
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Voriconazole (VCZ), a triazole with a large spectrum of action is one of the most recommended antifungal agents as the first line therapy against several clinically important systemic fungal infections, including those by Candida albicans. This antifungal has moderate water solubility and exhibits a nonlinear pharmacokinetic (PK) profile. By entrapping VCZ into liposomes, it is possible to circumvent certain downsides of the currently available product such as a reduction in the rate of its metabolization into an inactive form, avoidance of the toxicity of the sulfobutyl ether-beta-cyclodextrin (SBECD), vehicle used to increase its solubility. PKs and biodistribution of VCZ modified by encapsulation into liposomes resulted in improved antifungal activity, due to increased specificity and tissue penetration. In this work, liposomal VCZ resulted in AUC0–24/MIC ratio of 53.51 ± 11.12, whereas VFEND® resulted in a 2.5-fold lower AUC0–24/MIC ratio (21.51 ± 2.88), indicating favorable antimicrobial systemic activity. VCZ accumulation in the liver and kidneys was significantly higher when the liposomal form was used. Protection of the drug from biological degradation and reduced rate of metabolism leads to a 30% reduction of AUC of the inactive metabolite voriconazole-N-oxide (VNO) when the liposomal drug was administered. Liposomal VCZ presents an alternative therapeutic platform, leading to a safe and effective treatment against systemic fungal infections.
Introduction
The whole world needs new medicines against infectious diseases. Thanks to antimicrobials, an increase in life expectancy, especially of immunosuppressed individuals (e.g. HIV, cancer, or transplanted patients, etc.), as well as of those living in areas lacking basic sanitation infrastructure and infection prevention programs became possible (Roemer & Krysan, Citation2014; Rossolini et al., Citation2014).
The antifungal voriconazole (VCZ), a second-generation triazole with a large spectrum of action is one of the most recommended systemic antifungal agents as the first line therapy against several types of systemic mycoses, including Candida albicans. C. albicans is the most common opportunistic fungal pathogen, which may lead to life-threatening infections once the initially localized mycosis become systemic and the immune system of the host organism is compromised (Dupont, Citation1995; Lopes et al., Citation2015). VCZ exhibits a nonlinear pharmacokinetic (PK)profile due to metabolic clearance saturation (Yamada et al., Citation2015). In addition, VCZ is extensively metabolized in the liver, into its main circulating metabolite, voriconazole-N-oxide (VNO), which has minimal antifungal efficacy. VNO also interferes in the metabolic activity of CYP3A4 and CYP2C19, thus interfering with the metabolism of VCZ, a substrate of the aforementioned enzymes (Martinez, Citation2006; Theuretzbacher et al., Citation2006).
The solubility of VCZ in water is limited, presenting a technological issue for the development of IV formulations. To date, the only commercially available formulation of VCZ (VFEND®) for injection contains sulfobutyl ether-beta-cyclodextrin (SBECD) to increase drug apparent solubility in water. Similar to other cyclodextrins, SBECD can cause several clinically relevant issues, such as nephrotoxicity, hemolysis, blood vessels congestion, and hepatic and renal toxicity due to potential accumulation (Ling et al., Citation2016; Maertens et al., Citation2016; Bellmann & Smuszkiewicz, Citation2017; Ledoux, et al., Citation2017). To overcome these issues, the use of liposomes as carriers of VCZ has been explored (de Sa et al., Citation2015), showing clinical advantages besides eliminating the use of beta-cyclodextrins and its associated toxicity (Roffey et al., Citation2003; Luke et al., Citation2010; Kiser et al., Citation2015).
These advantages include targeting to infectious sites with higher local bioavailability, since the liposomes may possibly fuse to the fungal cell membrane and facilitate transference of VCZ, in an internalization mechanism already elucidated using AmBisome®, a liposomal formulation that encapsulates amphotericin B (Shimizu et al., Citation2010; Soo Hoo, Citation2017).
Although the PK and pharmacodynamics (PD) parameters are highly dependent on the physical-chemical characteristics of the drug, once encapsulated into liposomes, these parameters can be modulated. Different blood circulation times, tissue distribution, and the prevention of biological degradation of the antifungal might influence the efficacy of the drug (Pinto-Alphandary et al., Citation2000; Pattni et al., Citation2015). As such, these parameters must be investigated and correlated to the proposed treatment.
In this study, we prepared and characterized a novel formulation for the antifungal VCZ entrapped into liposomes (LVCZ) for intravenous delivery. In addition, PK and tissue distribution profiles were measured. In vitro susceptibility analysis of fungal strains, both standard and isolated in the clinic – (C. Albicans and Aspergillus sp.) was conducted as well as in vivo efficacy evaluation of LVCZ using a mouse model of systemic candidiasis.
Materials and methods
Materials
Cholesterol and soybean phosphatidylcholine (PC) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Voriconazole (VOR) was purchased from Hangzhou Dayangchem Co., Limited (Hangzhou, China). and voriconazole-N-oxide (VNO) was purchased from Genone Biotechnologies (Rio de Janeiro, RJ, Brazil) Alpha-tocopherol and Sabouraud’s dextrose agar were purchased from Sigma Aldrich (St. Louis, MO, USA). Acetonitrile was purchased from J. T. Baker (Phillipsburg, NJ, USA). Water was purified using a Milli-Q system (Millipore, Billerica, MA, USA) with a 0.22 µm pore end-filter. All other chemicals and reagents were of analytical grade or superior.
Fungal strains and culture conditions
For the in vitro susceptibility assay, a total of eight fungal strains were tested as following: two American Type Culture Collection (ATCC) strains, C. albicans ATCC 90028 and Candida parapsilosis ATCC 22019, and six clinical isolates, including two C. albicans (77 and 11 U) and four Aspergillus sp. (L, V1F2, V2, and V1F1) strains. The clinical isolates were obtained from the fungal library of the Laboratory of Mycology from Federal University of Goiás and originated from samples of patients with onychomycosis and hematologic malignancies from works previously performed at the Hospital das Clínicas da Universidade Federal de Goiás and Hospital Araújo Jorge/Associação de Combate ao Câncer em Goiás (ACCG) of Goiânia-GO. C. albicans ATCC 90028 strain was used to establish the mouse model of systemic candidiasis. All strains were aerobically cultured on Sabouraud agar and maintained on Sabouraud 4%-glucose agar at 37 °C overnight before the assay.
Animal care
The in vivo studies were carried out on Balb/c male mice (age: 7–8 weeks; weight: 25–30 g), obtained from the Animal Facilities of the Institute of Tropical Pathology and Public Health of Federal University of Goiás (Goiânia, Brazil). Before beginning the experiments, the animals were acclimatized for a week in the laboratory. Considering that animal experimentation in infectious diseases research remains essential to understand the fundamental mechanisms of antifungal drugs as well as to meet regulatory requirements, the in vivo analyses conducted in this study were focused on animal welfare. The experimental design considered the minimum number of animals without reducing the scientific integrity of data generated. In additions, procedures such as reduced total amount of blood collected, small volume of injections, anesthesia, and analgesia were observed.
Animals were kept under constant environmental conditions with light-dark (12:12 h) cycles and controlled temperature (23 ± 2 °C). Water and food (standard granulated chow) were provided ad libitum. At the end of each experiment, the mice were anesthetized with a solution containing xylazine (10 mg/kg) and ketamine hydrochloride (100 mg/kg) administered intraperitoneally and euthanized by cervical dislocation (Hubrecht & Kirkwood, Citation2010). The procedures and protocols were reviewed and approved by the Research Ethics Committee of the Federal University of Goiás (UFG N° 108/2014 and 095/2016).
Preparation and characterization of the liposomal voriconazole for intravenous delivery
A liposomal formulation was prepared by lipid-film hydration followed by extrusion. PC, cholesterol, VCZ, and alpha-tocopherol, were dissolved in chloroform (1.0:0.5:0.11:0.05 molar ratio PC:cholesterol:VCZ:alpha-tocopherol, respectively). Solvent was evaporated at 25 °C and 200 rpm under reduced pressure leading to the formation of a thin layer on the inner side of a round-bottomed flask. Lipid film was hydrated with water for a final volume reaching the concentration of 2 mg/mL of VCZ. Hydrated vesicles were reduced in size by extrusion through 200-nm-pore polycarbonate filters followed by 100-nm-pore filters, in a Lipex® Extruder device (Northern Lipids Inc., Burnaby, Canada) under nitrogen pressure.
Formulations were lyophilized using sucrose as cryoprotectant (4:1 of molar ratio sucrose:PC) and reconstituted in sterile saline immediately before use. Particle size and polydispersity index (PdI) were evaluated by dynamic light scattering (DLS) in a Zetasizer Nano S (Malvern Instruments, Worcestershire, UK) and the morphology by Transmission Electron Microscopy (TEM) examined in a Jeol JEM-2100 Electron Microscope (Jeol, Tokyo, Japan) operating at 100 kV, following negative staining. Drug-free liposomes were prepared as controls.
VCZ was quantified by High Performance Liquid Chromatography (HPLC) with UV detection at 255 nm. Zeta potential was assessed via electrophoretic mobility in a ZetaPlus (Brookhaven Instruments, Holtsville, NY, USA). Liposome Entrapment Efficiency (EE%) was determined after ultrafiltration (Vivaspin® 20 kDa membrane, SIGMA, Gillingham, UK) by the following equation:
where TD is the total drug content of the formulation and FD is the non-encapsulated drug (free drug, filtered through the ultrafiltration device).
In vitro antifungal activity of LVCZ against Candida sp. and Aspergillus sp. yeasts
In vitro susceptibility of yeast and filamentous fungi was performed using the microdilution method in broth following M27-A3, M27-S4 (2008, 2012), and M38-A2 (2008), proposed by the Clinical and Laboratory Standards Institute (CLSI). All the strains used were diluted in saline at the concentration of 0.5−2.5 × 103 and 0.4−5×104 CFU/mL for Candida and Aspergillus, respectively. VFEND® and LVCZ were diluted in the range of 0.0312−32 μg/mL.
Plates were incubated at 35 °C for 48 h for Candida species and for 72 h for Aspergillus species.
Minimal inhibitory concentration (MIC) was determined as the lower concentration of the compound capable of inhibiting the total growth of the microorganism. The experiment was performed in triplicate, and C. parapsilosis ATCC 22019 was used as control of MIC, according to CLSI 2008 (John, Citation2008).
For minimum fungicidal concentration (MFC) determination, 10 μL of antifungal MIC and four immediately higher concentrations were seeded in Petri dishes containing Sabouraud dextrose agar (SDA). Plates were incubated at 35 °C for 48 h for Candida species and for 72 h for Aspergillus species. MFC was defined as the lowest concentration of antifungal that resulted in the growth of up to two colonies, which represent the death of more than 99% of the initial inoculum (John, Citation2008).
Pharmacokinetic and biodistribution studies
PK studies were carried out in healthy male Balb/c mice weighing 25–30 g (7–8 weeks of age) randomly assigned to two groups (n = 12 for each group) according to the formulation received. Lyophilized powders of LVCZ and VFEND® were reconstituted with sterile saline to 2 mg/mL. An intravenous (I.V) bolus injection of 10 mg/kg of each formulation/group was administered via tail vein. Using a non-mortality model (Rauzi et al., Citation2017), blood samples were withdrawn from the saphenous vein at the following predetermined times: 0.25; 0.50; 1.00; 2.00; 4.00; 6.00; 16.0, and 24.0 h after intravenous administration (Diehl et al., Citation2001).
For the biodistribution study, mice were euthanized by cervical dislocation 4 h after receiving 10 mg/kg of LVCZ or VFEND®. Then, heart, spleen, kidney, lungs, liver, brain, and eyes/optic nerves, were rapidly excised, washed with ice-cold saline, blotted dry, weighed, and stored at −80 °C until analysis.
VCZ and its metabolite, VNO concentrations in whole blood and tissues were determined by HPLC using an Agilent 1200 system (Santa Clara, CA, USA), composed of a quaternary pump, an auto-sampler, a column oven, with a hyphenated API 3200 triple quadrupole mass spectrometer (MS/MS) detector (MDS-SCIEX, Concord, Ontario, Canada). Drugs and internal standard (ketoconazole) were partitioned through liquid–liquid extraction using methyl tert-butyl ether. Concentrations of VCZ and VNO in blood were determined simultaneously from blood samples collected at 0.5 and 1 h following injection.
VCZ blood concentration versus time curves were plotted, and key PK parameters were calculated by the non-compartmental analysis using Microsoft Excel® V.15.12.3 and the add in: PKSolver (Zhang et al., Citation2010b). Results were reported as means ± standard deviation (SD).
Mouse model of systemic candidiasis
Infection model was based on protocols previously described (Courjol et al., Citation2016; MacCallum, Citation2013). In brief, mice were first rendered neutropenic by intraperitoneal injection of cyclophosphamide (75 mg/kg/d) for 2 d. To confirm neutropenia status of each animal, blood samples were obtained by retro-orbital bleeding before (day 0) and 24 h after (day 3) cyclophosphamide treatment for hematology analysis uses ABX Micros 60 (HORIBA, Montpellier, France). On day 4, each mouse was injected intravenously by tail vein with 100 μL of saline containing C. albicans ATCC 90028 (1 × 107 cells/mL). After 2 h of infection, the animals were treated with 150 μL of either sterile saline, VFEND® or LVCZ, both at 10 mg/kg of VCZ, for 3 d. Treatment groups (n = 5/group) were distributed as follows: Group I – infected and treated with placebo (Placebo, control group); Group II – infected and treated with VFEND® (IT-VFEND® group); Group III – infected and treated with LVCZ (IT-LVCZ group). On day 7, after 24 h of treatment, mice were previously anesthetized for blood collection by cardiac puncture. After that, animals were euthanized and kidneys and liver were removed for fungal burden and histopathology analyzes. For the fungal burden evaluation, liver and kidneys sections were macerated in 3 and 5 mL of sterile saline, respectively, using a glass tissue macerator. Liver and kidneys homogenates were then diluted at 1/100 and 1/1000, respectively, and 100 μL were plated on sterile Petri dishes containing SDA. After incubation for 48 h at 35 °C, colony forming units were counted. The experimental schedule of mouse model of systemic candidiasis is shown in Supplementary data 1.
Histopathological evaluation
Parts of liver and kidneys from each animal were kept in 10% phosphate buffered formalin (pH 7.4) at room temperature for the histopathological analysis. In brief, two sagittal macroscopic cross sections were dehydrated in graded ethanol (70–100%), cleared in xylene and then paraffin-embedded. After that, paraffin-embedded tissue sections of organs (5 μm thickness) were obtained using a microtome (Leica RM 2155, Heidelberg, Germany). After mounting, the slides were dewaxed in xylene, hydrated using graded ethanol and stained with hematoxylin and eosin (HE) or periodic acid-Schiff (PAS). The slides were observed using a light microscope (Axio Scope A1 Carl Zeiss, Jena, Germany) using a 40× objective. The images were taken using an AxioCam MRc Carl Zeiss camera and AxioVs40 version 4.7.2.0 Carl Zeis software (Carl Zeiss, Jena, Germany).
Statistical analysis
Data were reported as mean ± SD. Statistical analysis was performed using GraphPad Prism version 5.01 software for Windows (San Diego, CA). The intergroup variation was carried out by the Student’s t-test or one-way Analysis of Variance (ANOVA) followed by Dunnett’s test. p <.05 was considered statistically significant.
Results
Preparation and characterization of liposomal voriconazole for intravenous delivery
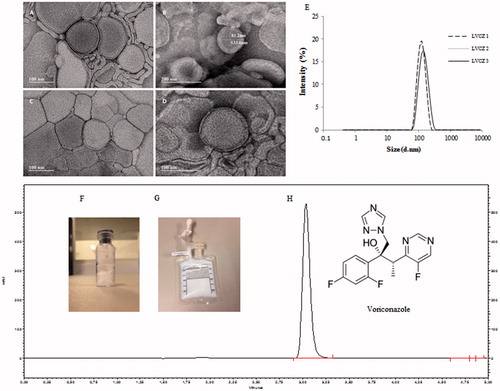
Intravenous liposomal formulation containing VCZ constituted by PC and cholesterol, herein called LVCZ, was developed for the treatment of C. albicans systemic infection by lipid-film hydration followed by extrusion. LVCZ exhibited a diameter of 95.3 ± 1.27 nm, very narrow size distribution (PdI =0.09 ± 0.01) and a relatively neutral charge. Entrapment efficiency was approximately 80%. Even though VCZ water solubility is low (<100 μg/mL), it is also only moderately lipophilic (log p ∼1.8). Drugs with intermediate partition coefficients (1.7 < log p < 4.0) can partition between the lipid membrane and the aqueous phase. (Gulati et al., Citation1998; Campbell et al., Citation2001; de Sa et al., Citation2015). TEM analysis confirmed liposomes diameter, exhibiting spherical morphology as uni- or oligo-lamellar vesicles. (more details of liposomal formulation are described in and also in the table of Supplementary data 2).
Figure 1. Voriconazole liposomal formulation characteristics. TEM micrograph of empty (blank) liposomes (A,B); VCZ liposomes (LVCZ) (C,D); VCZ liposomes (LVCZ) size distribution profiles by intensity obtained by DLS from three independent batches (E); freeze-dried formulation (F); redispersed freeze-dried LVCZ in a transparent infusion bag (G); and representative chromatogram from VCZ liposomal formulation at 2.0 mg/mL by HPLC with UV–VIS detector (H).
In vitro antifungal activity of VCZ
Activities of VFEND® and LVCZ against Candida sp. and Aspergillus sp. yeasts were carried out after 48 and 72 h of incubation, respectively, using the broth microdilution method. MIC and MFC values are summarized in and show that Candida sp. strains were more susceptible than Aspergillus sp. strains for both formulations. For Candida sp. strains, VFEND® and LVCZ have similar potency, although LVCZ triggered higher fungicide activity against C. albicans 77 U strain since the MFC value (0.12 μg/mL) was about 4-fold lower than that found for VFEND® (0.5 μg/mL). Among Aspergillus sp. strains, LVCZ showed higher inhibitory/fungicide activity against Aspergillus Flavus. For the A. Flavus V2 strain, MIC and MCF values were 2- and 4-fold lower (both at 0.25 μg/mL), respectively, than those found for VFEND® (MIC =0.5 μg/mL and MCF =1.0 μg/mL); while for V1F1 strain these values were 2-fold lower (MIC =0.5 μg/mL; MCF =1.0 μg/mL) in comparison to VFEND® values (MIC =1.0 μg/mL and MCF =2.0 μg/mL).
Table 1. Minimum inhibitory (MIC) and fungicide (MFC) concentrations (μg/mL) of voriconazole and liposomal voriconazole on Candida species sp. and Aspergillus sp.
Pharmacokinetic and tissue distribution
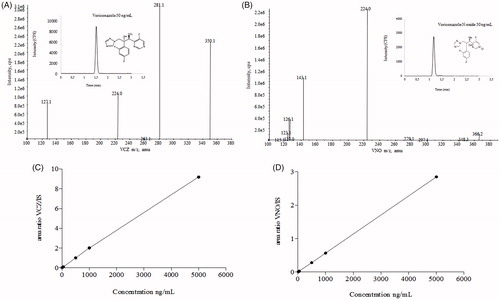
The bioanalytical quantitation method by HPLC-MS/MS was sensitive and selective enough to allow the simultaneous quantification of the antifungal VCZ, and its inactive metabolite VNO, even from tissue homogenates or in the whole blood samples from the animals. Chromatograms, mass spectra, and calibration curves of the above-mentioned substances are given in .
Figure 2. HPLC-MS/MS and analytical conditions. Full scan mass spectrum of (A) voriconazole and (B) voriconazole N-oxide by electrospray ionization in positive mode of the transitions of m/z 350.2/127.0 and m/z 366.2/224.2, respectively. Chromatographic profiles obtained under following conditions: ACE C18 column (100 × 4.6 mm ×5 µm), at 20 °C; mobile phase consisted of formic acid at 0.025% + ammonium acetate 2 mM and acetonitrile (25:75 v/v), flow rate of 1.0 mL/min. Run time was 2.0 min, and the injection volume was 5.0 µL, with an elution time for VCZ and VNO of 1.43 and 1.19 min, respectively. Calibration curves, ranging 5–5000 and 20–5000 ng/mL for VCZ (C) and VNO (D), respectively.
Differences in PK parameters of VCZ from both formulations were observed. When compared to VFEND®, liposomes reduced VCZ clearance by half; increased AUC0–24 by a 2.5-fold and reduced the drug volume of distribution. Main PK parameters and data for predicting the magnitude of the best PK/PD index, which were calculated by the ratio AUC0–24/MIC and Cmax/MIC also are shown in .
Table 2. Pharmacokinetic and pharmacodynamic parameters after 10 mg/kg IV administration of two different voriconazole formulations to Balb/c mice.
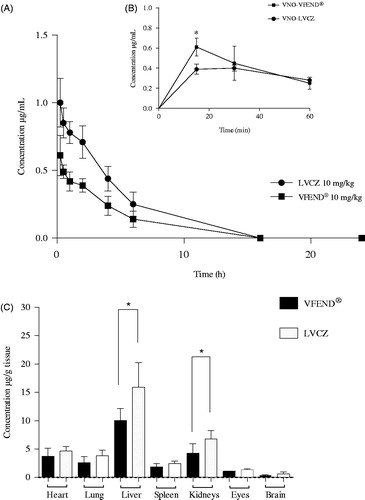
Blood concentration profiles following I.V injections of VFEND® or LVCZ formulations in Balb/c mice are shown in . Blood levels of VCZ delivered through liposomes was higher than VFEND® at any time point. shows that during the initial hour following administration of the formulations, the metabolism of VCZ when entrapped into liposomes is decelerated and thus slowing the formation of the inactive metabolite (VNO) by 30%. The initial blood concentration of VNO expressed as the AUC0–1 h was 0.49 µg/mL.h for VFEND® and 0.35 µg/mL.h for LVCZ.
Figure 3. Pharmacokinetic and biodistribution profile of voriconazole from two different formulations. (A) Blood levels of intravenously injected voriconazole versus time 0–24 h (AUC0–24) delivered from liposomal formulation (circles) and VFEND® (squares) at 10 mg/kg. (B) Concentrations versus time of the metabolite (voriconazole-N-oxide) measured within the hour following intravenous administration of the formulations. Data are expressed as mean ± SD (n = 12 animals per group). (C) Tissue distribution of voriconazole 4 h after I.V. administration of liposomal formulation (white bars) and voriconazole complexed in sulfobutyl ether-beta-cyclodextrin so + dium, commercially available as VFEND® (black bars). Bars represent the standard deviation (n = 6). (*indicates p<.05).
In order to avoid bias caused by the occurrence of hysteresis (disagreement between values of tissue distribution, determined only by approximation of antifungal blood concentrations obtained from PK), the quantification of VCZ in several tissues was performed.
Tissue distribution of VCZ from both formulations was evaluated in various tissues, including several organs of the reticuloendothelial system ().
Our PK assays using healthy animals have shown that blood concentrations of VCZ remained for 12 h above the MIC, following the administration of 10 mg/kg of the liposomal formulation. Conversely, VFEND® was able to maintain concentrations above MIC for only 8 h after a single bolus dose.
In vivo antifungal activity of LVCZ using a mouse model of systemic candidiasis
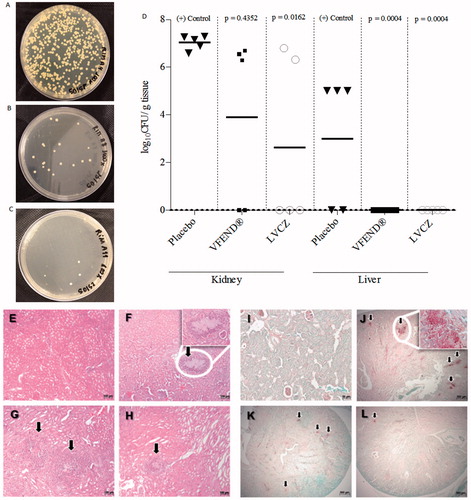
Antifungal activity measured by fungal burden demonstrated that treatment with LVCZ reduced significantly the number of fungal cells in the kidneys. LVCZ and VFEND® formulations showed inhibitory activity against fungal growth () when compared with the positive control (, placebo group). Kidney fungal burden for each group expressed numerically was 2.62 ± 3.60 and 3.91 ± 3.57 log10 CFU/g for LVCZ and VFEND®, respectively, after 3d of treatment. Infected animals that have not undergone any treatment (positive control group) exhibited 7.03 ± 0.29 log10 CFU/g. Livers of the animals from both groups were cleared from fungal cells (), while livers of animals from the control group had a 2.96 ± 2.71 log10 CFU/g.
Figure 4. Fungal growth of C. albicans from kidneys of imunossupressed and infected mice treated with placebo (A), VFEND® (B) and LVCZ (C). (D) Effects of the VCZ treatments on the tissue burden of C. albicans in infected mice and controls (n = 5 per group). Infection was completely resolved in the liver using VFEND® or LVCZ formulation. Representative micrographs of histological analysis of H&E stained kidney sections of (E) control group, healthy Balb/C mice, (F) infected mice, placebo group, (G) infected mice, treated with VFEND® and (H) infected mice, treated with LVCZ. PAS stained kidney sections showing fungal invasion in (I) control group, healthy Balb/C mice, (J) infected mice, placebo group, (K) infected mice, treated with VFEND® and (L) infected mice, treated with LVCZ. Inserts are higher magnification of fungal invasion areas in the kidney tissue, also indicated by black arrows.
All animals presented different degrees of weight loss (Supplementary data 3(B)), strikingly evident for the placebo group.
Histopathological evaluation
shows representative HE (4E–H) and PAS (4I–L) staining photomicrographs of kidney sections of healthy and C. albicans-infected mice following treatment with VFEND® or LVCZ for 3 d. In contrast to the normal kidney structure found in the healthy control group, infected mice from the placebo group showed extensive fungal foci with necrotic tissue damage in the areas around the infection. On the other hand, treated animals, particularly with LVCZ, showed minimal damages, with inflammatory response predominant due to neutrophil infiltration.
Discussion
Understanding the failures in the therapies that use drugs widely employed for the treatment of fungal systemic infections open new perspectives for the development of drug delivery systems. As such, the design of a liposomal formulation potentially able to overcome pitfalls of the current treatments used for C. albicans infections seems to be a viable alternative.
Inherent flaws in therapies that use VCZ (e.g. accelerated VCZ metabolism, low selectivity of the antimicrobial in its complexed form to the site of infection), may leave gaps for microbial resistance (Graybill et al., Citation2003; Voltan et al., Citation2016). For this reason, new formulations with higher specificities or capabilities to overcome these issues are in demand. When developing a new formulation, in particular, new drug delivery systems such as the liposomal VCZ used in this study, the knowledge of the PK s and biodistribution is crucial to predict in vivo antimicrobial activity. Susceptibility data against fungal infections such as C. albicans, one of the most prevalent fungal pathogen in lethal bloodstream infections of immunocompromised patients (Andes, Citation2006; Seneviratne et al., Citation2011; Das et al., Citation2015) are also crucial information.
In vitro antifungal effect shows good antifungal efficacy, both in commercial and liposomal formulation. Meanwhile, LVCZ showed superior performance than VFEND, suggesting that the liposomal formulation allowed superiority in inhibition of fungal growth even at lower VCZ concentrations. Which may be attributed to the effect of liposomes to across fungal cell wall, reach and enter in the cell membrane of the microorganism and release high quantities of VCZ also from the inside out (Shimizu et al., Citation2010; Ling et al., Citation2016; Soo Hoo Citation2017).
PK and biodistribution results indicated a higher renal and hepatic drug accumulation, supporting additional mechanisms of the LVCZ to enhance antifungal activity (Peer et al., Citation2007; Das et al., Citation2015). Tissue inflammation caused by C. albicans colonization, results in a remarkable change in blood vessels permeability forming fenestrations and allowing liposomes to accumulate by passive targeting (Maeda, Citation1996; Bazak et al., Citation2014; Sercombe et al., Citation2015). This accumulation, indicated by a higher drug concentration, was shown in liver and kidneys (important target organs of the colonization of this fungal species) (Calderone & Fonzi, Citation2001; Lionakis et al., Citation2011; Hohl, Citation2014). The release of the antifungal at these sites of infection, might contribute to an enhanced VCZ action, also reducing its toxicity to non-target tissues (Groll et al., Citation2000).
All PK/PD parameters of the antimicrobial were modulated by liposomal entrapment, leading to a more effective formulation. The use of liposomes as VCZ carriers enhanced all relevant parameters of the treatment against C. albicans infection (Zhang et al., 2010; Huh & Kwon, Citation2011; Merian et al., Citation2015).
Reduced conversion of VCZ into VNO during the metabolism phase indicates that liposomal entrapment protects the drug from metabolism in the initial stages of circulation and distribution. Decelerating the metabolism of VCZ thus slowing the formation of the inactive metabolite (VNO), might be a mechanism of improving the existing drug therapy (Peer et al., Citation2007; Li et al., Citation2017).
AUC0–24 h obtained of VCZ unbound plasma protein (Roffey et al., Citation2003) (33% of the AUC0–24 h originally obtained in the PK assay using healthy animals) remained for approximately 12 h above the MIC (i.e. a T > MIC =12 h), following the administration of 10 mg/kg of the LVCZ, ensuring the maximization of antifungal exposure and also avoiding toxicity by exposure to unnecessary high concentrations of the drug (Goodwin & Drew, 2008; Decosterd et al., Citation2010).
Moreover, in studies where VCZ was used in the treatment of C. albicans, it was well known that the fungistatic activity of the triazole is not related to its concentration, conversely, VCZ activity is time-dependent (Klepser, et al., Citation2000; Theuretzbacher, et al., Citation2006). Previous literature reports that best antifungal activity for VCZ is reached when AUC0–24/MIC ratio >32 (Andes, Citation2006). In this work, using the liposomal formulation AUC0–24/MIC was 53.51 ± 11.12, whereas VFEND® resulted in a 2.5-fold lower AUC0–24/MIC ratio (21.51 ± 2.88). In clinical trials using azole antimicrobials for the treatment of invasive candidiasis, when this AUC/MIC ratio is >25, treatment success is at least 91% (Lepak & Andes, Citation2014).
Histopathological findings showed that, in contrast to the normal kidney framework found in healthy animals (), the non-treated C. albicans-infected mice showed extensive fungal foci with necrotic tissue damage in areas around the infection (). On the other hand, VCZ treated mice showed fewer alterations, along with an inflammatory response predominantly with neutrophil infiltration and significant reduction of fungal load ().
Histological findings support antifungal effect of both formulations, although PK/PD data indicate a certain degree of superiority of the liposomal formulation.
Despite the short post-antifungal effect (PAFE) of VCZ (0.2–0.4 h for C. albicans) (Manavathu et al., Citation1998a; Manavathu et al., Citation1998b; Andes, Citation2003; Theuretzbacher et al., Citation2006), liposomal formulation proved to be highly effective in vivo, when treating C. albicans infected mice. This efficacy was probably due to increased renal accumulation and superiority in the PK parameters of the antifungal when encapsulated in liposomes.
Improvements in PK s and a tissue distribution more selective to the organs most affected by the infection might increase the efficacy of LVCZ, when compared directly with VFEND® in its capacity to reduce fungal burden from kidneys of leukopenic mice (Supplementary data 3(A) shows the decay of basal leukocyte levels, over cyclophosphamide dosing period, confirming leukopenia of animals).
Since the efficacy of antifungals cannot be estimated based merely on their blood concentration, it is important to correlate this with tissue distribution, which relates to the VCZ/fungal burden colocalization (“drug and bug”) (Theuretzbacher, Citation2007; Felton et al., Citation2014).
Our biodistribution results indicate that the liposome can penetrate into the tissues most affected by the infection, delivering the drug in deeper renal subcompartments, suffering less impact of the glomerular filtration and minimizing the conversion of the VCZ into its inactive metabolite (VNO). The extravasation of the liposomes to infected tissues was probably increased due the more permeable endothelium found in the infection sites (Maeda et al., Citation2000; Allen & Cullis, Citation2004; Phillips et al., Citation2010), achieving rapid and sustained levels of VCZ locally, contributing to a better resolution of the infection, as shown in liver and kidneys (). Avoiding suboptimal target site concentrations might enhance antifungal effects of VCZ and lead to a better reduction of the fungal burden in these tissues (Liu et al., Citation2002).
A high AUC/MIC correlation obtained in our PK/PD assays, suggests a more pronounced antifungal response when the liposomal formulation was used, highlighting a gain in the efficacy of the LVCZ formulation over VFEND®.
Nevertheless, limitations of murine models using this triazole may hinder further clinical correlation. Accelerated clearance and even genetic polymorphism in humans require thorough investigations involving different animal species or even patients of different age, gender, stage of disease, or ethnicity. Previous literature reports suggest the oral use of grapefruit concomitant with VCZ or the use of guinea pigs lineages instead of mice in order to reduce enzymatic activity that can lead to irrelevant serum VCZ concentrations due to their extremely rapid conversion to the inactive metabolite, VNO (Sugar & Liu, Citation2000; Eiden et al., Citation2010).
Conclusions
Liposomal formulation entrapping VCZ is an alternative formulation for intravenous antifungal treatment, avoiding the use of potentially toxic adjuvants and showing significant improvements in antifungal activity to target systemic C. albicans infection in a murine model, compared to current therapy.
The ability of VCZ to exert fungicidal activity might be enhanced by its entrapment within liposomes. Liposomal encapsulation showed a certain degree of protection from premature metabolism, contributing to more effective antimicrobial blood concentrations, allowing for deeper and more selective penetration and accumulation into the tissues more severely affected by the pathogen.
We have successfully obtained an effective, biocompatible, biodegradable, targeted and safe antifungal liposomal formulation for VCZ. Liposomal VCZ can be used for intravenous delivery of the drug, resulting in improved PK s parameters with positive impact in the antifungal activity of VCZ.
Our contribution encourages the development of an alternative formulation to the commercially available VCZ for intravenous delivery. PK data also reinforce the evidence that, by encapsulating VCZ in liposomes, it is possible to slow down the metabolic depletion of the antifungal. Additionally, a more selective tissue accumulation may be an effective strategy to deliver the antifungal into deeper regions of organs severely affected by the fungal infection.
Supplementary data 3
Download MS Word (83.2 KB)Supplementary data 2
Download MS Word (13.9 KB)Supplementary data 1
Download MS Word (422.1 KB)Acknowledgments
The authors thank the Instituto de Ciências Farmacêuticas (ICF) for analytical support, Dr. Rodrigo Saar Gomes for technical assistance and HalexIstar Ind. Farmacêutica.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Allen TM, Cullis PR. (2004). Drug delivery systems: entering the mainstream. Science 303:1818–22.
- Andes D. (2003). In vivo pharmacodynamics of antifungal drugs in treatment of candidiasis. Antimicrob Agents Chemother 47:1179–86.
- Andes D. (2006). Pharmacokinetics and pharmacodynamics of antifungals. Infect Dis Clin North Am 20:679–97.
- Bazak R, Houri M, Achy SE, et al. (2014). Passive targeting of nanoparticles to cancer: a comprehensive review of the literature. Mol Clin Oncol 2:904–8.
- Bellmann R, Smuszkiewicz P. (2017). Pharmacokinetics of antifungal drugs: practical implications for optimized treatment of patients. Infection 45:737–79.
- Calderone RA, Fonzi WA. (2001). Virulence factors of Candida albicans. Trends Microbiol 9:327–35.
- Campbell RB, Balasubramanian SV, Straubinger RM. (2001). Influence of cationic lipids on the stability and membrane properties of paclitaxel-containing liposomes. J Pharm Sci 90:1091–105.
- Courjol F, Mille C, Hall RA, et al. (2016). Initiation of phospholipomannan beta-1,2 mannosylation involves Bmts with redundant activity, influences its cell wall location and regulates beta-glucans homeostasis but is dispensable for Candida albicans systemic infection. Biochimie 120:96–104.
- Das PJ, Paul P, Mukherjee B, et al. (2015). Pulmonary delivery of voriconazole loaded nanoparticles providing a prolonged drug level in lungs: a promise for treating fungal infection. Mol Pharm 12:2651–64.
- de Sa FA, Taveira SF, Gelfuso GM, et al. (2015). Liposomal voriconazole (VOR) formulation for improved ocular delivery. Colloids Surf B Biointerfaces 133:331–8.
- Decosterd LA, Rochat B, Pesse B, et al. (2010). Multiplex ultra-performance liquid chromatography-tandem mass spectrometry method for simultaneous quantification in human plasma of fluconazole, itraconazole, hydroxyitraconazole, posaconazole, voriconazole, voriconazole-N-oxide, anidulafungin, and caspofungin. Antimicrob Agents Chemother 54:5303–15.
- Diehl KH, Hull R, Morton D, et al. (2001). A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxicol 21:15–23.
- Dupont PF. (1995). Candida albicans, the opportunist. A cellular and molecular perspective. J Am Podiatr Med Assoc 85:104–15.
- Eiden C, Cociglio M, Hillaire-Buys D, et al. (2010). Pharmacokinetic variability of voriconazole and N-oxide voriconazole measured as therapeutic drug monitoring. Xenobiotica 40:701–6.
- Felton T, Troke PF, Hope WW. (2014). Tissue penetration of antifungal agents. Clin Microbiol Rev 27:68–88.
- Goodwin ML, Drew RH. (2007). Antifungal serum concentration monitoring: an update. J Antimicrob Chemother 61:17–25.
- Graybill JR, Najvar LK, Gonzalez GM, et al. (2003). Improving the mouse model for studying the efficacy of voriconazole. J Antimicrob Chemother 51:1373–6.
- Groll AH, Giri N, Petraitis V, et al. (2000). Comparative efficacy and distribution of lipid formulations of amphotericin B in experimental Candida albicans infection of the central nervous system. J Infect Dis 182:274–82.
- Gulati M, Grover M, Singh S, Singh M. (1998). Lipophilic drug derivatives in liposomes. Int J Pharm 165:129–68.
- Hohl TM. (2014). Overview of vertebrate animal models of fungal infection. J Immunol Methods 410:100–12.
- Hubrecht RC, Kirkwood J. (2010). The UFAW handbook on the care and management of laboratory and other research animals. 8th ed. Hoboken (NJ): Wiley-Blackwell.
- Huh AJ, Kwon YJ. (2011). “Nanoantibiotics”: a new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J Control Release 156:128–45.
- John HR. (2008). Reference method for broth dilution antifungal susceptibility testing of yeasts: approved standard. 3rd ed. Wayne (PA): Clinical and Laboratory Standards Institute.
- Kiser TH, Fish DN, Aquilante CL, et al. (2015). Evaluation of sulfobutylether-beta-cyclodextrin (SBECD) accumulation and voriconazole pharmacokinetics in critically ill patients undergoing continuous renal replacement therapy. Crit Care 19:32.
- Klepser ME, Malone D, Lewis RE, et al. (2000). Evaluation of voriconazole pharmacodynamics using time-kill methodology. Antimicrob Agents Chemother 44:1917–20.
- Ledoux MP, Toussaint E, Denis J, Herbrecht R. (2017). New pharmacological opportunities for the treatment of invasive mould diseases. J Antimicrob Chemother 72:i48–58.
- Lepak AJ, Andes DR. (2014). Antifungal pharmacokinetics and pharmacodynamics. Cold Spring Harb Perspect Med 5:a019653.
- Li M, Zou P, Tyner K, Lee S. (2017). Physiologically based pharmacokinetic (PBPK) modeling of pharmaceutical nanoparticles. AAPS J 19:26–42.
- Ling X, Huang Z, Wang J, et al. (2016). Development of an itraconazole encapsulated polymeric nanoparticle platform for effective antifungal therapy. [10.1039/C5TB02453F]. J Mater Chem B 4:1787–96.
- Lionakis MS, Lim JK, Lee CC, Murphy PM. (2011). Organ-specific innate immune responses in a mouse model of invasive candidiasis. J Innate Immun 3:180–99.
- Liu P, Muller M, Derendorf H. (2002). Rational dosing of antibiotics: the use of plasma concentrations versus tissue concentrations. Int J Antimicrob Agents 19:285–90.
- Lopes JP, Stylianou M, Nilsson G, Urban CF. (2015). Opportunistic pathogen Candida albicans elicits a temporal response in primary human mast cells. Sci Rep 5:12287.
- Luke DR, Tomaszewski K, Damle B, Schlamm HT. (2010). Review of the basic and clinical pharmacology of sulfobutylether-beta-cyclodextrin (SBECD). J Pharm Sci 99:3291–301.
- MacCallum DM. (2013). Mouse model of invasive fungal infection. Methods Mol Biol 1031:145–53.
- Maeda H. (1996). Role of microbial proteases in pathogenesis. Microbiol Immunol 40:685–99.
- Maeda H, Wu J, Sawa T, et al. (2000). Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release 65:271–84.
- Maertens JA, Raad II, Marr KA, et al. (2016). Isavuconazole versus voriconazole for primary treatment of invasive mould disease caused by Aspergillus and other filamentous fungi (SECURE): a phase 3, randomised-controlled, non-inferiority trial. Lancet 387:760–9.
- Manavathu EK, Cutright JL, Chandrasekar PH. (1998a). Organism-dependent fungicidal activities of azoles. Antimicrob Agents Chemother 42:3018–21.
- Manavathu EK, Dimmock JR, Vashishtha SC, et al. (1998b). In-vitro and in-vivo susceptibility of Aspergillus fumigatus to a novel conjugated styryl ketone. J Antimicrob Chemother 42:585–90.
- Martinez R. (2006). An update on the use of antifungal agents. J Bras Pneumol 32:449–60.
- Merian J, De Souza R, Dou Y, et al. (2015). Development of a liposome formulation for improved biodistribution and tumor accumulation of pentamidine for oncology applications. Int J Pharm 488:154–64.
- Pattni BS, Chupin VV, Torchilin VP. (2015). New developments in liposomal drug delivery. Chem Rev 115:10938–66.
- Peer D, Karp JM, Hong S, et al. (2007). Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol 2:751–60.
- Phillips MA, Gran ML, Peppas NA. (2010). Targeted nanodelivery of drugs and diagnostics. Nano Today 5:143–59.
- Pinto-Alphandary H, Andremont A, Couvreur P. (2000). Targeted delivery of antibiotics using liposomes and nanoparticles: research and applications. Int J Antimicrob Agents 13:155–68.
- Rauzi F, Smyth E, Emerson M. (2017). Refinement of mouse protocols for the study of platelet thromboembolic responses in vivo. Thromb Haemost 117:2283–90.
- Roemer T, Krysan DJ. (2014). Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harb Perspect Med 4:a019703.
- Roffey SJ, Cole S, Comby P, et al. (2003). The disposition of voriconazole in mouse, rat, rabbit, guinea pig, dog, and human. Drug Metab Dispos 31:731–41.
- Rossolini GM, Arena F, Pecile P, Pollini S. (2014). Update on the antibiotic resistance crisis. Curr Opin Pharmacol 18:56–60.
- Seneviratne CJ, Wong SS, Yuen KY, et al. (2011). Antifungal susceptibility and virulence attributes of bloodstream isolates of Candida from Hong Kong and Finland. Mycopathologia 172:389–95.
- Sercombe L, Veerati T, Moheimani F, et al. (2015). Advances and challenges of liposome assisted drug delivery. Front Pharmacol 6:286.
- Shimizu K, Osada M, Takemoto K, et al. (2010). Temperature-dependent transfer of amphotericin B from liposomal membrane of AmBisome to fungal cell membrane. J Control Release 141:208–15.
- Soo Hoo L. (2017). Fungal fatal attraction: a mechanistic review on targeting liposomal amphotericin B (AmBisome(R)) to the fungal membrane. J Liposome Res 27:180–5.
- Sugar A, M, Liu, X, P. (2000). Effect of grapefruit juice on serum voriconazole concentrations in the mouse. Med Mycol 38:209–12.
- Theuretzbacher U. (2007). Tissue penetration of antibacterial agents: how should this be incorporated into pharmacodynamic analyses? Curr Opin Pharmacol 7:498–504.
- Theuretzbacher U, Ihle F, Derendorf H. (2006). Pharmacokinetic/pharmacodynamic profile of voriconazole. Clin Pharmacokinet 45:649–63.
- Voltan AR, Quindos G, Alarcon KP, et al. (2016). Fungal diseases: could nanostructured drug delivery systems be a novel paradigm for therapy? Int J Nanomedicine 11:3715–30.
- Yamada T, Mino Y, Yagi T, et al. (2015). Saturated metabolism of voriconazole N-Oxidation resulting in nonlinearity of pharmacokinetics of voriconazole at clinical doses. Biol Pharm Bull 38:1496–503.
- Zhang L, Pornpattananangku D, Hu CM, Huang CM. (2010a). Development of nanoparticles for antimicrobial drug delivery. Curr Med Chem 17:585–94.
- Zhang Y, Huo M, Zhou J, Xie S. (2010b). PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed 99:306–14.