
Abstract
The efficacy of traditional chemotherapy often suffers from rapid clearance and off-target toxicity. Drug delivery systems and controlled release are applied to improve the therapeutic efficiencies of small-molecule drugs. In this work, two novel oxidative/reductive (Ox/Re) -sensitive and one non-sensitive Paclitaxel (PTX) prodrugs were synthesized with a maleimide group, which rapidly conjugates with albumin in vivo. Albumin serves as a good vehicle to deliver more prodrug to tumors due to the enhanced permeation and retention (EPR) effect. PTX was then released from the prodrugs in glutathione(GSH)/ reactive oxygen species(ROS)-rich tumor microenvironments. This bioresponsive prodrug strategy demonstrates potent chemotherapeutic efficiency in vivo and may be utilized in clinical cancer therapy.
Introduction
Cancer is one of the most common life-threatening diseases in the world. Chemotherapy is the first choice for the treatment of most cancers but has some limitations, such as poor bioavailability, rapid blood clearance, non-selectivity, and high toxicity to normal cells and tissues. Drug delivery systems (DDS; Jahangirian et al., Citation2017; Ramasamy et al., Citation2017) are designed for improving the therapeutic efficiencies of anticancer drugs. Chemotherapeutic drugs are loaded or covalently conjugated to the delivery systems and expected to release only at specific tumor sites. In DDS, chemotherapeutic agents can be triggered by a unique tumor microenvironment (TME), a low pH value, a high concentration of reactive oxygen species (ROS), or glutathione (GSH), etc. The overproduced GSH creates a strongly reductive environment in tumor cells, for which prodrug, DDS, and disulfide bond strategies have been developed to facilitate an efficient intracellular release of anticancer drugs. The intramolecular disulfide bond is most likely mediated by thiol-disulfide exchange reactions with GSH. Moreover, most cancer cells simultaneously exhibit elevated amounts of ROS and various ROS-responsive DDS have been developed and investigated for therapeutic purposes. The monosulfide/thioether functional groups impart the intracellular ROS-responsiveness. A thioether group can be oxidized to a sulfone to induce ester bond hydrolysis.
Along with DDS, prodrug strategies are also applied to enhance the anticancer efficiencies of small-molecule drugs. Prodrugs are inactive compounds that can be triggered to release the parent drug in a controlled manner.
Paclitaxel (Taxol®, PTX), an acylated diterpene with strong antitumor activity isolated from yew tree bark, is one of the most effective chemotherapeutic drugs and is mainly used for the treatment of lung, ovarian, prostatic, and breast cancers (Malonga et al., Citation2005; Gordaliza, Citation2007). The mechanism of PTX involves the stabilization of microtubules in cells, which counteracts their depolymerization (Jordan & Wilson, Citation2004). Despite its excellent antitumor activity in vitro, the therapeutic efficacy of Taxol® is greatly limited by poor efficiency of drug delivery and serious systemic toxicity to normal tissues. To avoid the side effects of Taxol®, various approaches including stimuli-responsive DDS (Chuan et al., Citation2014; Xiao et al., Citation2017), targeted therapy (Zhong et al., Citation2016), nanoparticles (Han et al., Citation2015; Colby et al., Citation2016), nanomicelles (Lian et al., Citation2011; He et al., Citation2016), and prodrug strategies (Satsangi et al., Citation2014; Wohl et al., Citation2014) have been attempted to improve the delivery and antitumor efficiency of PTX.
Our group has investigated a series of taxane DDS, including redox-sensitive PTX-prodrug nanoparticles (Liu et al., Citation2011; Luo et al., Citation2016b), a dual-sensitive nanosystem prodrug (Luo et al., Citation2016a), and nanomicelles (Lian et al., Citation2011; Liu et al., Citation2014), and the oxidative/reductive (Ox/Re) release of drugs at the tumor site has proven to be a powerful strategy to improve taxane delivery and distribution (Wang et al., Citation2014).
Human serum albumin (HSA) is an abundant, multifunctional, and negatively charged plasma protein (Elsadek & Kratz, Citation2012; Kratz & Elsadek, Citation2012). Albumin is the most abundant serum protein and serves as a transport vehicle for several endogenous compounds with high affinity. The thiol (HS) group of cysteine-34 in HSA is the most reactive thiol group in human plasma because of the low pKa of Cys-34 in HSA, which is approximately seven compared to 8.5 and 8.9 for Cys and GSH, respectively (Kratz et al., Citation2002). The free thiol group in Cys-34 can covalently conjugate to a variety of nucleophile acceptors. (the [6-maleimidocaproyl]-hydrazine derivative of doxorubicin [DOXO-EMCH])(Kratz, Citation2011), an albumin-binding prodrug of doxorubicin invented by Kratz, is now under development for phase II studies. Compared to doxorubicin, INNO-206 showed a large area under the curve (AUC) and a low clearance (Kratz et al., Citation2013). However, the albumin-covalent conjugation strategy for taxane DDS has not been reported yet.
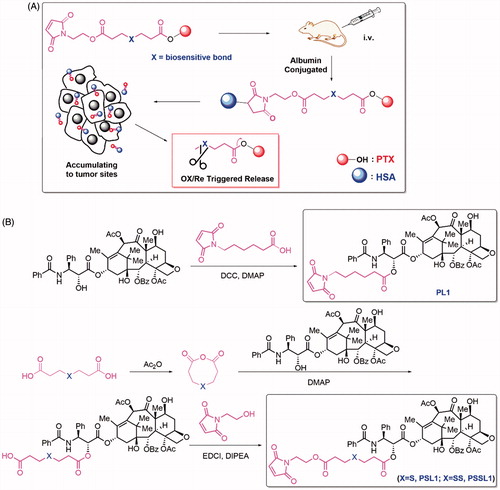
In this work, albumin is employed as an ideal drug cargo for covalent conjugation. As shown in , PTX was linked to an N-substituted maleimide group by a biosensitive, cleavable linker. After intravenous administration, the prodrugs bind rapidly to the Cys-34 position of circulating albumin and accumulate in solid tumors due to passive targeting and the enhanced permeation and retention (EPR) effect. The hyperpermeability of tumor vessels and the absence of functional lymphatic circulation allows the prodrug-albumin conjugates to remain in tumors for a long time. The binding of prodrugs to albumin and their EPR-mediated accumulation and distribution in tumors differ from the diffusion-mediated distribution of free PTX in a significant way.
Figure 1. (A) Design of the albumin-conjugated PTX prodrugs; (B) Synthetic routes of PL1, PSL1, and PSSL1.
Materials and methods
Materials
PTX was obtained from Jingzhu Biotech Corporation (Nanjing, China); N,N’-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI), N,N-diisopropylethylamine (DIPEA), 3,3′-thiodipropionic acid, 3,3′-dithiodipropionic acid, hydrogen peroxide (H2O2), and dithiothreitol (DTT) were purchased from Aladdin Industrial Corporation (Shanghai, China). Cell culture reagents were purchased from Gibco, Invitrogen Corp. (Carlsbad, CA). 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) and trypsin-EDTA were purchased from Meilun Biotech Co., Ltd. (Dalian, China). 6-Maleimidohexanoic acid and N-(2-hydroxyethyl) maleimide were synthesized according to previously reported literature.
Synthesis of PTX prodrugs
A solution of 6-maleimidohexanoic acid (211 mg, 1 mmol), DCC (412 mg, 2 mmol), and DMAP (24 mg, 0.2 mmol) in dry DCM (20 mL) was stirred for 0.5 h under an argon atmosphere and a solution of PTX (854 mg, 1 mmol) in dry DCM (10 mL) was added dropwise. After 12 h, the unsolvable solid was filtered off and the solvent was concentrated under reduced pressure. The residue was purified by silica gel chromatography to afford the PTX-prodrug PL1 () as a white powder (633 mg, 59%). Nuclear magnetic resonance (1 H NMR; 600 MHz, chloroform-d [CDCl3]) δ 8.16–8.12 (m, 2 H), 7.77–7.72 (m, 2 H), 7.61 (t, J = 7.4 Hz, 1 H), 7.51 (dt, J = 11.8, 7.6 Hz, 3 H), 7.45–7.32 (m, 7 H), 7.01 (d, J = 9.3 Hz, 1 H), 6.64 (s, 2 H), 6.34–6.21 (m, 2 H), 5.97 (dd, J = 9.2, 3.4 Hz, 1 H), 5.69 (d, J = 7.0 Hz, 1 H), 5.50 (d, J = 3.4 Hz, 1 H), 5.01–4.96 (m, 1 H), 4.46 (dt, J = 10.9, 6.0 Hz, 1 H), 4.32 (d, J = 8.5 Hz, 1 H), 4.21 (d, J = 8.4 Hz, 1 H), 4.05 (s, 1 H), 3.82 (d, J = 7.0 Hz, 1 H), 3.59–3.49 (m, 1 H), 3.49–3.42 (m, 3 H), 2.61–2.51 (m, 2 H), 2.47 (s, 3 H), 2.43–2.34 (m, 3 H), 2.23 (s, 3 H), 2.17 (s, 8 H), 1.94 (d, J = 1.4 Hz, 4 H), 1.88 (d, J = 12.7 Hz, 1 H), 1.80 (s, 1 H), 1.70 (s, 1 H), 1.68 (s, 4 H), 1.65–1.59 (m, 10 H), 1.57–1.52 (m, 2 H), 1.38–1.31 (m, 3 H), 1.26 (d, J = 4.9 Hz, 3 H), 1.24 (s, 3 H), 1.14 (s, 3 H), 1.09 (d, J = 11.9 Hz, 2 H), and 0.88 (t, J = 6.8 Hz, 1 H). 13 Carbon NMR (13 C NMR ;151 MHz, CDCl3) δ 207.02, 203.85, 172.42, 171.27, 170.84, 169.80, 168.07, 167.10, 167.04, 156.66, 142.87, 136.97, 134.03, 133.69, 132.70, 131.98, 130.23, 129.17, 129.04, 128.74, 128.69, 128.46, 127.14, 126.54, 84.46, 81.03, 79.17, 75.61, 75.09, 73.92, 72.13, 71.72, 58.51, 52.78, 51.28, 49.16, 46.97, 45.56, 45.00, 43.16, 37.36, 35.56, 35.51, 33.96, 33.55, 32.77, 30.95, 28.33, 27.97, 26.80, 26.33, 25.80, 25.61, 24.95, 24.76, 24.08, 22.71, 22.15, 20.85, 14.86 and 9.61. Electrospray mass spectrometry (MS (ESI)) m/z: 1069 [M + Na]+.
A solution of 3,3′-thiodipropionic acid (0.89 g, 5 mmol) in acetic anhydride was stirred at room temperature for 4 h under an argon atmosphere then the acetic anhydride was evaporated under vacuum followed by the addition of PTX (4.27 g, 5 mmol) and DMAP (244 mg, 2 mmol) in dry DCM (50 mL). The reaction was stirred for 16 h at room temperature under an argon atmosphere then concentrated under reduced pressure. The residue was purified by silica gel chromatography to afford PS () as a white powder (3.62 g, 69%).
To a solution of PS (1.02 g, 1 mmol), EDCI (384 mg, 2 mmol), DIPEA (129 mg, 1 mmol), and N-(2-hydroxyethyl) maleimide (212 mg, 1 mmol) in dry DCM were added at room temperature under argon atmosphere for 12 h. The solvent was then evaporated under vacuum and the residue was purified by silica gel chromatography to afford the PTX-prodrug PSL1 () as a white powder (733 mg, 68%). 1 H NMR (600 MHz, CDCl3) δ 8.17–8.12 (m, 2 H), 7.78–7.72 (m, 2 H), 7.64–7.58 (m, 1 H), 7.51 (dt, J = 14.2, 7.6 Hz, 3 H), 7.45–7.37 (m, 6 H), 7.37–7.31 (m, 1 H), 7.09 (d, J = 9.1 Hz, 1 H), 6.69 (s, 2 H), 6.30 (s, 1 H), 6.28–6.21 (m, 1 H), 5.98 (dd, J = 9.2, 3.4 Hz, 1 H), 5.69 (d, J = 7.1 Hz, 1 H), 5.51 (d, J = 3.4 Hz, 1 H), 4.98 (dd, J = 9.6, 2.3 Hz, 1 H), 4.48–4.42 (m, 1 H), 4.32 (d, J = 8.5 Hz, 1 H), 4.26–4.18 (m, 3 H), 3.81 (d, J = 7.0 Hz, 1 H), 3.77 (t, J = 5.2 Hz, 2 H), 2.79–2.65 (m, 6 H), 2.57 (td, J = 9.1, 8.7, 4.9 Hz, 1 H), 2.55–2.50 (m, 3 H), 2.45 (s, 3 H), 2.36 (dd, J = 15.4, 9.3 Hz, 1 H), 2.23 (s, 3 H), 2.17 (s, 8 H), 2.16–2.13 (m, 1 H), 1.94 (d, J = 1.4 Hz, 3 H), 1.92–1.86 (m, 1 H), 1.83 (d, J = 2.6 Hz, 1 H), 1.68 (s, 3 H), 1.68–1.64 (m, 5 H), 1.35 (d, J = 12.2 Hz, 1 H), 1.31–1.27 (m, 2 H), 1.27–1.25 (m, 1 H), 1.24 (s, 3 H), 1.18 (d, J = 6.1 Hz, 1 H), 1.14 (s, 3 H), 1.12–1.05 (m, 1 H), and 0.88 (t, J = 6.8 Hz, 1 H). 13 C NMR (151 MHz, CDCl3) δ 207.03, 203.82, 171.49, 171.26, 170.85, 170.47, 169.80, 167.96, 167.16, 167.02, 142.75, 136.91, 134.22, 133.68, 132.77, 132.00, 130.23, 129.17, 129.08, 128.74, 128.67, 128.51, 127.19, 126.59, 84.44, 81.05, 79.14, 75.59, 75.07, 74.27, 72.13, 71.87, 68.20, 61.79, 58.50, 52.80, 45.56, 43.17, 39.37, 36.80, 35.53, 34.42, 34.30, 33.95, 31.83, 30.95, 29.31, 26.81, 26.74, 26.69, 25.74, 25.61, 24.95, 23.48, 22.71, 22.61, 22.14, 20.85, 14.85, 14.10 and 9.61. MS (ESI) m/z: 1159 [M + Na]+.
The PTX-prodrug PSSL1 () (white powder, 865 mg, 74%) was obtained via the same protocol used to obtain PSL1. 1 H NMR (600 MHz, CDCl3) δ 8.17–8.12 (m, 2 H), 7.77–7.72 (m, 2 H), 7.61 (t, J = 7.4 Hz, 1 H), 7.51 (dt, J = 14.1, 7.5 Hz, 3 H), 7.46–7.37 (m, 6 H), 7.35 (t, J = 7.2 Hz, 1 H), 7.09 (d, J = 9.2 Hz, 1 H), 6.69 (s, 2 H), 6.30 (s, 1 H), 6.25 (t, J = 9.1 Hz, 1 H), 5.99 (dd, J = 9.2, 3.2 Hz, 1 H), 5.69 (d, J = 7.1 Hz, 1 H), 5.53 (d, J = 3.2 Hz, 1 H), 5.01–4.95 (m, 1 H), 4.45 (dt, J = 11.0, 5.8 Hz, 1 H), 4.32 (d, J = 8.6 Hz, 1 H), 4.26–4.17 (m, 3 H), 4.04 (d, J = 8.0 Hz, 1 H), 3.82 (d, J = 7.0 Hz, 1 H), 3.76 (dd, J = 6.0, 4.6 Hz, 2 H), 3.48 (d, J = 4.6 Hz, 1 H), 2.87 (ddd, J = 9.4, 6.3, 3.9 Hz, 3 H), 2.85–2.79 (m, 2 H), 2.66 (t, J = 7.0 Hz, 2 H), 2.56 (ddd, J = 14.6, 9.6, 6.5 Hz, 1 H), 2.52 (d, J = 4.0 Hz, 1 H), 2.46 (s, 3 H), 2.37 (dd, J = 15.4, 9.4 Hz, 1 H), 2.23 (s, 3 H), 2.17 (s, 6 H), 1.97–1.85 (m, 6 H), 1.79 (s, 1 H), 1.69 (s, 4 H), 1.34 (t, J = 10.8 Hz, 2 H), 1.31–1.28 (m, 2 H), 1.27 (d, J = 17.0 Hz, 3 H), 1.24 (s, 2 H), 1.18 (d, J = 6.1 Hz, 1 H), 1.14 (s, 4 H), 1.11–1.05 (m, 1 H) and 0.88 (t, J = 6.8 Hz, 1 H). 13 C NMR (151 MHz, CDCl3) δ 203.82, 171.42, 171.27, 170.78, 170.45, 169.81, 167.93, 167.18, 167.03, 142.76, 136.88, 134.23, 133.69, 133.66, 132.77, 132.00, 130.23, 129.16, 129.11, 128.74, 128.67, 128.52, 127.20, 126.57, 84.44, 81.05, 79.15, 75.60, 75.06, 74.28, 72.14, 71.89, 68.21, 61.86, 58.51, 52.74, 49.17, 45.56, 43.18, 39.38, 36.76, 35.53, 33.96, 33.87, 33.54, 33.00, 32.55, 31.84, 30.96, 29.32, 26.82, 25.74, 25.61, 24.95, 23.49, 22.72, 22.62, 22.14, 20.86, 14.86, 14.10 and 9.61. MS (ESI) m/z: 1192 [M + Na]+.
PTX release in vitro
The release of PTX from the prodrugs was investigated at 37 °C in PBS (pH 7.4) containing 20% ethanol (EtOH) with different concentrations of DTT or H2O2. Briefly, 1 mg/mL equivalent PTX prodrug/EtOH solution was incubated with bovine serum albumin (BSA) solution (10 mg/mL) to generate prodrug-albumin conjugates. Prodrug-albumin conjugates were sealed in a dialysis bag and incubated in 30 mL of release medium: PL1 in 0/10 mM H2O2/10 mM DTT/PBS; PSL1 in 0/10 µM/10 mM H2O2/PBS; and PSSL1 in 0/10 µM/10 mM DTT/PBS. At timed intervals, 1 mL of sample solution was withdrawn and analyzed by HPLC spectrometry (Hitachi HPLC system ,Tokyo, Japan; mobile phase: acetonitrile: water (70:30, v/v); flow rate: 1 mL/min; chromatographic column: 150 mm × 4.6 mm and SHISEIDO C18; wavelength: 230 nm; injection volume: 10 µL).
Cell culture
The PC-3 human prostatic carcinoma cell line was maintained in F-12 K medium containing 1% antibiotics and 10% fetal bovine serum (FBS) at 37 °C under 5% Carbon di oxide (CO2) and the KB human oral epidermoid carcinoma cell line and the 4T1 mouse breast cancer cell line were maintained in Roswell Park Memorial Institute medium (RPMI)-1640 medium containing 1% antibiotics and 10% FBS at 37 °C under 5% CO2.
Cytotoxicity analysis
The cytotoxicity of the PTX prodrugs and PTX was evaluated by MTT assays in accordance with the manufacturer’s instructions. Briefly, approximately 2000 carcinoma cells were seeded in each well of a 96-well plate. The original medium was removed after 12 h of attachment and exchanged with 200 µL of fresh medium containing different equivalent PTX concentrations (from 0 to 1000 ng) of PTX, PL1, PSL1, or PSSL1. The cells were incubated at 37 °C under 5% CO2 for 48 h. Then, the medium was replaced with a fresh medium without FBS followed by the addition of 20 µL of MTT (2 mg/mL) to each well. After 4 h of incubation, the solution was removed and 100 µL of DMSO was added. The absorbance was measured at 570 nm using a Thermo Scientific Microplate Reader (Thermo Scientific, Waltham, MA).
Cell uptake
4T1 breast tumor cells were seeded in each well (150,000 cells/well) in a 24-well plate and after 12 h of attachment, the original medium was removed and replaced with fresh medium containing different equivalent PTX concentrations (1 and 5 µg) of PTX, PL1, PSL1, or PSSL1 for another 1, 4 and 8 h incubation period. Then, the cells were washed three times with 4 °C PBS, harvested and collected by centrifugation (3000 rpm, 6 min) and the precipitate was resuspended in 300 µL of PBS. The residue was crushed in an ultrasonic cell crusher. The concentration of protein in the cells was measured using a bicinchoninic acid (BCA) protein assay kit in accordance with the manufacturer’s instructions. The intracellular PTX concentrations were measured by a Ultra performance liquid chromatography - tandem mass spectrometer (UPLC-MS/MS).system.
Formulation of the prodrugs
The formulation of the prodrugs was the same as the formulation used for PTX. 6 mg of equivalent PTX dose of each prodrug was dissolved in 0.5 mL of EtOH and were later mixed with 0.68 g of Cremophor. The solution was diluted with saline to obtain a 1 mg/mL equivalent PTX concentration solution. All the formulations were prepared before injection.
Biodistribution and fluorescence imaging
4T1 cells (2.5 × 105) were injected into the right flank of female BALB/c mice. When the tumor volume reached ∼200 mm3, 10 mg/kg equivalent PTX dose of the three prodrugs and PTX were injected into the tail veins, 4 and 24 h later, the mice were sacrificed and the main organs and tumors were harvested. All the samples were washed with PBS, weighed, and homogenized in 1 mL of 50% EtOH. The PTX concentrations in these tissues and tumors were measured by a UPLC-MS system.
The biodistribution properties of the PTX-prodrug-albumin conjugates were also assessed using an IVIS Lumina Imaging System (PerkinElmer, Waltham, MA). IR820-maleimide dye was synthesized according to reported literature. 10 µM/kg dose of free IR820 or IR820-maleimide was intravenously injected into the BALB/c mice bearing 4T1 cells. After 2, 8, 24 and 48 h, all the mice were sacrificed and the images and fluorescence intensities of tumors and organs (heart, liver, spleen, lung, and kidney) were visualized and calculated (for free IR820: excitation and emission wavelength was 780 and 845 nm; for IR820-maleimide: excitation and emission wavelength was 680 and 720 nm, respectively).
Antitumor efficiency
4T1 cells (approximately 5 × 105) were injected into the right flank of female BALB/c mice. When the tumor volume reached ∼100 mm3, saline, PTX, PL1, PSL1 and PSSL1 (10 mg/kg equivalent PTX) were intravenously injected every 2 days for a total five injections. The tumor volume and body weight of each mouse was measured every day. After the fifth injection, all the mice were sacrificed and the organs and tumors were weighed, fixed in 4% formaldehyde, cut into 5 mm sections and stained with hematoxylin and eosin (H&E) for the histopathological evaluation.
Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL) assay
The unstained tumor sections were dewaxed and further analyzed with a TUNEL assay kit in accordance with the manufacturer’s instructions to identify the apoptotic cells.
Hematological parameters
After the last treatment in the tumor efficiency assay, all the mice were sacrificed and the blood was collected. Serum (0.2 mL) was prepared to determine the blood urea nitrogen (BUN), creatinine (CREA), aspartate transaminase (AST), alanine transaminase (ALT), and uric acid (UA) levels.
Results and discussion
Design and synthesis of the PTX prodrugs
The synthetic routes of the prodrugs are shown in . PL1 was synthesized by a simple esterification of 6-maleimidohexanoic acid and PTX. The synthesis of PSL1 was initiated by the dehydration of 3,3′-thiodipropionic acid then PTX was added to afford the monoester intermediate PS. PSL1 was obtain by esterification of PS and N-(2-hydroxyethyl) maleimide. The structures of PL1, PSL1 and PSSL1 were confirmed by 1 H NMR, 13 C NMR and MS.
PTX release in vitro
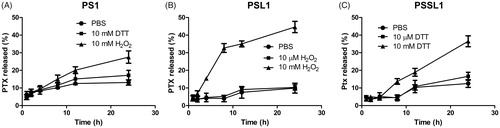
Hydrogen peroxide and DTT were utilized to investigate the Ox/Re responsiveness of the prodrugs. As shown in , less than 30% of PTX was released in 10 mM H2O2 or DTT after 24 h, which suggested that PL1 was inactive in release behavior due to the lack of a biosensitive bond. In contrast, the PSL1 prodrug, which was designed to be triggered by ROS with a monosulfide bond, showed a higher accumulated release rate in 10 mM H2O2 (ROS concentration in tumor cells) than 10 µM H2O2 (normal cells) and in PBS. More than 40% of the PTX was released from PSL1 in 10 mM H2O2 over 24 h and only ∼10% of the PTX was released in 10 µM H2O2 or PBS. The PSSL1 prodrug was successfully trigged by 10 mM DTT to release more PTX. The bioresponsive monosulfide/disulfide linker facilitated the hydrolysis of the prodrug and the ROS/GSH-induced release of PTX might enhance the PTX’s accumulation in tumors. The relatively low release rate (40–50%) of these prodrugs might be due to the non-covalent conjugate of albumin and free PTX. The PTX-albumin conjugate was prevented from escaping the dialysis bag.
Figure 2. Cumulative release of PTX from the three prodrugs under different conditions. (A) PL1 in PBS, 10 mM H2O2, and 10 mM DTT; (B) PSL1 in PBS, 10 µM H2O2, and 10 mM H2O2; (C) PSSL1 in PBS, 10 µM DTT, and 10 mM DTT.
Cytotoxicity assay
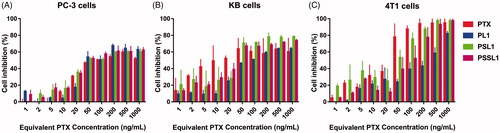
The cytotoxicity of the prodrugs was evaluated in PC-3, KB and 4T1 cells by MTT assays. As shown in , the cytotoxicity of PTX and the prodrugs exhibited concentration-dependent cytotoxic profiles. After 48 h of incubation, the half maximal inhibitory concentration (IC50) value of PTX in PC-3 cells was 16.36 ng, while the IC50 values of PL1, PSL1 and PSSL1 were 1.86-, 1.10- and 1.28-fold the PTX IC50 value, respectively. For the KB cells, the IC50 value of PTX was 32.21 ng, while those of the prodrugs were 4.38-, 1.04- and 1.23-fold the PTX value. The IC50 value of PTX in the 4T1 cells was determined to be 28.23 ng, but the prodrugs exhibited prominent increases in the IC50 value at 11.88-, 2.40- and 3.67-fold the PTX value. The decreased cytotoxicity of prodrugs might be attributed to the incomplete release of PTX or the stable conjugation between the prodrugs and albumin. The overproduced ROS/GSH in tumor cells induced a rapid and selective PTX release from the prodrugs. In contrast, PL1 presented a weaker cytotoxicity than PSL1 and PSSL1 due to its inactive release behavior in the TME.
Figure 3. Cell inhibition with various concentrations of PTX solution, PL1, PSL1, and PSSL1 in (A) PC-3 cells, (B) KB cells, and (C) 4T1 cells after 48 h of treatment.
Cell uptake
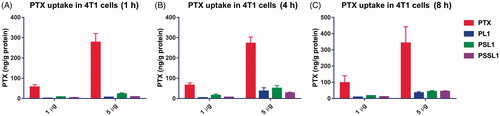
The cellular uptake behaviors of PTX and its prodrugs were investigated in 4T1 cells. The cells were incubated with PTX solution and the prodrugs for 1, 4 and 8 h, respectively. As shown in , the PTX solution was maintained at a high level in the cells through a simple concentration-mediated diffusion, while the prodrug-albumin conjugates passed through the bilayer membrane via an active transport system and the release in cells was a time-dependent process. The PTX concentration in the cells in the prodrug groups was gradually increased after an 8 h incubation.
Figure 4. Cellular uptake of PTX after treatment with 1 and 5 µg concentration PTX, PL1, PSL1, and PSSL1 at (A) 1 (B) 4, and (C) 8 h, respectively.
Biodistributions
The biodistribution of the prodrugs in 4T1 xenograft-bearing mice was investigated. When the tumor volume reached 200 mm3, the mice were intravenously administered PTX solution and the prodrugs. As shown in ), the PTX solutions rapidly distributed into the main organs but very quick cleared in vivo. In contrast, the PTX concentration in the tumors reached 229.10 ng/mL in the PSSL1 group at 24 h, while the PTX concentration dropped down to 39.50 ng/mL in the PTX solution group. This suggested that the PSSL1-albumin conjugate could successfully accumulate in tumors via passive targeting (the EPR effect) and the long retention at the tumor site and sustained release maintained the tumor concentration of PTX at an excellent high level.
Figure 5. Biodistribution of PTX in 4T1 xenograft tumor-bearing BALB/c mice at (A) 4 and (B) 24 h (n = 3). (C) Fluorescent distribution of IR-820 and IR820-maleimide at 2, 8, 24, and 48 h, respectively. (D) Calculated fluorescent efficiencies of IR820-maleimide in the main organs and tumors at 2, 8, 24, and 48 h, respectively.
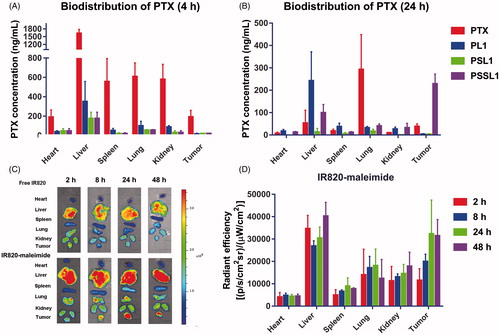
IR820-maleimide was then employed to simulate the biodistribution behavior of the PTX-maleimide-albumin conjugates in vivo. As shown in , free IR820 accumulated rapidly in the liver and tumors after 2 h, but the tumor fluorescence intensities were significantly decreased after 24 and 48 h. In contrast, IR820-maleimide demonstrated strong fluorescent signals at the tumor sites, even at 48 h, indicating that a maleimide group conjugated with albumin greatly enhanced the PTX-prodrug accumulation/retention in tumors. The fluorescence intensities of IR820-maleimide in the tumors and organs were calculated. As shown in , IR820-maleimide exhibited a strong and constant fluorescence in the tumors after 24 and 48 h.
Antitumor efficiency
The in vivo antitumor efficacy of the PTX prodrugs was further investigated using a xenograft model of BALB/c mice bearing 4T1 breast cancer cells. The mice were injected intravenously every other day with saline, PTX, PL1, PSL1 and PSSL1 (10 mg/kg equivalent PTX concentration) for five consecutive injections. The body weights and tumor volumes were measured every day and the results are presented in ). The PL1 prodrug showed a similar level of antitumor efficiency to that of the PTX solution, indicating that the PL1-albumin conjugate could accumulate in tumors with EPR effects, but the slow release rate limited its efficiency. The PSL1 and PSSL1 prodrugs had a long retention time in tumors and released more PTX triggered by high concentrations of ROS or GSH.
Figure 6. In vivo antitumor efficiencies of PTX, PL1, PSL1, and PSSL1 in 4T1 xenograft tumor-bearing BALB/c mice. (A) Changes in tumor volume; (B) Photographs of tumors after the last treatment; (C) Body weight variations; (D) Yumor weights and inhibition rates of tumor growth at the end of experiment; (E) H&E staining results of the main organs and tumors after treatment with saline, PTX, PL1, PSL1, and PSSL1; (F) TUNEL assay of tumor sections after treatment with different drug formulations.
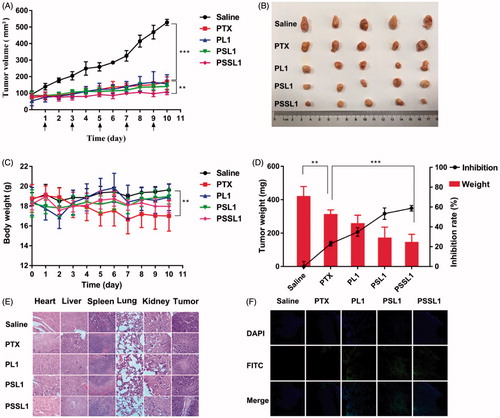
A notable weight loss was observed in the mice treated with PTX, as shown in , but there was no significant change in body weight observed in the prodrug groups. The excised tumor tissues were weighed and the tumor inhibition rates were calculated and are presented in .
H&E histopathological evaluation and TUNEL assay
To investigate the toxicity of the three prodrugs on different tissue sections, we further performed a histopathological analysis of tissues and tumors by H&E staining assays. As shown in , no noticeable histological changes were observed in H&E-stained tissue sections of the organs, which suggested that our prodrugs exhibited potent antitumor activity and no significant off-target toxicities to major organs or tissues. The degree of tumor cell necrosis in the prodrug groups, especially in the PSSL1 group, was higher than that in the PTX group, indicating that necrosis was greatly enhanced by the prodrugs compared with PTX group at the same dose.
Tumor sections from each group were then stained with TUNEL staining to evaluated the number of apoptotic cells in tumors. shows that the tumors treated with PSL1 and PSSL1 exhibited the strongest green fluorescence, which indicated the most abundant cell apoptosis. The significantly enhanced therapeutic efficiency of PSL1 and PSSL1 could be attributed to the EPR effect and the Ox/Re-trigged release of PTX in the tumor sites. The hematological parameters showed no significant difference among the three prodrugs and PTX in hepatic and renal function (Supplementary Figure S10).
Conclusion
In this study, bioresponsive albumin-conjugated prodrugs were designed and synthesized to improve the antitumor efficiency of PTX. A maleimide group was covalently conjugated to the HS group of Cys-34 in HSA after intravenous administration. The albumin was employed as a vehicle for PTX-prodrug delivery in vivo. PL1 with a non-sensitive bond was inactive in release behavior under Ox/Re conditions, while the Ox/Re-sensitive PSL1 and PSSL1 prodrugs were trigged to release more PTX in the GSH/ROS-rich TME. The prodrug-albumin conjugates displayed inferior antitumor efficiencies in the in vitro cytotoxicity and uptake assays compared with the concentration-mediated-diffusion of the PTX solution. However, the IR-maleimide-albumin conjugate indicated that the albumin conjugation strategy significantly improved the biodistribution and tumor accumulation of PTX due to its passive targeting (EPR effect). The Ox/Re-sensitive PSL1 and PSSL1 prodrugs exhibited a potent antitumor activity in 4T1 breast cancer-bearing BALB/c mice.
IDRD_Xu_et_al_Supplemental_Content.docx
Download MS Word (1.6 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Chuan X, Song Q, Lin J, et al. (2014). Novel free-paclitaxel-loaded redox-responsive nanoparticles based on a disulfide-linked poly(ethylene glycol)–drug conjugate for intracellular drug delivery: synthesis, characterization, and antitumor activity in vitro and in vivo. Mol Pharmaceutics 11:3656–70.
- Colby AH, Liu R, Schulz MD, et al. (2016). Two-step delivery: exploiting the partition coefficient concept to increase intratumoral paclitaxel concentrations in vivo using responsive nanoparticles. Sci Rep 6:18720.
- Elsadek B, Kratz F. (2012). Impact of albumin on drug delivery-new applications on the horizon. J Control Release 157:4–28.
- Gordaliza M. (2007). Natural products as leads to anticancer drugs. Clin Transl Oncol 9:767–76.
- Han J, Michel AR, Lee HS, et al. (2015). Nanoparticles containing high loads of paclitaxel-silicate prodrugs: formulation, drug release, and anticancer efficacy. Mol Pharmaceutics 12:4329–35.
- He Z, Wan X, Schulz A, et al. (2016). A high capacity polymeric micelle of paclitaxel: implication of high dose drug therapy to safety and in vivo anti-cancer activity. Biomaterials 101:296–309.
- Jahangirian H, Lemraski EG, Webster TJ, et al. (2017). A review of drug delivery systems based on nanotechnology and green chemistry: green nanomedicine. Int J Nanomedicine 12:2957–78.
- Jordan MA, Wilson L. (2004). Microtubules as a target for anticancer drugs. Nat Rev Cancer 4:253–65.
- Kratz F. (2011). INNO-206 (DOXO-EMCH), an albumin-binding prodrug of doxorubicin under development for phase II studies. Curr Bioact Compd 7:33–8.
- Kratz F, Azab S, Zeisig R, et al. (2013). Evaluation of combination therapy schedules of doxorubicin and an acid-sensitive albumin-binding prodrug of doxorubicin in the MIA PaCa-2 pancreatic xenograft model. Int J Pharm 441:499–506.
- Kratz F, Elsadek B. (2012). Clinical impact of serum proteins on drug delivery. J Control Release 161:429–45.
- Kratz F, Warnecke A, Scheuermann K, et al. (2002). Probing the cysteine-34 position of endogenous serum albumin with thiol-binding doxorubicin derivatives. improved efficacy of an acid-sensitive doxorubicin derivative with specific albumin-binding properties compared to that of the parent compound. J Med Chem 45:5523–33.
- Lian H, Sun J, Yu YP, et al. (2011). Supramolecular micellar nanoaggregates based on a novel chitosan/vitamin E succinate copolymer for paclitaxel selective delivery. Int J Nanomedicine 6:3323.
- Liu Y, Sun J, Cao W, et al. (2011). Dual targeting folate-conjugated hyaluronic acid polymeric micelles for paclitaxel delivery. Int J Pharm. 421:160–9.
- Liu Y, Sun J, Lian H, et al. (2014). Folate and CD44 receptors dual‐targeting hydrophobized hyaluronic acid paclitaxel‐loaded polymeric micelles for overcoming multidrug resistance and improving tumor distribution. J Pharm Sci 103:1538–47.
- Luo C, Sun J, Liu D, et al. (2016a). Self-assembled redox dual-responsive prodrug-nanosystem formed by single thioether-bridged paclitaxel-fatty acid conjugate for cancer chemotherapy. Nano Lett 16:5401–8.
- Luo C, Sun J, Sun B, et al. (2016b). Facile fabrication of tumor redox‐sensitive nanoassemblies of small‐molecule oleate prodrug as potent chemotherapeutic nanomedicine. Small 12:6353–62.
- Malonga H, Neault J, Diamantoglou S, et al. (2005). Taxol anticancer activity and DNA binding. Mini-Rev Med Chem 5:307–11.
- Ramasamy T, Ruttala HB, Gupta B, et al. (2017). Smart chemistry-based nanosized drug delivery systems for systemic applications: a comprehensive review. J Control Release 258:226–53.
- Satsangi A, Roy SS, Satsangi RK, et al. (2014). Design of a paclitaxel prodrug conjugate for active targeting of an enzyme upregulated in breast cancer cells. Mol Pharmaceutics 11:1906–18.
- Wang Y, Liu D, Zheng Q, et al. (2014). Disulfide bond bridge insertion turns hydrophobic anticancer prodrugs into self-assembled nanomedicines. Nano Lett 14:5577–83.
- Wohl AR, Michel AR, Kalscheuer S, et al. (2014). Silicate esters of paclitaxel and docetaxel: synthesis, hydrophobicity, hydrolytic stability, cytotoxicity, and prodrug potential. J Med Chem 57:2368–79.
- Xiao W, Suby N, Xiao K, et al. (2017). Extremely long tumor retention, multi-responsive boronate crosslinked micelles with superior therapeutic efficacy for ovarian cancer. J Control Release 264:169–79.
- Zhong Y, Goltsche K, Cheng L, et al. (2016). Hyaluronic acid-shelled acid-activatable paclitaxel prodrug micelles effectively target and treat CD44-overexpressing human breast tumor xenografts in vivo. Biomaterials 84:250–61.