
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Self-nanoemulsifying drug delivery system (SNEDDS) have been considered as a promising platform for oral delivery of many BCS (biopharmaceutics classification system) class IV drugs, such as docetaxel (DTX). However, oral chemotherapy with DTX is also restricted by its active P-glycoprotein (P-gp) efflux and hepatic first-pass metabolism. To address these challenges, we developed a novel SNEDDS co-loaded with DTX and cyclosporine A (CsA) to achieve effective inhibition of P-gp efflux and P450 enzyme metabolization, improving oral bioavailability of DTX. The SNEDDS showed uniform droplet size of about 30 nm. Additionally, the prepared SNEDDS exhibited a sequential drug release trend of CsA prior to DTX. The intestinal experiments confirmed that the membrane permeability of DTX was significantly increased in the whole intestinal tract, especially in the jejunum segment. Furthermore, the oral bioavailability of co-loaded SNEDDS was 9.2-fold and 3.4-fold higher than DTX solution and DTX SNEDDS, respectively. More importantly, it exhibited a remarkable antitumor efficacy with a reduced toxicity compared with intravenously administered DTX solution. In summary, DTX-CsA co-loaded SNEDDS is a promising platform to facilitate oral docetaxel-based chemotherapy.
Introduction
The global incidence of cancer is rising, causing approximately 8.2 million deaths worldwide each year, posing a huge threat to human health (Sohail et al., Citation2018). In recent years, oral chemotherapy has become increasingly attractive with many advantages over intravenous chemotherapy, including easy administration, good patient acceptance, reduced financial cost and none of the adverse risks associated with injections (Sun et al., Citation2016). However, the clinical application of oral chemotherapy is still greatly limited by the inferior oral absorption efficiency, especially for these poorly soluble drugs.
Docetaxel (DTX) is a taxane-based antineoplastic agent that exerts twice the potency of microtubule depolymerization inhibition as paclitaxel (Lavelle et al., Citation1995). DTX has been widely used to treat a variety of cancers, including nonsmall-cell lung cancer, prostate cancer, advanced or metastatic breast cancer, malignant melanoma and ovarian cancer (Lavelle et al., Citation1995; Sun et al., Citation2016). Currently, parenteral formulations of DTX are available for its clinical use (MacConnachie, Citation1997); however, severe systemic toxicities including hypersensitivity reactions, peripheral neuropathy, bone marrow suppression and fluid retention have become major obstacles to clinical treatment (Hu et al., Citation2012; Seo et al., Citation2013). In order to overcome these disadvantages, oral administration is the best solution. Unfortunately, DTX has a very low oral bioavailability (∼5%) (Hu et al., Citation2012), and several challenges remain to be addressed in oral DTX delivery, including poor aqueous solubility, easier chemical and enzymatic degradation in the gastrointestinal tract, limited intestinal permeability, P-glycoprotein (P-gp) mediated efflux in the intestine and rapid hepatic first pass metabolism (Ahmad et al., Citation2016; Ghadi & Dand, Citation2017). To address the challenges, various approaches including polymer prodrugs, polymer conjugates, liposomes, solid lipid nanoparticles, polymeric nanoparticles and self-nanoemulsifying drug delivery system (SNEDDS) have been adopted for oral delivery of DTX (Feng et al., Citation2009; Luo et al., Citation2014; Sun et al., Citation2016; Ghadi & Dand, Citation2017; Ahmad et al., Citation2018a, Citation2018b). Among these approaches, SNEDDS exhibits plenty of distinct advantages over other strategies, such as desired physical or chemical stability, ease of manufacture and scale-up and favorable biocompatibility (Dokania & Joshi, Citation2015).
SNEDDS, emulsion-based drug delivery nanosystem, which spontaneously forms nanoemulsions upon being exposed to water, is well known for its potential to solubilize lipophilic drugs. Besides, the encapsulation of drug molecules within SNEDDS shield them from the degradation of harsh gastrointestinal (GI) environment, improve diffusion across the unstirred aqueous layer, enhance mucosal permeability and biological membrane permeability, facilitate lymphoid-mediated absorption, and thus increase oral bioavailability (Zhao et al., Citation2010; Gao et al., Citation2011; Ahmad et al., Citation2018c).
The encapsulation of drug molecules within NPs shield them from the effect of efflux transporters on one side and the small particle size help facilitate the easy access of drug across the biological membrane on the other hand
Nevertheless, oral treatment with DTX SNEDDS is still severely hampered by two main factors. Firstly, DTX is a high-affinity substrate for the efflux multidrug transporter P-glycoprotein (P-gp), highly expressed in the lumen side of the intestinal tract. Secondly, DTX undergoes extensive first-pass metabolism by cytochrome P450 (CYP) enzymes (CYP 2C8 and CYP 3A4) distributed in enterocytes and hepatocytes (Paolini et al., Citation2017; Veltkamp et al., Citation2006).
In response to the problems, synchronous administration of efficacious inhibitors of both P-gp and CYP, e.g. cyclosporin A (CsA), have been successfully utilized to improve the systemic exposure of taxane formulations after oral administration through enhancing oral absorption and decreasing elimination (Malingre et al., Citation2001; McEntee et al., Citation2003; Yang et al., Citation2004; Oostendorp et al., Citation2011). Yang et al. developed a novel SMEDDS for the oral delivery of paclitaxel and demonstrated the enhancement of oral absorption of paclitaxel, especially when incorporated with an effective P-gp inhibitor and CYP3A4, such as CsA (Yang et al., Citation2004). Oostendorp et al. evaluated various self-microemulsifying oily formulations (SMEOF’s) of paclitaxel for oral application using mice and CsA as P-gp and CYP450 inhibitor. They confirmed that SMEOF’s may be a useful vehicle for oral delivery of paclitaxel in combination with CsA and enhanced bioavailability (Oostendorp et al., Citation2011). McEntee et al. demonstrated that coadministration of oral CsA strongly enhanced the oral bioavailability of docetaxel (McEntee et al., Citation2003). Besides, CsA has a high lipophilicity (logP 4.15), and the clinically available oral formulation of CsA is in the form of a self-microemulsion, Sandimmun Neoral® (Zhang et al., Citation2010). Thus, we hypothesized that co-incorporation of DTX and CsA into SNEDDS could further increase the oral bioavailability and therapeutic efficacy.
Aiming at this goal, we developed a unique DTX-CsA co-loaded SNEDDS for efficiently oral delivery of DTX, with DTX SNEDDS as a control formulation. The prepared SNEDDSs were characterized according to their droplet size, morphology and drug loading. The release behaviors of DTX and CsA from the SNEDDS in simulated gastrointestinal environment were investigated. The effects of SNEDDS on intestinal permeability and biodistribution of DTX were studied by in situ single-pass intestinal perfusion and confocal laser scanning microscopy. Moreover, pharmacokinetic study was conducted in rats to evaluate the oral bioavailability of co-loaded SNEDDS. Finally, in vivo antitumor efficacy of co-loaded SNEDDS was compared with that of DTX-solution and DTX SNEDDS.
Materials and methods
Materials
Docetaxel (DTX) and cyclosporine A (CsA) were obtained from Dalian Meilun Biotech Co., Ltd, China. Tween-80, isopropyl myristate, Cremophor EL and Cremophor RH40 were purchased from Aladdin Industrial Corporation, Shanghai, China. Soybean oil was bought from Tieling North Asia Medicinal Oil Co., Ltd. 2, 2′-thiobisacetic anhydride was obtained from Alfa Aesar (China) Chemicals Co., Ltd. Transcutol HP, Labrasol, Capryol 90, Labrafil M1944 CS, Maisin 35-1 and Plurol Oleique CC 497 were received as gifts from Gattefossé Co. (Saint Priest, Cedex, France). PEG 400 and 1, 2-propanediol were bought from Tianjin Bodi Chemical Co., Ltd. Egg phosphatidylcholines (PC) was generous gift from Lipoid Company (Ludwigshafen, Germany). All other reagents used in this study were of analytical grade.
Solubility study
The solubilities of DTX and CsA in various oils that are generally recognized as safe (GRAS) were determined using shake flask method. Briefly, excess amount of drug was added to 0.5 mL of each excipient in the centrifugal tube (in triplicate) and the cover was sealed with sealing film. Then the mixtures were vortexed and shaken in a water bath at 25 °C for 48 h to achieve the equilibrium. The mixtures were centrifuged at 13,000 rpm for 20 min to remove the excess drug and filtered through the millipore filter (0.22 μm), after which the concentrations of drugs were measured by high performance liquid chromatography (HPLC, Waters e2695, USA) after appropriate dilution with acetonitrile.
Determination of drug loading capacity of SNEDDS
To determine the maximum drug loading in the SNEDDS formulation, excess amounts of DTX and CsA were added to SNEDDS preconcentrate. It was vortex for 1 min and maintained mixing in a thermostatically controlled shaking incubator at 25 °C for 24 h. The concentrations of DTX and CsA were measured as described in the section of solubility study.
Preparation of SNEDDS formulations and solutions
The co-loaded SNEDDS was prepared as follows: Capryol 90, Cremophor EL and Transcutol HP were homogenously mixed at a ratio of 43:43:14 (w/w/w). Weighed quantity of DTX (10 mg/g drug loading) and CsA (13 mg/g drug loading) were dissolved in the resulting mixture and stirred to obtain a homogenous solution. The DTX SNEDDS was prepared as above except that no CsA was added. The prepared liquid SNEDDS was stored in sealed transparent vial at room temperature until used. DTX solution was prepared in accordance with the formulation of Taxotere®.
Characterization of SNEDDS
Droplet size and distribution
The droplet size and polydispersity index (PDI) of co-loaded SNEDDS was measured by dynamic light scattering (DLS) using a Nano ZS Zetasizer instrument (Nano ZS90, Malvern instruments Ltd., UK). 0.1 ml of formulation was dispersed into 10 ml of water with gentle stirring in a vial. Then a 1 ml aliquot was withdrawn and transferred to a sample cell for size measurement.
Transmission electron microscopy (TEM) of SNEDDS
The morphology of nanoemulsion was observed using Transmission electron microscopy (H-600, Hitachi, Japan). SNEDDS samples were diluted 100-fold with water to form an emulsion before the analysis. Then the samples were stained by 2% (w/v) phosphotungstic acid for 3 min on a copper grid stabilized with carbon support film.
In vitro drug release study
The in vitro release experiments of DTX and CsA from SNEDDS were performed using a dialysis method. Simulated gastric fluid (SGF, 0.1 M HCl, pH 1.2, enzyme-free) and simulated intestinal fluid (SIF, phosphate buffer, pH 6.8, enzyme free) were employed as release media, containing 30% ethanol (v/v) to attain sink conditions. The dialysis bags (MW cutoff 12-14 kDa) were soaked in the boiling water for 30 min before use. The SNEDDS (containing 0.200 mg of DTX and 0.067) was dispersed in 1 mL of distilled water and then sealed in the dialysis bags. The dialysis bags were incubated in conical flasks with 30 mL of release media under orbital shaking at 37 °C. At designated intervals, samples (1.0 mL) of dialyzed solution were withdrawn and the same volume of fresh media was added to maintain the volume. The drug content was determined by HPLC as described above.
Animals
BALB/c mice (18–22 g) and Sprague-Dawley (SD) rats (200–240 g) were obtained from the Laboratory Animal Center of Shenyang Pharmaceutical University. All the animal experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals Approved by the Institutional Animal Ethical Care Committee (IAEC) of Shenyang Pharmaceutical University. The rats were fasted overnight for approximately 12 h with free access to water before the experiments.
In situ single-pass intestinal perfusion (SPIP)
To evaluate the intestinal permeability of DTX in different formulations, the SPIP study was performed as previously described with slight modifications (Zhang et al., Citation2015). Sprague − Dawley (SD) rats fasted overnight were anesthetized by intraperitoneal injection with 20% ethyl carbamate and placed under an infrared lamp to maintain a normal body temperature. Then the abdomen was opened along the ventral midline. The intestinal segments (around 10 cm) including the duodenum, jejunum, ileum and colon were gently separated, cannulated and rinsed with warm saline solution. Before the perfusion, DTX solution, DTX SNEDDS and co-loaded SNEDDS were diluted to 20 μg/mL with Krebs Ringer’s (KR) buffer solution. Then the intestinal segments were infused with the perfusates at a flow rate of 0.2 mL/min. The perfusion experiment continued for 105 min, during which the outflowing perfusate was consecutively collected in a preweighed receptor vial at 15 min intervals. In the meantime, preweighed donor flask and the receptor vial were synchronously replaced and accurately weighed again. Finally, the rats were executed, and the length and radius of every infused intestinal segment were precisely measured. The samples were diluted five times with methanol and centrifuged at 13000 rpm for 10 min to determine the DTX content using the HPLC method described above. The absorption rate (Ka) and apparent permeability (Papp) were calculated as follows:
where Cout is the concentration of DTX in the outlet perfusate; Cin is the concentration of DTX in inlet perfusate; Vout is the volume of perfusate collected in the receptor vial; Vin is the volume of perfusate pumped from the donor flask; Q is perfusion flow rate (mL/min); r is the intestinal radius (cm); and l is the length of intestine for perfusion (cm).
Biodistribution of SNEDDS in the intestinal tract
To study the biodistribution of prepared SNEDDS in different intestinal segments, coumarin-6-solution and coumarin-6-SNEDDS were orally administrated to SD rats at a dose of 1 mg/kg, which was prepared according to the method described in Preparation of SNEDDS formulations and solutions, except that coumarin-6 was used instead of DTX. After 40 min, the rats were sacrificed. Isolated by incision and frozen in cryoembedding agent at −80, the everted segments of duodenum, jejunum, ileum, and colon were sliced into 10-micron-thick slices. The intestinal tissue was fixed on cationic resinous slides, incubated with 4% paraformaldehyde and washed with cold PBS (pH 7.4) three times. Then the tissue slide was incubated with Triton X-100 (0.5%) and subsequently rinsed with PBS. After staining with rhodamine-labeled phalloidin and DAPI for visualization of cytoskeleton (red) and cell nuclei (blue), the enterocytes were visualized under the confocal laser scanning microscopy (CLSM, TCS SP2/AOBS, LEICA, Germany).
Pharmacokinetic profiles
The oral absorption of DTX was systematically assessed through a pharmacokinetic study. The SD rats were fasted overnight and divided into four groups (n = 5) randomly. For the intravenous injection group, DTX solution (5 mg/kg) was administered via caudal vein. For oral administration groups, DTX solution, DTX SNEDDS and co-loaded SNEDDS (30 mg/kg) were orally administrated. At predetermined time, 300 μL of blood samples were collected from the orbital and transferred to heparinized tubes. The blood samples were centrifuged at 13,000 rpm immediately for 5 min, and then the separated plasma was stored at −20 °C for analysis using liquid chouromatography-mass spectrometry (LC−MS/MS).
The plasma concentrations of DTX were determined using a validated LC–MS/MS method after protein precipitation with acetonitrile. Diazepam was used as internal standard. The analysis was performed by Shimadzu LCMS-8060 system (Shimadzu Corporation, Japan). An ACE UltraCore 2.5 SuperC18 column (50 mm × 2.1 mm, 2.5 μm) was applied to carry out the chromatographic separation with mobile phase comprised of acetonitrile (A) and water (B) containing 0.2% formic acid at a flow rate of 0.2 ml/min. A gradient elution was employed to measure DTX concentration: 0 ∼ 0.8 min, 30% A; 0.81 ∼ 3.0 min, 95% A; 3.1 ∼ 5 min, 30% A. The injection volume was 10 μL, and the column and autosampler temperatures were maintained at 40 and 4 °C, respectively. The mass spectrometer was operated in positive ESI mode.
The pharmacokinetic parameters including peak time (Tmax), maximum plasma concentration (Cmax), area under curve (AUC) and half-life (t1/2) were calculated by the DAS 2.0 software. The relative bioavailability (Frel) and absolute bioavailability (Fabs) were calculated as follows:
where T, R and i.v are test product (oral DTX solution), reference product (other oral preparation), and intravenous product (intravenous DTX solution), respectively.
In vivo antitumor efficacy
4T1 cells (5 × 106 cells per 100 μL) were subcutaneously injected into the flank of female BALB/c mice (18–22 g). When tumor volume reached approximately 125 mm3, DTX solution, DTX SNEDDS and co-loaded SNEDDS were administered orally at a dose of 30 mg/kg equivalent to DTX, while DTX solution (10 mg/kg) was administered intravenously (n = 5 for each group). Normal saline (NS) was used as a control. Preparations were administrated every other day for a total of five injections. Body weight and tumor volume were monitored every day. The mice were sacrificed after two days of the last treatment. Tumors were weighed to calculate the tumor burden. The major organs (heart, liver, spleen, lung, and kidney) and tumor tissues were collected and fixed by 4% paraformaldehyde for hematoxylin and eosin (H&E).
Statistical analysis
Experimental results were shown as mean ± standard deviation (SD). Student’s t-test or one-way ANOVA was utilized to identify any significant difference in the experiments. Statistical difference was set at p < .05, very significant difference was defined as P < 0.01, and extremely significant was set as p < .001
Results and discussion
Solubility studies and formulation determination
The SNEDDS formulation was prepared to improve the solubility and bioavailability of drug upon oral administration. Hence, the oil utilized in the system should possess high drug solubility. In this work, solubility studies were conducted to identify appropriate oil for the development of desired SNEDDS with optimum drug loading and emulsifying efficiency. As presented in Figure S1, DTX exhibited the highest solubility of 131.62 ± 17.68 in Capryol 90 among the oils employed in the experiment. CsA exhibited the highest solubility in ethyl oleate, following the Capryol 90. Hence, under comprehensive consideration, Capryol 90 was designated as the oil phase for further study. The chemical name of Capryol 90 is propylene glycol monocaprylate. It is composed of 90% caprylic acid monoester and can be employed in self-emulsifying formulations.(Yin et al., Citation2009; Seo et al., Citation2013) According to our previous study, Cremophor EL and Transcutol HP not only provided strong solubilization capability for DTX, but they also possessed excellent emulsifying capacity. Therefore, among various excipients, Capryol 90, Cremophor EL and Transcutol HP were selected as the oil, surfactant and co-surfactant. A systematic method was adopted to identify the specific self-emulsifying region and the proportion of the three constituents within a ternary phase diagram (Supporting information).
Transcutol HP and 1,2-propanediol were selected as co-surfactants for further investigation. Regarding surfactants, there were no significant difference among Tween 80, Cremophor EL and Labrasol. Since the selection of surfactants and co-surfactants should depend not only on their drug solubilizing ability but also especially on their emulsifying efficiency, it is necessary to screen their emulsifying capacity. According to previous research of ternary phase diagram, Capryol 90/Cremophor EL/Transcutol HP at a weight ratio of 43/42/15 was selected as the optimized SNEDDS formulation for further studies.
Preparation and characterization of SNEDDS
The droplet size, PDI, measured equilibrium solubility and drug content of co-loaded SNEDDS are included in Table S1. Yang et al. demonstrated that Cmax and AUC values were substantially increased in rats only when paclitaxel was co-administrated with 40 mg/kg CsA (Yang et al., Citation2004). And the administration dosage of DTX was set as 30 mg/kg in animal experiments. Hence, the content ratio of DTX to CsA was determined to be 3:4. The drug loadings of DTX and CsA were 10 mg/g and 13 mg/g, respectively. The optimal SNEDDS had a limpid and slightly bluish appearance, fairly small droplet size (∼30 nm) and PDI when dispersed into water, indicating that prepared SNEDDS had an excellent self-nanoemulsifying ability ().
Figure 1. Appearance, size distribution and TEM images of co-loaded SNEDDS.
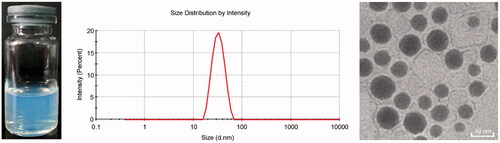
As shown in the TEM image (), the diluted co-loaded SNEDDS (nanoemulsions) appeared spheroid-like in shape with relatively smooth surface. TEM analysis revealed a droplet size range (20–40 nm). Furthermore, DLS measurements exhibited an average droplet size of 30.12 ± 1.29 nm with a narrow polydispersity index of 0.051 ± 0.01, which was in good agreement with the observation of TEM. Surfactants and co-surfactants can reduce the interfacial tension to an ultra-low value, resulting in the spontaneous formation of nanoemulsions (Rahman et al., Citation2013). Therefore, the droplet size can be reduced to about 30 nm by optimizing the formulation of surfactants and co-surfactants. Additionally, it was reported that nanoparticles with a small size of less than 300 nm are considered more appropriate for cellular internalization via endocytosis (Ahmad et al., Citation2018c). Thus, optimized SNEDDS with a size of about 30 nm could promote internalization of intestinal epithelial cells.
In vitro drug release
The cumulative release profiles of DTX and CsA SNEDDS in different media are exhibited in . In SGF, 43.0% of DTX and 65.0% of CsA were released from co-loaded SNEDDS after 4 h. In contrast, 62.1% of DTX and 90.0% of CsA were released from co-loaded SNEDDS after 12 h in SIF. It was revealed from the release patterns that DTX had a strong interaction with the oil core of the SNEDDS and exhibited a steady release (Ahmad et al., Citation2018b).
Figure 2. In vitro drug release of DTX SNEDDS and Co-loaded SNEDDS in pH 1.2 hydrochloric acid solution (A) and pH 6.8 PBS (B) (n = 3).
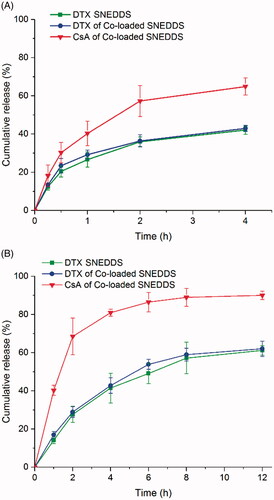
In addition, DTX of co-loaded SNEDDS exhibited similar drug release behavior as DTX SNEDDS, indicating that the co-loaded system did not interfere with DTX release behavior. As the release rate of DTX in SGF was low, SNEDDS may provide the protective effects for drug molecules under the severe gastric juice environments, enabling more drug molecules to enter the intestinal tract. Interestingly, the release rate of CsA from the co-loaded SNEDDS was higher than that of DTX under simulated intestinal juice conditions, and CsA was released prior to DTX, presenting a sequential drug release behavior. The rapid release of CsA prior to DTX was expected to inhibit intestinal P-gp, which may be beneficial for the absorption of DTX released subsequently, thereby improving oral bioavailability.
In situ single-pass intestinal perfusion
The absorption of DTX solution, DTX SNEDDS and co-loaded SNEDDS in four intestinal segments was investigated by SPIP. As indicated in , Ka and Papp values of co-loaded SNEDDS were obviously higher compared with DTX solution and DTX SNEDDS in every intestinal segment (P < 0.05). Ka values of DTX SNEDDS showed 1.3-, 1.7-, 1.6- and 1.6-fold improvement compared with DTX solution in the duodenum, jejunum, ileum and colon, while Ka values of co-loaded SNEDDS showed 1.8-, 2.8-, 2.6- and 1.7-fold improvement compared with DTX solution in the duodenum, jejunum, ileum and colon, respectively. These results showed that SNEDDS can increase the intestinal absorption of DTX to some extent, probably due to increasing dissolution rate of drugs, membrane permeability and inhibiting the P-gp mediated efflux (Porter et al., Citation2007; Singh et al., Citation2013). Nevertheless, it was not enough to increase DTX absorption merely by using SNEDDS. Co-loaded SNEDDS exhibited better absorption potential and apparent permeability than DTX SNEDDS, demonstrating co-delivery of CsA can effectively inhibit P-gp efflux to improve oral absorption in the intestine. In contrast, SNEDDS cannot overcome the efflux effect of the P-gp.
Figure 3. In situ single-pass intestinal perfusion (n = 3). Ka (A) and Papp (B) of duodenum, jejunum, ileum and colon. (*p < .05, **p < .01, *p < .001).
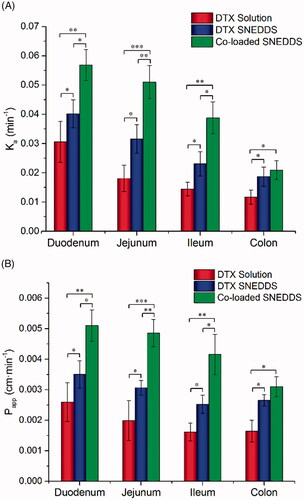
Intriguingly, the Papp of DTX solution was shown to be the highest in duodenum obviously, while the Papp of co-loaded SNEDDS was the highest in both of duodenum and jejunum. In addition, it was further found that permeability of SNEDDS increased most remarkably in jejunum and ileum compared to DTX solution. The reason for this may be that P-gp efflux pump has an increased distribution from proximal to the distal intestine segments (Chan et al., Citation2004). A large amount of P-gp in jejunum and ileum was sufficiently inhibited by CsA, so DTX absorption was improved most significantly in the jejunum and ileum.
Biodistribution of SNEDDS in the intestinal tract
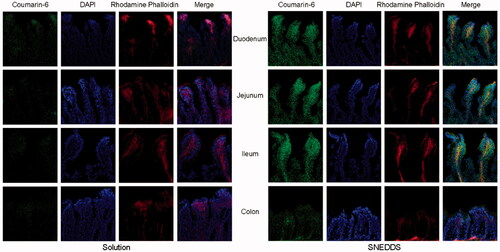
In order to investigate the biodistribution of SNEDDS in rat intestinal tract and visualize the results, coumarin-6-SNEDDS and coumarin-6 solution (negative control) were prepared to replace DTX formulations and orally administrated. As is shown in , a much stronger fluorescence of coumarin-6-SNEDDS could be observed in every intestinal segment compared to that of coumarin-6 solution. Additionally, the absorbed coumarin-6-SNEDDS was localized intracellularly inside the deep intestine villi with stronger fluorescence intensity. The results demonstrated that SNEDDS can enhance drug permeation through the intestinal epithelial cells. More specifically, the strongest green fluorescence was observed in the duodenum followed by the jejunum, consistent with the results in the SPIP study.
Figure 4. Intestinal fluorescence micrographs of coumarin-6 solution and coumarin-6-SNEDDS (green fluorescence). DAPI (blue) and rhodamine phalloidin (red) were used to label the cell nuclei and cytoskeleton respectively.
Pharmacokinetic study
The pharmacokinetic profiles of DTX solution, DTX SNEDDS and co-loaded SNEDDS were studied in SD rats. For intuitive comparison of different formulations, the DTX molar concentration − time curves were exhibited in The main pharmacokinetic parameters were calculated and presented in Table S2.
Figure 5. Concentration of plasma drug (DTX)-time profiles of DTX solution, DTX SNEDDS and Co-loaded SNEDDS following oral administration. (n = 5).
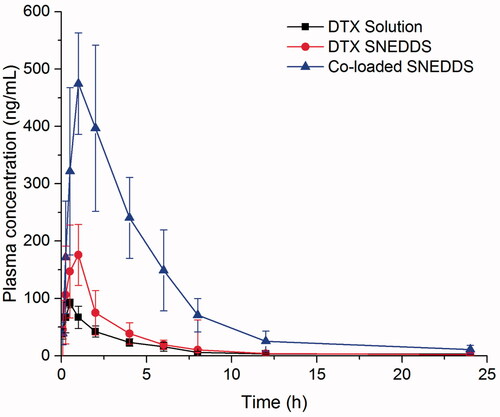
The oral absolute bioavailability of DTX SNEDDS and co-loaded SNEDDS were 13.2%, and 45.2%, respectively, while the oral absolute bioavailability of DTX solution was only 4.9%. Furthermore, the bioavailability of DTX SNEDDS and co-loaded SNEDDS exhibited a 2.7-fold and 9.2-fold increase significantly compared with DTX solution, respectively. These results indicated that SNEDDS can enhance the oral absorption to some extent, probably due to increasing solubility of DTX, protecting against degradation in gastrointestinal tract, enhancing enterocytic endocytosis and Payer's patches uptake and facilitating lymphatic transport (Yin et al., Citation2009; Seo et al., Citation2013; Ahmad et al., Citation2018a; Ahmad et al., Citation2019). However, SNEDDS alone cannot sufficiently overcome the efflux effect of the P-gp in the case of DTX. In contrast, there was a substantial increase in the AUC and Cmax values of co-loaded SNEDDS. The results indicated that co-delivery CsA could completely inhibit the efflux of the intestinal P-gp to ensure adequate absorption of DTX.
The low plasma concentration and poor oral bioavailability of DTX solution were due not only to the overexpression of P-gp by the intestinal cells but also to the significant first-pass extraction by CYP-dependent process (Gursoy & Benita, Citation2004). As shown in Table S2, the t1/2 (13.2 h) of co-loaded SNEDDS was also significantly extended compared with DTX solution (4.9 h) and DTX SNEDDS (7.5 h). This indicated that co-delivery CsA could diminish the hepatic CYP metabolism to DTX. Furthermore, SNEDDS might have an enduring positive effect on the P-gp inhibitory effect of CsA either through increasing its intestinal absorption or enhancing the interaction of CsA with CYP at the level of the villus tip enterocytes of the small intestine, resulting further improvement in DTX oral bioavailability (Zhang & Benet, Citation2001).
Notably, co-loaded SNEDDS showed a long-term high plasma concentration. As reflected in , high plasma level above 80 ng/mL was maintained for 8 h. Drug concentration of long-term therapeutic level will contribute to exerting the antitumor efficacy of DTX.
In vivo antitumor efficacy
The in vivo antitumor efficiency of different formulations was evaluated in 4T1 tumor bearing BALB/c mice. As shown in , NS did not have any measurable effect on tumor growth, and tumor volumes increased rapidly and reached about 1100–1300 mm3 at day 10. A similar trend was observed for the orally administered DTX solution. In comparison, DTX SNEDDS and co-loaded SNEDDS showed potent antitumor efficiency, especially co-loaded SNEDDS, with significantly smaller tumor volumes than the other oral administration groups, which was ascribed to its high oral bioavailability. Notably, the tumor burden efficiency of co-loaded SNEDDS was comparable to that of the DTX solution (i.v., 10 mg/kg). The results corroborated that in vivo antitumor efficiency is closely related to the pharmacokinetic behavior of the drug.
Figure 6. In vivo antitumor efficacy against 4T1 xenograft tumors after the intravenous and oral administration of DTX solution, DTX SNEDDS and Co-loaded SNEDDS. (A) Tumor volume (n = 5). (B) Images of tumors. (C) Tumor burden (n = 5). (D) Body weight changes (n = 5). (E) H&E staining of the major organs and tumor acquired from mice bearing 4T1 tumors at the tenth day after various treatments. Scale bars represent 100 μm. (* p< .05, **p < .01).
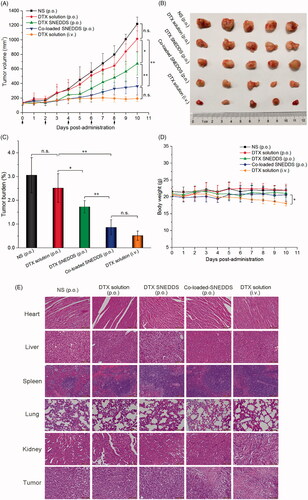
Although DTX solution (i.v.) demonstrated good antitumor effect among all the formulations studied in terms of the suppression of tumor growth, the body weight of the mice treated with DTX solution (i.v.) dropped significantly compared with the other oral administration groups. Besides, there were no obvious histological damages in H&E-stained tissue sections of major organs (heart, liver, spleen, lung and kidney. These results suggested that oral DTX formulations were well tolerated with negligible nonspecific toxicity to major organs and tissues. The tumor tissues of mice treated with DTX solution (i.v.) and co-loaded SNEDDS groups exhibited widespread apoptosis and necrosis, suggesting a potent antitumor efficacy.
Conclusions
In summary, DTX and CsA were successfully co-incorporated into SNEDDS with excellent self-nanoemulsifying capacity. DTX-CsA co-loaded SNEDDS showed spherical shape with smooth surface and homogeneous droplet size distribution (∼30 nm). Additionally, co-loaded SNEDDS exhibited a sequential drug release behavior. The co-loaded SNEDDS could significantly improve intestinal permeability of DTX when compared with DTX solution and DTX SNEDDS. Importantly, the co-loaded SNEDDS showed 9.2-fold and 3.4-fold higher oral bioavailability than DTX solution and DTX SNEDDS, respectively. As a result, it exhibited a remarkable antitumor efficacy with a reduced toxicity. The current study suggests SNEDDS co-loaded with DTX and CsA showed a great potential for enabling oral delivery of challenging chemotherapeutic drugs.
Supplemental_File.docx
Download MS Word (298.5 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Ahmad N, Ahmad R, Alam MA, et al. (2019). Daunorubicin oral bioavailability enhancement by surface coated natural biodegradable macromolecule chitosan based polymeric nanoparticles. Int J Biol Macromol 128:825–38.
- Ahmad N, Ahmad R, Naqvi AA, et al. (2016). Quantification of rutin in rat's brain by UHPLC/ESI-Q-TOF-MS/MS after intranasal administration of rutin loaded chitosan nanoparticles. EXCLI J 15:518–31.
- Ahmad N, Ahmad R, Alam MA, et al. (2018a). Enhancement of oral bioavailability of doxorubicin through surface modified biodegradable polymeric nanoparticles. Chem Central J 12:65.
- Ahmad N, Alam MA, Ahmad R, et al. (2018b). Preparation and characterization of surface-modified PLGA-polymeric nanoparticles used to target treatment of intestinal cancer. Artif Cells Nanomed Biotechnol 46:432–46.
- Ahmad N, Alam MA, Ahmad R, et al. (2018c). Improvement of oral efficacy of Irinotecan through biodegradable polymeric nanoparticles through in vitro and in vivo investigations. J Microencapsul 35:327–43.
- Chan LMS, Lowes S, Hirst BH. (2004). The ABCs of drug transport in intestine and liver: efflux proteins limiting drug absorption and bioavailability. Eur J Pharma Sci 21:25–51.
- Dokania S, Joshi AK. (2015). Self-microemulsifying drug delivery system (SMEDDS)-challenges and road ahead. Drug Deliv 22:675–90.
- Feng SS, Mei L, Anitha P, et al. (2009). Poly(lactide)-vitamin E derivative/montmorillonite nanoparticle formulations for the oral delivery of Docetaxel. Biomaterials 30:3297–306.
- Gao F, Zhang Z, Bu H, et al. (2011). Nanoemulsion improves the oral absorption of candesartan cilexetil in rats: Performance and mechanism. J Control Release 149:168–74.
- Ghadi R, Dand N. (2017). BCS class IV drugs: Highly notorious candidates for formulation development. J Controlled Release 248:71–95.
- Gursoy RN, Benita S. (2004). Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother 58:173–82.
- Hu K, Cao S, Hu F, et al. (2012). Enhanced oral bioavailability of docetaxel by lecithin nanoparticles: preparation, in vitro, and in vivo evaluation. Int J Nanomedicine 7:3537–45.
- Lavelle F, Bissery MC, Combeau C, et al. (1995). Preclinical evaluation of docetaxel (Taxotere). Semin Oncol 22:3–16.
- Luo C, Sun J, Du Y, et al. (2014). Emerging integrated nanohybrid drug delivery systems to facilitate the intravenous-to-oral switch in cancer chemotherapy. J Control Release 176:94–103.
- MacConnachie AM. (1997). Docetaxel (taxotere, Rhone-Poulenc Rorer). Intensive Crit Care Nurs 13:119–20.
- Malingre MM, Ten Bokkel Huinink WW, Mackay M, et al. (2001). Pharmacokinetics of oral cyclosporin A when co-administered to enhance the absorption of orally administered docetaxel. Eur J Clin Pharmacol 57:305–7.
- McEntee M, Silverman JA, Rassnick K, et al. (2003). Enhanced bioavailability of oral docetaxel by co-administration of cyclosporin A in dogs and rats. Vet Comp Oncol 1:105–12.
- Oostendorp RL, Buckle T, Lambert G, et al. (2011). Paclitaxel in self-micro emulsifying formulations: oral bioavailability study in mice. Invest New Drugs 29:768–76.
- Paolini M, Poul L, Berjaud C, et al. (2017). Nano-sized cytochrome P450 3A4 inhibitors to block hepatic metabolism of docetaxel. Int J Nanomedicine 12:5537–56.
- Porter CJ, Trevaskis NL, Charman WN. (2007). Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov 6:231–48.
- Rahman A, Hussain A, Hussain S, et al. (2013). Role of excipients in successful development of self-emulsifying/microemulsifying drug delivery system (SEDDS/SMEDDS). Drug Dev Ind Pharm 39:1–19.
- Seo YG, Kim DH, Ramasamy T, et al. (2013). Development of docetaxel-loaded solid self-nanoemulsifying drug delivery system (SNEDDS) for enhanced chemotherapeutic effect. Int J Pharm 452:412–20.
- Singh B, Singh R, Bandyopadhyay S, et al. (2013). Optimized nanoemulsifying systems with enhanced bioavailability of carvedilol. Colloids Surf B Biointerfaces 101:465–74.
- Sohail MF, Rehman M, Sarwar HS, et al. (2018). Advancements in the oral delivery of docetaxel: challenges, current state-of-the-art and future trends. Int J Nanomed 13:3145–61.
- Sun B, Luo C, Li L, et al. (2016). Core-matched encapsulation of an oleate prodrug into nanostructured lipid carriers with high drug loading capability to facilitate the oral delivery of docetaxel. Colloids Surf B Biointerfaces 143:47–55.
- Veltkamp SA, Thijssen B, Garrigue JS, et al. (2006). A novel self-microemulsifying formulation of paclitaxel for oral administration to patients with advanced cancer. Br J Cancer 95:729–34.
- Yang S, Gursoy RN, Lambert G, et al. (2004). Enhanced oral absorption of paclitaxel in a novel self-microemulsifying drug delivery system with or without concomitant use of P-glycoprotein inhibitors. Pharm Res 21:261–70.
- Yin YM, Cui FD, Mu CF, et al. (2009). Docetaxel microemulsion for enhanced oral bioavailability: preparation and in vitro and in vivo evaluation. J Control Release 140:86–94.
- Zhang Y, Benet LZ. (2001). The gut as a barrier to drug absorption: combined role of cytochrome P450 3A and P-glycoprotein. Clin Pharma 40:159–68.
- Zhang Y, Li X, Zhou Y, et al. (2010). Cyclosporin A-loaded poly(ethylene glycol)-b-poly(d,l-lactic acid) micelles: preparation, in vitro and in vivo characterization and transport mechanism across the intestinal barrier. Mol Pharm 7:1169–82.
- Zhang D, Pan X, Wang S, et al. (2015). Multifunctional Poly(methyl vinyl ether-co-maleic anhydride)-graft-hydroxypropyl-β-cyclodextrin Amphiphilic Copolymer as an Oral High-Performance Delivery Carrier of Tacrolimus. Mol Pharm 12:2337–51.
- Zhao Y, Wang C, Chow AH, et al. (2010). Self-nanoemulsifying drug delivery system (SNEDDS) for oral delivery of Zedoary essential oil: formulation and bioavailability studies. Int J Pharm 383:170–7.