
Abstract
Context: The role of hepatocyte growth factor (HGF) in diabetic kidney damage remains controversial.
Objective: To test the hypothesis that high glucose levels activate pathways related to HGF and its receptor Met and that this could participate in glucose-induced renal cell damage.
Materials and methods: HK2 cells, a human proximal tubule epithelial cell line, were stimulated with high glucose for 48 hours. Levels of pMet/Met, pEGFR/EGFR, pSTAT3/STAT3, pAkt/Akt and pERK1/2/ERK1/2 were studied by immunoblotting. Absence of HGF was verified by qRT-PCR and ELISA.
Results: High glucose level activated Met and its downstream pathways STAT3, Akt and ERK independently of HGF. High glucose induced an integrin ligand fibronectin. HGF-independent Met phosphorylation was prevented by inhibition of integrin α5β1, Met inhibitor crizotinib, Src inhibitors PP2 and SU5565, but not by EGFR inhibitor AG1478. High glucose increased the expression of TGFβ-1, CTGF and the tubular damage marker KIM-1 and increased apoptosis of HK2 cells, effects inhibited by crizotinib.
Conclusion: High glucose activated Met receptor in HK2 cells independently of HGF, via induction of integrin α5β1 and downstream signaling. This mode of Met activation was associated with tubular cell damage and apoptosis and it may represent a novel pathogenic mechanism and a treatment target in diabetic nephropathy.
Introduction
Diabetic nephropathy is a consequence of hyperglycemia and the main cause of chronic renal failure and end-stage renal disease, which is usually diagnosed at later stages [Citation1,Citation2]. Differential sensitivity of individual cell types to high glucose leads to an altered expression and activity of factors responsible for damage and progression in different tissues [Citation3,Citation4]. Hepatocyte growth factor (HGF) was originally identified as a motility factor and a scatter factor for hepatocytes [Citation5–7]. It has mitogenic, motogenic, morphogenic and anti-apoptotic effects. In addition, HGF has a role in construction, remodeling and protection of tissue structures during development and regeneration and plays a protective role in several diseases, including nephropathies [Citation8–10]. Several studies suggest that HGF plays an important role in renal regeneration, and has mitogenic and anti-apoptotic effects on renal tubular cells, podocytes and endothelial cells of the kidney [Citation8]. However, it was also reported that diabetes-related glomerular ultrafiltration of growth factors including HGF may lead to renal interstitial fibrosis [Citation11]. The effects of HGF are mediated by the Met receptor (Met), a member of the tyrosine kinase receptor superfamily, which is expressed in epithelial cells of many organs, including the liver, pancreas, prostate, kidney, muscle and bone marrow [Citation12,Citation13]. Met activates signaling pathways including the ERK1/2 and PI3K/Akt pathway, which are responsible for the cell survival response to Met signaling. Direct binding of transcription factor signal transducer and activator of transcription (STAT3) to Met results in STAT3 phosphorylation, dimerization and translocation to the nucleus. STAT3 activation by Met has been shown to be important in tubulogenesis [Citation13]. High glucose levels induce fibronectin overexpression in the kidney of diabetic rats directly without the influence of other extracellular matrix proteins [Citation14]. Fibronectin is important for cell adhesion, is a potential ligand for integrins. All integrins are composed of different α and β subunits, the combination of which determines the specificity for extracellular ligands and activation of intracellular signaling events [Citation15]. Integrin α5β1 is a prototype specific fibronectin receptor [Citation16]. Cell adhesion to integrins was shown to induce ligand-independent activation of Met in cancer cells [Citation17], however, such a pathway was not reported for diabetic nephropathy. Hyperglycemia could thus be a trigger of progressive molecular changes causing end-stage organ damage which are activated independently from further glucose regulation and fibronectin induction might be one of such pathways [Citation18]. In search of novel pathogenic mechanisms in diabetic nephropathy, the aim of our present study was to determine whether and how high glucose levels can induce fibronectin with subsequent integrin and HGF/Met signaling in human renal tubular epithelial cells.
Methods
Reagents and cell culture
Renal tubular epithelial human cell line HK2 (ATCC, LGC Standards, Germany, mycoplasma negative) was grown at 37 °C/5% CO2 in DMEM/F12 medium (Gibco, Life Technologies, Bleiswijk, Netherlands) supplemented with 5% fetal bovine serum (FBS), 100 IU/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, ITS supplement (all Life Technologies, Bleiswijk, Netherlands), tri-iodothyronine 20 ng/ml, 50 ng/ml hydrocortisone and 5 ng/ml prostaglandin E1 (all Sigma-Aldrich, Zwijndrecht, The Netherlands). The culturing wells were randomly assigned to specific experimental conditions, including control groups (stimulated with vehicle, mannitol or glucose, respectively) and experimental groups incubated with the corresponding inhibitors. In each experiment, two wells per condition represented technical replicates. All experiments were repeated three times. For details, see the Electronic Supplementary material.
Immunoblotting
Ninety percent confluent HK2 cells were serum-starved 24 h and grown for 48 h in DMEM/F12 medium supplemented with glucose (40 mM) or mannitol (40 mM). For inhibition studies, HK2 cells were treated with the corresponding inhibitors for 2 h before and throughout the incubation under high glucose conditions. In additional experiments, cells were seeded in six-well plates precoated with fibronectin and cultured for 48 h with or without the corresponding inhibitors. In acute experiments, the cells were stimulated with HGF or EGF for 5–30 min. For details of cell harvesting and immunoblotting, see the Electronic Supplementary material.
Real time-PCR
Real-time PCR was performed on cDNA obtained by reverse transcription of total RNA extracted using Trizol LS reagent (Invitrogen, Carlsbad, CA). For primer sequences and further details, see the Electronic Supplementary material.
Human HGF ELISA
The conditioned cell culture medium and the corresponding cell lysate were harvested and assayed in triplicates for HGF protein concentration using the DuoSet ELISA (R&D Systems) according to the manufacturer’s instructions. Conditioned medium from the cells treated with human recombinant HGF was used as a positive control.
Fibronectin immunofluorescence and microscopy
Following stimulation, paraformaldehyde-fixed cells were incubated with anti-fibronectin primary antibody and subsequently FITC-labeled secondary antibody. For further details, see the Electronic Supplementary material.
TUNEL assay
For detection of apoptotic cells during different conditions, we used In Situ Cell Death Detection Kit, Fluorescein (Sigma-Aldrich, Zwijndrecht, The Netherlands). For details, see the Electronic Supplementary material.
Statistical analysis
Results are shown as mean of three independent experiments with two technical replicates per each experimental condition. The results are reported as mean ± standard error of the mean (SEM). Statistical analysis was performed using the GraphPad Prism 5 software (La Jolla, CA). The data were first evaluated by the Shapiro–Wilk test for normality of data distribution. Subsequently, the appropriate statistical method was selected, either ANOVA with the Tukey post hoc test or the Kruskal–Wallis nonparametric test with Dunn’s post hoc comparison. p Values <.05 were considered significant.
Results
HGF independent activation of Met receptor in tubular epithelial cell line (HK2) following incubation with high glucose medium
Incubation of cells in high glucose medium for two days led to the phosphorylation of Met receptor at Y1234/1235 (). This activation started already after 3 h in high glucose medium (Figure S3). Phosphorylation of Met was significantly higher (*p < .05) after 48 h in high glucose conditions compared with controls and osmotic control (mannitol medium). Protein and mRNA levels of total Met were not changed (). mRNA expression of HGF, a Met ligand, was undetectable (). HGF was also undetected by ELISA in cell lysates except if cells were treated with exogenous HGF (*p < .001 vs. all groups) (). Short-term stimulation of HK2 cells with HGF activated Met within 5 min of stimulation at least for 60 min (Figure S1A). Previous reports indicated that Met could also be activated via EGFR [Citation19]. HK2 were therefore treated with EGF for 5, 30, 60 min and two days. Although EGF-dependent activation of Met was observed after 48 h (Figure S2A), neither the protein level of total EGFR nor phosphorylated EGFR was changed during 48 h of incubation in medium with high glucose level (Figure S2B). Moreover, addition of the EGFR inhibitor AG1478 did not result in changes in protein levels of either Met or pMet after 48 h of incubation in high glucose medium (Figure S2C).
Figure 1. HGF independent activation of Met receptor in tubular epithelium cell line (HK2) following the incubation in high glucose medium. HK2 cells were incubated in control medium (Veh), medium supplemented with mannitol (Man, 40 mM) or glucose (Glu, 40 mM) for 48 h. (A) Met phosphorylation was induced upon high glucose (but not mannitol) incubation without any changes in total Met protein expression. (B) Met receptor mRNA was not altered by high glucose incubation. (C) HGF was undetectable in lysates or conditioned media of HK2 cells by ELISA. Additional stimulation with HGF (20 ng/ml) was used as a positive control (renal endothelial cells). (D) HK2 did not express HGF mRNA. PCR products were visualized in agarose gel, PC – positive control (human endothelial cells).
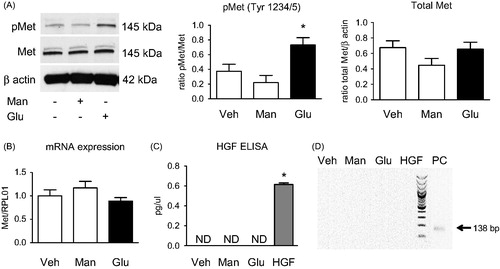
Crizotinib blocked the high-glucose induced phosphorylation of Met receptor in HK2
Protein levels of pMet and total Met were studied by using immunoblot after treatment of HK2 cells with crizotinib, a Met inhibitor, in high glucose medium. The results showed a significantly higher phosphorylation of Met in high glucose medium for 48 h and consequently also that crizotinib could inhibit this phosphorylation in comparison to control (*p < .05) as well as to high glucose group (+p < .01). The protein levels of total Met remained unchanged (). The inhibitory effect of crizotinib was also observed after short term stimulation of HK2 cells with HGF (Figure S1B). Phosphorylation of Met subsequently led to activation of multiple downstream signal pathways like STAT3, Akt or ERK 1/2 (Figure S1A). Crizotinib was observed to have an inhibitory effect on phosphorylation of STAT3 ().
Figure 2. Crizotinib blocks the high-glucose induced phosphorylation of Met receptor in HK2. Cells were treated with control medium (Veh), medium supplemented with high glucose (Glu, 40 mM) or high glucose medium with crizotinib (100 nM) (Glu + crizo) for 48 h. (A) Representative immunoblots of phosphorylated Met (pMet) and total Met receptors, Crizotinib markedly reduces Met phosphorylation without affecting total Met expression. (B) Representative immunoblots of downstream pathways of activated Met receptor stimulation. High glucose medium induces phosphorylation of STAT3, Akt and ERK1/2. The phosphorylation is prevented by crizotinib incubation.
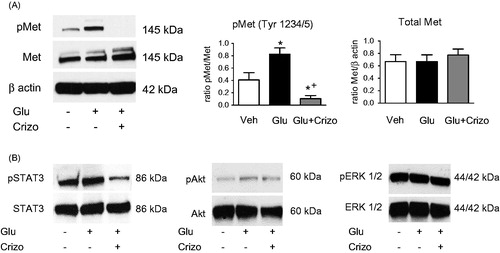
High glucose induced synthesis of fibronectin in HK2, fibronectin induced HGF-independent Met receptor phosphorylation
Significantly higher fibronectin mRNA levels were observed in HK2 cells under high glucose conditions by RT-PCR (*p < .05) (). Overexpression of fibronectin was also observed by immunofluorescence, quantified as a positive area per total area (*p < .05 vs. control) (). The antioxidant N-acetylcysteine (NAC) was used to prevent ROS which arise during high glucose levels. After treatment of cells with NAC, we observed high phosphorylation of Met receptor in cells which were stimulated with high glucose and phosphorylation of Met was decreased after pretreatment with NAC (). When cells were seeded into fibronectin-precoated plates, we observed a higher phosphorylation of Met as a result of high level of fibronectin which was inhibited by 2-h pretreatment with crizotinib ().
Figure 3. High glucose induces synthesis of fibronectin in HK2, and fibronectin induces ligand-independent Met receptor phosphorylation. (A) Fibronectin mRNA induction upon high glucose incubation (Glu) was partially reduced by antioxidant N-acetyl-cysteine (NAC, 5 mM), suggesting involvement of reactive oxygen species. (B) Immunofluorescent staining for cellular fibronectin in HK2 incubated in control (Veh), high glucose medium, (Glu) or Glu with NAC (5 mM). (C) Representative blots showing high glucose-induced phosphorylation of Met receptor is partially blocked by co-incubation with antioxidant N-acetyl-cysteine (NAC, 5 mM). (D) Fibronectin induced Met receptor phosphorylation in HK2, which could be blocked by crizotinib. Representative blots show Met receptor phosphorylation in cells seeded in uncoated, fibronectin-coated (0,1% solution) and fibronectin-coated plates incubated with crizotinib (100 nM) for 48 h. (B–D) The plates were coated with fibronectin and then treated with NAC (5 mM) or SU6656 (5 μM), a selective inhibitor of SRC, for 48 h.
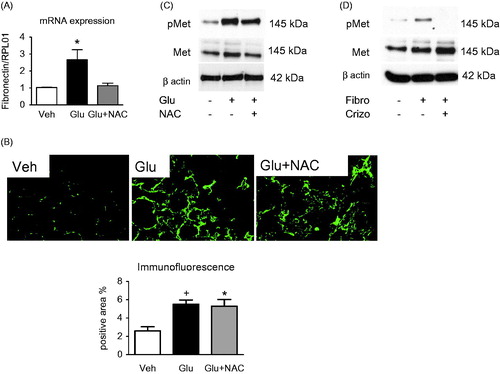
High-glucose-induced phosphorylation of Met receptor is mediated by activation of integrin α5β1 and Src tyrosine kinase
To elucidate signaling pathways leading to Met activation by high glucose, we treated HK2 cells for 48 h with an anti-integrin antibody as an inhibitor of integrin β1subunit in the presence or absence of high glucose medium. Inhibition of the integrin β1subunit led to a decreased phosphorylation of Met (). Additionally, we used a specific antibody against integrin α5β1 as a specific inhibitor of both subunits of integrin α5β1 for 48 h in the presence or absence of high glucose level. Immunoblots showed that inhibition of α5β1 subunits of integrin led to a reduction of phosphorylation of Met (). The results suggest a crosstalk between integrin α5β1 and activation of Met and one of the pathways is mediated by Src. Accordingly, Src inhibitor PP2 inhibited phosphorylation of Src, which was higher during high glucose conditions, as well as the phosphorylation of Met, while total Src and total Met were not changed (). To further confirm this, a more specific Src inhibitor SU5565 was used. HK2 cells were treated with the inhibitor for 2 h before being reseeded on fibronecting-precoated plates for 48 h. The stimulation by fibronectin led to phosphorylation of Met which could be inhibited by SU5565 (). Similar results were obtained with plates pre-coated with collagen 1 (Figure S4).
Figure 4. High-glucose-induced phosphorylation of Met receptor is mediated by activation of integrin α5β1 and Src tyrosine kinase. Representative immunoblots show Met phosphorylation in HK2 incubated for 48 h in control, high glucose medium (Glu) or in high glucose medium coincubated with (A) anti-integrin β1 (10 μg/ml) blocking antibody, (B) anti-integrin α5β1 (10 μg/ml) blocking antibody or (C) Src tyrosine kinase inhibitor PP2 (10 μM) (D). Fibronectin-induced Met phosphorylation is inhibited by Src tyrosine kinase inhibitor SU6656 (5 μM). Representative blots show Met receptor phosphorylation in cells seeded in uncoated, fibronectin-coated (0.1% solution) and fibronectin-coated plates incubated with Src tyrosine kinase inhibitor SU6656 (5 μM) for 48 h.
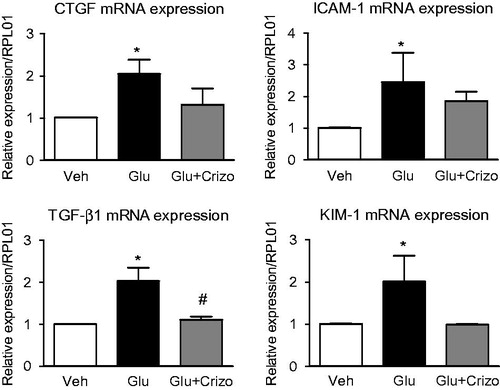
Effect of crizotinib on the expression of damage markers in the tubular epithelium cell line (HK2) during the incubation in high glucose medium
Additionally, we examined the effect of high glucose level on the gene expression of CTGF, ICAM-1, TGF-β1 and KIM-1, known markers of renal damage. Their expressions were increased upon high glucose conditions in comparison to control group (*p < .05). This overexpression was reduced by co-incubation with crizotinib. We observed a significantly lower expression of TGF-β1 in the group treated with crizotinib (#p < .05) in comparison to the group incubated in high glucose (*p < .01) ().
Figure 5. Effect of crizotinib on the expression of damage markers in tubular epithelium cell line (HK2) during incubation in high glucose medium. (A) mRNA levels of CTGF, ICAM-1, TGF-β1 and KIM were increased upon high glucose incubation (Glu) as determined by RT-PCR analysis. Such an induction was reduced by co-incubation with crizotinib (Crizo, 100 nM).
Effect of crizotinib on apoptosis of the HK2 cells
To determine apoptotic cell death, we used In Situ Cell Death Detection Kit, Fluorescein. TUNEL positive cells were detected under the fluorescence microscope in control cultures as well as in cultures stimulated in high glucose medium and high glucose medium with crizotinib. The percentage of positive cells was reduced in the group which was stimulated by high glucose medium with crizotinib (*p < .05) (Figure S5).
Discussion
We provide novel evidence showing that long-term stimulation of HK2, a human renal tubular epithelial cell line, with high glucose levels leads to HGF-independent activation of Met and its downstream signaling pathways. Our results indicate that high glucose levels increase fibronectin expression, which interacts with integrin α5β1 in HK2 cells to activate Src and subsequently Met and concomitantly, high glucose increased markers of renal damage, an effect abrogated by Met inhibitor. These findings could be of importance as a potential novel mechanism of hyperglycemia-induced renal damage.
HGF, first identified in 1984 [Citation20], is a ligand of Met receptor and has multiple roles, including mitogenic, motogenic, anti-apoptotic, angiogenic, angioprotective and morphogenic effects [Citation21]. Our results showed that high glucose level stimulated phosphorylation of Met at Y1234/1235 already after 3 h of stimulation, lasting for at least 48 h. Although HGF release from the cells or HGF presence in the media could be a suspected mechanism, the cytokine was not detected in cell lysates on mRNA or protein level, nor was it present in culture media. Thus, high glucose leads to the activation of Met in HK2 cells independently of HGF. We demonstrated that Met can be trans-activated by EGFR or can directly interact with EGFR, and in addition can be activated by using EGFR ligand EGF [Citation19,Citation22]. Our results showed that neither total EGFR nor phosphorylated EGFR were changed during 48 h of incubation in medium with high glucose level or after treatment with the EGFR inhibitor AG1478. We observed that stimulation of HK2 cells with EGF can lead to phosphorylation of Met after 48 h. However, it was suggested that stimulation with EGF markedly increased mRNA as well as protein expression of fibronectin after 24 h [Citation23]. As we have shown, overexpression of fibronectin can lead to the activation of Met receptor via integrin. This may explain activation of Met after treatment with EGF for 48 h.
HGF is expressed in tubular epithelial cells (interstitial cells, endothelial cells and macrophages) and in mesangial cells and endothelial cells of the glomerulus. HGF that is produced in the kidney, has paracrine and autocrine effects on renal cells, whereas HGF expressed in distant organs acts through an endocrine mechanism [Citation8]. As HGF is not produced by renal epithelial cells, we stimulated cells with recombinant HGF. Phosphorylation of Met was detected within 5 min, this effect declined after 1 h, suggesting that HGF-dependent activation of Met in HK2 cells is transient and does not persist for a long time. Earlier studies suggest that HGF has a pleiotropic role in tubular repair and regeneration after acute renal injury [Citation24]. HGF stimulates cell proliferation and differentiation during acute renal failure, thereby contributing to repair and regeneration. Similar protective effect of HGF was observed on cell death and renal dysfunction of tubular epithelial cells after acute renal failure induced by a folic acid [Citation24,Citation25]. HGF binding to Met results in receptor homodimerization and phosphorylation of the tyrosine kinase domain on tyrosine residue (Y1234/1235) [Citation26]. Multiple downstream signaling pathways are activated, including ERK/MAPK, PI3K/Akt pathway, which is responsible for cell survival, direct binding of STAT3 to Met resulting in STAT3 phosphorylation, dimerization and its translocation to the nucleus [Citation27–30]. These signaling molecules were phosphorylated during short term exogenous HGF stimulation, as well as HGF-independent activation of Met following incubation with high glucose. Crizotinib is an ATP-competitive receptor kinase inhibitor, highly specific for Met. Crizotinib inhibited Met phosphorylation and Met-dependent proliferation, migration, and invasion of human tumor cells in vitro [Citation31,Citation32]. We proved that crizotinib blocked HGF-dependent activation of Met as well as the high-glucose induced phosphorylation of Met in HK2 cells. Phosphorylation of Met subsequently led to the activation of multiple downstream signal pathways like STAT3, ERK 1/2 or AKT and crizotinib was observed to have an inhibitory effect on phosphorylation of STAT3.
Inflammatory processes can lead to the accumulation of extracellular matrix proteins, including fibronectin that was reported to be increased in the glomerular basement membrane and mesangial cells in the early stages of diabetic nephropathy. This effect was also observed by using high glucose level as a mimicking diabetic condition [Citation14,Citation33,Citation34]. We showed significantly higher levels of fibronectin mRNA and protein in the HK2 cells. It was shown that in mesangial cells, high glucose-induced matrix assembly and collagen IV deposition depended on fibronectin [Citation35]. Together with increased fibronectin, we observed higher phosphorylation of Met in HK2 cells stimulated with high glucose medium and mimicked stimulation of cells in fibronectin-precoated plates. We propose that overexpression of fibronectin in HK2 cells during hyperglycemia leads to ligand-independent Met phosphorylation and that this phosphorylation can be inhibited by crizotinib as a specific inhibitor for Met or NAC as an inhibitor upstream in the pathway of activation of Met. To date, 24 isoforms of integrins, cell surface receptors with α and β subunits, have been identified, with multiple signaling effects. Recent studies showed that integrin α5β1 is expressed in mesangial cells in the glomerulus [Citation36,Citation37]. We detected it also in HK2 cells. Pre-treatment of HK2 cells with anti-integrin β1 or anti-integrin α5β1 blocking antibodies in normal or high glucose medium markedly reduced the phosphorylation of Met. HGF/Met signaling can crosstalk with integrins by different pathways. First, Met can directly associate with integrins at the plasma membrane. Second, HGF/Met and integrin signaling pathways lead to the activation of the same signaling pathways, including FAK and Src. Third, HGF/Met signaling leads to alteration in the expression of integrins [Citation16]. Our results suggest that there is crosstalk between integrin α5β1 and activation of Met and that Src kinase could be the linking element. Treatment of HK2 cells with Src inhibitors PP2 or the highly specific SU5565 reduced Met as well as Src phosphorylation.
Several signaling molecules and growth factors including fibronectin, TGF-β1, CTGF (connective tissue growth factor), ICAM-1 (intercellular adhesion molecule-1) and KIM-1 (kidney injury molecule-1) play a role in the pathogenesis of diabetic nephropathy and are used as renal damage markers. Deposition of fibronectin can lead to the loss of renal function [Citation14,Citation38–40]. Expression on mRNA level of all other damage markers such as CTGF, TGF-β1, ICAM-1 and KIM-1 was significantly higher after stimulation in high glucose medium and we observed a significantly lower level of TGF-β1 and a trend of decline in the expression of CTGF, ICAM-1 and KIM-1 by crizotinib. TGF-β1 is known as a main pro-fibrotic signal molecule in diabetic nephropathy, especially in the processes of glomerulosclerosis and intestitial fibrosis. During hyperglycemia, TGF-β1 is increased mainly in mesangial cells, podocytes and tubular epithelial cells. It was suggested that inhibition of TGF-β1 was associated with a reduction of mRNA level of collagen IV and fibronectin [Citation38]. The damaging effect of hyperglycemia on the kidney includes also tubular atrophy, tubular necrosis and tubular dysfunction. High glucose level can lead to the generation of free radicals as well as oxidative stress in tubular cells, which can influence several biological processes such as proliferation, extracellular matrix deposition and apoptosis [Citation41]. In our study, we observed higher apoptosis after two days stimulation with high glucose, an effect inhibited by Met blocker crizotinib. Reduction of high glucose–induced apoptosis was observed in HK2 cells. Apoptosis of renal tubular cells during diabetes is the result of hyperglycemia but the exact mechanism behind this is still unclear. HGF was suggested to have a protective effect observed as a decrease in the level of stress fibers, focal contacts, and level of extracellular fibronectin. HGF/Met signaling was also suggested to have an anti-apoptotic effect on renal epithelial cells, which was caused by higher expression of Bcl-xL mediated by HGF. Unilateral nephrectomy was used as a model to study the effect of HGF/Met signaling during the regeneration processes [Citation42]. Expression of HGF markedly increased in the remaining kidney after nephrectomy. A positive effect was based on the high level of renal and plasma HGF and Met, which was induced by acute renal injury [Citation43]. Chronic renal diseases are characterized by fibrosis as a result of changes in the extracellular matrix deposition. Prolonged exposure of cells to hyperglycemia leads to the reduction of HGF level but on the other hand to overexpression of TGF-β1. As TGF-β1 is profibrotic, the supplementation of HGF could be a novel therapeutical option for chronic renal diseases [Citation8]. In db/db mice, HGF gene therapy decreased fibronectin accumulation and enhanced tissue repair mediated by bone marrow stem cells [Citation44]. In contrast, our results demonstrate that inhibition of Met signaling reduces high glucose related overexpression of damage markers and apoptosis in renal tubular epithelial cells.
In summary, we identified a signaling pathway in the human renal tubular epithelial cell line HK2 during stimulation of high glucose medium, which leads to HGF-independent phosphorylation of Met (Figure S6). Our study demonstrated that a high glucose level alone led to phosphorylation of Met and activation of downstream pathways and signaling molecules including the phosphorylation of STAT3, AKT and ERK 1/2. High glucose level stimulated the expression of fibronectin, the main integrin ligand. Through binding of fibronectin to integrin α5β1, followed by Src activation, phosphorylation of Met occurs without the presence of HGF. Although it still remains to be proven, our findings strongly indicate this as a novel signaling pathway constituting a new pathogenic mechanism and possibly a target for the therapy of diabetic nephropathy.
Supplementary Material
Download PDF (600.3 KB)Disclosure statement
The authors have nothing to disclose.
Additional information
Funding
Related Research Data
References
- Remuzzi G, Schieppati A, Ruggenenti P. Clinical practice. Nephropathy in patients with type 2 diabetes. N Engl J Med. 2002;346:1145–1151.
- Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol. 2003;14:1358–1373.
- Parving HH, Tarnow L, Rossing P. Genetics of diabetic nephropathy. J Am Soc Nephrol. 1996;7:2509–2517.
- Kiencke S, Handschin R, von Dahlen R, et al. Pre-clinical diabetic cardiomyopathy: prevalence, screening, and outcome. Eur J Heart Fail. 2010;12:951–957.
- Weidner KM, Arakaki N, Hartmann G, et al. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc Natl Acad Sci USA. 1991;88:7001–7005.
- Nakamura T, Nishizawa T, Hagiya M, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–443.
- Stoker M, Gherardi E, Perryman M, et al. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327:239–242.
- Matsumoto K, Nakamura T. Hepatocyte growth factor: renotropic role and potential therapeutics for renal diseases. Kidney Int. 2001;59:2023–2038.
- Liu Y, Yang J. Hepatocyte growth factor: new arsenal in the fights against renal fibrosis? Kidney Int. 2006;70:238–240.
- Okada H, Kalluri R. Cellular and molecular pathways that lead to progression and regression of renal fibrogenesis. Curr Mol Med. 2005;5:467–474.
- Wang S-N, Hirschberg R. Growth factor ultrafiltration in experimental diabetic nephropathy contributes to interstitial fibrosis. Am J Physiol-Ren Physiol. 2000;278:F554–F560.
- Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516.
- Organ SL, Tsao M-S. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3:S7–S19.
- Falk RJ, Scheinman JI, Mauer SM, et al. Polyantigenic expansion of basement membrane constituents in diabetic nephropathy. Diabetes. 1983;32 Suppl 2:34–39.
- Steffes MW, Brown DM, Basgen JM, et al. Glomerular basement membrane thickness following islet transplantation in the diabetic rat. Lab Investig J Tech Methods Pathol. 1979;41:116–118.
- Chan P-C, Chen S-Y, Chen C-H, et al. Crosstalk between hepatocyte growth factor and integrin signaling pathways. J Biomed Sci. 2006;13:215–223.
- Wang R, Kobayashi R, Bishop JM. Cellular adherence elicits ligand-independent activation of the Met cell-surface receptor. Proc Natl Acad Sci USA. 1996;93:8425–8430.
- Roy S, Sala R, Cagliero E, et al. Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. Proc Natl Acad Sci USA. 1990;87:404–408.
- Presnell SC, Stolz DB, Mars WM, et al. Modifications of the hepatocyte growth factor/c-met pathway by constitutive expression of transforming growth factor-alpha in rat liver epithelial cells. Mol Carcinog. 1997;18:244–255.
- Nakamura T, Nawa K, Ichihara A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun. 1984;122:1450–1459.
- Nakamura T. Structure and function of hepatocyte growth factor. Prog Growth Factor Res. 1991;3:67–85.
- Jo M, Stolz DB, Esplen JE, et al. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem. 2000;275:8806–8811.
- Mimura Y, Ihn H, Jinnin M, et al. Epidermal growth factor induces fibronectin expression in human dermal fibroblasts via protein kinase C delta signaling pathway. J Invest Dermatol. 2004;122:1390–1398.
- Kawaida K, Matsumoto K, Shimazu H, et al. Hepatocyte growth factor prevents acute renal failure and accelerates renal regeneration in mice. Proc Natl Acad Sci USA. 1994;91:4357–4361.
- Dai C, Yang J, Liu Y. Single injection of naked plasmid encoding hepatocyte growth factor prevents cell death and ameliorates acute renal failure in mice. J Am Soc Nephrol. 2002;13:411–422.
- Rodrigues GA, Park M. Autophosphorylation modulates the kinase activity and oncogenic potential of the Met receptor tyrosine kinase. Oncogene. 1994;9:2019–2027.
- Graziani A, Gramaglia D, dalla Zonca P, et al. Hepatocyte growth factor/scatter factor stimulates the Ras-guanine nucleotide exchanger. J Biol Chem. 1993;268:9165–9168.
- Xiao GH, Jeffers M, Bellacosa A, et al. Anti-apoptotic signaling by hepatocyte growth factor/Met via the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase pathways. Proc Natl Acad Sci USA. 2001;98:247–252.
- Boccaccio C, Andò M, Tamagnone L, et al. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature. 1998;391:285–288.
- Syed ZA, Yin W, Hughes K, et al. HGF/c-met/Stat3 signaling during skin tumor cell invasion: indications for a positive feedback loop. BMC Cancer. 2011;11:180.
- Zou HY, Li Q, Lee JH, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67:4408–4417.
- Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6:3314–3322.
- Dixon AJ, Burns J, Dunnill MS, et al. Distribution of fibronectin in normal and diseased human kidneys. J Clin Pathol. 1980;33:1021–1028.
- Weiss MA, Ooi BS, Ooi YM, et al. Immunofluorescent localization of fibronectin in the human kidney. Lab Invest. 1979;41:340–347.
- Miller CG, Pozzi A, Zent R, et al. Effects of high glucose on integrin activity and fibronectin matrix assembly by mesangial cells. Mol Biol Cell. 2014;25:2342–2350.
- Kerjaschki D, Ojha PP, Susani M, et al. A beta 1-integrin receptor for fibronectin in human kidney glomeruli. Am J Pathol. 1989;134:481–489.
- Kagami S, Border WA, Ruoslahti E, et al. Coordinated expression of beta 1 integrins and transforming growth factor-beta-induced matrix proteins in glomerulonephritis. Lab Investig J Tech Methods Pathol. 1993;69:68–76.
- Ban CR, Twigg SM. Fibrosis in diabetes complications: pathogenic mechanisms and circulating and urinary markers. VHRM. 2008;4:575–596.
- Sugimoto H, Shikata K, Hirata K, et al. Increased expression of intercellular adhesion molecule-1 (ICAM-1) in diabetic rat glomeruli: glomerular hyperfiltration is a potential mechanism of ICAM-1 upregulation. Diabetes. 1997;46:2075–2081.
- Zhao X, Zhang Y, Li L, et al. Glomerular expression of kidney injury molecule-1 and podocytopenia in diabetic glomerulopathy. Am J Nephrol. 2011;34:268–280.
- Habib SL. Diabetes and renal tubular cell apoptosis. World J Diabetes. 2013;4:27–30.
- Nagaike M, Hirao S, Tajima H, et al. Renotropic functions of hepatocyte growth factor in renal regeneration after unilateral nephrectomy. J Biol Chem. 1991;266:22781–22784.
- Liu Y, Tolbert EM, Lin L, et al. Up-regulation of hepatocyte growth factor receptor: an amplification and targeting mechanism for hepatocyte growth factor action in acute renal failure. Kidney Int. 1999;55:442–453.
- Flaquer M, Franquesa M, Vidal A, et al. Hepatocyte growth factor gene therapy enhances infiltration of macrophages and may induce kidney repair in db/db mice as a model of diabetes. Diabetologia. 2012;55:2059–2068.