:2157–66.
- Mcelroy MC , Kasper M. (2004). The use of alveolar epithelial type I cell-selective markers to investigate lung injury and repair. Eur Respir J 24:664–73.
- Meindl C , Kueznik T , Bosch M , et al . (2015a). Intracellular calcium levels as screening tool for nanoparticle toxicity. J Appl Toxicol 35:1150–9.
- Meindl C , Stranzinger S , Dzidic N , et al . (2015b). Permeation of therapeutic drugs in different formulations across the airway epithelium in vitro . PLoS One 10:e0135690.
- Mercuri A , Wu S , Stranzinger S , et al . (2016). In vitro and in silico characterisation of Tacrolimus released under biorelevant conditions. Int J Pharm 515:271–80.
- Mia S , Warnecke A , Zhang XM , et al . (2014). An optimized protocol for human M2 macrophages using M-CSF and IL-4/IL-10/TGF-beta yields a dominant immunosuppressive phenotype. Scand J Immunol 79:305–14.
- Miller FJ. (2000). Dosimetry of particles in laboratory animals and humans in relationship to issues surrounding lung overload and human health risk assessment: a critical review. Inhal Toxicol 12:19–57.
- Molina SA , Stauffer B , Moriarty HK , et al . (2015). Junctional abnormalities in human airway epithelial cells expressing F508del CFTR. Am J Physiol Lung Cell Mol Physiol 309:L475–87.
- Morrow PE. (1988). Possible mechanisms to explain dust overloading of the lungs. Fundam Appl Toxicol 10:369–84.
- Moss OR. (1979). Simulants of lung interstitial fluid. Health Phys 36:447–8.
- Moss OR , Wong VA. (2006). When nanoparticles get in the way: impact of projected area on in vivo and in vitro macrophage function. Inhal Toxicol 18:711–6.
- Murty VL , Sarosiek J , Slomiany A , Slomiany BL. (1984). Effect of lipids and proteins on the viscosity of gastric mucus glycoprotein. Biochem Biophys Res Commun 121:521–9.
- Myrdal PB , Sheth P , Stein SW. (2014). Advances in metered dose inhaler technology: formulation development. AAPS PharmSciTech 15:434–55.
- Nacca N , Bhamidipati CM , Yuhico LS , et al . (2012). Severe amiodarone induced pulmonary toxicity. J Thorac Dis 4:667–70.
- National Research Council U. 1977. Respiratory transport and absorption. In: National Academy of Science, ed. Ozone and other photochemical oxidants. Washington, DC: National Research Council.
- Ng SF , Rouse J , Sanderson D , Eccleston G. (2010). A comparative study of transmembrane diffusion and permeation of Ibuprofen across synthetic membranes using Franz diffusion cells. Pharmaceutics 2:209–23.
- Nichols A. (2007). High content screening as a screening tool in drug discovery. Methods Mol Biol 356:379–87.
- Niles AL , Moravec RA , Riss TL. (2009). In vitro viability and cytotoxicity testing and same-well multi-parametric combinations for high throughput screening. Curr Chem Genomics 3:33–41.
- Nonoyama T , Fukuda R. (2008). Drug-induced phospholipidosis pathological aspects andiIts Prediction. J Toxicol Pathol 21:9–24.
- Okumura K , Iwakawa S , Yoshida T , et al . (1992). Intratracheal delivery of insulin Absorption from solution and aerosol by rat lung. Int J Pharm 88:63–73.
- Olsson B , Bondesson E , Borgström L , et al . 2011. Pulmonary Drug Metabolism, Clearence, and Absorption. In: Smyth H & Hickey A , eds. Controlled pulmonary drug delivery. New York: Springer; 21–50.
- Parra E , Perez-Gil J. (2015). Composition, structure and mechanical properties define performance of pulmonary surfactant membranes and films. Chem Phys Lipids 185:153–75.
- Patil JS , Sarasija S. (2012). Pulmonary drug delivery strategies: a concise, systematic review. Lung India 29:44–9.
- Patton JS , Byron PR. (2007). Inhaling medicines: delivering drugs to the body through the lungs. Nat Rev Drug Discov 6:67–74.
- Patton JS , Fishburn CS , Weers JG. (2004). The lungs as a portal of entry for systemic drug delivery. Proc Am Thorac Soc 1:338–44.
- Patton JS , Mccabe JG , Hansen SE , Daugherty AL. (1989). Absorption of human growth hormone from the rat lung. Biotechnol Ther 1:213–28.
- Pollmacher J , Figge MT. (2015). Deciphering chemokine properties by a hybrid agent-based model of Aspergillus fumigatus infection in human alveoli. Front Microbiol 6:503.
- Prietl B , Meindl C , Roblegg E , et al . (2014). Nano-sized and micro-sized polystyrene particles affect phagocyte function. Cell Biol Toxicol 30:1–16.
- Ramsey BW , Pepe MS , Quan JM , et al . (1999). Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 340:23–30.
- Reasor MJ , Mccloud CM , Dimatteo M , et al . (1996). Effects of amiodarone-induced phospholipidosis on pulmonary host defense functions in rats. Proc Soc Exp Biol Med 211:346–52.
- Renwick LC , Donaldson K , Clouter A. (2001). Impairment of alveolar macrophage phagocytosis by ultrafine particles. Toxicol Appl Pharmacol 172:119–27.
- Ribero R , Ferreira I , Figueiredo I , Vericimo M. (2015). Access to the tracheal pulmonary pathway in small rodents. J Bras Patol Med Lab 51:183–8.
- Riley T , Christopher D , Arp J , et al . (2012). Challenges with developing in vitro dissolution tests for orally inhaled products (OIPs). AAPS PharmSciTech 13:978–89.
- Rodero MP , Poupel L , Loyher PL , et al . (2015). Immune surveillance of the lung by migrating tissue monocytes. Elife 4:e07847.
- Salama RO , Traini D , Chan HK , et al . (2009). Preparation and evaluation of controlled release microparticles for respiratory protein therapy. J Pharm Sci 98:2709–17.
- Salama RO , Traini D , Chan HK , Young PM. (2008). Preparation and characterisation of controlled release co-spray dried drug-polymer microparticles for inhalation 2: evaluation of in vitro release profiling methodologies for controlled release respiratory aerosols. Eur J Pharm Biopharm 70:145–52.
- Satoh T , Kidoya H , Naito H , et al . (2013). Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature 495:524–8.
- Schurch S , Qanbar R , Bachofen H , Possmayer F. (1995). The surface-associated surfactant reservoir in the alveolar lining. Biol Neonate 67:61–76.
- Schwaiblmair M , Behr W , Haeckel T , et al . (2012). Drug induced interstitial lung disease. Open Respir Med J 6:63–74.
- Schwaiblmair M , Berghaus T , Haeckel T , et al . (2010). Amiodarone-induced pulmonary toxicity: an under-recognized and severe adverse effect? Clin Res Cardiol 99:693–700.
- Sdraulig S , Franich R , Tinker RA , et al . (2008). In vitro dissolution studies of uranium bearing material in simulated lung fluid. J Environ Radioact 99:527–38.
- Seymour S. 2006. Pulmonary Consultation [online]. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021868s000_MedR_P2.pdf [Accessed 11/02/2017]
- Shapiro DL , Nardone LL , Rooney SA , et al . (1978). Phospholipid biosynthesis and secretion by a cell line (A549) which resembles type II aleveolar epithelial cells. Biochim Biophys Acta 530:197–207.
- Shayman JA , Abe A. (2013). Drug induced phospholipidosis: an acquired lysosomal storage disorder. Biochim Biophys Acta 1831:602–11.
- Simon R. 1992. The biology and biochemistry of pulmonary alveolar epithelial cells. In Parent R , ed. Treatise on pumonary toxicology comparative biology of the normal lung. Boca Raton: CRC Press; 545–64.
- Somers GI , Lindsay N , Lowdon BM , et al . (2007). A comparison of the expression and metabolizing activities of phase I and II enzymes in freshly isolated human lung parenchymal cells and cryopreserved human hepatocytes. Drug Metab Dispos 35:1797–805.
- Son Y , Horng M , Copley M , Mcconville J. (2010). Optimization of an in vitro dissolution test method for inhalation formulations. Dissolution Technol 17:6–13.
- Son YJ , Mcconville JT. (2009). Development of a standardized dissolution test method for inhaled pharmaceutical formulations. Int J Pharm 382:15–22.
- Stone KC , Mercer RR , Freeman BA , et al . (1992). Distribution of lung cell numbers and volumes between alveolar and nonalveolar tissue. Am Rev Respir Dis 146:454–6.
- Stopford W , Turner J , Cappellini D , Brock T. (2003). Bioaccessibility testing of cobalt compounds. J Environ Monit 5:675–80.
- Takaya M , Shinohara Y , Serita F , et al . (2006). Dissolution of functional materials and rare earth oxides into pseudo alveolar fluid. Ind Health 44:639–44.
- Taunton A , Gunter M , Druschel G , Wood S. (2010). Geochemistry in the lung: reaction-path modeling and experimental examination of rock-forming minerals under physiologic conditions. Am Min 95:1624–35.
- Taylor MK , Hickey AJ , Vanoort M. (2006). Manufacture, characterization, and pharmacodynamic evaluation of engineered ipratropium bromide particles. Pharm Dev Technol 11:321–36.
- Tiwari G , Tiwari R , Sriwastawa B , et al . (2012). Drug delivery systems: an updated review. Int J Pharm Investig 2:2–11.
- Tomashefski J , Farver C. 2008. Anatomy and histology of the lung. In: Tomashefski J , Cagle P , Farver C & Fraire A , eds. Dail and hammar’s pulmonary pathology: Volume I: Nonneoplastic lung disease. New York: Springer Science + Business Media LLC, 19–48.
- Ungaro F , D’emmanuele Di Villa Bianca R , Giovino C , et al . (2009). Insulin-loaded PLGA/cyclodextrin large porous particles with improved aerosolization properties: in vivo deposition and hypoglycaemic activity after delivery to rat lungs. J Control Release 135:25–34.
- Ursini CL , Cavallo D , Fresegna AM , et al . (2014). Differences in cytotoxic, genotoxic, and inflammatory response of bronchial and alveolar human lung epithelial cells to pristine and COOH-functionalized multiwalled carbon nanotubes. Biomed Res Int 2014:359506.
- Van Der Deen M , De Vries EG , Timens W , et al . (2005). ATP-binding cassette (ABC) transporters in normal and pathological lung. Respir Res 6:59.
- Vickers AE , Jimenez RM , Spaans MC , et al . (1997). Human and rat lung biotransformation of cyclosporin A and its derivatives using slices and bronchial epithelial cells. Drug Metab Dispos 25:873–80.
- Vlahos R , Bozinovski S. (2014). Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol 5:435.
- Vrbanac J , Slauter R. 2013. ADME in drug discovery. In Faqi A , ed. A comprehensive guide to toxicology in preclinical drug development. London: Academic Press; 3–31.
- Walski M , Pokorski M , Antosiewicz J , et al . (2009). Pulmonary surfactant: ultrastructural features and putative mechanisms of aging. J Physiol Pharmacol 60:121–5.
- Wang W , Zhou QT , Sun SP , et al . (2016). Effects of surface composition on the aerosolisation and dissolution of inhaled antibiotic combination powders consisting of colistin and rifampicin. AAPS J 18:372–84.
- Wauthoz N , Amighi K. 2015. Formulation Strategies for Pulmonary Delivery of Poorly Soluble Drugs. In Nokhodchi A & Martin G , eds. Pulmonary drug delivery advances and challenges. Chichester: John Wiley & Sons.
- Weagel E , Smith C , Liu P , et al . (2015). Macrophage polarization and its role in cancer. J Clin Cell Immunol 6:338.
- Weidauer E , Lehmann T , Ramisch A , et al . (2004). Response of rat alveolar type II cells and human lung tumor cells towards oxidative stress induced by hydrogen peroxide and paraquat. Toxicol Lett 151:69–78.
- Welty-Wolf KE , Simonson SG , Huang YC , et al . (1997). Aerosolized manganese SOD decreases hyperoxic pulmonary injury in primates. II. Morphometric analysis. J Appl Physiol 83:559–68.
- Winterbottom C , Shah R , Patterson K , et al . 2014. Ambient Particulate Matter Is Associated With Functional Decline In Idiopathic Pulmonary Fibrosis. http://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2014.189.1_MeetingAbstracts.A1501.
- Wragg J , Klinck B. (2007). The bioaccessibility of lead from Welsh mine waste using a respiratory uptake test. J Environ Sci Health A Tox Hazard Subst Environ Eng 42:1223–31.
- Yang W , Tam J , Miller DA , et al . (2008). High bioavailability from nebulized itraconazole nanoparticle dispersions with biocompatible stabilizers. Int J Pharm 361:177–88.
- Yao W , Li K , Liao K. (2009). Macropinocytosis contributes to the macrophage foam cell formation in RAW264.7 cells. Acta Biochim Biophys Sin (Shanghai) 41:773–80.
- Yayoi Y , Ohsawa Y , Koike M , et al . (2001). Specific localization of lysosomal aminopeptidases in type II alveolar epithelial cells of the rat lung. Arch Histol Cytol 64:89–97.
- Yin L , Stearns R , Gonzalez-Flecha B. (2005). Lysosomal and mitochondrial pathways in H2O2-induced apoptosis of alveolar type II cells. J Cell Biochem 94:433–45.