
Abstract
Anti-mPEG/anti-human epidermal growth factor receptor 2 (HER2) bispecific antibodies (BsAbs) non-covalently bound to a docetaxel (DTX)-loaded mPEGylated lecithin-stabilized micellar drug delivery system (LsbMDDs) were endowed with active targetability to improve the chemotherapeutic efficacy of DTX. DTX-loaded mPEGylated LsbMDDs formulations were prepared using lecithin/DSPE-PEG(2K or 5K) nanosuspensions to hydrate the thin film, and then they were subjected to ultrasonication. Two BsAbs (anti-mPEG/anti-DNS or anti-HER2) were simply mixed with the LsbMDDs to form BsAbs-LsbMDDs formulations, respectively, referred as the DNS-LsbMDDs and HER2-LsbMDDs. Results demonstrated that the physical characteristics of the BsAbs-LsbMDDs were similar to those of the plain LsbMDDs but more slowly released DTX than that from the LsbMDDs. Results also showed that the HER2-LsbMDDs suppressed the growth of HER2-expressing MCF-7/HER2 tumors, increasing the amount taken up via an endocytosis pathway leading to high drug accumulation and longer retention in the tumor. In conclusion, the BsAbs-LsbMDDs preserved the physical properties of the LsbMDDs and actively targeted tumors with a drug cargo to enhance drug accumulation in tumors leading to greater antitumor activity against antigen-positive tumors.
1. Introduction
In the past few decades of rapidly evolving drug research, numerous high-potency chemotherapeutic drugs have been discovered. However, notwithstanding the rapid progress in drug innovations, cancer drugs have gained a reputation for having high risks with little chance of efficacy. These are mainly attributed to the following causes: (i) many potent drugs are highly hydrophobic which keeps them from being used in the clinic; (ii) a lack of specificity of chemotherapeutic drugs also causes high toxicity to normal cells; (iii) an unsuitable biodistribution following an intravenous (IV) injection for most drugs results in low therapeutic efficacy and adverse effects; and (iv) excipients used to enhance the solubility of the drug formulations might cause additional toxicities in patients (Chabner & Roberts, Citation2005). For the success of chemotherapeutic agents in clinical applications, a durable and specific drug delivery system is required to carry and release the drugs into the right pathological site (Peer et al., Citation2007; Brigger et al., Citation2012). For this purpose, numerous nanocarrier (NC) types like liposomes, micelles, polymeric nanoparticles (NPs), dendrimers, solid-lipid nanoparticles (SLNs), and gold NPs have been investigated for controlled drug release applications (Davis et al., Citation2008; Wang et al., Citation2012). These NCs delivery systems can through a leaky tumor blood vasculature via an enhanced permeability and retention (EPR) effect. Also, further modification of stealthy decorations on the surface with polyethylene glycol (PEG) offers to reduce immunogenicity and prolong the circulation times. Those advances in nanomedicine have demonstrated obvious advantages, including preferential drug accumulation in tumor sites, decreased side effects, better drug tolerance, and improved patient compliance in clinical practice (Zhong et al., Citation2014; Hare et al., Citation2017).
Recently, robust and emerging drug delivery systems known as lipid-polymer hybrid nanoparticles (LPHNs) which take advantage of the unique strengths of liposomes and polymeric NPs have successfully application in anti-cancer drug, anti-microbial agent, and nucleic acid (Krishnamurthy et al., Citation2015; Dave et al., Citation2017). However, LPHNs still have two problems of low encapsulation efficiency (EE) and drug loading (DL) needed to be overcome (Li et al., Citation2017). In our previous study, we have established high EE and drug-loaded lecithin-stabilized micellar drug delivery system (LsbMDDs) that has a polymeric core and a lipid shell to delivery hydrophobic drug to enhance antitumor efficacy and reduce systemic side effect (Su et al., Citation2018).
For additionally improving drug accumulation and high tumor cell specificity, the surface of the NCs can be modified by specific tumor-targeted ligands such as monoclonal antibodies (Abs; mAbs), aptamers, peptides, small molecules, and so on, to achieve active tumor targeting (Allen, Citation2002). Ligand-directed active targeting of NP drug formulations present improved therapeutic performances compared to their passive targeting counterparts in preclinical study (Koo et al., Citation2011; Nicolas et al., Citation2013). Among actively targeted ligands, various forms of Abs such as mAbs, antigen-binding fragment (Fab), and single-chain variable fragment (scFv)) are frequently used as efficient targeting moieties due to its nanomolar affinity and high specificity to tumor antigens (Kamaly et al., Citation2012; Tietze et al., Citation2017). A number of NCs conjugated with Abs have been developed to target tumor which highly expressing a particular surface marker, such as human epidermal growth factor receptor 2 (HER2), epidermal growth factor receptor (EGFR), and vascular endothelial growth factor (VEGF). Nonetheless, chemical conjugation of Abs to NCs might have the disadvantages of heterogeneous coupling orientations, antigen-binding function damage, altered physical properties of NCs can be altered, and it is laborious and time-consuming (Manjappa et al., Citation2011; Zhong et al., Citation2014). To avoid the problems of chemical conjugation, Kao et al. offered a simple one-step method to confer tumor specificity to methoxyl PEG (mPEG)-NCs by non-covalently bound with anti-mPEG/anti-tumor bispecific antibodies (BsAbs). BsAbs-PEG-NC showed increased drug accumulation and enhanced therapeutic efficacy in EGFR+ colon tumor-bearing mice (Kao et al., Citation2014). Thus, the mPEGylated NCs modification with anti-mPEG/anti-tumor BsAb can acquire specific tumor-targeting without further chemical coupling reactions.
Herein we established active HER2 targeting mPEGylated lecithin-stabilized micellar drug delivery system (HER2-LsbMDDs) loading with docetaxel (DTX), a first line chemotherapeutic agent for breast cancer (BC). Humanized anti-mPEG/anti-HER2 BsAbs non-covalently bound to DTX-loaded LsbMDDs to enhance tumor accumulation and improve the chemotherapeutic efficacy against HER2-positive BC is schematically shown in . The LsbMDDs was incorporated with 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol) (DSPE-PEG) with different molecular weights (2K or 5K) by ultrasonication in a lecithin nanosuspension to fuse the supported lipid layers onto the micellar core. The anti-mPEG/anti-HER2 BsAbs consisted of a Fab fragment to attach to the methoxy ends of the mPEG on LsbMDDs surface, and a scFv targeting the HER2 on tumor cells. In this study, uptake mechanisms of the anti-mPEG/anti-HER2 BsAb-unbound/bound DTX-loaded LsbMDDs were examined, and their physical characteristics were evaluated, including the particle size and distribution, morphology, optimal BsAbs/mPEG molar ratio, cell viability, in vitro drug release, and biopharmaceutical characteristics of the tumor proliferation inhibition, pharmacokinetics (PK), and biodistribution.
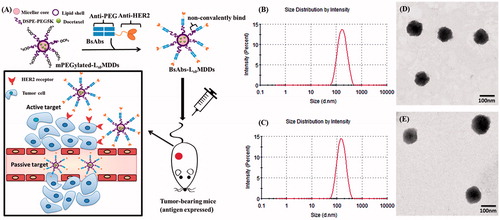
Figure 1. (A) Strategy of bispecific antibodies (BsAbs and anti-mPEG/anti-HER2) non-covalently bound to a mPEGylated LsbMDDs to form the BsAbs-LsbMDDs, which can specifically target antigen-expressing cancer cells by passive and active targeting. (B) Size distributions of LsbMDDs(2K) and (C) LsbMDDs(5K) were measured using dynamic light scattering (DLS) and TEM micrographs of (D) the LsbMDDs(2K) and (E) LsbMDDs(5K) (scale bars =100 nm).
2. Materials and methods
2.1. Materials
DTX was supplied by Qilu Pharmaceutical (Jinan, China). DSPE-PEG2K was purchased from NOF (Tokyo, Japan). DSPE-PEG5K was obtained from Avanti Polar Lipids. (Alabaster, AL). 3,3′-Dioctadecyloxacarbocyanine perchlorate (DIO), nystatin, and sucrose were obtained from Sigma-Aldrich (St. Louis, MO). 1,1’-Dioctadecyltetramethyl indotricarbocyanine iodide) (DIR) was purchased from Perkin-Elmer (Waltham, MA). Soybean lecithin (Lipoid S-100) was obtained from Lipoid GmbH (Ludwigshafen, Germany). Chlorpromazine and cytochalasin D were supplied by Cayman Chemicals (Ann Arbor, MI). Dynasore was obtained from MedChem Express (Monmouth Junction, NJ). LysoTracker Red DND-99 and Hoechst3342 were obtained from Thermo Fisher Scientific (Waltham, MA). The anti-PEG backbone mAb AGP4 was provided by Dr. Steve R. Roffler (Academia Sinica, Taipei, Taiwan). Tynen® (solvent-based DTX) is a generic product of DTX manufactured by TYY Pharmaceutical (lot no: STW1407, Taipei, Taiwan). All reagents were of analytical grade and solvents used in the high-performance liquid chromatography (HPLC) or ultra-performance liquid chromatography (UPLC)/mass spectrometry (MS/MS) analysis was of HPLC or MS grade.
2.2. Animals and cell lines
A BC cell line (MCF-7) that poorly expresses HER2 (HER2-) and HER2-overexpressing cell lines (HER2+), were cultured in Dulbecco’s modified Eagle’s medium Ham’s F12 (DMEM/F12) containing 10% fetal bovine serum (FBS) and 1% antibiotics (penicillin/streptomycin). Nu/nu mice (females, 5 ∼ 7 weeks old) and Sprague-Dawley (SD) rats (males, 8 ∼ 10 weeks old) were used for the animal studies. The nu/nu mice were purchased from the National Laboratory Animal Center (Taipei, Taiwan), and SD rats were purchased from BioLASCO Taiwan (Taipei, Taiwan). All animal experiments were carried out in accordance with a protocol approved by the Laboratory Animal Center of Taipei Medical University (approval no: LAC-2014-0253), and all experiments were performed in accordance with animal care guidelines.
2.3. Preparation of the mPEGylated LsbMDDs
The DTX-loaded LsbMDDs was prepared following previously reported procedures with minor modifications to introduce mPEGylation with DSPE-PEG2K or DSPE-PEG5K into the outer shell portion of micelles (Chen et al., Citation2015; Su et al., Citation2018). In brief, a fixed drug (DTX)/amphiphilic polymer (DSPE-PEG2K) ratio of 1:5 with the addition of an appropriate amount of TPGS as an antioxidant was first dissolved in methanol, and a thin film was formed after evaporation (Rotavapor R124; Buchi, Flawil, Switzerland) of the organic solvent. Soybean lecithin (S100) at 1000 and 375 mg of DSPE-PEG5K (or DSPE-PEG2K) were suspended in 25 mL of deionized water and then subjected to ultrasonication (VCX 750, 20 kHz, Sonics and Materials, Market Harborough, United Kingdom) to form a lecithin/DSPE-PEG (2K or 5K) nanosuspension. Then 1 mL of the lecithin/DSPE-PEG (2K or 5K) nanosuspension was used to hydrate the thin film obtained above, and the reconstituted mixture was further subjected to ultrasonication at full power for at least 5 min while maintaining a constant temperature to form lecithin-stabilized NCs in the solution. Any unencapsulated drug was discarded by filtering this NC solution through a 0.22-μm membrane (Millipore, Billerica, MA). An appropriate amount of an anti-freeze agent was added to the filtrate and then freeze-dried to obtain the dry powder form of NCs. Two amphiphilic polymers of DSPE-PEG2K and DSPE-PEG5K with different PEG chain lengths were used to formulate the DTX-loaded mPEGylated LsbMDDs, respectively, designated DTX-loaded LsbMDDs(2K) and DTX-loaded LsbMDDs(5K). To prepare the DIO (green fluorescence for in vitro assays)-loaded or DIR (near-infrared fluorescence for in vivo imaging)-loaded LsbMDDs, we followed the same procedure as that for the DTX-loaded LsbMDDs except that the DTX was replaced with either DIO or DIR.
2.4. Physical characterization of the DTX-loaded LsbMDDs and BsAbs-LsbMDDs
Characteristics of the DTX-loaded LsbMDDs and BsAbs-LsbMDDs including the particle size, zeta potentials (ZPs), binding activity, morphology, EE, DL are described in Supplemental information.
2.5. Construction and expression of BsAbs and non-covalent modification of LsbMDDs with BsAbs
The anti-mPEG/anti-HER2 BsAbs were composed of a Fab of a humanized anti-mPEG (clone 15-2b) Ab and an scFv of trastuzumab (humanized anti-HER2 Ab) as previously described with minor modifications (Kao et al., Citation2014). The light and heavy chains of the anti-mPEG Fab gene were linked by the internal ribosome entry site (IRES) sequences (Chuang et al., Citation2010). A (Gly4Ser)3 linker, the anti-HER2 scFv, and an 6xHis tag genes were ligated after the heavy chain of the anti-mPEG Fab. To evaluate the tumor-binding specificity, negative control (anti-mPEG/anti-dansyl (DNS)) BsAbs were generated by replacing the anti-HER2 scFv with an anti-DNS scFv that binds the small chemical hapten of DNS, which is not present in cell or body (Kao et al., Citation2014). BsAb genes were then inserted into the pLNCX vector. To mass-produce the BsAbs, plasmids were transiently transfected into the Expi293 cells, according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA). After 6 d, BsAbs in the supernatant were purified using HisTrap HP columns (GE Healthcare Life Sciences, Little Chalfont, United Kingdom), dialyzed against phosphate-buffered saline (PBS, pH 7.4, 10 mM), and sterilized by 0.2-μm filtration. Concentrations of BsAbs were determined by a bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, Waltham, MA).
Non-covalent modification of LsbMDDs with BsAbs to form BsAbs-LsbMDDs were simply and freshly prepared by incubation of LsbMDDs with anti-mPEG/anti-HER2 BsAbs in bovine serum albumin (BSA)/PBS buffer (0.05% w/v) at RT for 1h. DNS-LsbMDDs were generated with anti-mPEG/anti-DNS BsAbs and LsbMDDs under the same procedure, and was used as a control.
2.6. Detection of the BsAbs (anti-mPEG/anti-HER2)non-covalently bound to the LsbMDDs(2K or 5K) by the sandwich enzyme-linked immunosorbent assay (ELISA)
The experimental protocol to assess the presence of anti-mPEG/anti-HER2 BsAbs on the LsbMDDs(2K or 5K) is illustrated in Scheme 1 (Supplemental information). First, 96-well plates were coated with 5 μg/mL of an anti-PEG backbone mAb (AGP4) in 50 μL of 0.1 M NaHCO3 per well at 37 °C for 2 h, and then this was blocked with a 200 μL/well of dilution buffer containing 5% w/v skim milk in PBS at 4 °C overnight. HER2-LsbMDDs were prepared at 0.01:1 of BsAbs/mPEG molar ratio. The LsbMDDs or HER2-LsbMDDs (2K or 5K) diluted with 2% skim milk to give different DSPE-PEG molar concentrations (36, 144, and 576 nM) was added to AGP4-coated wells. After incubation for 1 h, each well was washed with PBS to remove unbound NCs. Then 0.1 μg/mL of goat anti-human immunoglobulin G (IgG) F(ab’)2-horseradish peroxidase (HRP) (Jackson ImmunoResearch Laboratories, West Grove, PA) in 50 μL of dilution buffer was added for another 1 h to detect the BsAbs on LsbMDDs. The plates were finally washed with PBS and then 150 μL/well of ABTS substrate was added for 30 min. Color development was measured at 405 nm (Bio-Tek, Winooski, VT).
2.7. Optimal ratio of BsAbs to mPEG5K on the LsbMDDs for preparation of HER2-LsbMDDs
To optimize the molar ratio of the anti-mPEG/anti-HER2 BsAbs to mPEG5K on the LsbMDDs, free BsAbs unbound to HER2-LsbMDDs was measured by ELISA method illustrates in Scheme 2 (Supplemental information). First, 96-well plates were coated with 20 μg/mL of mPEG5K-NH2 ligand (with a methoxy end group in the PEG chain) in 50 μL of 0.1 M NaHCO3 per well at 37 °C for 2 h and then blocked with 200 μL/well of dilution buffer containing 5% w/v skimmed milk in the PBS at 4 °C overnight. The HER2-LsbMDDs(5K) prepared at three different BsAbs/mPEG molar ratios (of 0.002:1, 0.01:1, and 0.02:1) with a fixed amount of the LsbMDDs(5K) was diluted to same concentrations of BsAbs, and were incubated in the wells for 1 h, followed by extensive washing each well with PBS, and then goat anti-human IgG F(ab’)2-HRP was added, and the procedure described in Section 2.6. The addition of only the LsbMDDs(5K) or free anti-mPEG/anti-HER2 BsAbs was, respectively, used as the negative control and positive control.
2.8. Tumor targeting of the mPEGylated LsbMDDs with non-covalently bound BsAbs
To optimize the PEG chain length (PEG2K or PEG5K) and the BsAbs to mPEG molar ratio for tumor targeting, the DIO-loaded LsbMDDs (2K or 5K), DIO-loaded DNS-LsbMDDs (2K or 5K), and DIO-loaded HER2-LsbMDDs (2K or 5K) were prepared as follows: two BsAbs (anti-mPEG/anti-HER2 and anti-mPEG/anti-DNS) at various BsAbs/mPEG molar ratios (of 0.001:1, 0.002:1, 0.01:1, and 0.02:1) were separately mixed with a fixed amount of the DIO-loaded LsbMDDs(2K or 5K) in BSA/PBS buffer (0.05% w/v) for 1 h to form the DIO-loaded HER2-LsbMDDs(2K or 5K) and DIO-loaded DNS-LsbMDDs(2K or 5K), respectively. The HER2-overexpressing BC cell line of MCF-7/HER2 was seeded onto 24-well plates at a density of 5 × 104 cells/well. The DIO-loaded BsAbs-LsbMDDs(2K or 5K) was added to the wells and incubated for 4 h. After removing the unbound NC, the cells were collected, washed, and resuspended in the PBS. Cellular uptake of the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs by the MCF-7/HER2 cells was quantitatively evaluated by flow cytometry. The presence of the DIO detected by excitation at 484 nm and emission at 501 nm was used as an indicator of the uptake amount of NCs.
2.9. Drug release of the optimal LsbMDDs(5K) and BsAbs-LsbMDDs(5K)
Amounts of drug released from Tynen® (solvent-based DTX), the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs were investigated in PBS (containing 0.5% Tween 80) by a dialysis method. Briefly, 1 mL of the three LsbMDDs solutions containing 0.25 mg DTX or 0.25 mg/mL Tynen® was loaded into a dialysis bag (MWCO 6000, Cellu-Sep® T1, Orange Scientific, Seguin, TX) against 25 mL of release medium with shaking at a speed of 100 rpm at 37 °C. At a predetermined time point, the release medium in the dialysis bag was replaced with a fresh medium to maintain the sink conditions. The drug concentration was analyzed by the HPLC method described above. All measurements were carried out in triplicate.
2.10. Cell viability
Cell viabilities of the three BC cell lines of MCF-7, MCF-7/HER2, and SKBR-3 treated with Tynen®, the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs were evaluated by an MTT assay. Cells were seeded at a density of 5 × 104 cells per well in 24-well plates and incubated for 24 h at 37 °C with 5% CO2. Then, they were treated with different concentrations of the formulations for 4 h under the same conditions. After incubation for 48 h, 50 μL of MTT (6 mg/mL) was added to each well for 3 h. The medium was removed and 200 μL of DMSO was added to each well and gently shaken to dissolve any purple formazan crystal formations. The absorbance of each well was measured at 550 nm (BioTek, Winooski, VT).
2.11. In vitro cellular uptake of the optimal LsbMDDs(5K) and elucidation of the cellular uptake mechanism
BC cell lines (MCF-7, MCF-7/HER2, and SKBR-3 cells) were seeded at a density of 5 × 104 cells per well on 12-well microplates. The DIO-loaded LsbMDDs, DIO-loaded DNS-LsbMDDs, and DIO-loaded HER2-LsbMDDs (with a molar ratio of BsAbs to mPEG of 0.01:1) were added to separate wells and incubated for 0.5, 2, and 8 h. After incubation for different times, cells were collected, and analyzed by flow cytometry quantitatively evaluate the intracellular uptake of the LsbMDDs and the BsAbs-LsbMDDs by MCF-7, MCF-7/HER2, and SKBR-3 cells. To further understand the cellular uptake mechanism of the HER2-LsbMDDs by MCF-7/HER2 cells, cells were incubated for 60 min separately with cytochalasin D (10 μg/mL) as an inhibitor of phagocytosis and micropinocytosis (Kuhn et al., Citation2014), with amiloride (50 μM) as an inhibitor of micropinocytosis (Dutta & Donaldson, Citation2012), with methyl-β-cyclodextrin (MBCD, 0.5 mM) as an inhibitor of lipid rafts involved in caveolae-mediated endocytosis (Itoh et al., Citation2008), with nystatin (50 μg/mL) as an inhibitor of caveolae-mediated endocytosis (Kuhn et al., Citation2014), with chlorpromazine (20 μg/mL), sucrose (450 mM), or dynasore (40 μM) as inhibitors of clathrin-mediated endocytosis (Sahay et al., Citation2010), and with herceptin (0.5 μg/mL) as an inhibitor of the HER2 receptor, and then were treated with formulations in the presence of the inhibitors for 2 h. After incubation, cells were treated as aforementioned. The fluorescence was measured using flow cytometry (SA3800, Sony, San Jose, CA).
2.12. Intracellular localization of DIO-loaded -LsbMDDs
MCF-7/HER2 cells were seeded in 3.5-cm glass bottom dishes for 24 h. After 2 h of treatment with the DIO-loaded LsbMDDs, DNS-LsbMDDs, or HER2-LsbMDDs formulation, cells were stained with LysoTracker Red DND-99 for 30 min and Hoechst for 10 min to indicate lysosomes and nuclei, respectively. Subcellular localization of each target signal was observed using the TCS SP5 Confocal Spectral Microscope Imaging System (Leica, Wetzlar, Germany).
2.13. In vivo PK studies of intravenous administration
SD rats at 8 ∼ 10 weeks old were used to study the PK profiles of DTX after administration of Tynen® (solvent-based DTX), the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs. Rats were given a single dosage of 8 mg/kg of each formulation via a jugular vein injection (three rats per group). Blood was collected from the jugular vein in heparinized tubes at 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, 10, 24, 48, and 72 h after administration. Blood samples were centrifuged at 3000 rpm for 10 min to obtain plasma and were stored at −80 °C prior to analysis by UPLC/MS/MS (detailed describe in Supplemental information). In order to examine the in vivo binding activity of HER2-LsbMDD, a supplemental PK study was conducted to measure the HER2 binding activity of HER2-LsbMDD along with the detection of plasma DTX concentration at the same predetermined time points. The HER2 binding activity of HER2-LsbMDD in this PK study was detected by a cell-based ELISA method and the details were described in Supplemental information.
2.14. Tumor inhibition studies
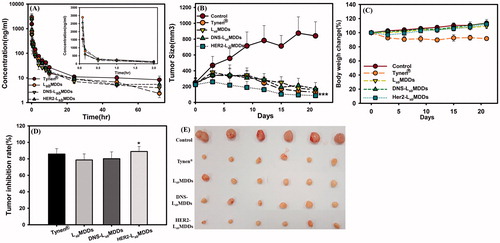
All female nu/nu mice received a subcutaneous injection of 100 μL (containing 5 × 106 cells) of the MCF-7/HER2 cell suspension in Matrigel into their right thighs of mice. Tumor growth was promoted by subcutaneously injecting 20 μg of estradiol valerate in 50 μL of sesame oil once a week near the neck. These tumor-bearing mice with around 200 mm3 tumor volumes were randomized into five groups: one control group (PBS) and four groups including Tynen®, the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs (5 mg DTX/kg, n = 6). Each formulation was injected once every 3 d for 12 d. The tumor volume was calculated by the modified ellipsoidal formula of 1/2 length × width2. Mice body weights and tumor volumes were measured every 3 d after the injection. The mice were sacrificed by CO2 and the tumors were harvested and weighed on day 21. The tumor inhibitory rate (%) was calculated as follows: (Wc − Wt)/Wc, where Wc is the tumor weight of the control group and Wt is the tumor weight of each formulation group.
2.15. In vivo biodistribution studies
The biodistribution study was evaluated in the MCF-7/HER2-bearing nu/nu mice. After tumor sizes had reached ∼200 mm3, Tynen®, the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs were administered at 40 mg DTX/kg through the tail vein. At the time points of 2 and 16 h, the mice were sacrificed by CO2 and perfused with a PBS buffer containing a 0.1% heparin solution to remove blood from the organs. The heart, liver, spleen, lung, kidney, and tumor were harvested. All organs were weighed and stored at −80 °C until being assayed for DTX. Organ extraction used a homogenization method. Briefly, a 5-fold volume of a PBS/0.1% heparin solution was added to each of the weighed tissues, and an SH-100 homogenizer (Kurabo Industries, Osaka, Japan) was used to homogenize the sample. The tissue homogenate (200 µL) was obtained, and the DTX concentration in the tissue was analyzed using the UPLC/MS/MS as the disposal method for plasma samples.
2.16. In vivo imaging of tumor-bearing mice
To compare the tumor-targeting efficiency of the HER2-LsbMDDs with those of the LsbMDDs and DNS-LsbMDDs, the DIR, a near-infrared fluorescent probe, was encapsulated into formulations, and then 200 µg/kg of the DIR was administered to mice via the tail vein. The mice were then monitored at 2, 8, and 24 h after the injection using an IVIS 200 Imaging System with an ICG emission filter (Perkin Elmer, Waltham, MA). For ex vivo imaging, the major organs of the heart, liver, spleen, lung, and kidney, as well as the tumor, were excised at 24 h post-injection.
2.17. Statistical analysis
Data are presented as the mean ± standard deviation (SD) of each group. The significance among samples was performed using a one-way analysis of variance (ANOVA). Significant differences between groups were indicated by *p < .05, **p < .01, and ***p < .001.
3. Results
3.1. Physical characterization of the LsbMDDs
To introduce mPEGylation to robust and previously developed promising delivery systems known as the LsbMDDs (Chen et al., Citation2015; Chen et al., Citation2016), the thin film of self-assembling micelles was hydrated with a lecithin/DSPE-PEG (2K or 5K) nanosuspension in this study. The micellar core of the so-obtained LsbMDDs was composed of DTX and DSPE-PEG2K, while the lipid shell consisted of lecithin and DSPE-PEG (2K or 5K) at a ratio of 40:15 (w/w). The average particle size and distribution pattern of the LsbMDDs(2K) and LsbMDDs(5K) are as shown in , respectively. Results showed that the average particle sizes of the LsbMDDs(2K) and LsbMDDs(5K) were 131.0 ± 1.9 and 152.5 ± 3.28 nm, polydispersity index (PDIs) were 0.29 ± 0.067 and 0.25 ± 0.01, EE were 91.3 and 95%, ZPs were −38.4 ± 0.2 and −34.2 ± 0.1 mV, and DL were 5.93 and 6.16%, respectively. The structures of both the LsbMDDs formulations as observed in the TEM images (, respectively) exhibited a spherical morphology and were well dispersed and separated.
3.2. Optimal modification of the LsbMDDs withanti-mPEG/anti-HER2 BsAbs via non-covalent binding
To investigate whether anti-mPEG/anti-HER2 BsAbs could be non-covalently bound to the LsbMDDs(2 K or 5 K), the BsAbs non-covalently bound to HER2-LsbMDDs(2 K or 5 K) were detected by a sandwich ELISA. The anti-PEG backbone mAbs (AGP4) which can specifically bind to the backbone of the PEG chain were coated in 96-well plates to capture LsbMDDs(2 K or 5 K) with or without BsAbs modification. The HRP-conjugated secondary antibody was added for detecting the BsAbs on LsbMDDs and HRP activity was determined by absorbance at 405 nm (OD value) due to the oxidation product of ABTS. The results in demonstrate that the binding concentration of HER2-LsbMDDs(2 K or 5 K) in the formulations increased leading to an increase in the resulting absorbance, indicating that BsAbs were non-covalently bound to LsbMDDs with different chain length of mPEG (2 K or 5 K) followed with a concentration-dependent manner at a BsAbs/mPEG molar ratio of 0.01:1.
Figure 2. (A) Detection of BsAbs on HER2-LsbMDDs(2K or 5K) by a sandwich ELISA method (n = 3). (B) The optimal molar ratio of BsAbs to mPEG5K on the LsbMDDs(5K) was assessed at three different molar ratios of BsAbs to mPEG5K (of 0.002:1, 0.01:1, and 0.02:1) by an ELISA method (n = 3). *p < .05 compared to the LsbMDDs(0.01:1). (C) Cellular uptake of the LsbMDDs(2K and 5K), DNS-LsbMDDs(2K and 5K), and HER2-LsbMDDs(2K and 5K) with various molar ratios of BsAbs to mPEG (of 0.001:1, 0.002:1, 0.01:1, and 0.02:1) were measured by flow cytometry (n = 3).
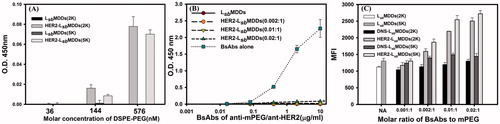
To determine the optimal ratio of the BsAbs (anti-mPEG/anti-HER2) to mPEG5K on the LsbMDDs, three HER2-LsbMDDs(5K) formulations prepared by mixing BsAbs at three different molar ratios of BsAbs to the mPEG-5K (of 0.002:1, 0.01:1, and 0.02:1) with a fixed amount of the LsbMDDs(5K). Then, all formulations were diluted to the indicated concentrations of BsAbs, and were added to the 96-well plates coated with the mPEG5K ligand (with a methoxy end group in the PEG chain) to assess the extent of unbound BsAbs to the LsbMDDs. shows that no absorbance with the molar ratios of BsAbs to mPEG of 0.002:1 and 0.01:1 was observed, indicating that all BsAbs were bound to the LsbMDDs with no detection of free BsAbs in the formulation. However, unbound BsAbs were detected in the higher BsAbs to mPEG molar ratio of 0.02:1. Further, no free BsAbs were detected when only the LsbMDDs was added as the negative control, whereas free BsAbs were detected at a concentration proportional to the amount of free BsAbs added as the positive control. This confirms that BsAbs could homogeneously non-covalent bind to the LsbMDDs with optimal molar ratios of BsAbs to mPEG-5K of 0.0 1 ∼ 0.02:1. The number of lipids in a 100 nm size liposome is about 80047 (Dennison et al., Citation2009; Mikhalin et al., Citation2014). At a molar ratio of lecithin: DSPE-PEG5K equal to 95:5, it is accordingly expected to have approximately 4002 molecules of DSPE-PEG5K molecules on the surface of the LsbMDDs with a similar size. Since the optimal ratio of BsAbs/mPEG was 0.01:1, there expected to have 40 molecules of BsAbs on one NC of LsbMDDs.
3.3. Optimization of tumor-targeting by BsAbsnon-covalently bound to the LsbMDDs
First, the bi-functional binding of the BsAbs (anti-mPEG/anti-HER2 and anti-mPEG/anti-DNS) for HER2-positive (MCF-7/HER2 and SKBR-3) and HER2-negative (MCF-7) cancer cells were examined and Trastuzumab was used as a positive control. Result of flow cytometry (Supplemental information Figure S1) showed that anti-mPEG/anti-HER2 BsAbs displays the binding activity to HER2 overexpressed cells of MCF-7/HER2 and SKBR-3 with the expression level of HER2 being greater for the former than the latter. Further, anti-mPEG/anti-HER2 BsAbs displays the similar results as that for Trastuzumab since the anti-HER2 portion of the former was constructed as a scFv according to a humanized anti-HER2 Ab (Trastuzumab). On the contrary, isotype BsAbs (anti-mPEG/anti-DNS) did not bind to any of three cancer cell lines examined in this study (MCF-7/HER2, SKBR-3, and MCF-7) since all three cancer cells did not express the ligand of DNS for binding.
Further, based on the uptake amount of the DIO-loaded LsbMDDs non-covalently bound with the BsAbs by the MCF-7/HER2, the optimal ratio of the two BsAbs (anti-mPEG/anti-DNS or anti-HER2) bound to the LsbMDDs composed of either mPEG2K or mPEG5K was examined, and referred to as the DNS-LsbMDDs(2K or 5K) and HER2-LsbMDDs(2K or 5K). Results in demonstrate that compared to the LsbMDDs, uptake amounts of both the HER2-LsbMDDs(2K) and the HER2-LsbMDDs(5K) increased with an increasing BsAbs to mPEG molar ratio, showing that the 0.01:1 ratio was optimal. Further, the uptake amount of the HER2-LsbMDDs(5K) at its optimal ratio was better than that of the HER2-LsbMDDs(2K). Contrarily, the uptake amounts of neither the LsbMDDs(2K or 5K) nor the DNS-LsbMDDs(2K or 5K) increased with an increasing BsAbs to mPEG molar ratio and were maintained at a similar uptake amounts as that for the LsbMDDs. This confirms that the HER2-LsbMDDs non-covalently bound of HER2 targeting BsAbs to the LsbMDDs was able to enhance the targetability to the cells that over-expressed HER2 on its cell membranes, resulting in an increase in cellular uptake, whereas those without the tumor targeting ligands (LsbMDDs) or with a non-expressing targeting ligand of DNS (DNS-LsbMDDs), were unable to enhance the cellular uptake. It was also concluded that the HER2-LsbMDDs(5K) incorporated the DSPE-PEG5K in the lipid shell of the LsbMDDs was more appropriate and was thus selected for subsequent studies with the abbreviated name of the HER2-LsbMDDs.
3.4. Physical characterizations of the optimal BsAbs-LsbMDDs
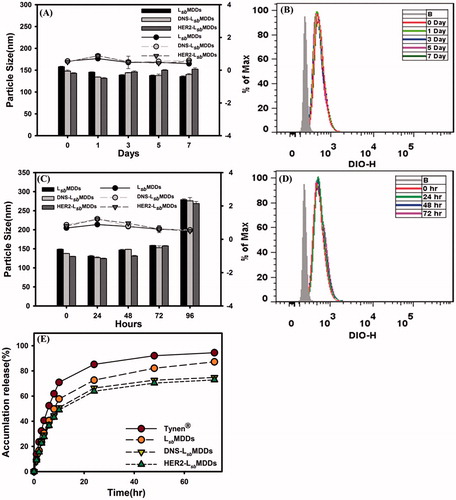
Physical characteristics were examined, including the average particle size, PDI, ZPs, and the storage stability of the optimal BsAbs-LsbMDDs composed of DSPE-PEG5K. Results showed that the resultant DNS-LsbMDDs and HER2-LsbMDDs were 148.4 ± 1.04 and 152.3 ± 0.94 nm in size, respectively, which were similar to the LsbMDDs alone (152.5 ± 2.88 nm; p > .05). The PDI values for all the particles were around 0.25. These results indicated that the attachment of both the BsAbs which were non-covalently bound to the LsbMDDs caused no significant change in the average particle size or distribution. Respective values of the ZPs of the DNS-LsbMDDs and HER2-LsbMDDs were −40.5 ± 0.62 and −40.1 ± 0.62 mA, which were more negative than that of the LsbMDDs. In another report, the ZPs consistently increased after BsAbs were incorporated into NPs (Gao et al., Citation2011). To examine the in vitro stability of the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs, they were incubated in PBS and FBS to monitor changes in the average particle sizes and PDI with respect to time, and the preservation of HER2 binding activity was determined as well. The results are as shown in that the average particle size and PDI of both BsAbs-LsbMDDs formulations were stable for at least 7 d in PBS, with no precipitation or significant changes observed. However, shows both BsAbs-LsbMDDs formulations incubated in FBS were observed to be stable for only 72 h with no significant changes in the average particle size or PDI. The particle size increased to >200 nm which means that the NPs became aggregated, and precipitation of the drug was observed at 96 h. To determine the HER2 binding activity, the cellular uptake of DIO-loaded HER2-LsbMDDs after different incubation time in PBS and FBS was examined on MCF7/HER2 which overexpressed HER2. shows that the similar extent of cellular uptake were observed for all samples compared to that at 0 h even after 7 d in the presence of PBS and 72 h in FBS indicating that HER2-LsbMDDs retained the same ability to bind to MCF-7/HER2 cells. These results indicate that BsAbs modified LsbMDDs was stable for at least 72 h under physiological conditions.
Figure 3. (A) The particles size of the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs during storage in PBS. (B) The binding activity of HER2-LsbMDDs during storage in PBS. (C) The particles size of the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs during storage in FBS. (D) The binding activity of HER2-LsbMDDs during storage in FBS. (E) Drug release profiles of Tynen®, the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs.
3.5. In vitro release profiles of DTX from the optimal BsAbs-LsbMDDs
Drug release profiles were calculated by plotting the release percentage of the drug from the optimal DTX-loaded BsAbs-LsbMDDs when compared to that for the solvent-based formulation (Tynen®), and the DTX-loaded LsbMDDs. Results in illustrate that the initial release of the DTX from Tynen® was the fastest among all the formulations examined with 80% of the DTX being released within 24 h. The release rate of the DTX from the LsbMDDs formulations was slower than that for Tynen® with 70% being released within 24 h. The release rates for both the BsAbs-LsbMDDs formulations were found to be similar and the slowest, with 60% being released in a 24 h period. These results indicated that a greater proportion of the DTX was entrapped in the micellar core of the LsbMDDs and BsAbs -LsbMDDs. The shielding effect of the non-covalently bound of both the BsAbs onto the outer shell of the LsbMDDs might have further impeded the diffusion of the DTX resulting in an even smaller portion of the DTX being released.
3.6. In vitro cytotoxicity
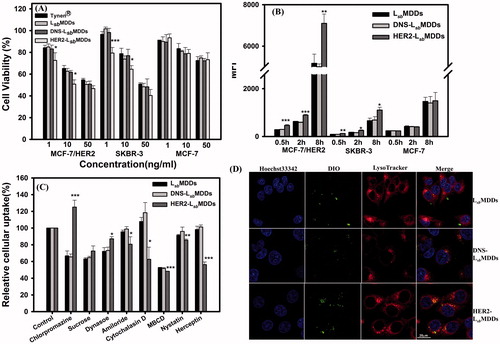
The cytotoxicity of Tynen®, the LsbMDDs, and BsAbs-LsbMDDs against the HER2-positive (MCF-7/HER2 and SKBR-3) and HER2-negative (MCF-7) cancer cells was examined. Result in and S2 (Supplemental information) shows that a dose-dependent effect is observed with all the formulations on HER2+ cells lines but not on the MCF-7 cells. The reason is that MCF-7 cells are less sensitive to DTX (the IC50 of MCF-7/HER2 is 52 ng/mL, while that of the MCF-7 is 763 ng/mL) resulting that a low dose range of DTX did not cause a significant difference in cell cytotoxicity. Both the LsbMDDs and DNS-LsbMDDs formulations exhibited similar cytotoxicity to that of Tynen® against the three cell lines (MCF-7/HER2, SKBR3, and MCF-7). Indeed, the HER2-LsbMDDs produced a significantly higher cytotoxicity to the MCF-7/HER2 and SKBR-3 cancer cell lines, both of which overexpressed HER2 on cell membranes, but displayed similar cytotoxicity to MCF-7 (HER2-) which poorly expresses HER2 as those for Tynen®, the LsbMDDs, and DNS-LsbMDDs. This demonstrates that the increase in cytotoxicity induced by the HER2-LsbMDDs requires the presence of the HER2 tumor antigen. It was concluded that the BsAbs confirmed the tumor specificity and enhanced the cytotoxicity of the LsbMDDs toward antigen-positive cancer cells.
Figure 4. (A) Cell viabilities of Tynen®, the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs in the MCF-7/HER2, SKBR-3, and MCF-7 cell lines (n= 4). (B) Cellular uptake of the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs was examined by flow cytometry after incubating DIO-loaded formulations with the MCF-7/HER2, SKBR-3, and MCF-7 cell lines at the time points of 0.5, 2, and 8 h (n= 3). (C) Cells were treated with 5 μM of the DIO-loaded LsbMDDs for 2 h in the presence of various inhibitors. Uptake is presented as the percentage of the control. (D) Confocal images of MCF-7/HER2 cells after treatment with the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs for 2 h after triple fluorescence-labeling experiments: red fluorescence from LysoTracker, green fluorescence from DIO, and blue fluorescence from Hoechst 33342 for nuclei. Colocalization of red and green fluorescence was observed in cells (scale bar =20 μm). *p < .05, **p < .01, and ***p < .001 compared to the LsbMDDs.
3.7. Cellular uptake of the optimal BsAbs-LsbMDDs
To examine the targetability of the optimal BsAbs-LsbMDDs to HER2-positive tumor cell lines, the cellular uptake of the DIO-loaded BsAbs-LsbMDDs was examined after incubating the DIO-loaded BsAbs-LsbMDDs at a 0.01:1 molar ratio of BsAbs to mPEG with the HER2-positive cell lines of MCF-7/HER2 and SKBR-3 and the HER2-negative cell line of MCF-7 at 37 °C. As shown in , cellular uptake amount of the HER2-LsbMDDs by the MCF-7/HER2 and SKBR-3 cells at all three time points (0.5, 2, and 8 h) were higher than those for the LsbMDDs and DNS-LsbMDDs, both of which had the same level of cellular uptake at all-time points in the three cell lines examined. also illustrates that uptake of the HER2-LsbMDDs was the same as those for the LsbMDDs and DNS-LsbMDDs in the antigen-negative MCF-7 cell line. This further confirmed that the optimal HER2-LsbMDDs non-covalently bound of anti-HER2/anti-mPEG BsAbs was able to enhance the cellular uptake by MCF-7/HER2 and SKBR-3 cells, both of which over-expressed HER2 on their cell membranes.
3.8. Cellular uptake mechanism
The general pathways of NCs internalized into cells are known to be phagocytosis, macropinocytosis, caveolae-dependent, and clathrin-mediated endocytosis (Zhao et al., Citation2011). Herein, we used cytochalasin D as a phagocytosis and macropinocytosis inhibitor, amiloride as a macropinocytosis inhibitor, MBCD as an inhibitor of lipid rafts involved in caveolae-dependent endocytosis, nystatin as a caveolae-dependent endocytosis inhibitor, chlorpromazine, sucrose, and dynasore as clathrin-mediated endocytosis inhibitors, and herceptin as a HER2 blocker, to determine which pathway participates in HER2-mediated cellular uptake. Results in show that the fluorescence intensities in the chlorpromazine, sucrose, and dynasore-treated LsbMDDs and DNS-LsbMDDs were significantly reduced when compared to that in the HER2-LsbMDDs. On the other hand, the relative uptake levels of the HER2-LsbMDDs in the presence of amiloride, cytochalasin D MBCD, and nystatin were considerably lower than those of the LsbMDDs in the MCF-7/HER2 cells. Results also show that the fluorescence intensity was notably reduced by the herceptin treatment in the HER2-LsbMDDs, but had no effect on the other two groups. These results demonstrate that the caveolae-mediated endocytosis and the macropinocytosis were more important for the cellular uptake of the HER2-LsbMDDs than for the LsbMDDs.
Promotion of the internalization of the LsbMDDs into tumor cells by BsAbs of HER2 was assessed. Confocal microscopic graphs in visualize green fluorescence from the DIO-loaded LsbMDDs, red fluorescence from the LysoTracker for lysosomes, and nuclear DNA labeled with Hoechst 33342 which emits blue fluorescence. The red and green fluorescence signals were imaged as being colocalized in MCF-7/HER2 cells treated with the HER2-LsbMDDs for 2 h, which demonstrated that anti-mPEG/anti-HER2 BsAbs could specifically deliver the HR2-LsbMDDs to MCF-7/HER2 cells, and when accompanied by HER2-LsbMDDs treatment, markedly increased colocalization of lysosomes with NCs of the HER2-LsbMDDs. It was concluded that anti-mPEG/anti-HER2 BsAbs can mediate selective binding and internalization of the HER2-LsbMDDs into the HER2+ cancer cells.
3.9. PK studies of the optimal BsAbs-LsbMDDs
PK profiles of the BsAbs-LsbMDDs were performed and compared to those with the LsbMDDs alone and Tynen®. The related PK parameters estimated by WinNonlin are listed in Supplemental information Table S1. All the PK profiles plotted in show a high initial DTX concentration after the injection, followed by a rapid decline to the terminal phase which gradually reached a steady-state concentration, and which was observed to be slightly higher for Tynen® than the other three formulations. Supplemental information Table S1 illustrates that a 3 ∼ 6-fold higher initial concentrations (C0) for the three LsbMDDs formulations were observed as compared to that for Tynen®. The AUC0–72 and AUC0–inf values for the LsbMDDs formulations were similar to those of Tynen®. The AUC0–inf for LsbMDDs (2276 ± 473 h*ng/mL) was slightly higher than those of the DNS-LsbMDDs and the HER2-LsbMDDs; nevertheless, the difference between them was insignificant (p > .05). CL and V values of the HER2-LsbMDDs were 1.08- and 1.2-times larger than those of the LsbMDDs, while there was no dramatic difference in the half-life of these three different LsbMDDs formulations. In a supplemental PK study, the HER2 binding activity of HER2-LsbMDDs along with the detection of plasma DTX concentration at the same predetermined time points was measured and results are shown in Figure S3 (in Supplemental information). As shown in Figure S3, the plasma PK profile of DTX after administration of HER2-LsbMDDs was similar to that revealed in confirming the reproducibility of the PK study. Along with the plasma DTX concentration illustrated by Figure S3 is the binding activity of HER2-LsbMDDs remained in the plasma at each time point. Since it has been confirmed that after administration of DTX-loaded LsbMDDs, most of DTX in the plasma was encapsulated in LsbMDDs (Sheu et al., Citation2017). Therefore, the binding activity of HER2-LsbMDDs that encapsulated DTX was measured at the same DTX concentration (all were diluted to 5 ng/mL DTX) as an indication of the same HER2-LsbMDDs concentration being loaded in the measurement of the binding activity. Using the binding amount of HER2-LsbMDDs at time point of 1 h as 100%, the binding activity expressed as percentage remained for each time point as compared to that at 1 h is illustrated in Figure S3. The results demonstrated that the binding activity of HER2-LsbMDDs remained in the plasma gradually decreased but maintained at a 50% binding activity at 72 h. The gradual loss of the binding activity of HER2-LsbMDDs means that the BsAbs was detached from HER2-LsbMDDs leading to the less amount of NCs being able to bind to HER2 receptor. Nevertheless, the 50% binding activity of DTX-loaded HER2-LsbMDDs remained in plasma was still stable and targetable even being subjected to vigorous blood flow for 72 h.
Figure 5. (A) Plasma concentration-time curves of docetaxel after intravenous administration of Tynen®, the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs at a dose of 8 mg/kg to rats (n= 3). (B) The MCF-7/HER2 tumor growth curve after intravenous administration at a dosing regimen of 5 mg/kg Q3D*4 (n= 6). (C) Body weight changes in tumor-bearing mice. (D) Tumor inhibition rates and (E) photographs showing the size of tumors after tumor-bearing mice were sacrificed on day 21. *p < .05 and ***p < .001 compared to the LsbMDDs on day 21.
3.10. In vivo antitumor efficacy of the HER2-LsbMDDs in tumor-bearing mice
The anti-tumor effects of Tynen® and the BsAbs-LsbMDDs were evaluated in a HER2-positive cancer cell (MCF-7/HER2) model. Results are shown in clearly demonstrate that the three LsbMDDs formulations and Tynen® all efficaciously inhibited the growth of MCF-7/HER2 tumors after treatment. The HER2-LsbMDDs showed the greatest anti-tumor effect among all the formulations in the MCF-7/HER2 tumor-bearing mice (). Tumor growth in the HER2-LsbMDDs treatment group was significantly suppressed as compared to those of Tynen® (p < .05 on day 21), the LsbMDDs alone (p < .001 on day 21), and the negative control of the DNS-LsbMDDs (p < .05 on day 21). The tumor inhibitory rate of the HER2-LsbMDDs was 88.9%, whereas they were 85.8, 78.4, and 80.1% for Tynen®, LsbMDDs, and DNS-LsbMDDs, respectively (). Nevertheless, the weight change profiles of all the treatments illustrated in demonstrate that there was greater weight loss in the Tynen® treatment group than for any of the three LsbMDDs formulations, indicating that the three LsbMDDs formulations induced less systemic toxicity than did Tynen®. Thus, it was concluded that the treatment with the HER2-LsbMDDs was more efficacious in inhibiting tumor growth than all of the other formulations while showing no signs of adverse side-effects.
3.11. Biodistribution assessment of the HER2-LsbMDDs
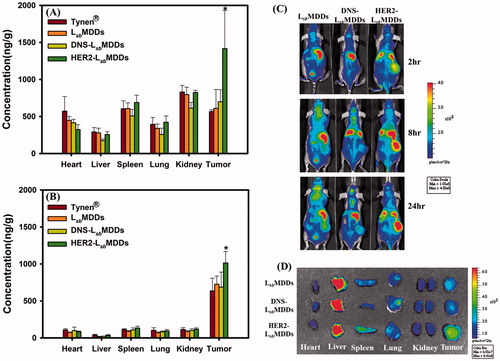
Biodistributions of the HER2-LsbMDDs in various organs after an IV administration were assessed compared to those for Tynen®, the LsbMDDs, and the DNS-LsbMDDs in MCF-7/HER2 tumor-bearing mice. At 2 and 16 h after the IV administration, organs, including the heart, liver, spleen, lung, and kidney, as well as the tumor were excised to analyze them for DTX. DTX concentrations in these tissues at 2 and 16 h are, respectively, shown in . DTX was observed to be mainly distributed in the heart, spleen, kidney, and tumor at 2 h after administration, and a higher concentration of DTX was only retained in the tumor at 16 h for all the four formulations. At both time points, the DTX concentrations following an injection of Tynen® were found to be slightly higher than those for the LsbMDDs and DNS-LsbMDDs formulations in all the tissues, except in the tumor tissues. Furthermore, a statistically significantly higher DTX concentration was only shown in tumor tissues for treatment at 2 and 16 h with the HER2-LsbMDDs respectively being 2.32 and 1.3-fold higher than the other three formulations. At 16 h after the injection, the DTX in the tumor site was still maintained at a 5 ∼ 10-fold higher concentration compared to that in other tissues for all the four formulations. These results indicated that the HER2-LsbMDDs not only enhanced the targeting to the tumor site resulting in a higher accumulation of DTX in the tumor, but also further retained the DTX in the tumor for a longer time to improve the therapeutic efficacy.
Figure 6. Tissue distributions of docetaxel (DTX) at (A) 2 and (B) 16 h after intravenous administration of Tynen®, the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs at a dose of 40 mg/kg into MCF-7/HER2 tumor-bearing nu/nu mice (n = 3). (C) Mice were imaged at 2, 8, and 24 h with an IVIS spectrum optical imaging system after being intravenously injected with the DIR-loaded LsbMDDs, DIR-loaded DNS-LsbMDDs, and DIR-loaded HER2-LsbMDDs (200 μg/kg of DIR). (D) Ex vivo fluorescence images of excised organs and tumors at 24 h post-injection of the LsbMDDs, DNS-LsbMDDs, and HER2-LsbMDDs in MCF-7/HER2 tumor-bearing nu/nu mice. *p < .05 compared to the LsbMDDs.
3.12. In vivo imaging of tumor-bearing mice
To investigate tumor targeting of the BsAbs-LsbMDDs in vivo, images of mice on an IVIS imaging system were captured at 2, 8, and 24 h post-injection of the DIR-loaded LsbMDDs and the DIR-loaded BsAbs-LsbMDDs. As shown in , the fluorescent intensity (total flux) of the HER2-LsbMDDs was primarily found at tumor sites (except in liver) and was 2 ∼ 3-fold higher than that for the LsbMDDs and DNS-LsbMDDs at 24 h, indicating higher accumulation of the HER2-LsbMDDs in the tumor. Additionally, ex vivo images in from harvested tumors also confirmed obviously higher fluorescence in the HER2-LsbMDDs group than those from the LsbMDDs and DNS-LsbMDDs groups, which was further evidence of the higher tumor targeting efficiency of the HER2-LsbMDDs. These data indicated that BsAbs of anti-mPEG/anti-HER2 were able to actively target the HER2+ cancer cells, thereby facilitating enhanced HER2-LsbMDDs accumulation in HER2+ tumors to improve the therapeutic efficacy and minimize potential side effects as a result of a preferable biodistribution in tumor cells. Both in vivo images in and ex vivo images in illustrate that the fluorescent signal was the highest detected in the liver, which was in contrary to what was observed in biodistribution study that demonstrated the accumulation of DTX was higher in tumor than that in liver. This discrepancy might be explained by that the fluorescent dye uptake via NCs could be retained longer in the liver in comparison to DTX resulting in the accumulation of fluorescent dye in the liver with showing the highest signal.
4. Discussion
To enable active tumor targeting of NCs, we established non-covalently bound of BsAbs (anti-mPEG/anti-tumor) to mPEGylated NCs of LsbMDDs designated LsbMDDs(2K and 5K), which were based on chemotherapeutic drug-loaded polymeric micelles stabilized by a lipid layer (lecithin/DEPSE-PEG) during the self-assembling hydration of micelles. By taking advantage of the unique strengths of the lecithin-based mixed polymeric micelles (LMPMs) and liposomes, the supported lipid shell composed an appropriate ratio of lecithin and DSPE-PEG was fused onto the polymeric micellar core of the LsbMDDs by ultrasonication. The physical characteristics of the LsbMDDs were a mean size of <200 nm, an EE of >90%, and DL of >5%. The surface of the LsbMDDs modification by PEG chains provides a steric barrier to prevent the opsonization and evade the reticuloendothelial system (RES). The outer structure of the LsbMDDs consisted of lecithin and DSPE-PEG at molar ratios of 95/5. When the PEG contents in the formulations were 5%, the structure was brush-like (Allen et al., Citation2002), which enabled the BsAbs to be non-covalently bound to methoxy terminus of the straight PEG chain via the anti-mPEG Fab fragments of BsAbs, to enhance active targeting of the mPEGylated NCs to the tumors via the anti-tumor scFv fragment of the BsAbs.
shows that the BsAbs could non-covalently bound to both the LsbMDDs(2K) and LsbMDDs(5K) via the methoxy end group of the mPEG chain that linked to the DSPE located on the surface of the LsbMDDs(2K) or LsbMDDs(5K). Further, the molar ratio of BsAbs to mPEG5K on the LsbMDDs(5K) was optimized based on the extents of the free BsAbs observed at the three different molar ratios of BsAbs to the LsbMDDs(5K). As shown in , no free BsAbs were detected when the molar ratio was 0.01:1, whereas some extent of the free BsAbs was observed at a higher molar ratio of 0.02:1. This confirmed that an optimal molar ratio of the BsAbs to LsbMDDs(5K) should be 0.01 ∼ 0.02:1.
Amounts of the mPEGylated LsbMDDs taken up with either the DSPE-PEG2K or the DSPE-PEG5K were compared after the BsAbs were non-covalently bound to the LsbMDDs. Results showed that the longer the PEG chain was, the higher the cellular uptake was observed. Contrary to our results, the study by Charmainne & Chithrani reported that the shorter PEG chain lengths (PEG2K vs. PEG5K) resulted in higher uptake for two grafting densities (Charmainne & Chithrani, Citation2014). It was attributed to that the shorter chain lengths have higher cancer cell uptake due to a greater probability of nonspecific protein adsorption, which mediates the entry of inorganic NPs by receptor-mediated endocytosis. Conforming to our results, the study on delivering small interfering (si)RNA with lipid-polymer hybrid NPs reported that the formulation with DSPE-PEG5K had higher tumor accumulation than did that with DSPE-PEG3K (Zhu et al., Citation2015). Sadzuka et al. also reported that the longer PEG chain length in the 1-monomethoxypolyethyleneglycol-2,3-dimyristoylglycerol(PEG-DMG) and 1-monomethoxypolyethyleneglycol-2,3-distearoylglycerol (DSG) groups increased tumor cell uptake of liposome, i.e. the value of PEG5K was better than that of PEG2K (Sadzuka et al., Citation2003). It was attributed to that a longer PEG chain more easily attached to the tumor cell membrane. Along with this, BsAbs non-covalently bound on the methoxy terminal end of longer PEG chains make BsAbs-LsbMDDs more easily attach to the tumor cells resulting in the increase of tumor cell uptake.
The isotype control of anti-mPEG/anti-DNS BsAbs non-covalently bound to the LsbMDDs did not alter the physical characteristics of the LsbMDDs or the non-specific targeting to the tumor sites. The ZPs of the LsbMDDs was observed to be negative, thus conferring the lower interactions with the plasma proteins than the positively charged ones, which are expected to strongly interact with the blood components causing a higher extent of the drug leakage (Tenzer et al., Citation2013). The ZPs of both the BsAbs-LsbMDDs formulations were even more negative, indicating that the BsAbs were bound to the termini of mPEG chains. Drug release profiles described in also shows that the BsAbs-LsbMDDs formulations released drugs more slowly than did the LsbMDDs, implying that the non-covalent binding of BsAbs to NCs might have retarded drug leakage from the NCs by providing a stabilization effect on the shell structure of the LsbMDDs and a shielding effect on the permeation of the DTX.
As shown in , enhanced cytotoxicity induced by HER2-LsbMDDs was only observed for the HER2-presenting tumor cell lines of the MCF-7/HER2 and the SKBR-3. Contrarily, Tynen® and those LsbMDDs formulations without the BsAbs bound or bound with the isotype control antibody of the anti-DNS expressed similar cytotoxicities to tumor cell lines regardless of whether or not they presented the corresponding antigen. The uptake amount of the DIO-loaded HER2-LsbMDDs as shown in at all-time points was obviously greater than that for the LsbMDDs without BsAbs bound or bound with the isotype control antibody of the anti-DNS in HER2 over-presenting tumor cells of MCF-7/HER2 and SKBR-3, but not in MCF-7 tumor cells poorly presenting HER2. The cellular uptake of the HER2-LsbMDDs is higher in MCF-7/HER2 than SKBR-3 cell lines, because the HER2 expressed at the surface of the MCF-7/HER2 is higher than the SKBR-3 cell lines (Supplemental information Figure S1). This further confirmed that BsAbs can selectively bind to a tumor and enhance the cytotoxicity of the BsAbs-LsbMDDs toward the antigen-positive cancer cells as a result of an increased amount taken up.
To understand the mechanism of the cellular uptake of the LsbMDDs, results in , show that most clathrin-mediated endocytosis inhibitors effectively reduced LsbMDDs uptake in the MCF-7/HER2 cells. However, chlorpromazine treatment increased cellular uptake of the HER2-LsbMDDs, but reduced uptake of the LsbMDDs, and the DNS-LsbMDDs. It was reported that the HER2 expression can inhibit the downregulation by having a negative effect on the formation of the clathrin-coated structures (Cortese et al., Citation2013). Since the inhibitory property of chlorpromazine acts through its ability to translocate clathrin from the cell surface to the intracellular endosomes (Dutta & Donaldson, Citation2012), this effect may indirectly increase the HER2 expression on the cell surfaces, thus causing more HER2-LsbMDDs to be transported into the cells. These results also indicated that the clathrin-mediated endocytosis is a more-preferable pathway for the cellular uptake of the LsbMDDs. Similar results can also be seen in the dynasore- and sucrose-treated groups. Nonetheless, when compared to the chlorpromazine-treated groups, dynasore and sucrose still had inhibitory effects, even with the HER2-LsbMDDs. Dynasore inhibits the clathrin-mediated endocytosis by blocking the GTPase activity of dynamin (Macia et al., Citation2006). Since dynamin is involved in new vesicle formation on the membranes (Henley et al., Citation1999), it might more directly influence the HER2-mediated transport. Overall results shown in suggest that the macropinocytosis and caveolae-mediated endocytosis were more-preferable pathways for HER2-LsbMDDs uptake than for LsbMDDs uptake. Furthermore, the BsAbs-LsbMDDs was taken up by receptor-mediated endocytosis, so it might be able to avoid the particles being pumped out by the P-glycoprotein (P-gp) or the breast cancer resistance protein (BCRP) in multiple drug-resistant cells.
demonstrates that the HER2-LsbMDDs significantly enhanced the in vivo anticancer activity, as shown by the higher growth inhibition rate in the HER2-overexpressing tumor cells. This likely can be attributed to the increased cytotoxicity of the HER2-LsbMDDs against the tumor cells over-expressing the HER2 anti-tumor antigen as shown in . Further, according to the biodistribution study and the IVIS images in , the DTX concentration after an IV injection of the HER2-LsbMDDs was highest at the tumor site, but with a similar DTX concentration to those for Tynen®, the LsbMDDs, and DNS-LsbMDDs in other organs of the heart, liver, spleen, lung, and kidney. Overall, the enhanced chemotherapeutic efficacy of the HER2-LsbMDDs can be attributed to the preferable biodistribution to tumor sites by active targeting of the HER2-LsbMDDs endowed with non-covalently bound of BsAbs and the enhanced DTX uptake amount via an endocytosis-mediated pathway leading to a greater accumulation and a longer retention of the DTX in the tumor.
Active targeting strategies facilitate NC internalization, binding, and homing to targeted cells. Antibody-drug conjugates (ADCs) consisting of highly potent cytotoxic agents covalently linked to a mAb are an emerging novel class of chemotherapeutics. However, the most commonly identified weaknesses limiting the effective uses of ADCs are the low anticancer drug potency, low antigen selectivity, and unstable linkers (Perez et al., Citation2014). Antibody-NP conjugates have great benefits in overcoming limitations in current approaches as NPs have the ability to release a drug at desirable sites, improve cell penetration, and cross biological barriers by conjugating Abs with high affinity. However, the method for Ab conjugation to NCs has the same challenges. Also, chemical modifications cause heterogeneous orientations, Ab lose their functions and are non-reproducible, and site-directed modifications cause complex operations and high costs (wasting large amounts of Abs) (Haberger et al., Citation2014). The primary goal of conjugating targeted ligands to NCs is to not lose the functionality of the ligand targeting. Using humanized BsAbs non-covalently bound to the LsbMDDs avoids direct, potentially denaturing interactions with NC surfaces, minimizing possible alterations of NC properties, and low immunogenicity. Our results showed no obvious changes in the physical properties of the LsbMDDs with the non-covalently bound of BsAbs. They also confirmed that the BsAbs were bound to the ends of mPEG molecules, thus orienting the anti-tumor scFv portion of the BsAbs outward and minimizing steric masking of the BsAbs by mPEG. This could be a simple and promising way to introduce active tumor targetability to the drug-loaded mPEGylated NCs to enhance their chemotherapeutic efficacy and minimize the systemic toxicities.
5. Conclusions
In conclusion, a BsAbs-modified LsbMDDs was established which minimized the changes in physical properties and structures and conferred the BsAbs target specificity to high drug-loaded NCs of the LsbMDDs. The BsAbs-modified LsbMDDs was effectively taken up by a combination of both passive targeting by the EPR and active targeting by internalization. The BsAbs non-covalently bound to the LsbMDDs can target antigen-overexpressing tumors with a drug cargo to enhance the drug uptake and the accumulation in tumors, leading to greater antitumor activities against antigen-positive tumors. This well-characterized platform can be applied to the BsAbs targeting other tumor markers, such as the prostate-specific membrane antigen (PSMA), EGFR, VEGF, and programed death-ligand 1 (PD-L1), non-covalently bound to mPEGylated NCs that can be loaded with chemotherapeutic drugs including paclitaxel, irinotecan, rapamycin, etc.
IDRD_Sheu_et_al_Supplemental_Content.docx
Download MS Word (1.8 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Allen TM. (2002). Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer 2:750–63.
- Allen C, Dos Santos N, Gallagher R, et al. (2002). Controlling the physical behavior and biological performance of liposome formulations through use of surface grafted poly(ethylene glycol). Bio Sci Rep 22:225–50.
- Brigger I, Dubernet C, Couvreur P. (2012). Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev 64:24–36.
- Chabner BA, Roberts TG. Jr. (2005). Timeline: chemotherapy and the war on cancer. Nat Rev Cancer 5:65–72.
- Charmainne C, Chithrani DB. (2014). Polyethylene glycol density and length affects nanoparticle uptake by cancer cells. J Nanomed Res 1:00006.
- Chen LC, Chen YC, Su CY, et al. (2016). Development and characterization of self-assembling lecithin-based mixed polymeric micelles containing quercetin in cancer treatment and an in vivo pharmacokinetic study. Int J Nanomed 11:1557–66.
- Chen YC, Su CY, Jhan HJ, et al. (2015). Physical characterization and in vivo pharmacokinetic study of self-assembling amphotericin B-loaded lecithin-based mixed polymeric micelles. Int J Nanomedicine 10:7265–74.
- Chuang KH, Tzou SC, Cheng TC, et al. (2010). Measurement of poly(ethylene glycol) by cell-based anti-poly(ethylene glycol) ELISA. Anal Chem 82:2355–62.
- Cortese K, Howes MT, Lundmark R, et al. (2013). The HSP90 inhibitor geldanamycin perturbs endosomal structure and drives recycling ErbB2 and transferrin to modified MVBs/lysosomal compartments. Mol Biol Cell 24:129–44.
- Dave V, Yadav RB, Kushwaha K, et al. (2017). Lipid-polymer hybrid nanoparticles: development & statistical optimization of norfloxacin for topical drug delivery system. Bioact Mater 2:269–80.
- Davis ME, Chen ZG, Shin DM. (2008). Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov 7:771–82.
- Dennison SM, Stewart SM, Stempel KC, et al. (2009). Stable docking of neutralizing human immunodeficiency virus type 1 gp41 membrane-proximal external region monoclonal antibodies 2F5 and 4E10 is dependent on the membrane immersion depth of their epitope regions. J Virol 83:10211–23.
- Dutta D, Donaldson JG. (2012). Search for inhibitors of endocytosis: intended specificity and unintended consequences. Cell Logist 2:203–8.
- Gao J, Liu W, Xia Y, et al. (2011). The promotion of siRNA delivery to breast cancer overexpressing epidermal growth factor receptor through anti-EGFR antibody conjugation by immunoliposomes. Biomaterials 32:3459–70.
- Haberger M, Bomans K, Diepold K, et al. (2014). Assessment of chemical modifications of sites in the CDRs of recombinant antibodies: susceptibility vs. functionality of critical quality attributes. MAbs 6:327–39.
- Hare JI, Lammers T, Ashford MB, et al. (2017). Challenges and strategies in anti-cancer nanomedicine development: an industry perspective. Adv Drug Deliv Rev 108:25–38.
- Henley JR, Cao H, Mcniven MA. (1999). Participation of dynamin in the biogenesis of cytoplasmic vesicles. FASEB J 13:S243–S7.
- Itoh K, Watanabe A, Funami K, et al. (2008). The clathrin-mediated endocytic pathway participates in dsRNA-induced IFN-β production. J Immunol 181:5522.
- Kamaly N, Xiao Z, Valencia PM, et al. (2012). Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev 41:2971–3010.
- Kao CH, Wang JY, Chuang KH, et al. (2014). One-step mixing with humanized anti-mPEG bispecific antibody enhances tumor accumulation and therapeutic efficacy of mPEGylated nanoparticles. Biomaterials 35:9930–40.
- Koo H, Huh MS, Sun IC, et al. (2011). In vivo targeted delivery of nanoparticles for theranosis. Acc Chem Res 44:1018–28.
- Krishnamurthy S, Vaiyapuri R, Zhang L, Chan JM. (2015). Lipid-coated polymeric nanoparticles for cancer drug delivery. Biomater Sci 3:923–36.
- Kuhn DA, Vanhecke D, Michen B, et al. (2014). Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J Nanotechnol 5:1625–36.
- Li Q, Xia D, Tao J, et al. (2017). Self-assembled core-shell-type lipid-polymer hybrid nanoparticles: intracellular trafficking and relevance for oral absorption. J Pharm Sci 106:3120–30.
- Macia E, Ehrlich M, Massol R, et al. (2006). Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10:839–50.
- Manjappa AS, Chaudhari KR, Venkataraju MP, et al. (2011). Antibody derivatization and conjugation strategies: application in preparation of stealth immunoliposome to target chemotherapeutics to tumor. J Control Release 150:2–22.
- Mikhalin AA, Evdokimov NM, Frolova LV, et al. (2014). Lipophilic prodrug conjugates allow facile and rapid synthesis of high loading capacity liposomes without the need for post-assembly purification. J Liposome Res 25:1–29.
- Nicolas J, Mura S, Brambilla D, et al. (2013). Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem Soc Rev 42:1147–235.
- Peer D, Karp JM, Hong S, et al. (2007). Nanocarriers as an emerging platform for cancer therapy. Nature Nanotech 2:751–60.
- Perez HL, Cardarelli PM, Deshpande S, et al. (2014). Antibody-drug conjugates: current status and future directions. Drug Discov Today 19:869–81.
- Sadzuka Y, Kishi K, Hirota S, Sonobe T. (2003). Effect of polyethyleneglycol (PEG) chain on cell uptake of PEG-modified liposomes. J Liposome Res 13:157–72.
- Sahay G, Alakhova DY, Kabanov AV. (2010). Endocytosis of nanomedicines. J Control Release 145:182–95.
- Sheu MT, Wu CY, Su CY, Ho HO. (2017). Determination of total and unbound docetaxel in plasma by ultrafiltration and UPLC-MS/MS: application to pharmacokinetic studies. Sci Rep 7:14609.
- Su CY, Liu JJ, Ho YS, et al. (2018). Development and characterization of docetaxel-loaded lecithin-stabilized micellar drug delivery system (LsbMDDs) for improving the therapeutic efficacy and reducing systemic toxicity. Eur J Pharm Biopharm 123:9–19.
- Tenzer S, Docter D, Kuharev J, et al. (2013). Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat Nanotechnol 8:772.
- Tietze S, Schau I, Michen S, et al. (2017). A poly(propyleneimine) dendrimer-based polyplex-system for single-chain antibody-mediated targeted delivery and cellular uptake of SiRNA. Small 13: 1700072.
- Wang AZ, Langer R, Farokhzad OC. (2012). Nanoparticle delivery of cancer drugs. Annu Rev Med 63:185–98.
- Zhao F, Zhao Y, Liu Y, et al. (2011). Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small 7:1322–37.
- Zhong Y, Meng F, Deng C, Zhong Z. (2014). Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules 15:1955–69.
- Zhu X, Xu Y, Solis LM, et al. (2015). Long-circulating siRNA nanoparticles for validating Prohibitin1-targeted non-small cell lung cancer treatment. Proc Natl Acad Sci USA 112:7779–84.