
Abstract
Vascular-targeted photodynamic therapy (PDT) is an important strategy for cancer therapy. Conventional vascular-targeted PDT has been achieved by passive photosensitizer (PS) delivery, which involves a high risk of adverse effects. Active PS delivery is urgently required for vascular-targeted PDT. Although endothelial cells and pericytes are major cellular components of tumor blood vessels, little attention has been paid to pericyte-targeted PDT for cancer therapy. PDGFRβ is abundantly expressed in the pericytes of various tumors. In this experiment, a dimeric ZPDGFRβ affibody with a 0.9 nM affinity for PDGFRβ was produced. The ZPDGFRβ affibody showed PDGFRβ-dependent pericyte binding. Intravenously injected ZPDGFRβ affibody was predominantly distributed on pericytes and thus accumulated in LS174T tumor grafts. The conjugate of the ZPDGFRβ affibody and IR700 dye, i.e. ZIR700, bound to PDGFRβ+ pericytes but not to PDGFRβ− LS174T tumor cells. Accordingly, ZIR700-mediated PDT in vitro induced the death of pericytes but not of LS174T tumor cells. In mice bearing LS174T tumor grafts, ZIR700-mediated PDT damaged tumor blood vessels, thus inducing tumor destruction by intensifying tissue hypoxia. The average mass of tumor grafts administered with ZIR700-mediated PDT was approximately 20–30% of that of the control, indicating that pericyte-targeted PDT is efficient for cancer therapy. In addition, ZIR700-mediated PDT increased the tumor uptake of TNF-related apoptosis-inducing ligand (TRAIL) injected post-illumination. Consequently, combination therapy of ZIR700-mediated PDT and TRAIL showed greater tumor suppression than ZIR700-mediated PDT- or TRAIL-based monotherapy. These results demonstrated that active vascular-targeted PDT could be achieved by using ZPDGFRβ affibody-directed delivery of PS.
Introduction
Photodynamic therapy (PDT) is a two-stage procedure involving the administration of a photosensitizer (PS) followed by exposure to light. In the presence of tissue oxygen, the PS, activated by light of a specific wavelength, could stimulate the production of reactive oxygen species (ROS), thus inducing cell death (Dolmans et al., Citation2003; Dabrowski & Arnaut, Citation2015). Compared to conventional surgical interventions, PDT is minimally invasive for patients. In addition, PS, without excitation by light, is usually non-toxic. Moreover, due to the very short lifespan (<40 ns in biological systems) and a limited radius (<20 nm) of action of ROS, PDT-mediated photodamage can be restricted in disease sites by topical illumination. Unlike conventional chemotherapy, PDT involves a low risk of systematic toxicity and has thus been considered an ideal modality for the clinical treatment of cancers of the skin, brain, head, and neck, lung, esophagus, pancreas, bile duct, breast, bladder, prostate, stomach, colon, female reproductive tract, etc. (Shafirstein et al., Citation2017; van Straten et al., Citation2017).
In the case of passive PS-delivery-based PDT, although topical illumination reduced the risk of systematic toxicity, photosensitive reactions were usually observed in normal tissues, such as the skin and eyes, that are exposed to daylight (Bugaj, Citation2011). These side effects were attributed to the unspecific accumulation of free PS in these tissues. Consequently, tumor-targeted delivery of PS is required for PDT in the future (Bugaj, Citation2011; Shirasu et al., Citation2013). To achieve tumor-targeted delivery, passive and active targeting strategies have been developed in recent years. Passive tumor targeting might be achieved by exploiting the enhanced permeability and retention (EPR) effect caused by the leaky vasculature and impaired lymphatic drainage of tumors. In fact, most nanoparticles loaded with PS can be selectively accumulated in tumors due to the EPR effect (Lowa & Lin, Citation2000). Active tumor targeting was developed by the conjugation of PS to molecules that specifically bind antigens or receptors that are overexpressed in tumors. Thus far, antibodies and their fragments, peptides and protein ligands, and small molecules have been widely used for tumor-targeted delivery of PS (Schmitt & Juillerat-Jeanneret, Citation2012). Since passive targeting relies on only the EPR effect of tumors, its specificity and efficacy are limited. In contrast, active targeting depends on both the EPR effect and molecule–molecule interactions, which lead to more specific and effective delivery of PS to tumors (Shirasu et al., Citation2013).
Tumor cell targeting and vascular cell targeting are important strategies for active tumor targeting. Compared to tumor cell-targeted PDT, vascular-targeted PDT takes advantage of restricting nutrients and the oxygen supply, thus leading to stronger tumor growth suppression (Krzykawska-Serda et al., Citation2014), suggesting that vascular-targeted PDT is more efficient for cancer therapy. It is known that the walls of tumor blood vessels predominantly consist of irregularly lined endothelial cells and pericytes covering the endothelial tubule (Chang et al., Citation2013; Geevarghese & Herman, Citation2014). In previous works, vascular-targeted PDT has been achieved by the conjugation of PS to molecules that specifically bind vascular endothelial growth factor receptor (VEGFR) or αvβ3 integrin, which are overexpressed in tumor-associated endothelial cells (Thomas et al., Citation2010; Srivatsan et al., Citation2011). However, it is not known whether tumor-associated pericytes could also be considered target cells for vascular-targeted PDT.
Platelet-derived growth factor receptor β (PDGFRβ) is overexpressed on the pericytes of many types of tumors (Paulsson et al., Citation2009), suggesting that conjugation to PDGFRβ-binding molecules might deliver PS to tumor blood vessels. In previous works, several PDGFRβ-binding peptides were identified (Prakash et al., Citation2010; Askoxylakis et al., Citation2013; Marr et al., Citation2013). However, these peptides were limited by their low affinity (μM level) for PDGFRβ. Recently, Lindborg et al. (Citation2011) identified numerous PDGFRβ-specific affibodies. Of these affibodies, Z02465 showed a nM affinity for human and mouse PDGFRβ. In addition, Z02465 did not bind PDGFRα, which is highly homologous to PDGFRβ. Due to its high affinity and specificity for PDGFRβ, affibody Z02465 and its derivative have been considered ideal tools for the delivery of anti-cancer drugs and contrast media. Tolmachev et al. (Citation2014) found that affibody Z09591, a derivative of Z02465, was tumor homing in mice bearing glioma tumor grafts, which triggered our interests in developing this affibody as an anti-cancer drug carrier. In our previous work, we prepared the monomeric Z09591 affibody, designated ZPDGFRβ, and found that it showed PDGFRβ-dependent pericyte binding and thus increased the tumor uptake of fused anti-cancer proteins (Tao et al., Citation2017). These results suggested that the ZPDGFRβ affibody might be used as a carrier of PS for pericyte-targeted PDT.
Since polymerization might improve the stability and avidity of the affibody (Kim et al., Citation2012), in this study, we first prepared the dimeric ZPDGFRβ affibody and determined its PDGFRβ-dependent pericyte-binding and tumor-homing capabilities in mice bearing tumor grafts. Since IR700 dye is a soluble PS, we subsequently produced ZIR700 by conjugation of IR700 to ZPDGFRβ and tested its photocytotoxicity and specificity in cultured cells. Finally, the antitumor effect of ZIR700-mediated PDT as a monotherapy or in combination with anti-cancer protein TRAIL was evaluated in mice bearing tumor grafts.
Materials and methods
Expression and purification of proteins
To produce the dimeric ZPDGFRβ affibody, an artificial gene encoding Z09591 containing a unique cysteine (Tolmachev et al., Citation2014) with an additional HE-tag (Hofstrom et al., Citation2011) at its N-terminus was designed according to the codon usage preference of E. coli. The optimized gene was synthesized by GenScripts (Nanjing, China) followed by subcloning into the pQE30 plasmid with a deleted His-tag to construct the expressive plasmid pQE30-ZPDGFRβ. Expression of the ZPDGFRβ affibody was induced by the addition of isopropyl β-D-thiogalactoside (IPTG, 0.1 mM) to a culture of E. coli M15 containing the pQE30-ZPDGFRβ plasmid. After induction at 24 °C overnight, the culture was centrifuged at 7000 × g at 4 °C for 10 min. The pellets were resuspended in lysis buffer (50 mM phosphate (pH 8.0), 300 mM NaCl, and 5 mM imidazole) followed by processing 3–4 times in a high-pressure homogenizer (60–70 MPa). The recombinant ZPDGFRβ affibody in the supernatant was recovered by using a Ni-NTA super flow column (Qiagen, Valencia, CA) according to the manual provided by the manufacturer. The recovered ZPDGFRβ affibody was further dialyzed against phosphate-buffered saline (PBS, 10 mM Na2HPO4, 137 mM NaCl, 2.68 mM KCl, and 2 mM KH2PO4, pH 7.4) overnight and stored at −80 °C until further use. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and size-exclusion chromatography in the absence or presence of β-mercaptoethanol (2-ME) were used to estimate the purity and molecular weight of the ZPDGFRβ affibody. The concentration of the gel for SDS-PAGE was 16%. In addition, a Superdex G-75 Increase 10/30 column (GE Healthcare, Anaheim, CA) was used for size-exclusion chromatography with PBS as the eluent. Protein concentration was measured by using a protein DC assay kit (Bio-Rad, Hercules, CA). Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) was prepared according to an earlier description (Li et al., Citation2016).
PDGFRβ-binding assays
PDGFRβ-expressing pericytes were incubated with FAM- or IR700-labeled ZPDGFRβ affibody at room temperature for 1 h. After two washes with PBS, the cells were analyzed by using a flow cytometer. To block the binding of ZPDGFRβ affibody to PDGFRβ, cells were pre-incubated with a goat anti-human PDGFRβ antibody for 1 h before the addition of the ZPDGFRβ affibody. In addition, PDGFRβ-binding of the ZPDGFRβ affibody was further determined by using protein interaction analysis performed on an OpenSPR system (Nicoya Life Sciences Inc., Kitchener, Canada) (McGurn et al., Citation2016). Briefly, PDGFRβ-Fc (R&D, MN) was immobilized on the COOH-sensor chips, and solutions containing increasing concentrations of the ZPDGFRβ affibody were introduced onto the chip, followed by surface plasmon resonance analysis on the OpenSPR system. The kinetic constants, including the association constant (Ka), dissociation constant (Kd) and affinity (KD, KD = Kd/Ka), were calculated using software according to a 1:1 binding model.
Bio-distribution assay
ZPDGFRβ affibody was labeled with CF™ 750 succinimidyl ester (CF750) or 5(6)-carboxyfluorescein (FAM) according to our previous description (Wei et al., Citation2015). Briefly, the pH value of the ZPDGFRβ affibody (1.5 mg/ml) solution was adjusted to 8.0 by the addition of 1 M NaHCO3. The fluorescent dye dissolved in dimethyl sulfoxide (DMSO) was added to the ZPDGFRβ affibody at a 6:1 (for CF750) or 20:1 (for FAM) molar ratio of dye to protein. After reaction at room temperature for 1 h in darkness, the mixture was dialyzed against PBS overnight. To monitor the tumor uptake of the ZPDGFRβ affibody, a single dose (3.2 mg/kg) of the CF750-labeled ZPDGFRβ affibody was intravenously injected into mice bearing LS174T tumor xenografts, followed by dynamic scanning with SPECTRAL Lago and Lago X Imaging Systems (Spectral, Tucson, AZ). At the end of this experiment, the organs/tissues of these mice were removed and scanned simultaneously. To determine the cellular distribution, the FAM-labeled ZPDGFRβ affibody was intravenously injected into the mice. Subsequently, the tumor grafts were removed at different times post-injection and sectioned under freezing conditions. The nuclei were visualized by using DAPI. In addition, the FAM-labeled ZPDGFRβ affibody was co-localized with CD31, PDGFRβ or NG2 by using immunofluorescence.
Cell culture
Cell lines, including human colorectal cancer cells (LS174T), human umbilical vein-derived endothelial cells (ECs), and human uterine-derived smooth muscle cells (SMCs) were purchased from American Type Culture Collection (ATCC, Manassas, VA) and cultured in DMEM containing 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Primary human brain vascular pericytes (PCs) were obtained from ScienCell (CA) and cultured in their specific media. All the cells were cultured at 37 °C in a 5% CO2 humidified atmosphere.
Cytotoxicity assay
Cells (1 × 104) were inoculated in the wells of a 96-well plate and cultured overnight. Subsequently, proteins diluted with media were added to the cells at different concentrations (0–10 μM). The same volume of PBS was used as a control. After treatment for 16–18 h, the viable cells were determined by the addition of Cell Counting Kit-8 (CCK-8) solution (Dojindo, Japan). The viability of PBS-treated cells was considered 100%.
Expression of biomarkers in cells and tissues
Expression of PDGFRβ, NG2, or α-SMA in cells was detected by flow cytometry. Cells were incubated with primary antibody for 1 h at room temperature followed by further incubation with secondary antibody for 30 min (if needed) prior to analysis with a flow cytometer. For immunofluorescence, the tumor grafts were sectioned under freezing conditions immediately after removal from the mice. To localize the expression of biomarkers in tumor blood vessels, tumor tissues were co-stained with antibody against PDGFRβ, NG2, or α-SMA and antibody against CD31, followed by observation under a fluorescence microscope. The primary antibodies included rat anti-mouse CD31 and PE-mouse anti-human PDGFRβ (Biolegend, San Diego, CA), goat anti-human PDGFRβ (R&D, MN), and rabbit anti-human PDGFRβ, rabbit anti-human NG2, and rabbit anti-human α-SMA (Abcam, Burlingame, CA). Secondary antibodies included donkey anti-rabbit IgG (DyLight 488) and goat anti-rat IgG (DyLight 550) (Abcam, Burlingame, CA). An isotype antibody was used as a control.
Conjugation of the photosensitizer to the ZPDGFRβ affibody
The photosensitizer IRDye 700DX N-hydroxysuccinimide ester (IR700) was obtained from LI-COR Biosciences (Lincoln, NE). Conjugation was performed as described by Mitsunaga et al. (Citation2011) with some modifications. Briefly, IR700 dissolved in DMSO was mixed with the ZPDGFRβ affibody (1.5 mg/ml, pH 8.0) at a molar ratio of 2:1 (dye to protein) followed by incubation at room temperature for 1 h in darkness. The conjugation of IR700 to the ZPDGFRβ affibody was verified by SDS-PAGE followed by scanning the gel with Odyssey CLx (LI-COR Biosciences, Lincoln, NE). The conjugate was designated ZIR700.
In vitro PDT
Cells (1 × 104/well) were inoculated in 96-well plates. After incubation overnight, ZIR700 (4 μM IR700 equivalent) was added to the wells followed by two washes with PBS 1 h later. Subsequently, 100 μl medium without phenol red was added to the wells, and the cells were illuminated at a fluence rate of 16 mW/cm2 for a total dose of 10 J/cm2 with a laser at a wavelength of 690 nm. The viability of the cells was examined by using a live/dead BacLight bacterial viability kit (Invitrogen, Carlsbad, CA) (Shirasu et al., Citation2014). Non-illuminated cells were used as a control.
Dichlorodihydrofluorescein diacetate (DCFH) and singlet oxygen sensor green (SOSG) were used to detect hydroxyl radicals (Setsukinai et al., Citation2003) and singlet oxygen (Gollmer et al., Citation2011; van Driel et al., Citation2016), respectively. DCFH or SOSG (10 μM) plus ZIR700 were pre-incubated with cells for 1 h followed by two washes with PBS. Subsequently, 100 μl medium without phenol red was added to the cells followed by illumination. Approximately 1–2 h later, the fluorescence was measured using a fluorescence microplate reader (λexc = 488 nm). Simultaneously, the cells were observed under a fluorescence microscope after the nuclei were visualized using Hoechst.
In vivo PDT
Approximately, 2 × 106 LS174T cells were subcutaneously implanted into the right hind leg of BALB/c nude mice (n = 3). Once the tumor size reached 150 mm3, ZIR700 (0.8 mg/kg ZPDGFRβ affibody equivalent) was intravenously injected into the mice. Approximately 4 h later, the tumor grafts were illuminated at a fluence rate of 100 mW/cm2 for a total dose of 120 J/cm2. The longitudinal (L) and transverse (W) diameters of the tumor grafts were measured every day. In addition, the tumor volume (V) was calculated according to the following formula: V = L × W2/2. At the end of experiment, all tumor grafts were removed and weighed.
For histochemistry, tumor grafts were removed at different times post-illumination. The paraffin-sectioned tumor tissues were stained by H&E. To assess the leakage of tumor blood vessels, fluorescein isothiocyanate (FITC)-labeled dextran (70 kDa, Sigma, St. Louis, MO) was intravenously injected into mice bearing LS174T tumor grafts at different times post-illumination (0–8 h). The dextran was allowed to circulate for 20 min. To perfuse the mouse, 60 ml PBS was injected from the ventriculus sinister and drained from the right atrium. Subsequently, the tumor grafts were removed and sectioned (50 μm) under freezing conditions. Blood vessels were visualized by immunofluorescence with anti-mouse CD31. In addition, the nuclei were visualized with DAPI. Finally, the tumor tissues were scanned using a multiphoton laser scanning confocal microscope (Nikon, Tokyo, Japan). To illustrate hypoxia in tumor grafts, expression of hypoxia-inducible factor 1α (HIF1α) was detected by using a rabbit anti-human HIF1α antibody (Novus Biologicals, San Diego, CA).
Combination therapy of ZIR700-mediated PDT and TRAIL
Selective cytotoxicity of TRAIL in tumor cells highlighted its potential as a candidate drug for cancer therapy (Holland, Citation2013). The increase of vessel permeability induced by vascular targeted PDT triggered our interest on the combination therapy of ZIR700-mediated PDT and TRAIL. To determine whether ZIR700-mediated PDT would increase the tumor uptake of TRAIL, a single dose (10 mg/kg) of FAM-labeled TRAIL was intravenously injected into the mice bearing LS174T tumor grafts prior to illumination. The tumor grafts were removed 1 h post-illumination and sectioned under freezing conditions. After the tumor tissues were stained with antibody against mouse CD31, they were observed under a fluorescence microscope. Since TRAIL induces apoptosis in tumor cells, the increase in the tumor uptake of TRAIL was also reflected by an increase in apoptotic cells in the tumor tissues. To visualize the apoptotic cells, tumor grafts were removed 16 h post-illumination and sectioned under freezing conditions, followed by TdT-mediated dUTP nick-end labeling (TUNEL).
To compare the antitumor effect of different regimens, mice (n = 6–7) bearing LS174T tumor grafts were treated with PBS, TRAIL (10 mg/kg), ZIR700 (0.8 mg/kg), and ZIR700 (0.8 mg/kg)/TRAIL (10 mg/kg). Additionally, mice treated with ZIR700 or ZIR700/TRAIL were illuminated at a fluence rate of 100 mW/cm2 for a total dose of 120 J/cm2 after each injection. The tumor volume was measured every day. At the end of the experiment, all tumor grafts were removed and weighed.
Statistical analysis
SPSS software (version 20) was used for one-way analysis of variance (ANOVA) for multiple comparisons. The significance level was defined as p < .05. The results are expressed as the mean ± standard deviation (SD).
Results
PDGFRβ-positive cells are predominantly located with the mural cells of tumor blood vessels
Tumor blood vessels can be composed of endothelial cells (ECs), pericytes (PCs), and smooth muscle cells (SMCs). Endothelial cells can be characterized by CD31 expression. However, there is no exclusive marker for pericytes or smooth muscle cells. Consequently, the identification of pericytes and smooth muscle cells depends on both cell surface markers and cell location (Geevarghese & Herman, Citation2014). As shown in , flow cytometry demonstrated that the PDGFRβ-positive (PDGFRβ+) rates of cultured pericytes and smooth muscle cells were 72.9 and 47.6% compared to 10.1% for that of endothelial cells, indicating that PDGFRβ was predominantly expressed on pericytes and smooth muscle cells. Expression of NG2 was detectable in 66.5% of pericytes compared to less than 10% for that of smooth muscle cells and endothelial cells. Approximately 70% of smooth muscle cells were α-SMA positive (α-SMA+), whereas both pericytes and endothelial cells were α-SMA negative (α-SMA−, positive rate <8%). These results demonstrated that pericytes were PDGFRβ+NG2+α-SMA−, whereas smooth muscle cells were PDGFRβ+NG2−α-SMA+. Consequently, of these three cells, NG2 might be considered an exclusive marker for pericytes. As shown in , most PDGFRβ+ cells in LS174T tumor tissues were proximate to CD31-positive (CD31+) endothelial cells, suggesting that these PDGFRβ+ cells are mural cells. Moreover, the distribution profile of PDGFRβ is identical to that of NG2, whereas few α-SMA+ cells were observed in tumor tissues, indicating that among the mural cells of tumor blood vessels, PDGFRβ+ cells were predominantly pericytes, not smooth muscle cells.
Figure 1. Expression of surface markers on mural cells. (A) Expression of PDGFRβ, NG2, and α-SMA in pericytes (PCs), smooth muscle cells (SMCs), and endothelial cells (ECs). (B) Co-localization of PDGFRβ, NG2, or α-SMA with CD31 in tumor tissues derived from LS174T tumor grafts. The nuclei were visualized by DAPI staining. Original magnification 200×.
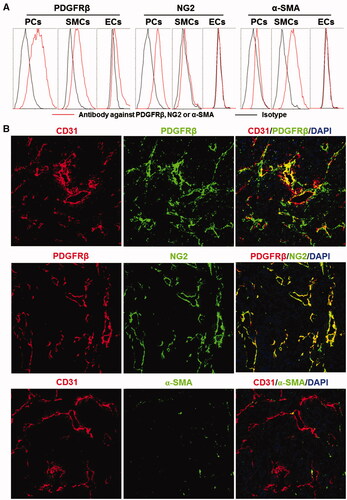
ZPDGFRβ affibody binds PDGFRβ with high affinity
As shown in , purified ZPDGFRβ affibody was visualized as a single band on an SDS-PAGE gel. The molecular weight of the ZPDGFRβ affibody under natural conditions (in the absence of 2-ME) was approximately 2-fold (∼15 kDa versus 7 kDa) that of the ZPDGFRβ affibody under reductive conditions (in the presence of 2-ME), indicating that the ZPDGFRβ affibody forms disulfide bond-containing dimers under natural conditions. Size-exclusion chromatography further revealed that over 90% of the ZPDGFRβ affibody exists as a dimer under natural conditions. The binding rate of the ZPDGFRβ affibody to pericytes was 71%, while that of the ZPDGFRβ affibody to smooth muscle cells was 28.4%. In addition, the binding of the ZPDGFRβ affibody to endothelial cells was undetectable (). The binding rate of the ZPDGFRβ affibody to pericytes, smooth muscle cells, and endothelial cells was closely related to the expression level of PDGFRβ in these cells. Moreover, the binding of the ZPDGFRβ affibody to pericytes was drastically (from 70% to 10%) reduced by pre-incubation of the cells with a blocking antibody against PDGFRβ (), indicating that the ZPDGFRβ affibody specifically binds PDGFRβ. Protein–protein interaction analysis demonstrated that the ZPDGFRβ affibody binds PDGFRβ with an affinity of 0.9 nM. However, even treatment with 10 μM of the ZPDGFRβ affibody overnight did not reduce the viability of these three cells (). These results indicated that the ZPDGFRβ affibody binds PDGFRβ with a high affinity but is not cytotoxic in the tested PDGFRβ-positive cells.
Figure 2. Preparation and characterization of the ZPDGFRβ affibody. (A) SDS-PAGE and size-exclusion chromatography of the purified ZPDGFRβ affibody in the presence or absence of 2-ME. (B) Binding of the ZPDGFRβ affibody to pericytes (PCs), smooth muscle cells (SMCs), and endothelial cells (ECs) as analyzed by flow cytometry. (C) Inhibition of pericyte binding of the ZPDGFRβ affibody by the PDGFRβ-specific antibody (left) and the PDGFRβ-binding of the ZPDGFRβ affibody analyzed using the OpenSPR system (right). (D) Cytotoxicity of the ZPDGFRβ affibody in pericytes (PCs), smooth muscle cells (SMCs), and endothelial cells (ECs).
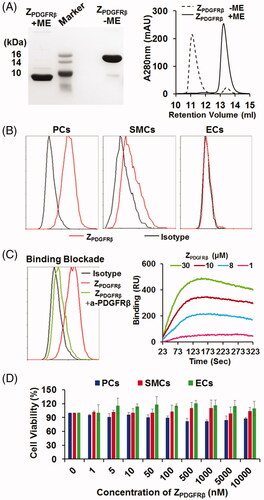
ZPDGFRβ affibody is tumor homing
To test its tumor-homing characteristics, CF750-labeled ZPDGFRβ affibody was intravenously injected into mice bearing LS174T tumor grafts followed by dynamic optical imaging. As shown in , optical images demonstrated that the ZPDGFRβ affibody accumulated in subcutaneous tumor grafts from 1 h post-injection and persisted till at least 4 h. Organ/tissue scanning at 4 h post-injection confirmed the accumulation of the ZPDGFRβ affibody in tumor grafts. Notably, the uptake of the ZPDGFRβ affibody by tumor grafts was even higher than that of the ZPDGFRβ affibody by livers and kidneys (). These results indicated that the ZPDGFRβ affibody is tumor homing. Moreover, cellular localization analysis demonstrated that FAM-labeled ZPDGFRβ affibody co-localized well with PDGFRβ and with NG2 ( and Supplementary Figure S1), suggesting that the ZPDGFRβ affibody was predominantly distributed on pericytes in the mural of tumor blood vessels.
Figure 3. Tumor-homing characteristics of the ZPDGFRβ affibody. (A) Tumor uptake of the ZPDGFRβ affibody. CF750-labeled ZPDGFRβ affibody was intravenously injected into mice (n = 3) bearing subcutaneous LS174T tumor grafts (arrow indicated), followed by scanning with SPECTRAL Lago and Lago X Imaging Systems at different times (0–4 h) post-injection. (B) Bio-distribution of the ZPDGFRβ affibody in mice bearing LS174T tumor grafts. Mice (n = 3) injected with CF750-labeled ZPDGFRβ affibody were sacrificed at 4 h post-injection for optical imaging of organs/tissues. (C) Co-localization of the ZPDGFRβ affibody with CD31, PDGFRβ, or NG2 in tumor tissues. FAM-labeled ZPDGFRβ affibody was intravenously injected into mice bearing LS174T tumor grafts. Tumor grafts were removed and sectioned under freezing conditions, followed by staining with antibody against CD31, PDGFRβ, or NG2. The nuclei were visualized by DAPI staining. Original magnification 200×.
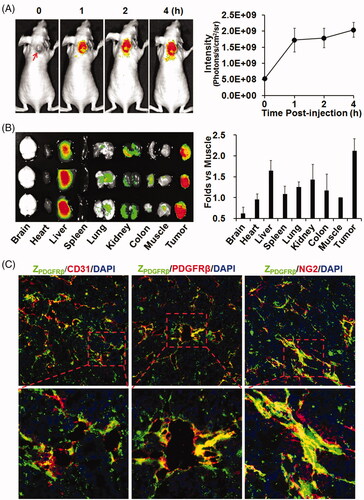
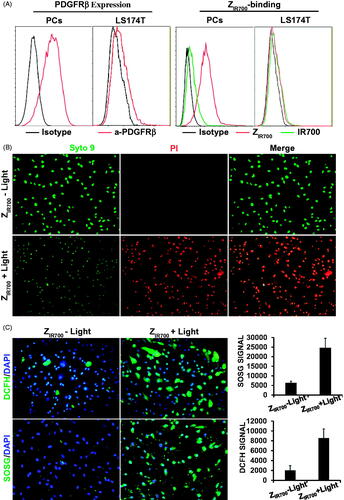
ZIR700-mediated PDT specifically kills PDGFRβ-expressing cells
To evaluate the specificity of ZIR700-mediated PDT, the photocytotoxicity of ZIR700 was determined in PDGFRβ+ and PDGFRβ− cells. As shown in , flow cytometry demonstrated that the 73% of the pericytes were PDGFRβ+ and that 8% of the LS174T tumor cells were PDGFRβ+. The binding rate of ZIR700 to pericytes was 75%, whereas its binding rate to LS174T tumor cells was 4%. The free IR700 dye showed little (<8%) binding to both cells. Illumination induced over 90% death of PDGFRβ+ pericytes, which were pre-incubated with ZIR700 and washed prior to illumination (). However, illumination did not induce obvious cell death in PDGFRβ− LS174T tumor cells under the same conditions (Supplementary Figure S2(A)). After incubation of PDGFRβ+ pericytes with free IR700 dye, illumination only induced obvious death of unwashed cells (Supplementary Figure S2(B)). These results demonstrated that the binding of ZIR700 to pericytes was mediated by the ZPDGFRβ affibody, which is specific for PDGFRβ. demonstrates that reactive oxygen species (ROS), including hydroxyl radicals (DCFH signal) and singlet oxygen (SOSG signal), accumulated in pericytes treated with ZIR700-mediated PDT. Since ROS contribute to the phototoxicity of PS (Dąbrowski, Citation2017), these results indicated that ZIR700-mediated PDT killed PDGFRβ+ pericytes by ROS-mediated phototoxicity.
Figure 4. ZIR700-mediated PDT kills PDGFRβ-expressing cells in vitro. (A) Flow cytometry analysis of PDGFRβ expression (left) and ZIR700 binding (right) to pericytes (PCs) and LS174T tumor cells. (B) Cytotoxicity of ZIR700 in PDGFRβ-expressing pericytes under different conditions. Cells were pre-incubated with ZIR700 for 1 h followed by washing with PBS and illumination with (ZIR700+Light) or without (ZIR700-Light) laser. The live/dead cells were visualized by SYTO9/PI staining. Original magnification 200×. (C) Production of ROS in pericytes treated with ZIR700-mediated PDT. Cells were incubated with DCFH or SOSG during ZIR700-mediated PDT and were observed under a fluorescence microscope (left) or measured with a fluorescence microplate reader (right). Original magnification 200×.
ZIR700-mediated PDT suppresses tumor growth by damaging blood vessels
Since ZIR700-mediated PDT induced the death of pericytes in vitro, it might damage tumor blood vessels, thus increasing their permeability once in vivo PDT is performed. To determine the change in the permeability of tumor blood vessels, FITC-labeled dextran was intravenously injected into mice bearing LS174T tumor grafts at different times post-illumination. As shown in and Supplementary Figure S3, little dextran was observed in tumor tissues derived from mice injected with ZIR700 without illumination. However, when injected at the beginning of illumination (0 h post-illumination), dextran was observed in a wide range of tumor tissues derived from mice treated with ZIR700-mediated PDT, indicating the leakage of tumor blood vessels. When dextran was injected at 1 or 8 h post-illumination, although the amount of dextran decreased drastically, dextran was still observed in tumor blood vessels and neighboring tissues. These results demonstrated that ZIR700-mediated PDT increases the permeability of tumor blood vessels.
Figure 5. ZIR700-mediated PDT suppresses LS174T tumor growth in mice. (A) Permeability of tumor blood vessels visualized by the leakage of dextran. After intravenous injection of ZIR700 followed by illumination (ZIR700 + Light) or not (ZIR700−Light), mice bearing LS174T tumor grafts were intravenously injected with FITC-dextran at different times (0–8 h) post-illumination. Approximately 20 min later, the tumor grafts were removed and sectioned under freezing conditions, followed by visualization of the blood vessels with antibody against CD31. DAPI was used to visualize the nuclei. Original magnification 100×. (B) Expression of HIF1α in tumor tissues at different times (1–8 h) post-illumination. Original magnification 200×. (C) H&E staining of tumor tissues derived from tumor grafts at different times (0–72 h) post-illumination. (D) Tumor growth suppression by ZIR700-mediated PDT. Mice (n = 3) bearing LS174T tumor grafts were intravenously injected with ZIR700, followed by illumination (ZIR700+Light) or not (ZIR700-Light) at day 0 and day 3, respectively. Mice in the control group were treated with PBS. The tumor volumes were measured every day, and the (1–3) growth curves of mice in each group are shown. At the end of this experiment, all tumor grafts were removed and weighed.
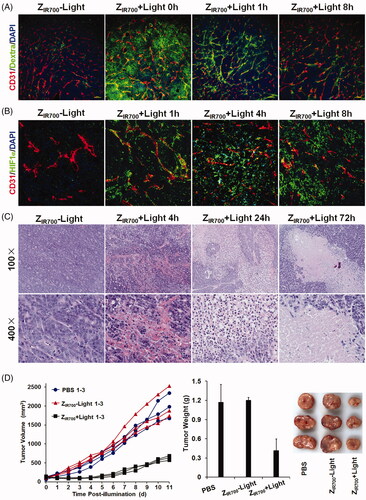
The level of HIF1α reflects tissue hypoxia. As shown in , few cells were HIF1α expressive in LS174T tumor grafts from mice injected with ZIR700. ZIR700-mediated PDT increased the number of HIF1α-expressing cells in tumor grafts over time, indicating the intensification of hypoxia in tumor grafts. In addition, a great number of red blood cells were observed in tumor tissues collected within 8 h post-illumination. Blood clots were visible in tumor tissues collected between 1 and 4 h post-illumination (). These results indicated that ZIR700-mediated PDT damages tumor blood vessels and induces hemorrhage and thrombosis, which would impair the delivery function of tumor blood vessels, thus intensifying hypoxia in tumor grafts. Approximately 24 h post-illumination, a wide range of tumor tissues became non-coherent. In addition, most cells in non-coherent tumor tissues disappeared at 72 h post-illumination (). Since hypoxia would induce necrosis/apoptosis of tumor cells, these results demonstrated that ZIR700-mediated PDT damaged tumor blood vessels, thereby inducing tumor destruction by the intensification of tissue hypoxia.
Moreover, the growth of LS174T tumor grafts in mice treated with ZIR700-mediated PDT was much slower than that in mice injected with ZIR700 without illumination ( and Supplementary Figure S4). At the end of this experiment, the average tumor mass of mice treated with ZIR700-mediated PDT was 0.41 ± 0.04 g, compared to 1.20 ± 0.18 g of mice injected with ZIR700 without illumination and 1.17 ± 0.28 g of PBS-treated mice. These results demonstrated that ZIR700-mediated PDT significantly (p < .05) suppresses tumor growth in mice.
ZIR700-mediated PDT and TRAIL combination therapy yields a better antitumor effect
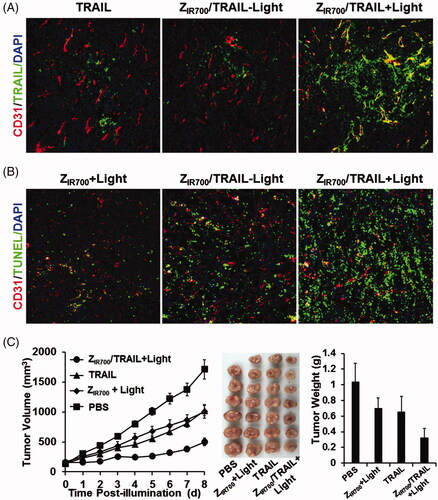
Since ZIR700-mediated PDT increased the permeability of tumor blood vessels, we attempted to investigate whether it would increase the tumor uptake of the protein drug TRAIL, thereby demonstrating a better antitumor effect. To monitor the tumor uptake, FAM-labeled TRAIL was intravenously injected into the mice at the beginning of illumination during ZIR700-mediated PDT. As shown in , ZIR700-mediated PDT obviously increased the tumor uptake of TRAIL. Since TRAIL could induce apoptosis in tumor cells, the numbers of apoptotic cells were used to reflect the uptake of TRAIL under different conditions. As shown in , compared to the monotherapy based on ZIR700-mediated PDT or TRAIL, combination therapy of ZIR700-mediated PDT and TRAIL induced greater apoptosis in tumor cells. Accordingly, the growth of tumor grafts treated with combination therapy was slower than that of tumor grafts treated with monotherapy. At the end of this experiment, the average tumor mass after combination therapy was 0.33 ± 0.11 g, compared to 0.65 ± 0.20 g (for TRAIL) and 0.69 ± 0.13 g (ZIR700-mediated PDT) after monotherapy. These results demonstrated that ZIR700-mediated PDT could increase tumor uptake of TRAIL, thus leading to greater tumor growth suppression.
Figure 6. ZIR700-mediated PDT and TRAIL combination therapy leads to a better antitumor effect. (A) ZIR700-mediated PDT increases the tumor uptake of TRAIL. Mice bearing LS174T tumor grafts were treated with FAM-labeled TRAIL (TRAIL) or ZIR700 combined with TRAIL followed (ZIR700/TRAIL + Light) or not followed by (ZIR700/TRAIL-Light) illumination. The tumor grafts were removed and sectioned under freezing conditions 1 h post-injection of TRAIL. The blood vessels were visualized with antibody against CD31. The nuclei were visualized by DAPI staining. Original magnification 200×. (B) ZIR700-mediated PDT and TRAIL combination therapy induced stronger apoptosis. Mice bearing LS174T tumor grafts were treated with ZIR700-mediated PDT alone (ZIR700+Light) or ZIR700 combined with TRAIL followed (ZIR700/TRAIL + Light) or not followed (ZIR700/TRAIL-Light) by illumination. Tumor grafts were removed 16 h post-injection of TRAIL and sectioned under freezing conditions. TUNEL was used to visualize apoptotic cells in tumor tissues. Original magnification 200×. (C) ZIR700-mediated PDT and TRAIL combination therapy shows greater tumor growth suppression. Mice (n = 6–7) were treated with TRAIL or ZIR700-mediated PDT (ZIR700+Light) or TRAIL and ZIR700-mediated PDT combination therapy (ZIR700/TRAIL + Light). PBS was used as a control.
Discussion
Conventional tumor vascular-targeted PDT is achieved by the injection of free PS followed by illumination within a short time (several minutes), when the majority of PS is retained in the blood vessels (Krzykawska-Serda et al., Citation2014; Azzouzi et al., Citation2017). The short drug (PS administration)-to-light (illumination) interval (DLI) and non-specific accumulation of PS would definitely limit the application of passive PS-delivery-based vascular-targeted PDT in the clinic. In recent years, increasing attention has been paid to active PS-delivery-based vascular-targeted PDT (Kamarulzaman et al., Citation2011). Theoretically, as the major vascular cells of tumors, both endothelial cells and pericytes could be considered target cells for PS delivery. Endothelial cell-targeted PS delivery has been achieved by the conjugation of PS to VEGFR- or αvβ3 integrin-binding molecules (Thomas et al., Citation2010; Srivatsan et al., Citation2011). However, tumor vascular-targeted PDT based on pericyte-targeted PS delivery has not been reported.
Endothelial cells compose the endothelium, whereas pericytes cover the endothelial tubule. In tumor blood vessels, endothelial cells irregularly line the walls of the vessel, which allows delivery of PS to pericytes (Chang et al., Citation2013; Geevarghese & Herman, Citation2014). However, the pericytes in normal blood vessels are shielded by the intact endothelial-cell barrier. Consequently, compared to the endothelial cell-targeted strategy, pericyte-targeted PS delivery involves a lower risk of accumulation of PS in normal tissues. Owing to the high expression of PDGFRβ in tumor-associated pericytes (Paulsson et al., Citation2009) and the high affinity and specificity of ZPDGFRβ affibody for PDGFRβ (Lindborg et al., Citation2011; Tao et al., Citation2017), pericyte-targeted delivery can be achieved by the conjugation of PS to the ZPDGFRβ affibody. It is noteworthy that among vascular cells, smooth muscle cells also express PDGFRβ. Unlike the normal mature blood vessels, most tumor blood vessels are immature, with cellular abnormalities (Baluk et al., Citation2005). In fact, few smooth muscle cells were observed in tumor blood vessels (). Consequently, pericytes are major PDGFRβ+ mural cells in tumor grafts, thus conjugation to the ZPDGFRβ affibody would deliver PS to pericytes.
Due to the ease of purification and reduced hepatic accumulation of the HE-tagged affibody (Hofstrom et al., Citation2011), a HE-tag, not a His-tag, was introduced at the N-terminus of Z09591 to produce the ZPDGFRβ affibody. In addition, the ZPDGFRβ affibody was designed to form a dimer upon the addition of a cysteine residue at the C-terminus. As expected, most ZPDGFRβ affibodies formed dimers under natural conditions (). In addition, the affinity (0.9 nM) of the dimeric ZPDGFRβ affibody for human PDGFRβ () was higher than that (4.5 nM) of the monomeric ZPDGFRβ affibody (Tao et al., Citation2017). In mice bearing LS174T tumor grafts, accumulation of the dimeric ZPDGFRβ affibody in tumor grafts was higher than that in the liver or the kidney at 4 h post-injection (), indicating the tumor-homing characteristic of the ZPDGFRβ affibody. Among vascular cells, NG2 is exclusively expressed on pericytes (). The ZPDGFRβ affibody co-localized well with NG2 in the mural cells of tumor blood vessels ( and Supplementary Figure S1), demonstrating that it was predominantly distributed on pericytes in tumor blood vessels. ZIR700, the conjugate of the ZPDGFRβ affibody and IR700, bound pericytes and induced obvious cell death in vitro after excitation by light ( and Supplementary Figure S2(A)). Since pericytes are major mural cells, ZIR700-mediated PDT in vivo might damage them, thus increasing the permeability of tumor blood vessels.
To determine the PDT-induced increase in permeability, dextran (∼70 kDa) was intravenously injected into mice bearing LS174T tumor grafts at different times post-illumination. As shown in , ZIR700-mediated PDT induced the leakage of dextran injected at the beginning (0 h) of illumination. Tumor uptake of TRAIL injected at the same time point was increased by ZIR700-mediated PDT (). In addition, numerous red blood cells were observed in tumor tissues collected at different times post-illumination (). These results demonstrated that ZIR700-mediated PDT rapidly damaged tumor blood vessels and induced hemorrhage. However, compared to tumor accumulation of the dextran injected at the beginning (0 h) of illumination, that of dextran injected at 1 and 8 h post-illumination decreased drastically (). However, HIF1α expression, reflecting hypoxia in tumor tissues, increased over time post-illumination (). Moreover, blood clots were observed in tumor tissues collected between 1 and 4 h post-illumination (). These results demonstrated that thrombosis, and even vessel occlusion, occurred after a short time post-illumination. Consequently, the delivery function of tumor blood vessels was impaired, and tissue hypoxia was intensified. It is known that intensified hypoxia can induce the necrosis/apoptosis of tumor cells (van Straten et al., Citation2017). In fact, tumor destruction was observed in tumor grafts collected at 24 and 72 h post-illumination (). Due to the inefficiency of ROS to kill bystander cells (Heukers et al., Citation2014), ZIR700-mediated pericyte-targeted PDT would not kill bystander LS174T tumor cells directly, suggesting that ZIR700-mediated PDT induced tumor destruction by intensifying hypoxia.
To improve the efficacy, the DLI for passive PS-delivery-based vascular-targeted PDT is usually shortened to several minutes. For example, long-term tumor suppression was observed when verteporfin was excited by light at 5 min post-injection. If the illumination was performed at 3 h post-injection of verteporfin, no apparent acute vascular responses were seen (Fingar et al., Citation1999). In this experiment, vascular responses and long-term tumor suppression were observed even when illumination was performed at 4 h post-injection of ZIR700 ( and ). These results suggested that DLI of the ZPDGFRβ affibody-based active vascular-targeted PDT might be considerably longer than that of passive vascular-targeted PDT, which would drastically reduce the adverse effects of PS. Compared to the big (∼150 kDa) antibody, the small (∼6–7 kDa) affibody has the advantage of superior tumor penetration (Sexton et al., Citation2013). In addition, high yield (20–30 mg/l culture) of the small affibody in E. coli would reduce the cost of PDT. These results highlighted the potential of ZPDGFRβ affibody-based vascular-targeted PDT for cancer therapy.
Conclusions
Abnormal structures of tumor blood vessels make PDGFRβ-positive pericytes attractive target cells for drug delivery. Due to the high affinity and specificity for PDGFRβ, the ZPDGFRβ affibody was predominantly distributed on PDGFRβ+ pericytes and accumulated in tumor grafts. The conjugate of the ZPDGFRβ affibody and IR700, i.e. ZIR700, specifically killed pericytes once excited by light. ZIR700-mediated PDT damaged tumor blood vessels thus induced tumor destruction in mice. In addition, ZIR700-mediated PDT enhanced the antitumor effect of TRAIL by increasing its tumor uptake. Taken together, these results demonstrate that the ZPDGFRβ affibody could be used as a PS carrier for active vascular-targeted PDT as a monotherapy as well as combination therapy for cancers with rich pericytes.
Ethical approval
All applicable institutional guidelines for the care and use of animals were followed.
IDRD_Lu_et_al_Supplemental_Content.docx
Download MS Word (10.3 MB)Acknowledgements
The authors thank Yi Zhang, Yan Liang (from the Experimental Histopathology Platform of West China Hospital) for their help in collecting and sectioning tumor tissues.
Disclosure statement
The authors declare that they have no conflicts of interest.
Additional information
Funding
Related Research Data
References
- Askoxylakis V, Marr A, Altmann A, et al. (2013). Peptide-based targeting of the platelet-derived growth factor receptor beta. Mol Imaging Biol 15:212–21.
- Azzouzi A-R, Vincendeau S, Barret E, et al. (2017). Padeliporfin vascular-targeted photodynamic therapy versus active surveillance in men with low-risk prostate cancer (CLIN1001 PCM301): an open-label, phase 3, randomised controlled trial. Lancet Oncol 18:181–91.
- Baluk P, Hashizume H, McDonald DM. (2005). Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev 15:102–11.
- Bugaj AM. (2011). Targeted photodynamic therapy-a promising strategy of tumor treatment. Photochem Photobiol Sci 10:1097–109.
- Chang WG, Andrejecsk JW, Kluger MS, et al. (2013). Pericytes modulate endothelial sprouting. Cardiovasc Res 100:492–500.
- Dąbrowski JM. (2017). Reactive oxygen species in photodynamic therapy: mechanisms of their generation and potentiation. Adv Inorgan Chem 70:343–94.
- Dabrowski JM, Arnaut LG. (2015). Photodynamic therapy (PDT) of cancer: from local to systemic treatment. Photochem Photobiol Sci 14:1765–80.
- Dolmans DEJGJ, Dai F, Jain RK. (2003). Photodynamic therapy for cancer. Nat Rev Cancer 3:380–5.
- Fingar VH, Kik PK, Haydon PS, et al. (1999). Analysis of acute vascular damage after photodynamic therapy using benzoporphyrin derivative (BPD). Br J Cancer 79:1702–8.
- Geevarghese A, Herman IM. (2014). Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Transl Res 163:296–306.
- Gollmer A, Arnbjerg J, Blaikie FH, et al. (2011). Singlet oxygen sensor green(R): photochemical behavior in solution and in a mammalian cell. Photochem Photobiol 87:671–9.
- Heukers R, van Bergen en Henegouwen PM, Oliveira S. (2014). Nanobody-photosensitizer conjugates for targeted photodynamic therapy. Nanomedicine 10:1441–51.
- Hofstrom C, Orlova A, Altai M, et al. (2011). Use of a HEHEHE purification tag instead of a hexahistidine tag improves biodistribution of affibody molecules site-specifically labeled with (99m)Tc, (111)In, and (125)I. J Med Chem 54:3817–26.
- Holland PM. (2013). Targeting Apo2L/TRAIL receptors by soluble Apo2L/TRAIL. Cancer Lett 332:156–62.
- Kamarulzaman EE, Benachour H, Barberi-Heyob M, et al. (2011) Vascular targeted photodynamic therapy (VTP). In: Gali-Muhtasib H, ed. Advances in cancer therapy. Croatia: InTech, 99–122.
- Kim D, Yan Y, Valencia CA, Liu R. (2012). Heptameric targeting ligands against EGFR and HER2 with high stability and avidity. PLoS One 7:e43077.
- Krzykawska-Serda M, Dabrowski JM, Arnaut LG, et al. (2014). The role of strong hypoxia in tumors after treatment in the outcome of bacteriochlorin-based photodynamic therapy. Free Radic Biol Med 73:239–51.
- Li R, Yang H, Jia D, et al. (2016). Fusion to an albumin-binding domain with a high affinity for albumin extends the circulatory half-life and enhances the in vivo antitumor effects of human TRAIL. J Control Release 228:96–106.
- Lindborg M, Cortez E, Hoiden-Guthenberg I, et al. (2011). Engineered high-affinity affibody molecules targeting platelet-derived growth factor receptor beta in vivo. J Mol Biol 407:298–315.
- Lowa SW, Lin AW. (2000). Apoptosis in cancer. Carcinogenesis 21:489–95.
- Marr A, Nissen F, Maisch D, et al. (2013). Peptide arrays for development of PDGFRβ Affine molecules. Mol Imaging Biol 15:391–400.
- McGurn LD, Moazami-Goudarzi M, White SA, et al. (2016). The structure, kinetics and interactions of the β-carboxysomal β-carbonic anhydrase, CcaA. Biochem J 473:4559–72.
- Mitsunaga M, Ogawa M, Kosaka N, et al. (2011). Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat Med 17:1685–91.
- Paulsson J, Sjoblom T, Micke P, et al. (2009). Prognostic significance of stromal platelet-derived growth factor beta-receptor expression in human breast cancer. Am J Pathol 175:334–41.
- Prakash J, de Jong E, Post E, et al. (2010). A novel approach to deliver anticancer drugs to key cell types in tumors using a PDGF receptor-binding cyclic peptide containing carrier. J Control Release 145:91–101.
- Schmitt F, Juillerat-Jeanneret L. (2012). Drug targeting strategies for photodynamic therapy. Anticancer Agents Med Chem 12:500–25.
- Setsukinai K, Urano Y, Kakinuma K, et al. (2003). Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J Biol Chem 278:3170–5.
- Sexton K, Tichauer K, Samkoe KS, et al. (2013). Fluorescent affibody peptide penetration in glioma margin is superior to full antibody. PLoS One 8:e60390,
- Shafirstein G, Bellnier D, Oakley E, et al. (2017). Interstitial photodynamic therapy – a focused review. Cancers 9:e12.
- Shirasu N, Nam SO, Kuroki M. (2013). Tumor-targeted photodynamic therapy. Anticancer Res 33:2823–32.
- Shirasu N, Yamada H, Shibaguchi H, et al. (2014). Potent and specific antitumor effect of CEA-targeted photoimmunotherapy. Int J Cancer 135:2697–710.
- Srivatsan A, Ethirajan M, Pandey SK, et al. (2011). Conjugation of cRGD peptide to chlorophyll a based photosensitizer (HPPH) alters its pharmacokinetics with enhanced tumor-imaging and photosensitizing (PDT) efficacy. Mol Pharm 8:1186–97.
- Tao Z, Yang H, Shi Q, et al. (2017). Targeted delivery to tumor-associated pericytes via an affibody with high affinity for PDGFRβ enhances the in vivo antitumor effects of human TRAIL. Theranostics 7:2261–76.
- Thomas N, Pernot M, Vanderesse R, et al. (2010). Photodynamic therapy targeting neuropilin-1: interest of pseudopeptides with improved stability properties. Biochem Pharmacol 80:226–35.
- Tolmachev V, Varasteh Z, Honarvar H, et al. (2014). Imaging of platelet-derived growth factor receptor beta expression in glioblastoma xenografts using affibody molecule 111In-DOTA-Z09591. J Nucl Med 55:294–300.
- van Driel PB, Boonstra MC, Slooter MD, et al. (2016). EGFR targeted nanobody-photosensitizer conjugates for photodynamic therapy in a pre-clinical model of head and neck cancer. J Control Release 229:93–105.
- van Straten D, Mashayekhi V, de Bruijn HS, et al. (2017). Oncologic photodynamic therapy: basic principles, current clinical status and future directions. Cancers 9:e19.
- Wei D, Fan Q, Cai H, et al. (2015). CF750-A33scFv-Fc-based optical imaging of subcutaneous and orthotopic xenografts of GPA33-positive colorectal cancer in mice. BioMed Res Int 2015:505183.