
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Galactose-modified selenium nanoparticles (GA-SeNPs) loading with doxorubicin (DOX) for hepatocellular carcinoma (HCC) therapy was investigated in this paper. Selenium nanoparticles (SeNPs) were modified with galactose as tumor targeting moiety to fabricate tumor-targeted delivery carrier GA-SeNPs, then doxorubicin was loaded onto the surface of GA-SeNPs for improving antitumor efficacy of DOX in HCC therapy. Chemical structure characterization of GA-Se@DOX showed that DOX was successfully loaded to the surface of GA-SeNPs to prepare functionalized antitumor drug delivery system GA-Se@DOX. GA-Se@DOX exhibited effective cellular uptake in HepG2 cells and entered HepG2 cells mainly by clathrin-mediated endocytosis pathway. GA-Se@DOX showed significant activity to induce the apoptosis of HepG2 cells in vitro. The western blotting result indicated that GA-Se@DOX induced HepG2 cells apoptosis via activating caspase signaling and Bcl-2 family proteins. Moreover, active targeting delivery system GA-Se@DOX exhibited excellent antitumor efficacy in vivo in comparison with passive targeting delivery system Se@DOX. Histology analysis showed that GA-Se@DOX exhibited no obvious damage to major organs including heart, liver, spleen, lung, and kidney under the experimental condition. Taken together, GA-Se@DOX may be one novel promising nanoscale drug candidate for HCC therapy.
Introduction
Hepatocellular carcinoma (HCC) is one of the most common lethal diseases worldwide (Hu et al., Citation2015; Jha et al., Citation2017). The present therapy option mainly includes liver transplantation, liver resection, and chemotherapy (He et al., Citation2017; Xu et al., Citation2017; Han et al., Citation2018). However, the therapeutic effect is not entirely satisfactory. Thus, more effective strategies are needed (Yang et al., Citation2017; Chowdhury et al., Citation2018; Liang et al., Citation2018; Wang et al., Citation2018b). Doxorubicin (DOX) is one very common and effective chemotherapeutic drugs for cancer therapy (Bi et al., Citation2018; Licciardello et al., Citation2018). Nevertheless, clinical applications of DOX have been limited by its poor water solubility and off-target side effects (Wu et al., Citation2018; Zhang et al., Citation2018). Nanomedicine exhibits many merits to overcome pharmaceutical challenge of traditional hydrophobic antitumor drug, for example nonspecific biodistribution, off-target toxicity and poor water solubility (Chinen et al., Citation2015; Yu et al., Citation2015; Cao et al., Citation2016; Xia et al., Citation2017; Kamegawa et al., Citation2018; Li et al., Citation2018a). Thus, a number of strategies using nanoparticles have been applied in the field of cancer therapy, and most of these therapies are based on the enhanced permeation and retention (EPR) effect (Ai et al., Citation2018; Bentz & Savin, Citation2018; Wang et al., Citation2018a). Nevertheless, uncontrolled release of drug and drug delivery to the unintended site may compromise treatment effect along with increased risk of toxicity and side effect (Le et al., Citation2015; Sun et al., Citation2018b). These side effects can be minimized by using controlled targeted drug delivery, such as nanocarriers (Li et al., Citation2017a; Kim et al., Citation2018).
Selenium nanoparticles (SeNPs) as drug carriers have received a large number of attention. For one thing, selenium (Se) as a trace element is very important to human biological process and involves many physiological functions (Zheng et al., Citation2016). For another, Se plays a key role in cancer prevention and immune response (Zhou et al., Citation2016). Maiyo reported that cancer prevention afforded by selenium is through protection of DNA damage by dimethylbenz(a)anthracene-induced adduct formation (Maiyo & Singh, 2017). Tan’s research showed that selenium deficiency has been associated with cardiovascular diseases and immune dysfunction (Tan et al., Citation2016). Moreover, SeNPs showed some other advantages, such as controlled size, potent drug loading capacity, improved antitumor effect and low cytotoxicity (Sun et al., Citation2014). SeNPs are considered superior to metal nanoparticles, such as gold, silver, and platinum nanoparticles, due to their superior biocompatibility and degradability in vivo (Maiyo & Singh, 2017). Various targeting ligands, chemotherapeutics, and gene can be attached to SeNPs to prepare functionalized nanoparticles which increase specificity for target cancer cells without causing substantial untoward effects on normal tissues (Liu et al., Citation2012; Xia et al., Citation2018). Thus, SeNPs gradually developed into one excellent anticancer drug carrier (Chen et al., Citation2015). However, some deficiency, especially the lack of active tumor-targeted capacity still existed in such delivery carrier (Du et al., Citation2018). To obtain high targeting ability, a lot of tumor-targeted molecules were used for decorating nanoparticles (Wang et al., Citation2016; Mohamed et al., Citation2017). Galactose (GA) has been investigated for its capacity to target HCC cells with expression of asialoglycoprotein receptor (ASGR) (Zhao et al., Citation2017; Jain et al., Citation2018; Sun et al., 2018a). ASGR is capable of recognizing galactose-terminated glycoproteins and glycoconjugates, therefore, many materials modified with galactose was used as a drug carrier for HCC target therapy (Mou et al., Citation2016; Zhang et al., Citation2016; Zheng et al., Citation2018).
In this paper, galactose was installed on the surfaces of SeNPs to prepare tumor targeting carrier GA-SeNPs. Then DOX was loaded onto GA-SeNPs to prepare functionalized anti-tumor nanoparticles GA-Se@DOX (). GA-Se@DOX could inhibit the proliferation of HepG2 cells and induce apoptosis of HepG2 cells in vitro. It is worth noting that GA-Se@DOX obtained excellent antitumor efficacy in vivo in comparison with Se@DOX or DOX, indicating that GA-Se@DOX holds great potential for HCC treatment.
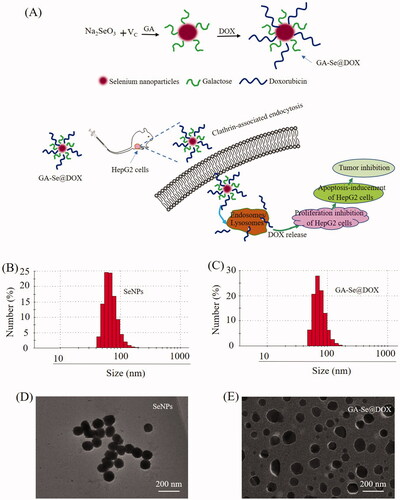
Figure 1. (A) Schematic illustration of the formation of GA-Se@DOX nanoparticles. (B) Particle size distribution of SeNPs nanoparticles. (C) Particle size distribution of GA-Se@DOX nanoparticles. (D) TEM image of SeNPs nanoparticles. (E) TEM image of GA-Se@DOX nanoparticles.
Materials and methods
Materials
Galactose, ascorbic acid (Vc), sodium selenite (Na2SeO3), doxorubicin hydrochloride (DOX⋅HCl), DAPI and MTT were obtained from Sigma-Aldrich Chemicals (Scotland, UK). Fetal bovine serum (FBS) and Dulbecco's modified eagle's medium (DMEM) medium were obtained from Gibco BRL/Life Technologies (Paisley, UK). Antibodies were obtained from Cell Signaling Technology (MA, USA).
Preparation and characterization of GA-Se@DOX nanoparticles
Selenium nanoparticles (SeNPs) were prepared as previously reported with partial modification (Li et al., Citation2017b). Briefly, 0.25 mL Na2SeO3 (0.2 M) solution and 2 mL vitamin C (Vc, 1 mM) solution were slowly added into 22.75 mL Milli-Q water in a 50 mL beaker. Solution mixtures were magnetically stirred for 30 min at room temperature to manufacture SeNPs. Then 1 mL 2 mg/mL galactose was dropped to SeNPs solution and mixed solutions were magnetically stirred for 4 h to fabricate GA-modified selenium nanoparticles (GA-SeNPs). After that, 2 mg DOX·HCl dissolved in 5 μL DMSO was added into GA-SeNPs solutions and mixed solutions were magnetically stirred for another 8 h. At last, high-purity GA-Se@DOX was obtained via dialyzing reaction solutions for 4 h. Chemical structure of GA-Se@DOX was characterized by dynamic light scattering (DLS) analysis, transmission electronic microscopy (TEM), energy dispersive X-ray (EDX) and Fourier transform infrared (FTIR). The size of nanoparticles in the water solution was continually observed during 16 days.
Cell culture
HepG2 cells were obtained from American Tissue Culture Collection (ATCC) and cultivated in DMEM containing 10% FBS at 37°C with 5% CO2.
Cellular uptake study
0.5 mL HepG2 cells suspension at a density of 4 × 104 cells/mL was assigned to 12-well plate and cultivated for 6 h at 37°C. Afterward, HepG2 cells were co-cultured with DOX, Se@DOX, and GA-Se@DOX at equivalent DOX dose of 4 μg/mL. The cells were washed and measured using flow cytometer. In order to research the cellular uptake mechanism, HepG2 cells were cultivated under the condition of different uptake inhibitor. Then HepG2 cells were treated with GA-Se@DOX for 4 h in the absence of inhibitor at 4°C, or with 50 mM 2-deoxy-d-glucose (DOG)+3 mg/mL NaN3 or various cellular uptake inhibitor chlorpromazine (2 μg/mL), amiloride (5 μg/mL), nystatin (4 μg/mL) at 37°C, respectively. Then HepG2 cells were tested using flow cytometry (BD Bioscience, CA, USA). To further verify uptake mechanism of GA-Se@DOX, HepG2 cells were pretreated with chlorpromazine and then co-cultured with GA-Se@DOX at equivalent DOX dose of 4 μg/mL for a different time. HepG2 cells were washed with PBS and stained with DAPI for 15 min. Then HepG2 cells were washed and observed by fluorescence microscope.
Colocalization study of GA-Se@DOX nanoparticles
The cellular colocalization of GA-Se@DOX in HepG2 cells was pretreated with lysosomal marker LysoTracker Green. Briefly, HepG2 cells were incubated in 12-well plate overnight until 65% confluence was reached. Then the cells were incubated with 50 nM LysoTracker Green for 30 min and subsequently stained with DAPI for 15 min. After washing with PBS for twice, the cells were incubated with 10 µg/mL GA-Se@DOX for various time. The cells were then washed twice using PBS and examined with a fluorescence microscope (Leica DMi8).
In vitro release of DOX from nanoparticles
For in vitro release detection, 5 mg GA-Se@DOX nanoparticles were dissolved in 5 mL of PBS solution and placed into a pre-swelled dialysis bag with 3.5 kDa molecular weight cutoff. Then the sealed dialysis bag was immersed into 40 mL of PBS (pH 7.4 or 5.4) with gentle agitation at 37 °C. 1 mL sample was withdrawn at different time intervals and then replaced with equal volume of PBS. The concentration of DOX was tested by measuring the fluorescence intensity of DOX at excitation/emission wavelengths of 535/590 nm by UV-vis spectroscopy.
MTT assay
4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide, thiazolyl blue tetrazolium bromide (MTT) assay was carried out to test the cellular cytotoxicity of nanoparticles (Li et al., Citation2018b). 200 µL HepG2 cells (5 × 104 cells/mL) were added to 96-well plate and cultured for 24 h. Then HepG2 cells were incubated with DOX or Se@DOX or GA-Se@DOX (equivalent DOX concentration of 0.25, 0.5, 1, 2, 4, 8 µg/mL) or GA-SeNPs for 48 h at 37°C. Then the previous medium was taken away and 100 µL medium containing 40 µL of MTT (0.5 mg/mL) was gently put in each well, followed by incubating for another 4 h. Then the medium was taken away and 200 µL of dimethyl sulfoxide was put in each well. Culture plate was incubated at 37°C for another 0.5 h. Absorbance at 570 nm was taken using a 96-well microplate reader.
Flow cytometry assay
Flow cytometry was used to test the apoptosis of HepG2 cells. Briefly, the cells were exposed to GA-Se@DOX, Se@DOX and free DOX at equivalent DOX dose of 4 µg/mL for 24 h, respectively. The cells were washed with PBS and collected cells were stained with PI or Annexin V-FITC/PI for 30 min. Finally, stained cells were examined by flow cytometry and data were analyzed by FlowJo software (Treestar, Ashland, OR, USA).
Western blot analysis
The protein expressions were tested by western blot assay. HepG2 cells were incubated in 6-well plate to reach about 70% confluence and then exposed to GA-Se@DOX at various equivalent concentration of DOX for 24 h. After that, the cells were harvested and prepared for further tests according to previous literature (Li et al., Citation2017b).
Xenograft mouse model
All animal experiments were carried out according to the guideline of Experimental Animal Center of Guangzhou Medical University and approved by the Ethics Committee of Guangzhou Medical University. BALB/c nude mice (about 7 weeks old) were applied to study in vivo antitumor efficacy of GA-Se@DOX. 1 × 107 HepG2 cells suspended in 150 µL saline and injected in abdomens of mice subcutaneously. The mice were randomly categorized into four groups after the volume of tumor reached ∼100 mm3. Subsequently, saline (control group), DOX, Se@DOX, and GA-Se@DOX (at equivalent DOX dose of 2 mg/kg) were intravenously injected to tumor-bearing mice once every other day, respectively. Tumor volume was reckoned up using formula as follows: Tumor volumes (mm3) = ×length × width2.
Histology and immunohistochemistry
Tumors and organs including heart, liver, spleen, lung, and kidney were fixed with 3.7% buffered paraformaldehyde for over 24 h, then paraffin embedded and sectioned into slices at 6 µm thickness. Histological sections of main organs (heart, liver, spleen, lung, and kidney) were used for hematoxylin-eosin (H&E) staining. The terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was carried out according to manufacturer’s instruction. The expression of Ki67, phosphorylated p53 (pp53) and caspase in tumors were measured via immunohistochemical method according to manufacturer’s instruction.
Statistical analysis
All the data represented mean ± standard deviations (S.D.). The statistical differences between two groups were analyzed via Student's t-test. The differences were judged to be significant and highly significant at *p < .05 and **p < .01, respectively.
Results and discussion
Preparation and characterizations of GA-Se@DOX
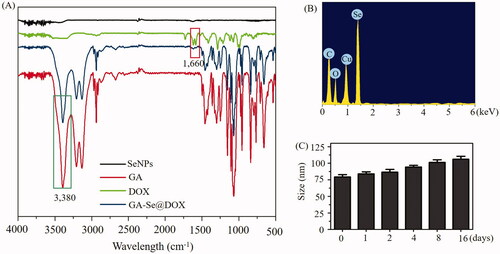
In this paper, one novel tumor-targeting delivery system GA-Se@DOX was synthesized. Tumor-targeting molecular galactose (GA) was linked to selenium nanoparticles (SeNPs) to fabricate tumor-targeting delivery carrier GA-SeNPs, then antitumor drug doxorubicin (DOX) was loaded to the surface of GA-SeNPs to prepare tumor-targeting delivery system GA-Se@DOX. As shown in , the average sizes of SeNPs and GA-Se@DOX were 91.3 nm and 95 nm, respectively. The morphology of nanoparticles was shown in TEM image (), unmodified SeNPs were uniform spherical particles with a size range of about 90 nm and they were prone to gathering together. However, SeNPs loaded with GA and DOX presented monodisperse spherical particles with a size range of 50–150 nm. FTIR spectrums of GA-Se@DOX, DOX, SeNPs, and GA are shown in , typical peak of SeNPs existed in the spectrum of GA-Se@DOX. GA showed telescopic vibration peak of O–H bond at 3380 cm−1. After loading GA onto SeNPs, typical characteristic peaks at ∼3380 cm−1 was also observed, indicating that GA was successfully loaded onto SeNPs. The peak at ∼1660 cm−1 from characteristic aldehyde group C=O bond of DOX existed in the spectrum of GA-Se@DOX, verifying effective linking between DOX and GA-SeNPs. The driving force of GA/DOX loading is that the lone pair of nitrogen and oxygen atoms tend to form covalent bonds with the outermost unoccupied orbit of selenium atom and the electrostatic interaction partially contributed to the loading of DOX onto the surface of GA-SeNPs. Obvious signals of carbon atom, oxygen atom, and selenium atom were observed in EDX analysis, indicating that GA and DOX were linked to the surface of SeNPs (). The Cu atom signal was due to the copper-mesh matrix that was used to suspend the particles before loading on to the machine. As shown in , the size distribution of GA-Se@DOX showed that GA-Se@DOX could keep stable with small sizes (<120 nm) for 16 days. This data indicated that GA-Se@DOX exhibited good stability in water solution. The zeta potentials of SeNPs, GA-SeNPs, and GA-Se@DOX were shown in Supplementary Figure S1. After loading with GA, the zeta potentials of SeNPs were slightly changed from −22.4 mV to −23.3, then increased to −15.6 mV after loading with DOX.
Figure 2. (A) FTIR spectra of selenium nanoparticles (SeNPs), galactose (GA), doxorubicin (DOX) and GA-Se@DOX. (B) EDX analysis of GA-Se@DOX. (C) Stability observation of GA-Se@DOX nanoparticles in aqueous solution.
GA-SeNPs enhances the cellular uptake of DOX
Drug delivery efficiency is closely related to cellular uptake (Kamegawa et al., Citation2018). High cellular uptake of the drug can result in effective treatment efficacy. To test cellular uptake efficiency of various DOX formulations, HepG2 cells were incubated with free DOX, Se@DOX, and GA-Se@DOX for 6 h, respectively, and then were analyzed by flow cytometer. As shown in , fluorescence signal of cells treated with PBS was set as a control group, and fluorescence signaling intensity of the cells exposed to GA-Se@DOX was more obvious in comparison with Se@DOX and DOX groups, suggesting that GA-Se@DOX exhibited higher uptake efficacy in comparison with Se@DOX and free DOX. This result indicated delivery of DOX by GA-SeNPs can enhance cellular uptake of DOX in HepG2 cells.
Figure 3. (A) Cellular uptake of DOX, Se@DOX, and GA-Se@DOX in HepG2 cells was tested by flow cytometry. (B) Effect of endocytosis inhibitors and temperature on the internalization of GA-Se@DOX. *p < .05, **p < .01 vs. control group. (C) Cellular uptake of GA-Se@DOX in HepG2 cells was observed by fluorescence microscope. Scale bar is 50 μm. (D) Cellular uptake of GA-Se@DOX pretreated with chlorpromazine in HepG2 cells was observed by fluorescence microscope. Scale bar is 50 μm.
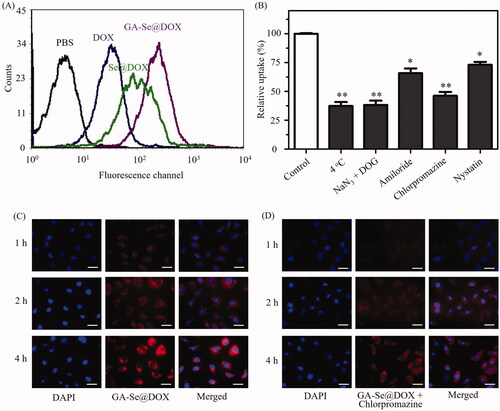
It has been reported that nanoparticles can enter cancer cell via energy-dependent endocytosis way (Kuhn et al., Citation2014). The incubation of HepG2 cells at 4 °C or pretreated with NaN3/DOG markedly reduced cellular uptake of nanoparticles (), indicating that endocytosis of GA-Se@DOX nanoparticles is an active energy-dependent process. The cells endocytosis mainly includes three pathways including, micropinocytosis, caveolae-mediated endocytosis, and clathrin-mediated endocytosis. To examine endocytosis mechanism of GA-Se@DOX in HepG2 cells, various endocytosis inhibitor was used to investigate the effect of GA-Se@DOX on cellular uptake. Nystatin, chlorpromazine, and amiloride are usually used to inhibit caveolae-mediated endocytosis, clathrin-associated endocytosis, and macropinocytosis, respectively. After pretreating with nystatin or amiloride, cellular uptake of GA-Se@DOX was obviously decreased by 27.2% and 34.5%, respectively. Nevertheless, chlorpromazine-pretreatment led to 53.8% decrease in cellular uptake of GA-Se@DOX, suggesting that clathrin-associated endocytosis played an important role in the internalization of GA-Se@DOX in HepG2 cells.
Fluorescence microscopy was used to further verify whether GA-Se@DOX entered HepG2 cells via clathrin-associated endocytosis pathway. As shown in , obvious red fluorescence of HepG2 cells from GA-Se@DOX was gradually increased with increasing incubation time, suggesting that GA-Se@DOX entered HepG2 cells in a time-dependent way. However, after pretreatment with chlorpromazine (clathrin-associated endocytosis inhibitor), the fluorescence intensity of HepG2 cells incubated with GA-Se@DOX in the same condition decreased dramatically (). This result verified that GA-Se@DOX entered HepG2 cells mainly through clathrin-associated endocytosis way.
Endosomal/lysosomal escape of GA-Se@DOX nanoparticles
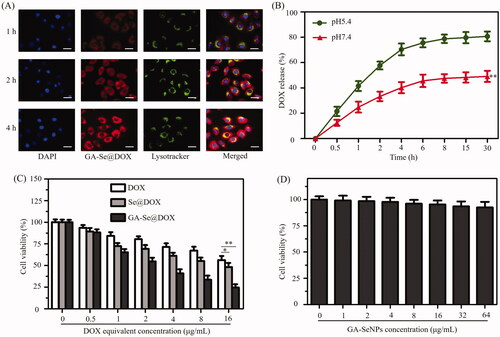
In order to exert antitumor activity, nanomedicine needs to cross cell membrane to internalize into the cells through endocytosis and efficaciously escape from endosomes/lysosomes into the cytoplasm (Yang et al., Citation2018). Herein, colocalization of GA-Se@DOX and endosomes/lysosomes was studied via fluorescence microscope. As shown in , GA-Se@DOX gradually accumulated in endosomes/lysosomes area during 1 h of incubation. After incubation for 2 h, a fraction of GA-Se@DOX diffused into the cytoplasm. At 4 h, red fluorescence of GA-Se@DOX could be observed in entire cells. These results indicated that GA-Se@DOX might escape from endosomes/lysosomes to cytoplasm after internalization.
Figure 4. (A) The observation of the escape of GA-Se@DOX from endosomes/lysosomes after 1, 2, and 4 h of incubation. Scale bar is 50 μm. (B) In vitro release of DOX from GA-Se@DOX nanoparticles. **p < .01 vs. pH5.4 group. (C) The cytotoxicity of DOX, Se@DOX, and GA-Se@DOX against HepG2 cells. *p < .05, **p < .01 vs free DOX group. (D) The cytotoxicity of GA-SeNPs against HepG2 cells.
In vitro release of DOX
Two types of pH values (pH 5.4 and 7.4) were used to simulate cancer cell microenvironment and normal physiological environment, respectively (Huo et al., Citation2015). DOX release profiles were shown in , there was a noteworthy burst drug release during initial 4 h in both pH values. It was worth noting that GA-Se@DOX presented a faster release of DOX in the acidic environment during initial 30 h, which was up to 80.7%. However, the release rate was just 49.1% in normal physiological environment (pH 7.4). The faster release in acidic environment may be due to the decrease of the surface negative charge of GA-SeNPs in acidic pH, which weakens the electrostatic attraction of DOX and facilitates the release of DOX from GA-SeNPs. Such an acid-dependent drug release feature of GA-Se@DOX is very beneficial for drug delivery system in cancer therapy.
GA-Se@DOX inhibits the proliferation of HepG2 cells
MTT experiment was used to study cytotoxicity of different DOX formulations against HepG2 cells. Free DOX and passive targeting nanoparticle Se@DOX were set as a negative control. showed that the viability of HepG2 cells exposed to various formulations of DOX gradually declined with increasing DOX concentrations. Free DOX, Se@DOX, and GA-Se@DOX at equivalent DOX dose of 8 μg/mL obviously suppressed the proliferation of HepG2 cells, and cell viability rates were 71.4%, 61.3%, and 40.8%, respectively, suggesting that GA-Se@DOX exhibited greater cytotoxicity against HepG2 cells compared with Se@DOX and free DOX, possibly because of enhanced cellular uptake of GA-Se@DOX. The proliferation inhibition of HepG2 cells treated with drug carrier GA-SeNPs at used dose was not obvious (), indicating good biocompatibility of GA-SeNPs in HepG2 cells. MTT results indicated that delivery of DOX using active tumor-targeted carrier GA-SeNPs could effectively enhance anticancer activity of DOX with low cytotoxicity.
GA-Se@DOX induces HepG2 cells apoptosis
DOX is a very effective antitumor drug and can induce cancer cell apoptosis (Tang et al., Citation2018). Herein, flow cytometry was used to test whether GA-Se@DOX showed stronger activity to induce HepG2 cells apoptosis in comparison with free DOX or Se@DOX. In this study, the apoptosis cells with DNA fragmentations were reflected as Sub-G1 peaks. showed that Sub-G1 apoptosis peak of cells in GA-Se@DOX-treatment group (35.3%) was stronger than that of DOX-treatment group (12.08%) and Se@DOX-treatment group (25.25%), indicating that GA-Se@DOX exhibited the stronger capacity to induce HepG2 cells apoptosis.
Figure 5. (A) Representative flow cytometry histograms of cell cycle analysis of HepG2 cells after incubation with various formulations of DOX for 24 h. (B) The apoptosis percentages analysis of HepG2 cells after incubation with various formulations of DOX for 24 h.
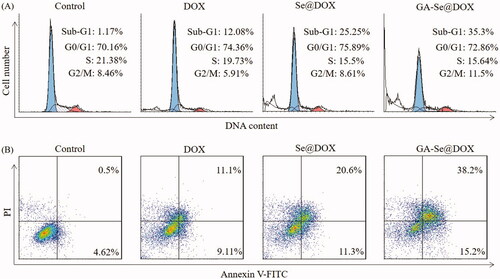
To further analyze apoptosis of HepG2 cells treated with various formulations of DOX, the cells were detected using Annexin V-FITC/PI staining. As shown in , GA-Se@DOX-treatment obviously induced apoptosis of HepG2 cells and resulted in higher cell apoptosis rates (53.4%) in comparison with the cells treated with DOX (20.21%) or Se@DOX (31.9%). These results indicated that GA-Se@DOX could enhance the anticancer activity of DOX to induce HepG2 cells apoptosis by loading DOX onto active tumor-targeting carrier GA-SeNPs.
Analysis of the expression levels of apoptosis-related proteins
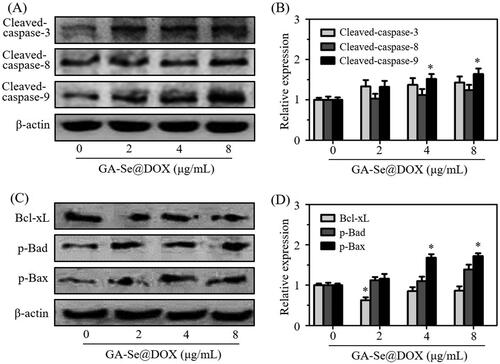
Caspase-3 is a main executioner which regulates the process of cell apoptosis due to its contribution to the cleavage of many proteins (Dreaden et al., Citation2012). Caspase-8 and caspase-9 also take part in the initiation of cell apoptosis (Thapa et al., Citation2015). Herein, expression levels of caspase-related proteins cleaved-caspase-3, cleaved-caspase-8, and cleaved-caspase-9 were analyzed. As shown in , treatment with GA-Se@DOX significantly increased expression levels of cleaved-caspase-3, cleaved-caspase-8, and cleaved-caspase-9 proteins, indicating that GA-Se@DOX might activate caspase-mediated apoptotic pathway to induce HepG2 cells apoptosis. Meanwhile, many anticancer drugs were also reported to induce apoptosis of cancer cells through a mitochondria-mediated death process which was mediated by apoptosis-related Bcl-2 family proteins, including anti-apoptotic protein Bcl-xL, and pro-apoptotic proteins Bad and Bax (Zhang et al., Citation2015). To research whether GA-Se@DOX activated Bcl-2 signaling pathway, expression levels of Bcl-xL, phosphorylated Bad and phosphorylated Bax proteins were tested via western blotting. As shown in , the expression level of antiapoptotic protein Bcl-xL was significantly inhibited by GA-Se@DOX while pro-apoptotic proteins phosphorylated Bad and phosphorylated Bax were up-regulated after 24 h of exposure to GA-Se@DOX at various DOX equivalent concentrations. These results indicated that GA-Se@DOX could induce apoptosis of HepG2 cells via activating caspase and Bcl-2 signaling pathways.
Figure 6. (A) The protein expression of cleaved-caspase-3, cleaved-caspase-8, and cleaved-caspase-9 in HepG2 cells after exposing to various concentrations of GA-Se@DOX. (B) The semi-quantitative analysis result of cleaved-caspase-3, cleaved-caspase-8, and cleaved-caspase-9 was shown by the histogram. *p < .05 vs. control group. (C) The protein expression of Bcl-xL, p-Bad, and p-Bax in HepG2 cells after exposing to various concentrations of GA-Se@DOX. (D) The semi-quantitative analysis result of Bcl-xL, p-Bad, and p-Bax was shown by the histogram. *p < .05 vs. control group.
In vivo anti-tumor efficacy of GA-Se@DOX
HepG2 tumor xenograft was used to assess anti-tumor efficacy of GA-Se@DOX. Mice were assigned to four groups randomly and intravenously injected with GA-Se@DOX, Se@DOX, free DOX, and saline, respectively. Tumor volume and body weight of mice were tested every other day up to 21 days. As shown in , compared to the saline-treated control group, GA-Se@DOX-treatment obviously suppress tumor growth during treatment period. Moreover, GA-Se@DOX was more effective in suppressing tumor growth in comparison with free DOX or Se@DOX at an equivalent dose of DOX, proving excellent anti-tumor efficacy of GA-Se@DOX. As shown in , tumor images and weight of GA-Se@DOX-treated mice further verified the significant antitumor activity of GA-Se@DOX. In addition, the body weight of mice kept slight increasing during treatment period, indicating GA-Se@DOX had no obvious side effects at a tested dose (). In vivo anti-tumor efficacy showed that tumor-targeted delivery system GA-Se@DOX exhibited a great potential for HCC therapy.
Figure 7. (A) Tumor growth curve of the xenograft nude mice bearing HepG2 cells after intravenous administration of saline and various formulations of DOX. (B) Morphology of tumors stripped from mice. (C) Tumor weight analysis of mice after 21 days of treatment. (D) Body weight change of mice during the treatment. *p < .05, **p < .01 vs. saline group. (E) Ki67, pp53, caspase-3, and TUNEL immunohistochemistry analysis of tumors treated with saline, DOX, Se@DOX, and GA-Se@DOX. Scale bar is 50 μm.
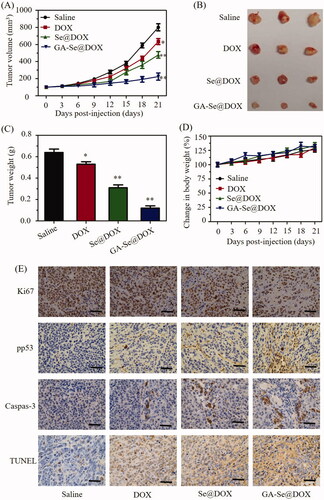
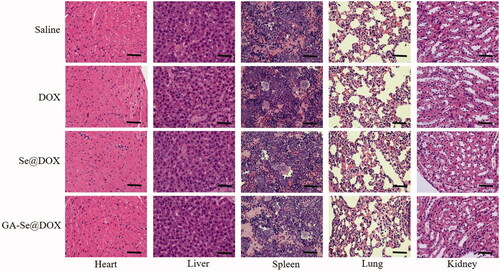
To further study in vivo antitumor mechanism of GA-Se@DOX, cell proliferation and apoptosis in tumors were analyzed by Ki67, caspase-3, pp53, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay after treatment with different formulations of DOX. Ki67 is a nuclear protein linked with cellular proliferation. As shown in , GA-Se@DOX-treatment reduced the percentage of Ki67-positive cancer cells and induced more caspase-3/pp53/TUNEL-positive cells in comparison with free DOX- or Se@DOX-treatment groups, indicating enhanced efficiency of GA-Se@DOX in inhibiting proliferation of tumor cells and inducing tumor cells apoptosis. This evidence also indicated that the active tumor-targeted carrier GA-SeNPs played a significant role for effective delivery of DOX to tumor sites for improved HCC therapy. Then hematological analysis was carried out to assess in vivo systemic cytotoxicity of GA-Se@DOX. As shown in , H&E staining results showed that no obvious pathological changes were observed in heart, liver, spleen, lung, and kidney in tumor-bearing mice after treated with GA-Se@DOX during treatment time, suggesting negligible in vivo toxicity of GA-Se@DOX.
Figure 8. Hematoxylin-eosin (H&E) analyses of heart, liver, spleen, lung, and kidney after treatment with saline, DOX, Se@DOX, and GA-Se@DOX, respectively. Scale bar is 50 μm.
Conclusion
In the present study, one novel active tumor-targeting selenium nanoparticles GA-Se@DOX was successfully synthesized to effectively deliver DOX for HCC therapy. GA-Se@DOX showed excellent cellular uptake in HepG2 cells through clathrin-mediated endocytosis pathway and exhibited faster release of DOX from nanoparticles at pH5.4 which simulate the acidic microenvironment of cancer cells. GA-Se@DOX was more superior to suppress the proliferation of HepG2 cells and induce HepG2 cells apoptosis in vitro in comparison with free DOX and passive tumor-targeting selenium nanoparticles Se@DOX. The anti-tumor mechanism analyzed from the western blotting result showed that GA-Se@DOX induced the apoptosis of HepG2 cells via activating caspase and Bcl-2 signaling pathways. Furthermore, GA-Se@DOX exhibited excellent anti-tumor efficacy in vivo in comparison with free DOX and Se@DOX. Taken together, active tumor-targeted delivery system GA-Se@DOX exhibits significant potential for HCC therapy.
Supplemental Material
Download Zip (122.5 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Ai T, Shang W, Yan H, et al. (2018). Near infrared-emitting persistent luminescent nanoparticles for hepatocellular carcinoma imaging and luminescence-guided surgery. Biomaterials 167:216–25.
- Bentz KC, Savin DA. (2018). Hollow polymer nanocapsules: synthesis, properties, and applications. Polym Chem 9:2059–81.
- Bi D, Zhao L, Yu R, et al. (2018). Surface modification of doxorubicin-loaded nanoparticles based on polydopamine with pH-sensitive property for tumor targeting therapy. Drug Deliv 25:564–75.
- Cao H, Dan Z, He X, et al. (2016). Liposomes coated with isolated macrophage membrane can target lung metastasis of breast cancer. ACS Nano 10:7738–48.
- Chen Q, Yu Q, Liu Y, et al. (2015). Multifunctional selenium nanoparticles: chiral selectivity of delivering MDR-siRNA for reversal of multidrug resistance and real-time biofluorescence imaging. Nanomedicine 11:1773–84.
- Chinen AB, Guan CM, Ferrer JR, et al. (2015). Nanoparticle probes for the detection of cancer biomarkers, cells, and tissues by fluorescence. Chem Rev 115:10530–74.
- Chowdhury SM, Lee T, Bachawal SV, et al. (2018). Longitudinal assessment of ultrasound-guided complementary microRNA therapy of hepatocellular carcinoma. J Control Release 281:19–28.
- Dreaden EC, Gryder BE, Austin LA, et al. (2012). Antiandrogen gold nanoparticles dual-target and overcome treatment resistance in hormone-insensitive prostate cancer cells. Bioconjugate Chem 23:1507–12.
- Du Y, Tian C, Wang M, et al. (2018). Dipeptide-modified nanoparticles to facilitate oral docetaxel delivery: new insights into PepT1-mediated targeting strategy. Drug Deliv 25:1403–13.
- Guo M, Li Y, Lin Z, et al. (2017). Surface decoration of selenium nanoparticles with curcumin induced HepG2 cell apoptosis through ROS mediated p53 and AKT signaling pathways. RSC Adv 7:52456–64.
- Han Y, An Y, Jia G, et al. (2018). Theranostic micelles based on upconversion nanoparticles for dual-modality imaging and photodynamic therapy in hepatocellular carcinoma. Nanoscale 10:6511–23.
- He XP, Hu XL, James TD, et al. (2017). Multiplexed photoluminescent sensors: towards improved disease diagnostics. Chem Soc Rev 46:6687–96.
- Hu Q, Wu M, Fang C, et al. (2015). Engineering nanoparticle-coated bacteria as oral DNA vaccines for cancer immunotherapy. Nano Lett 15:2732–9.
- Huo ZJ, Wang SJ, Wang ZQ, et al. (2015). Novel nanosystem to enhance the antitumor activity of lapatinib in breast cancer treatment: therapeutic efficacy evaluation. Cancer Sci 106:1429–37.
- Jain A, Jain A, Parajuli P, et al. (2018). Recent advances in galactose-engineered nanocarriers for the site-specific delivery of siRNA and anticancer drugs. Drug Discov Today 23:960–73.
- Jha S, Ramadori F, Quarta S, et al. (2017). Binding and uptake into human hepatocellular carcinoma cells of peptide-functionalized gold nanoparticles. Bioconjugate Chem 28:222–9.
- Kamegawa R, Naito M, Miyata K. (2018). Functionalization of silica nanoparticles for nucleic acid delivery. Nano Res 11:5219–39.
- Kim GH, Won JE, Byeon Y, et al. (2018). Selective delivery of PLXDC1 small interfering RNA to endothelial cells for anti-angiogenesis tumor therapy using CD44-targeted chitosan nanoparticles for epithelial ovarian cancer. Drug Deliv 25:1394–402.
- Kuhn DA, Vanhecke D, Michen B, et al. (2014). Different endocytotic uptake mechanisms for nanoparticles in epithelial cells and macrophages. Beilstein J Nanotechnol 5:1625–36.
- Le TC, Yan B, Winkler DA. (2015). Robust prediction of personalized cell recognition from a cancer population by a dual targeting nanoparticle library. Adv Funct Mater 25:6927–35.
- Li F, Lu J, Kong X, et al. (2017a). Dynamic nanoparticle assemblies for biomedical applications. Adv Mater 29:1605897.
- Li M, Su Y, Zhang F, et al. (2018a). A dual-targeting reconstituted high density lipoprotein leveraging the synergy of sorafenib and antimiRNA21 for enhanced hepatocellular carcinoma therapy. Acta Biomater 75:413–26.
- Li Y, Lin Z, Guo M, et al. (2017b). Inhibitory activity of selenium nanoparticles functionalized with oseltamivir on H1N1 influenza virus. IJN 12:5733–43.
- Li Y, Lin Z, Guo M, et al. (2018b). Inhibition of H1N1 influenza virus-induced apoptosis by functionalized selenium nanoparticles with amantadine through ROS-mediated AKT signaling pathways. IJN 13:2005–16.
- Liang Y, Huang W, Zeng D, et al. (2018). Cancer-targeted design of bioresponsive prodrug with enhanced cellular uptake to achieve precise cancer therapy. Drug Deliv 25:1350–61.
- Licciardello N, Hunoldt S, Bergmann R, et al. (2018). Biodistribution studies of ultrasmall silicon nanoparticles and carbon dots in experimental rats and tumor mice. Nanoscale 10:9880–91.
- Liu W, Li X, Wong Y, et al. (2012). Selenium nanoparticles as a carrier of 5-fluorouracil to achieve anticancer synergism. ACS Nano 6:6578–91.
- Maiyo F, Singh M. (2017). Selenium nanoparticles: potential in cancer gene and drug delivery. Nanomedicine (Lond) 12:1075–89.
- Mohamed NK, Hamad MA, Hafez MZ, et al. (2017). Nanomedicine in management of hepatocellular carcinoma: challenges and opportunities. Int J Cancer 140:1475–84.
- Mou Q, Ma Y, Zhu X, Yan D. (2016). A small molecule nanodrug consisting of amphiphilic targeting ligand-chemotherapy drug conjugate for targeted cancer therapy. J Control Release 230:34–44.
- Sun D, Liu Y, Yu Q, et al. (2014). Inhibition of tumor growth and vasculature and fluorescence imaging using functionalized ruthenium-thiol protected selenium nanoparticles. Biomaterials 35:1572–83.
- Sun H, Zhang Y, Zhong Z. (2018a). Reduction-sensitive polymeric nanomedicines: an emerging multifunctional platform for targeted cancer therapy. Adv Drug Deliv Rev 132:16–32.
- Sun X, Chen Y, Zhao H, et al. (2018b). Dual-modified cationic liposomes loaded with paclitaxel and survivin siRNA for targeted imaging and therapy of cancer stem cells in brain glioma. Drug Deliv 25:1718–27.
- Tan L, Nancharaiah Y, Hullebusch E, Lens P. (2016). Selenium: environmental significance, pollution, and biological treatment technologies. Biotechnol Adv 34:886–907.
- Tang J, Zhang R, Guo M, et al. (2018). Nucleosome-inspired nanocarrier obtains encapsulation efficiency enhancement and side effects reduction in chemotherapy by using fullerenol assembled with doxorubicin. Biomaterials 167:205–15.
- Thapa RK, Choi JY, Poudel BK, et al. (2015). Multilayer-coated liquid crystalline nanoparticles for effective sorafenib delivery to hepatocellular carcinoma. ACS Appl Mater Interfaces 7:20360–8.
- Wang J, Hu X, Xiang D. (2018a). Nanoparticle drug delivery systems: an excellent carrier for tumor peptide vaccines. Drug Deliv 25:1319–27.
- Wang K, Kievit FM, Sham JG, et al. (2016). Iron-oxide-based nanovector for tumor targeted siRNA delivery in an orthotopic hepatocellular carcinoma xenograft mouse model. Small 12:477–87.
- Wang Q, Zhang X, Liao H, et al. (2018b). Multifunctional shell-core nanoparticles for treatment of multidrug resistance hepatocellular carcinoma. Adv Funct Mater 28:1706124.
- Wu D, Wang H, Hou X, et al. (2018). Effects of gold core size on regulating the performance of doxorubicin-conjugated gold nanoparticles. Nano Res 11:3396–410.
- Xia Y, Guo M, Xu T, et al. (2018). siRNA-loaded selenium nanoparticle modified with hyaluronic acid for enhanced hepatocellular carcinoma therapy. IJN 13:1539–52.
- Xia Y, Lin Z, Li Y, et al. (2017). Targeted delivery of siRNA using RGDfC-conjugated functionalized selenium nanoparticles for anticancer therapy. J Mater Chem B 5:6941–52.
- Xu X, Wu J, Liu Y, et al. (2017). Multifunctional envelope-type siRNA delivery nanoparticle platform for prostate cancer therapy. ACS Nano 11:2618–27.
- Yang S, Tang Z, Zhang D, et al. (2017). pH and redox dual-sensitive polysaccharide nanoparticles for the efficient delivery of doxorubicin. Biomater Sci 5:2169–78.
- Yang T, Chen Y, Zhao P, et al. (2018). Enhancing the therapeutic effect via elimination of hepatocellular carcinoma stem cells using Bmi1 siRNA delivered by cationic cisplatin Nanocapsules. Nanomedicine 14:2009–21.
- Yu C, Qian L, Uttamchandani M, et al. (2015). Single-vehicular delivery of antagomir and small molecules to inhibit mir-122 function in hepatocellular carcinoma cells by using “smart” mesoporous silica nanoparticles. Angew Chem Int Ed Engi 54:10574–8.
- Zhang B-F, Xing L, Cui P-F, et al. (2015). Mitochondria apoptosis pathway synergistically activated by hierarchical targeted nanoparticles co-delivering siRNA and lonidamine. Biomaterials 61:178–89.
- Zhang X, Yang X, Ji J, et al. (2016). Tumor targeting strategies for chitosan-based nanoparticles. Colloid Surface B 148:460–73.
- Zhang Z, Wang X, Li B, et al. (2018). Development of a novel morphological paclitaxel-loaded PLGA microspheres for effective cancer therapy: in vitro and in vivo evaluations. Drug Deliv 25:166–77.
- Zhao R, Li T, Zheng G, et al. (2017). Simultaneous inhibition of growth and metastasis of hepatocellular carcinoma by co-delivery of ursolic acid and sorafenib using lactobionic acid modified and pH-sensitive chitosan-conjugated mesoporous silica nanocomplex. Biomaterials 143:1–16.
- Zheng G, Zhao R, Xu A, et al. (2018). Co-delivery of sorafenib and siVEGF based on mesoporous silica nanoparticles for ASGPR mediated targeted HCC therapy. Eur J Pharm Sci 111:492–502.
- Zheng W, Yin T, Chen Q, et al. (2016). Co-delivery of Se nanoparticles and pooled SiRNAs for overcoming drug resistance mediated by P-glycoprotein and class III β-tubulin in drug-resistant breast cancers. Acta Biomater 31:197–210.
- Zhou Y, Yu Q, Qin X, et al. (2016). Improving the anticancer efficacy of laminin receptor-specific therapeutic ruthenium nanoparticles (RuBB-loaded EGCG-RuNPs) via ROS-dependent apoptosis in SMMC-7721 cells. ACS Appl Mater Inter 8:15000–12.