Abstract
A rapid, highly sensitive and specific high-performance liquid chromatography coupled with tandem mass spectrometry (HPLC-MS-MS) quantitation method was developed and validated for the determination of hydrocodone in human plasma. Sample was extracted from 0.5mL heparinized plasma by a simple liquid-liquid extraction method and analyzed on a C18 column with a mobile phase of acetonitrile-water (78:22,v/v,0.1% acetic acid). Detection was carried out by positive elevtrospray ionization (ESI) in multiple reactions monitoring (MRM) mode of 300.3→199.2 (m/z) for hydrocodone and 341.2→107.2 (m/z) for canrenone (I.S.), respectively. A good linearity was obtained from 0.5 to 60 ng·mL−1 and the lower limit of quantification (LLOQ) was 0.1ng·mL−1. Compared to an existing method, the extraction method, internal standard and chromatographic conditions were modified and the cost of a large amount of samples determination was decreased obviously. The method was successfully applied to the pharmacokinetic and bioequivalence studies in healthy Chinese volunteers.
INTRODUCTION
Hydrocodone, (4,5α-epoxy-3-methoxy-17-methyl-morphinan-6-one) is a semi- synthetic opioid analgesic and is frequently used for the relief of moderate pain. It exerts effects on the central nervous system (CNS) and smooth muscle through the center opioid receptor Citation[1].
Only a few quantitative methods have been reported. It was necessary to develop a highly sensitive and specific bioanalytical method to quantify the low concentrations of hydrocodone in human plasma. A GC-MS method Citation[2] was used to determine hydrocodone and its metabolite in human urine, but was not suitable for plasma sample. Yu-Luan Chen et al. Citation[3] developed a liquid chromatography with tandem mass spectrometry method to determine the hydrocodone and hydromorphone in human plasma, but no pharmacokinetic study was reported and its cost was too large for pharmacokinetic and bioequivalence studies. In this research, we modified some conditions and procedures based on the existing method, such as chromatographic conditions, internal standard and extraction method, and so on. The method was validated and applied to the pharmacokinetic and bioequivalence studies of hydrocodone in humans successfully.
MATERIALS
Chemicals and reagents: Hycodan and Acetaminophen Tablets (5mg Hycodan per tablet) for testing was provided by Mallinckrodt Inc., Batch No: 0357H02598, and the reference was supplied by Huasheng Pharmaceutical Factory, USA, Batch No: 34966C05. Hycodan (purity 99.5%) was provided by the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Canrenone (purity 99.8%) was obtained from Shenzhou Pharmaceutical Factory, Zhejiang Province.
Acetonitrile, ethyl acetate and acetic acid (HPLC-grade) were purchased from Tedia Company (USA). Ammonia solution (analytical grade) was from Chemical Industry Institute of Shandong Province. Distilled water was purified using a Milli-Q system (Milford, USA).
Instrumentation and HPLC-MS-MS condition: A 1200 series HPLC system (including Degasser, Binary pump, HiP-ALS, TCC and DAD), coupled with a 6410 triple quad mass spectrometer, a C18 column (2.1×100 mm, 3.5µm) and an on-line filter, were from Agilent Technologies, USA. The AX-205 Electronic Balance was from METTLER TOLEDO Instrument, the XW-80A Vortex was from Shanghai Jingke Industry Company and the SORVALL Biofuge PRIMO centrifuge was from Kendro Company, USA.
Separation of hydrocodone and I.S. was achieved on a 1200 series HPLC using a C18 column (2.1×100mm,3.5µm) and isocratically eluted with mobile phase of acetonitrile-water (78:22, v/v, 0.1% acetic acid) at a flow-rate of 0.2 mL·min−1.
Agilent 6410 Triple Quad Mass Spectrometer was operated using ESI spray in positive ionization mode with nebulizer gas of 30 psi. The gas temperature was set at 300°C and gas flow was 8 L·min−1. The capillary voltage was 4000V. The fragmentor energy was 110V. Nitrogen was used as collision gas at an energy of 40V for hydrocodone and 30V for canrenone. The determination was performed by multiple reaction monitoring (MRM) mode, and ion transitions of m/z 300.3 →199.2 for hydrocodone and m/z 341.2→107.2 for canrenone (I.S.) were selected.
METHODS
Preparation of Stock and Working Standard Solutions
The 1.0 mg·mL−1 stock solutions were prepared by dissolving appropriate amounts of hycodan (equal to 1.0 mg·mL−1 hydrocodone) standard or I.S. in mobile phase or methonal, respectively. Stock solutions were diluted appropriately to obtain working solutions of 1000, 250 and 25.0 ng·mL−1 for hydrocodone and 1.0 µg·mL−1 for I.S. All solutions were stored at 4°C until use.
Preparation of Calibration Standards and Quality Control Samples (QC)
Blank plasma was spiked by appropriate amounts of working solutions to 0.5 mL for standards in concentrations of 0.5, 1.0, 5.0, 10.0, 20.0, 40.0 and 60.0 ng·mL−1. Quality control samples were prepared in the same way at concentrations of 1.0, 10.0 and 40.0 ng·mL−1.
Sample Preparation
0.5mL samples spiked with 30µL of I.S. and 100µL of 1M ammonia solution were mixed with 5mL ethyl acetate, vortexed for 2 min, and centrifuged at 2739g for 10 min. The supernatant was transferred and evaporated to dryness under a gentle nitrogen stream in water bath at 40°C. Residue was reconstituted with 100µL of mobile phase and 10µL was injected into HPLC-MS/MS system for analysis.
Data Treatment
Data were acquired and manipulated on the software of Analyst (Agilent HPLC-MS-MS, USA). Standard calibration curves were plotted by chromatographic ratio of hydrocodone-I.S versus the corresponding nominal plasma hydrocodone concentrations (0.5-60.0ng·mL−1). A1/concentration2 weighted regression analysis was used to determine the slope, intercept and coefficient of determination (c2).
Pharmacokinetic and Bioequivalence Studies
Twenty healthy Chinese volunteers, aged 22.75±1.37 years, were involved and all obtained written informed consents. The protocol has been approved by the Ethic Committee of Qilu Hospital.
A two-way crossover design study with one-week washout period was carried out. Volunteers were divided into two groups randomly and orally given test or reference Hycodan and Acetaminophen tablets (two tablets, containing 10mg hycodan) in the morning after 10h fasting, respectively. 3mL of blood was collected before and 0.17, 0.33, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 24h after administration; plasma were separated and stored at −20°C for analysis. The second experimental period was carried out as mentioned above one week later with the exchanging of test and reference.
The main pharmacokinetic parameters and the bioequivalence of hydrocodone test/reference were calculated and evaluated by Drug and Statistic software (DAS, version 2.0.1, by Sun et al., China).
RESULTS AND DISCUSSIONS
Optimization of HPLC-MS/MS Condition and Sample Preparation
A 1200 series HPLC system was a new product of Agilent. Its advantage was rapid separation with a low flow-rate. When the chromatographic conditions were optimized, the trifluoroacetic acid (TFA) Citation[3] was replaced by acetic acid, which was more commonly used. The mobile phase was acetonitrile-water (78:22, v/v, 0.1% acetic acid) and flow-rate was 0.2 mL·min−1, which was lower than 0.7mL·min−1 of the existing method.
Hydrocodone had stronger signal responses in the positive ion mode than the negative ion mode and higher sensitivity was achieved by ESI. The protonated molecular at 300.3 ([M + H]+) for hydrocodone and 341.3 ([M + H]+) for canrenone appeared in the full scan spectra, respectively. Under the selected fragmentation conditions, several product ions were observed. For hydrocodone, the most abundant fragment was at m/z 199.2 when the collision voltage was 40V, and for canrenone the fragment at m/z 107.2 was strongest with a collision voltage of 30V. To obtain the best sensitivity for the quantification, the transition patterns of 300.3→199.2 and 341.2→107.2 were chosen to monitor hydrocodone and canrenone, respectively. The mass-spectrogram of hydrocodone and I.S. are shown in .
Figure 1. The mass spectra of hydrocodone (A) and I.S. (B).
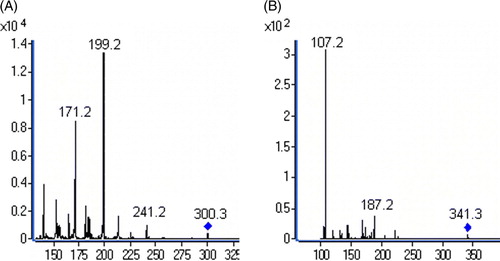
In the existing method, solid-phase extraction method was used Citation[3]. In order to decrease the cost for the quantitation of a large amount of samples, we tried to use other methods instead of solid-phase extraction. In this study, protein precipitation and liquid-liquid extraction methods were tested. The prior method were discarded because of high interference and low extraction recovery. Ethyl acetate, dichlormethane and chloroform were tested as the extraction solvents; it was found that ethyl acetate was better than the others, and the addition of appropriate amounts of ammonia solution improved extraction recovery. The procedure of extraction was simple and rapid.
Select of Internal Standard
Isotope-labeled hydrocodone was expensive and difficult to obtain, so several substances were tested as an internal standard. Canrenone was selected because it was stable, high responding and its retention time was short enough to reduce the analysis time.
Validation Procedure
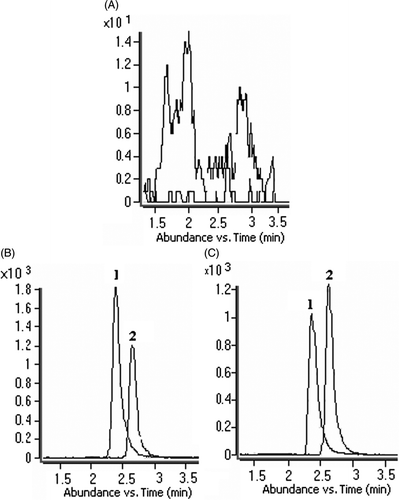
Specificity: Five batches of blank plasma were analyzed on HPLC-MS-MS to assess specificity. The retention times of hydrocodone and I.S. were about 2.4min and 2.6min, respectively. No distinguished interfering peaks for hydrocodone and I.S. were found. The chromatograms of blank plasma, standards solution and a sample of volunteers are shown in . Matrix effects were evaluated by comparison of blank extracted samples with three concentration levels of QCs. No matrix effect was detected.
Figure 2. The chromatograms of blank plasma (A), standard solution (B), and sample (C). 1: hydocodone, 2: I.S.
Calibration and LLOQ
Five sets of calibration curves were prepared over the concentration range of 0.5∼60.0 ng·mL−1. A good linearity was obtained; the regression coefficient was 0.9928. The lower limit of quantification (LLOQ) was 0.1ng·mL−1, which wasdetermined on five replicates of samples.
Extraction Recovery
The recovery was calculated at three concentration levels: 1.0, 10.0 and 40.0 ng·mL−1 of hydrocodone standards. Two series of five replicates of samples were prepared as “Sample Preparation,” one with blank plasma and another with water. The extraction recovery was obtained from the area ratio of spiked plasma samples to correspond to the sample without plasma. The average extraction recovery of hydrocodone of 1.0, 10.0 and 40.0 ng·mL−1 was 72.50%.
Precision and Accuracy
The precision and accuracy of the method were assessed by intra-day and inter-day RSD (relative standard deviation) and RE (relative error). The intra-day average accuracy was 3.33% with a precision of 2.43%. The inter-day average accuracy was 4.16% with a precision of 4.64%. The results are shown in .
Table 1. Precision, accuracy and stabilities of hydrocodone in plasma (n = 5, x±sd)
Stability
To investigate the stability of hydrocodone during preparation and storage processes, QC samples were subjected to room temperature (25°C) for 4h, freezed and thawed three cycles and stored at −20°C for 1 day and 7 days. The results are shown in . No significant degradation occurred during preparation and storage processes.
Application in Pharmacokinetic Study
The developed analytical method was successfully applied to a pharmacokinetic study and bioequivalent evaluation of hydrocodone in humans. The procedure of hydrocodone in humans was fitted to a double-compartmental model with a lag time. The pharmacokinetic parameters of hydrocodone test and reference were as follows: the time to peak concentration (Tmax) was 1.44±0.59h and 1.51±0.60h; the peak concentration (Cmax) was 21.72±6.10ng·mL−1 and 20.57±5.38 ng·mL−1; the half-life (T1/2) was 5.37±0.86h and 5.32±1.38h; the mean area under the concentration-time curve from zero to 24.0h (AUC0−24) and infinite time (AUC0−∞) were 135.49±33.84, 142.38±35.77ng·mL−1·h, 133.01±30.81, 139.24ng·mL−1·h, respectively. The relative bioavailability of hydrocodone in test was 102.9±6.9%. The test and reference Hycodan and Acetaminophen tablets were bioequivalent for hydrocodone.
CONCLUSIONS
A rapid, highly sensitive and specific high-performance liquid chromatography, coupled with tandem mass spectrometry (HPLC-MS-MS) quantitation method, was developed and validated for the determination of hydrocodone in human plasma. It was successfully applied to the pharmacokinetic and bioequivalence studies of hydrocodone. In comparison with the other methods previously reported, the method is easy to handle, more convenient and low cost for analysis of large amount of biological samples.
Acknowledgement
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Zheng H. Medicinal Chemistry [M]5th, ed. People's Health Press, Beijing 2004; 40–41
- Wu J. Studies on the analysis of hydrocodone and its metabolite in human urine by GC/MS. Acta Pharmaceutica Sinica 1997; 32(4)305–309
- Chen, Yu-Luan, Hanson, Glenn D., Jiang, Xiangyu,, et al. 2002. Simultaneous determination of hydrocodone and hydromorphone in human plasma by liquid chromatography with tandem mass spectrometric detection. Journal of Chromatography B, 769: 55–64.