
Abstract
Purpose
MicroRNA (miRNA) is known to be involved in the pathological process of congenital heart disease (CHD), and nodal modulator1 (NOMO1) is a critical determinant of heart formation. The present study aims to discover the effect of miR-33a-5p and NOMO1 on CHD.
Methods
Quantitative real-time polymerase chain reaction (qRT-PCR) was used to detect expressions of miR-33a-5p mimic or inhibitor and overexpressed NOMO1 plasmid orNOMO1 knockdown. Human cardiomyocyte progenitor cells (hCMPCs) proliferation was measured by cell counting kit-8 (CCK-8) at 24, 48 and 72 h. Flow cytometry was applied to investigate hCMPCs cell cycle progression and apoptosis. Expressions of cell apoptotic proteins Bax, Cleaved(C) caspase-3 and Bcl-2, and expressions of cardiomyocyte differentiation markers GATA4, troponin T (cTnT) and myocyte enhancer factor2C (MEF2C) in hCMPCs were identified by qRT-PCR and western blot. Target genes and potential binding sites of NOMO1 and miR-33a-5p were predicted with Targetscan 7.2, and was confirmed through dual-luciferase reporter assay.
Results
Up-regulation of miR-33a-5p inhibited hCMPCs proliferation, cell cycle G0/S transition but promoted hCMPCs apoptosis, which was partially mitigated by overexpressed NOMO1. NOMO1 was the target gene of miR-33a-5p. Expressions of Bax and C caspase-3 were enhanced but expressions of Bcl-2, GATA4, cTnT and MEF2C were reduced by up-regulation of miR-33a-5p, which was partially mitigated by overexpressed NOMO1.
Conclusion
Up-regulation of miR-33a-5p inhibited hCMPCs proliferation, cell cycle G0/S transition and differentiation into cardiomyocytes but promoted apoptosis via targeting NOMO1.
Introduction
Congenital heart disease (CHD) is a significant cause of neonatal morbidity and mortality occurring in just over 1% of neonates with heart murmurs [Citation1]. Cardiomyocytes proliferation and apoptosis are two important processes of cardiac morphogenesis [Citation2]. And CHD is often accompanied by accumulation of cell apoptosis [Citation3], low proliferation and dysregulation of differentiation [Citation4]. Nevertheless, neonates with CHD continue to increase in number [Citation5], which has created a huge burden to those families. Therefore, it is necessary to develop a novel therapeutic method for CHD.
MicroRNAs (miRNAs; miR), which are small non-coding RNAs, ∼22 bp in length that plays a key role in regulating gene expression and the regulatory function is on account of targeting mRNAs for cleavage or translational repression which contributes to a down-regulating of the corresponding miRNA targets [Citation6]. Over the past few decades, emerging evidence has unveiled the important functions of miRNAs that are important for the development of various organs [Citation7,Citation8], including the human heart [Citation9]. Of note, significant overexpression of miR-486 has been found in children with CHD, suggesting that miR-486 may participate in a common molecular pathway that regulates the pathogenesis of CHD [Citation10]. Importantly, Wu and colleagues [Citation11] discovered that human cardiomyocyte progenitor cells (hCMPCs) proliferation was enhanced by overexpression of miR-134 via targeting Meis2, indicating that miRNA-targeting techniques could be used as an alternative therapeutic approach to the exogenous application of gene on hCMPCs proliferation. As for miR-33a-5p, it has been reported that functions as a key regulator in multiple cancers. For example, up-regulation of miR-33a-5p has been demonstrated to restrain epithelial-mesenchymal transition (EMT) and metastasis of prostate cancer cells [Citation12]. And miR-33a-5p has been proved to inhibit the growth and migration of colorectal cancer by targeting MTHFD2 [Citation13]. MiR-33a-5p also plays a role in cardiovascular diseases, according to Liwei Diao et al. [Citation14] increased expression of miR-33a-5p was found in cardiac microvascular endothelial cell injury. Another study [Citation15] found that inhibition of miR-33a could inhibit cardiac fibrosis. These findings suggest that miR-33a-5p may play a role in promoting heart disease. However, there are rarely studies that research the effect of miR-33a-5p on CHD or heart-related diseases.
Through the biological information website targetscan v7.2, we predicted the target genes of the miR-33a-5p and Nodal modulator1 (NOMO1) aroused our attention. According to the published literature, H19 gene could suppress human trophoblast cell proliferation through encoding miR-675 which was targeted with NOMO1 [Citation16]. Moreover, the NOMO1 gene has also been identified that it is evidently down-regulated in human ventricular septal defect myocardium, and it was found that knockdown of NOMO1 gene inhibited the differentiation of P19 cells into cardiomyocytes [Citation17]. Nodal is a member of transforming growth factor-β (TGF-β), which acts as an essential role on heart development and formation of other visceral organs [Citation18]. Nodal is also important for the maintenance of pluripotency of embryonic stem cells and germ layer specification [Citation19]. NOMO1 gene has been proved that it positively regulates the expression of the master regulator and other key development genes involved in bone formation and cartilage development [Citation20]. And NOMO1 has also been proved to targeted with miR-675 in regulating trophoblast cell growth [Citation16]. But there are rarely studies introduced the relationship between miR-33a-5p and NOMO1. Therefore, we hypothesized for the first time that miR-33a-5p regulated the differentiation of hCMPCs into cardiomyocytes by targeting NOMO1.
The present study was designed to discover the effect of miR-33a-5p and NOMO1 on CHD, and further uncover whether NOMO1 was directly targeted with miR-33a-5p.
Materials and methods
Ethical statement
For executing the study, the approval was obtained from the ethics committee of Gansu Provincial Maternity and Child-care Hospital (approval number: XNK20190805), and the heart tissue of fetus after elective abortion was used in this study under written informed consent.
Cell isolation, culture and transfection
Human cardiomyocyte progenitor cells (hCMPCs) were acquired and cultured as the previous study described [Citation21]. Specifically, fetal heart tissue was collected after elective abortion and digested by collagenase to obtain a single cell suspension. The cardiomyocyte progenitor cells were isolated by magnetic cell sorting (MACS, 130-090-312; Miltenyi Biotech, Bergisch Gladbach, Germany) using the Sca-1 antibody (eBioscience, San Diego, CA, USA). hCMPCs were cultured in Dulbecco’s modified eagle medium (DMEM; PM150210, Procell Life Science & Technology Co., Ltd., Wuhan, China) containing 1% penicillin, 22% endothelial cell growth medium-2 (EGM-2), 10% fetal bovine serum (FBS), 1% minimal essential medium (MEM) nonessential amino acids and 66% medium 199, and the medium was changed every other day. The follow-up experiments would be conducted when the growth confluence of cell was about 80%∼90%.hsa-miR-33a-5p mimics (M; #B01001, sense: 5′-GUGCAUUGUAGUUGCAUUGCA-3′), inhibitor (I; #B04004, sense: 5′-UGCAAUGCAACUACAAUGCAC-3′) and mimic negative control (MC; #B04001, sense: 5′-UGAAUGUUGGAUCGCUUCAUG-3′) was synthesized by Genepharma (Shanghai, China), and inhibitor negative control (IC; sense: 5′-GCACUAUACAUGAACUCGCAA-3′) was synthesized by Thermo Fisher Scientific (AM17010, Waltham, MA, USA). pcDNA3.1 plasmid was purchased (VT1010, YouBio, China) and NOMO1 overexpression plasmid construction (pcDNA3.1-NOMO1) was accomplished by Genomeditech. Small interfering RNA for NOMO1 (siNOMO1, sense: 5′-AUGCUUAUUUUGUAUUUUCCA-3′; antisense: 5′-GAAAAUACAAAAUAAGCAUCA-3′) and its negative control (siNC, sense: 5′-UUCUCCGAACGUGUCACGUTT-3′; antisense: 5′-ACGUGACACGUUCGGAGAATT-3′) was synthesized by RiboBio Co., Ltd. (Guangzhou, China). The hCMPCs (4 × 105 cells/well) were transfected with 110 pmol of siNOMO1 or overexpressed NOMO1 plasmid or mimics or inhibitor in Opti-MEM medium (11058021, Invitrogen, USA) containing Lipofectamine® 2000 (11668019, Invitrogen, USA) at 37 °C. Cells were harvested after transfection for 48 h.
RNA extraction and real-time quantitative PCR (qRT-PCR)
Total RNA was isolated from cultured cells using TRIzol reagent (93289, Sigma-Aldrich, USA). RNA concentration was identified by a biological spectrometer (Nano Drop 2000; Thermo Fisher Scientific, USA). A total of 2 μl of cDNA was synthesized with the iScript cDNA synthesis kit (Bio-Rad Laboratories, Inc., Hercules, California, USA). QRT-PCR experiment was performed using the FastStart Universal SYBR Green Master mix (Roche, Switzerland) in Touch real-time PCR Detection system (CFX96, Bio-Rad, Inc., Hercules, CA, USA). Parameters: 95 °C for 5 min, 40 cycles of 95 °C for 15 s, 60 °C for 30 s, and 70 °C for 10 s. GAPDH and U6 were chosen as internal references. Relative gene expression was quantified by the comparative Ct method formula 2−ΔΔCt [Citation22] and the real-time PCR primer sequences in the present study were listed in .
Table 1. Primer for qRT-PCR.
Cell counting kit-8 (CCK-8) assay
Cell Counting Kit-8 kit (HY-K0301; MedChemExpress, Monmouth Junction, NJ, USA) was employed to investigate hCMPCs proliferative capacity. Specifically, cells were seeded in a 96-well plate (1 × 104 cells/well) in 100 μL of culture medium followed by being cultured at 37 °C for 24 h, 48 h and 72 h in an incubator containing CO2 at 37 °C. And then 10 μL of Cell Counting Kit-8 solution was added to each well of the plate, later the cells were incubated for 4 h at 37 °C in the incubator. A microplate reader (PHERAstar FSX, BMG LABTECH Inc., Cary, NC, USA) was applied to measure the absorbance at 450 nm. The results in the Blank group were standardized to 100%.
Detection of cell cycle progression and cell apoptosis
ANNEXIN V- FITC/PI kit (640914, BioLegend, Inc., San Diego, CA, USA) was used to detect cell apoptosis and the process was strictly executed in line with the manufacturer’s instructions. Briefly, the cells were firstly washed twice with cold BioLegend's Cell Staining Buffer followed by being resuspended in Annexin V Binding Buffer at a concentration of 0.5 × 107 cell/ml. Next, transfer 100 µl of cell suspension in a 5 ml test tube and then it was applied with 5 µl of FITC Annexin V and 10 µl of Propidium Iodide Solution. Subsequently, the cells were gently shaken and incubated at 25 °C without light for 15 min. Finally, 400 µl of Annexin V Binding Buffer was added to each tube and the staining was identified after 48 h by CytoFLEX S flow cytometer (Beckman Coulter, Inc., Miami, Florida, USA).
The detection of cell cycle progression was conducted by the DNA Content Quantitation Assay kit (CA1510, Solarbio Science & Technology Co., Ltd., Beijing, China) following the manufacturer’s instructions. Specially, the cells were collected and the cell concentration was adjusted to 1 × 106/ml, 1 ml cell suspension was used in the follow-up steps. Pre-cold ethanol (70%, 500 µl) was added into cells overnight and stored at 4 °C, the stationary liquid was washed off by PBS before staining. Subsequently, 100 μl RNase A solution and 400 µl PI solution was added to each sample. Cells were then incubated from the light at 37˚C for 30 min. The staining was identified by CytoFLEX S flow cytometer (Beckman Coulter, Inc., Miami, Florida, USA) at wavelength of 488 nm and the percentage of HUVECs in G0/G1, S, and G2/M phases was calculated by ModFit LT analysis program (Verity Software House, USA).
Western blot
Total protein was solubilized from cell lysates using RIPA buffer (20-188, Millipore Sigma, Burlington, Massachusetts, USA) and quantified using a bicinchoninic protein assay (BCA) kit (BL521A, Biosharp, China). A total of 40 µg proteins were electrophpresed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA) which were blocked in 5% skimmed milk dissolved in TBS (20mMTris–HCl, pH7.4,150mMNaCl) with 0.5% Tween-20 for 1 h and then incubated overnight at 4 °C with specific primary antibodies: anti-MFE2C antibody (rabbit, #5030, 1:1000, Cell Signaling Technology, USA), anti-GAPDH antibody (rabbit, ab181602, 1:10000, Abcam, UK), anti- GATA4 antibody (rabbit, ab84593, 1 µg/ml, Abcam, UK), anti- Cleaved(C) caspase-3 antibody (rabbit, #9661, 1:1000, Cell Signaling Technology, USA), anti-Bax antibody (rabbit, ab32503, 1:1000, Abcam, UK), anti-cTnT antibody (rabbit, ab209813, 1:5000, Abcam, UK) and anti-Bcl-2 antibody (rabbit, ab182858, 1:2000, Abcam, UK). GAPDH was employed as the normalization reference. Membranes were then incubated with secondary antibody conjugated to horseradish peroxidase (HRP): goat anti-rabbit IgG, HRP-linked antibody (#7074, Cell Signaling Technology, USA). Immunopositive bands were investigated by the FluorChem M System (ProteinSimple, San Jose, CA, USA) [Citation23].
Prediction on target gene and dual-luciferase reporter assay
Biological information website TargetScan V7.2 (www.targetscan.org/) was employed to predict target gene and potential binding sites of miR-33a-5p and NOMO1 in the present study and confirmed by dual-luciferase reporter assay.
PMIR-REPORT Luciferase vector (AM5795; Thermo Fisher Scientific, USA) which contained NOMO1 sequences (wild-type or mutated) were cloned into the pMirGLO reporter vector (E1330, Promega, USA) to form NOMO1-WT and NOMO1-MUT. hCMPCs cells (5 × 103 cells/well) were maintained in 96-well plates and co-transfected with luciferase reporter plasmids and mimic or inhibitor by Lipofectamine 2000 Transfection reagent (Thermo Fisher Scientific, USA). After 48-h incubation at 37 °C, the firefly luciferase activity was measured through the Dual-Glo luciferase report system (E6120; Promega, USA) and calculated by normalization to the Renilla luciferase control.
Statistical analysis
The quantitative data are expressed mean ± standard deviation (SD) and each experiment was conducted at least for 3 times. It would be considered statistically significant when P-value below 0.05. SPSS 22.0 software (SPSS Inc, Chicago, IL, USA) were employed for statistical analysis. Comparisons between quantitative variables were realized by One-Way ANOVA followed by Dunnet’s post hoc test.
Results
hCMPCs proliferation and cell cycle G0/S transition were regulated by miR-33a-5p mimic and inhibitor
With the aim to discover the effect of miR-33a-5p on hCMPCs proliferation and cell cycle progression, miR-33a-5p mimic or miR-33a-5p inhibitor was successfully transfected into hCMPCs and their negative controls were established at the same time (, p < 0.001). As the results of CCK-8 exhibited in , the proliferation of hCMPCs was down-regulated by mimic transfection compared with MC group, while it was up-regulated by inhibitor transfection as in comparison with IC group over time, suggesting that hCMPCs proliferation was suppressed by up-regulation of miR-33a-5p (p < 0.05).
Figure 1. hCMPCs proliferation and cell cycle G0/S transition were regulated by miR-33a-5p mimic and inhibitor. (A) Transfection efficiency of miR-33a-5p mimic or miR-33a-5p inhibitor was identified by quantitative real-time polymerase chain reaction (qRT-PCR). U6 was used as the internal reference. (B) After transfection, hCMPCs proliferation was detected by cell counting kit-8 (CCK-8) assay at 24, 48 and 72 h. (C,D) After transfection, hCMPCs cell cycle progression was investigated by flow cytometry. (E-F) After transfection, hCMPCs cell apoptosis was detected by flow cytometry at 48 h. All experiments have been performed in triplicate and experimental data were expressed as mean ± standard deviation (SD). (**p < 0.01, ***p < 0.001, vs. MC; ∧p < 0.05, ∧∧p < 0.01, ∧∧∧p < 0.001, vs. IC) hCMPCs: human cardiomyocyte progenitor cells, MC: mimic control, IC: inhibitor control, M: miR-33a-5p mimic. I: miR-33a-5p inhibitor.
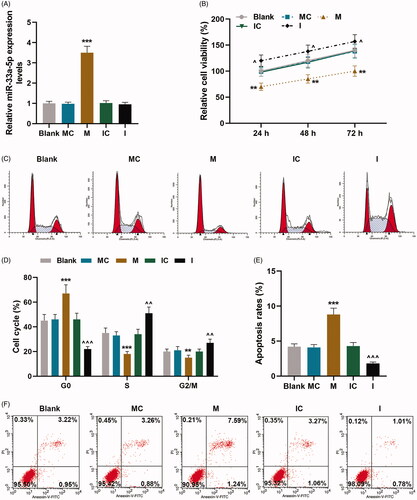
On the other hand, hCMPCs cell cycle progression after transfection was identified under flow cytometry, and the results shown in demonstrated that when compared with MC group, cell percentage at G0 phase of cells transfected with mimic was increased but it was decreased at S and G2/M phases (p < 0.01). By contrast, as in comparison with IC group, cell percentage at G0 phase of cells transfected with inhibitor was reduced but it was promoted at S and G2/M phases (p < 0.01). These findings unveiled that hCMPCs cell cycle G0/S transition was inhibited via up-regulation of miR-33a-5p.
hCMPCs apoptosis was regulated by miR-33a-5p mimic and inhibitor
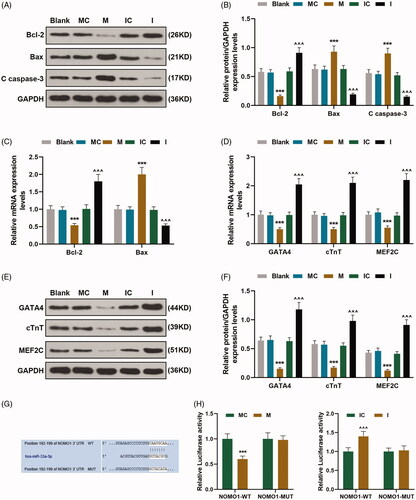
To further uncover the effect of miR-33a-5p on hCMPCs apoptosis, flow cytometry was employed to investigate cell apoptosis and expressions of cell apoptotic-related proteins Bcl-2, Bax and Cleaved(C) caspase-3 in hCMPCs were measured through qRT-PCR and western blot. The outcomes of flow cytometry were exhibited in which uncovered that apoptosis rate of cells transfected with mimic was evidently enhanced as in comparison with its control value but was obviously decreased after inhibitor transfection compared with IC group (p < 0.001). And the results of qRT-PCR and western blot in uncovered that expressions of pro-apoptotic proteins Bax and C caspase-3 were up-regulated in cells transfected with mimic compared with its control value but down-regulated after inhibitor transfection as in comparison with IC group (p < 0.001). On the contrary, anti-apoptotic protein Bcl-2 expression in cells transfected with mimic was decreased compared with cells in MC group but was increased when cells were transfected with inhibitor as in comparison with its control value (, p < 0.001).
Figure 2. hCMPCs apoptosis was regulated by miR-33a-5p mimic and inhibitor, and NOMO1 was the target gene of miR-33a-5p. (A–C) After transfection, expressions of Bax, Bcl-2 and Cleaved(C) caspase-3 in hCMPCs were measured by qRT-PCR and western blot. GAPDH was used as the internal reference. (D–F) After transfection, expressions of GATA4, cardiac troponin T (cTnT) and myocyte enhancer factor2C (MEF2C) in hCMPCs were detected by qRT-PCR and western blot. GAPDH was used as the internal reference. (G)TargetScan V7.2 was used to predict the binding sites of miR-33a-5p and NOMO1. (H-I) The hCMPCs were co-transfected with wild-type or mutant of NOMO1 (NOMO1-WT; NOMO1-MUT), miR-33a-5p mimic and miR-33a-5p inhibitor, and luciferase activity was measured by dual-luciferase reporter assay. All experiments have been performed in triplicate and experimental data were expressed as mean ± standard deviation (SD). (***p < 0.001, vs. MC; ∧∧∧p < 0.001, vs. IC) hCMPCs: human cardiomyocyte progenitor cells, MC: miR-33a-5p mimic control, IC: miR-33a-5p inhibitor control, NOMO1: nodal modulator 1; Bcl-2: B-cell lymphoma 2; Bax: Bcl-2-associated X protein.
Expressions of cardiomyocyte differentiation markers was regulated by miR-33a-5p mimic and inhibitor
In this phase, in order to discover the effect of miR-33a-5p on the degree of hCMPCs differentiation into cardiomyocytes, qRT-PCR and western blot were used to detected the expressions of GATA4, troponin T (cTnT) and myocyte enhancer factor2C (MEF2C) which were involved in the differentiation of cardiomyocytes after transfection. As shown in , it was clear that expressions of GATA4, cTnT and MEF2C in cells transfected with mimic were enhanced compared with its control value but reduced after cells transfected with inhibitor as in comparison with IC group (p < 0.001). These outcomes demonstrated that the degree of hCMPCs differentiation into cardiomyocytes was inhibited by up-regulation of miR-33a-5p.
NOMO1 was targeted with miR-33a-5p, and miR-33a-5p regulated hCMPCs proliferation and cell cycle G0/S transition via overexpressed NOMO1 and siNOMO1
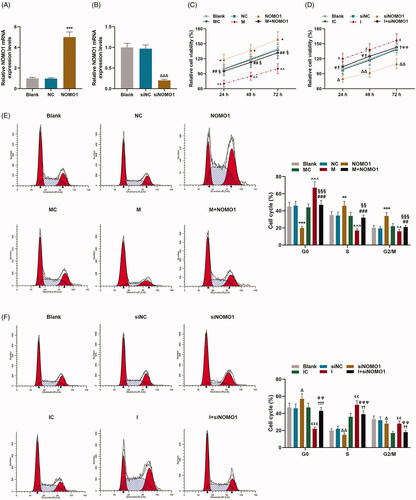
The prediction software TargetScan V7.2 demonstrated that NOMO1 was a target gene of miR-33a-5p, which contained complementary binding sites in 3′-UTR (). Dual luciferase reporter assay was performed to further validate that miR-33a-5p directly binds to NOMO1. As revealed in dual-luciferase reporter assay that luciferase activity in NOMO1-WT-mimic group was down-regulated compared with MC group while it in NOMO1-WT-inhibitor was up-regulated as in comparison with IC (, p < 0.001), suggesting that NOMO1 was the target gene of miR-33a-5p.
To further unveil the correlation between miR-33a-5p and NOMO1, overexpressed NOMO1 plasmid and small interfering RNA for NOMO1 (siNOMO1) were successfully transfected into hCMPCs (, p < 0.001). As shown in , CCK-8 assay analysis proved that hCMPCs proliferation was up-regulated by overexpressed NOMO1 (p < 0.05). And the inhibitory effect of miR-33a-5p mimic on hCMPCs proliferation was partially rescued by overexpressed NOMO1 (, p < 0.05). Besides, the promotive effect of miR-33a-5p inhibitor on cell proliferation was partially mitigated by knockdown of NOMO1. (, p < 0.05). At the same time, the results of flow cytometry shown in demonstrated that cell percentage at G0 phase was inhibited but promoted at S and G2/M phases by overexpressed NOMO1(, p < 0.01). And the positive effect of miR-33a-5p mimic on cell percentage at G0 phase as well as the inhibitory effect of mimic on cell percentage at S and G2/M phases were both partially rescued by overexpressed NOMO1. (, p < 0.01). In addition, as exhibited in , cell percentage was decreased at G0 phase but increased at S and G2/M phases by miR-335-5p inhibitor, while siNOMO1 posed the opposite function (, p < 0.05). Furthermore, the inhibitory effect of miR-33a-5p inhibitor on cell percentage at G0 phase as well as the encouraging effect of inhibitor on cell percentage at S and G2/M phases were both partially rescued by knockdown of NOMO1 (, p < 0.01).
Figure 3. MiR-33a-5p regulated hCMPCs proliferation and cell cycle G0/S transition via overexpressed NOMO1 and siNOMO1. (A,B) Transfection efficiency of overexpressed NOMO1 plasmid and small interfering RNA for NOMO1 (siNOMO) was identified by qRT-PCR. GAPDH was used as the internal reference. (C,D) After transfection, hCMPCs proliferation was detected by cell counting kit-8 (CCK-8) assay at 24, 48 and 72 h. (E,F) After transfection, hCMPCs cell cycle progression was investigated by flow cytometry. All experiments have been performed in triplicate and experimental data were expressed as mean ± standard deviation (SD). (*p < 0.01, **p < 0.05, ***p < 0.001, vs. NC; ∧∧p < 0.01, ∧∧∧p < 0.001, vs. MC; ##p < 0.01, ###p < 0.001, vs. M; §p < 0.05, §§p < 0.01, §§§p < 0.001, vs. NOMO1; Δp < 0.01, ΔΔp < 0.05, ΔΔΔp < 0.001, vs. siNC; εp < 0.01, εεp < 0.05, εεεp < 0.001, vs. IC; †p < 0.01, ††p < 0.05, †††p < 0.001, vs. I; φp < 0.01, φφp < 0.05, φφφp < 0.001, vs. siNOMO1) hCMPCs: human cardiomyocyte progenitor cells, NC: negative control, siNC: negative control for small interfering RNA, siNOMO1: small interfering RNA for NOMO1, M: miR-33a-5p mimic, I: miR-33a-5p inhibitor, MC: mimic control, IC: inhibitor control, NOMO1: nodal modulator 1.
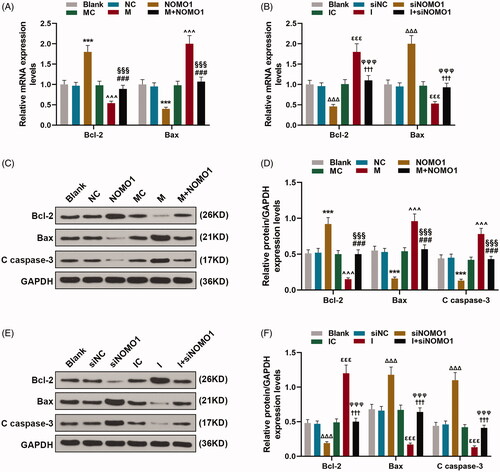
MiR-33a-5p regulated the apoptosis of hCMPCs via overexpressed NOMO1 and siNOMO1
As shown in , the results of flow cytometry uncovered that hCMPCs apoptosis rates was decreased by overexpressed NOMO1 compared with un-treated cells(p < 0.001). And the promotive effect of miR-33a-5p on cell apoptosis was partially mitigated by overexpressed NOMO1(, p < 0.001). By contrast, as revealed in , hCMPCs apoptosis rates were increased by siNOMO1 as in comparison with cells in siNC group, while miR-33a-5p inhibitor decreased the apoptosis rates compared with cells in IC group (p < 0.01). Meanwhile, the suppressive effect of miR-33a-5p inhibitor on cell apoptosis was partially disarmed by knockdown of NOMO1 (, p < 0.01). Moreover, the qRT-PCR and western blot analysis shown in demonstrated that expressions of pro-apoptotic proteins Bax and C caspase-3 were up-regulated by overexpressed NOMO1 as in comparison with the control value (p < 0.001). And the promotive effect of miR-33a-5p mimic on expressions of Bax and C caspase-3 was partially alleviated by overexpressed NOMO1 (, p < 0.001). Besides, expression of anti-apoptotic protein Bcl-2 was up-regulated by overexpressed NOMO1 compared with the control value (, p < 0.001). Moreover, the inhibitory effect of miR-33a-5p mimic on Bcl-2 expression was partially rescued by overexpressed NOMO1 (, p < 0.001). Conversely, miR-33a-5p inhibitor reduced the expressions of Bax and C caspase-3 but increased Bcl-2 expression, while knockdown of NOMO1 exerted the opposite function (, p < 0.001). Besides, the suppressive effect of miR-33a-5p inhibitor on expressions of Bax and C caspase-3 but the promotive effect of miR-33a-5p inhibitor on Bcl-2 expression were both partially mitigated by knockdown of NOMO1 (, p < 0.001).
Figure 4. MiR-33a-5p regulated hCMPCs apoptosis via overexpressed NOMO1 and siNOMO1. (A–D) After transfection, hCMPCs cell apoptosis was detected by flow cytometry at 48 h. All experiments have been performed in triplicate and experimental data were expressed as mean ± standard deviation (SD). (***p < 0.001, vs. NC; ∧∧∧p < 0.001, vs. MC; ###p < 0.001, vs. M; §§§p < 0.001, vs. NOMO1; ΔΔΔp < 0.001, vs. siNC; εεp < 0.05, vs. IC; ††p < 0.05, vs. I; φφφp < 0.001, vs. siNOMO1) hCMPCs: human cardiomyocyte progenitor cells, NC: negative control, siNC: negative control for small interfering RNA, siNOMO1: small interfering RNA for NOMO1, M: miR-33a-5p mimic, I: miR-33a-5p inhibitor, MC: mimic control, IC: inhibitor control, NOMO1: nodal modulator 1.
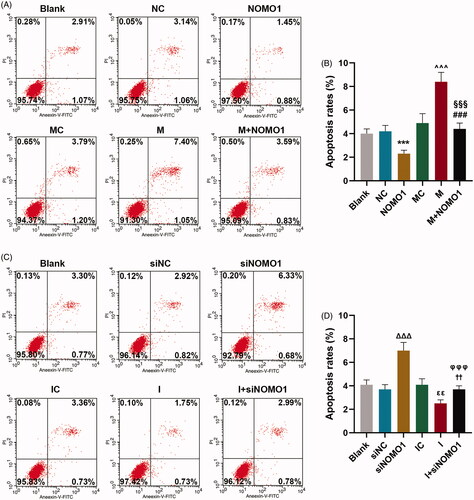
Figure 5. MiR-33a-5p regulated hCMPCs apoptosis-related proteins via overexpressed NOMO1 and siNOMO1. (A–F) After transfection, expressions of Bax, Bcl-2 and Cleaved(C) caspase-3 in hCMPCs were measured by qRT-PCR and western blot. GAPDH was used as the internal reference. All experiments have been performed in triplicate and experimental data were expressed as mean ± standard deviation (SD). (***p < 0.001, vs. NC; ∧∧∧p < 0.001, vs. MC; ###p < 0.001, vs. M; §§§p < 0.001, vs. NOMO1; ΔΔΔp < 0.001, vs. siNC; εεεp < 0.001, vs. IC; †††p < 0.001, vs. I; φφφp < 0.001, vs. siNOMO1) hCMPCs: human cardiomyocyte progenitor cells, NC: negative control, siNC: negative control for small interfering RNA, siNOMO1: small interfering RNA for NOMO1, M: miR-33a-5p mimic, I: miR-33a-5p inhibitor, MC: mimic control, IC: inhibitor control, NOMO1: nodal modulator 1.
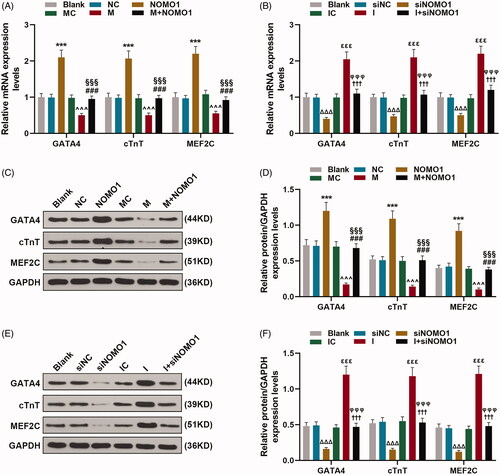
MiR-33a-5p regulated expressions of cardiomyocyte differentiation markers of hCMPCs via overexpressed NOMO1 and siNOMO1
As exhibited in , expressions of GATA4, cTnT and MEF2C were enhanced by overexpressed NOMO1 compared with untreated cells(p < 0.001). And the suppressive effect of miR-33a-5p mimic on expressions of these cardiomyocyte differentiation markers was partially disarmed by overexpressed NOMO1 (, p < 0.001). Meanwhile, expressions of GATA4, cTnT and MEF2C were reduced by miR-33a-5p inhibitor, while knockdown of NOMO1 posed the opposite result (, p < 0.001). In addition, the promotive effect of miR-33a-5p inhibitor on expressions of GATA4, cTnT and MEF2C was partially attenuated by siNOMO1 (, p < 0.001).
Figure 6. MiR-33a-5p regulated expressions of cardiomyocyte differentiation markers of hCMPCs via overexpressed NOMO1 and siNOMO1. (A–F) After transfection, expressions of GATA4, cardiac troponin T (cTnT) and myocyte enhancer factor2C (MEF2C) in hCMPCs were detected by qRT-PCR and western blot. GAPDH was used as the internal reference. All experiments have been performed in triplicate and experimental data were expressed as mean ± standard deviation (SD). (***p < 0.001, vs. NC; ∧∧∧p < 0.001, vs. MC; ###p < 0.001, vs. M; §§§p < 0.001, vs. NOMO1; ΔΔΔp < 0.001, vs. siNC; εεεp < 0.001, vs. IC; †††p < 0.001, vs. I; φφφp < 0.001, vs. siNOMO1) hCMPCs: human cardiomyocyte progenitor cells, NC: negative control, siNC: negative control for small interfering RNA, siNOMO1: small interfering RNA for NOMO1, M: miR-33a-5p mimic, I: miR-33a-5p inhibitor, MC: mimic control, IC: inhibitor control, NOMO1: nodal modulator 1.
Discussion
During cardiac morphogenesis, both progenitor cells and differentiated cardiomyocytes are subjected to strict spatiotemporal regulation to ensure that the heart achieves the correct size, structure and function [Citation11]. Human cardiomyocyte progenitor cells (hCMPCs) are pluripotent cells isolated from human fetal heart tissue, which can be differentiated into cardiomyocytes in vitro from electrophysiology, immunology and other aspects [Citation21,Citation24]. And in the present study, hCMPCs were successfully isolated from fetal heart tissue and cultured for investigating the effect of miR-33a-5p and nodal modulator 1 (NOMO1) on congenital heart disease (CHD).
Several studies have shown the effect of different miRNAs in cardiac tissue. For example, it has been reported that down-regulation of endogenous miR-320 supplies protection against ischemia/reperfusion (I/R)-induced cardiomyocyte death and apoptosis via targeting Hsp20 [Citation25]. And overexpression of miR-1 could facilitate H2O2-induced apoptosis in cardiomyocytes by targeting Bcl-2 in accordance with the previous study [Citation26]. Meanwhile, there are complex molecular networks based on the proliferation and differentiation of hCMPCs, among which miRNA plays a critical role. For instance, Liang et al. [Citation27] found that hCMPCs proliferation was inhibited by miR-10a via restraining its cell cycle progression, and they also discovered that decreased hCMPCs proliferation by miR-10a is not involved in cell apoptosis. However, in line with the experimental data in the present study, hCMPCs proliferation was restrained by up-regulation of miR-33a-5p but apoptosis was enhanced by up-regulation of miR-33a-5p, and down-regulation of miR-33a-5p did the opposite function. Besides, the Bcl-2 family can be categorized into the anti-apoptotic proteins, and the pro-apoptotic proteins such as Bax, and Caspase is a family of single-chain synthesized zymogens playing central roles in apoptotic signaling and execution [Citation28]. And in the present study, expressions of Bax and Cleaved(C) caspase-3 were promoted while Bcl-2 expression was inhibited by up-regulation of miR-33a-5p. Progression through the cell cycle is tightly regulated and checkpoints at phase transitions during the cell cycle ensure that only healthy cells progress and proliferate [Citation29]. And the data in the present study uncovered that hCMPCs cell cycle G0/S transition was suppressed by up-regulation of miR-33a-5p.
Transcription factor GATA4 has become a the nuclear effector of several cardiac signaling pathways that regulate critical cardiac cascades through post-translational modifications and protein-protein interactions [Citation30], and deletion of GATA4 from mesodermal progenitors during early development would lead to absence of cardiomyocytes [Citation31]. Troponin T (TnT), the tropomyosin-binding and thin filament anchoring subunit, of the troponin complex in regulating of muscle contraction [Citation32]. Cardiac troponin T (cTnT) is considered to be a reliable biomarker of myocardial injury in humans [Citation33]. And during the differentiation process of cardiomyocytes, cTnT expression was increased [Citation34]. Myocyte enhancer factor2C (MEF2C) is a member of the MEF2 family, functions as a key role in activating cardiac-specific embryonic genes expressions [Citation35]. It was worth noting that expressions of GATA4, cTnT and MEF2C were down-regulated by up-regualtion of miR-33a-5p in the present study, indicating that degree of hCMPCs differentiation into cardiomyocytes was inhibited by up-regulation of miR-33a-5p.
Nodal is a member of the transforming growth factor-β (TGF-β) superfamily and induces β-cell apoptosis through the activation of the ALK7-Smad3-caspase-3 signaling pathway [Citation36]. Nomo (Nodal modulator) and its binding partner, Nicalin, are components of a novel protein complex involved in Nodal signaling [Citation37], which is important to the formation of the early embryo during mesoderm and endoderm induction and the specification of left-right asymmetry [Citation38]. It has been found that Nodal modulator1 (NOMO1) significantly expressed low in human ventricular septal defect versus non- ventricular septal defect myocardium [Citation39]. Of note, NOMO1 is identified to be a target gene of miRNAs, for example, it can targeted with miR-675, and the suppressive effect of miR-675 on trophoblast cell growth was rescued by overexpressed NOMO1 [Citation16]. In the present study, we firstly found that NOMO1 was the target gene of miR-33a-5p by the bioinformatic prediction. And after transfection of overexpressed NOMO1 plasmid into hCMPCs, the suppressive effects of up-regulation of miR-33a-5p on hCMPCs proliferation, cell cycle G0/S transition and the degree of differentiation into cardiomyocytes, and the promotive effect of up-regulation of miR-33a-5p on hCMPCs apoptosis were partially mitigated by overexpressed NOMO1.
Nevertheless, this paper lacked equivalent experimental identification in an animal in vivo model and our findings required to be verified in more cell lines. Hence, further researches in vivo were needed. Furthermore, the localization of NOMO1 in cells and whether it changes according to the cell cycle of CMPCs differentiation state will be further investigated in future studies. Myocardial ischemia is also a common manifestation of heart disease, it is worth further exploring whether NOMO1 and miR-33a-5p can regulate ischemia-induced apoptosis in the future.
Taken together, the experimental data in the present study unveiled that NOMO1 was the target gene of miR-33a-5p, and up-regulation of miR-33a-5p inhibited hCMPCs proliferation, cell cycle G0/S transition and differentiation into cardiomyocytes but promoted apoptosis via targeting NOMO1. These discoveries may offer novel molecular therapeutic method for CHD.
Disclosure of interest
No potential conflict of interest was reported by the author(s).
Data availability statement
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.
References
- Mandalenakis Z, Skoglund K, Dellborg M. Congenital heart disease: the children will become elderly. Aging (Albany NY)). 2019;11(3):851–852.
- Lévy M, Maurey C, Celermajer DS, et al. Impaired apoptosis of pulmonary endothelial cells is associated with intimal proliferation and irreversibility of pulmonary hypertension in congenital heart disease. J Am Coll Cardiol. 2007;49(7):803–810.
- Wang J, Wang F, Gui Y-H. [Research advances in the mechanism of congenital heart disease induced by pregestational diabetes mellitus. Zhongguo Dang Dai Er Ke Za Zhi. 2017;19(12):1297–1300.
- Yester JW, Kühn B. Mechanisms of Cardiomyocyte Proliferation and Differentiation in Development and Regeneration. Curr Cardiol Rep. 2017;19(2):13.
- Lüscher TF. Frontiers in congenital heart disease: pulmonary hypertension, heart failure, and arrhythmias. Eur Heart J. 2016;37(18):1407–1409.
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297.
- Chen X-M. MicroRNA signatures in liver diseases. World J Gastroenterol. 2009;15(14):1665–1672.
- Cattani AA, Allene C, Seifert V, et al. Involvement of microRNAs in epileptogenesis. Epilepsia. 2016;57(7):1015–1026.
- Wong LL, Wang J, Liew OW, et al. MicroRNA and Heart Failure. Int J Mol Sci. 2016;17(4):502.
- Song Y, Higgins H, Guo J, et al. Clinical significance of circulating microRNAs as markers in detecting and predicting congenital heart defects in children. J Transl Med. 2018;16(1):42.
- Wu Y-H, Zhao H, Zhou L-P, et al. miR-134 Modulates the Proliferation of Human Cardiomyocyte Progenitor Cells by Targeting Meis2. Int J Mol Sci. 2015;16(10):25199–25213.
- Dai Y, Wu Z, Lang C, et al. Copy number gain of ZEB1 mediates a double-negative feedback loop with miR-33a-5p that regulates EMT and bone metastasis of prostate cancer dependent on TGF-β signaling. Theranostics. 2019;9(21):6063–6079.
- Yan Y, Zhang D, Lei T, et al. MicroRNA-33a-5p suppresses colorectal cancer cell growth by inhibiting MTHFD2. Clin Exp Pharmacol Physiol. 2019;46(10):928–936.
- Diao L, Bai L, Jiang X, et al. Long-chain noncoding RNA GAS5 mediates oxidative stress in cardiac microvascular endothelial cells injury. J Cell Physiol. 2019;234(10):17649–17662.
- Chen Z, Ding HS, Guo X, et al. MiR-33 promotes myocardial fibrosis by inhibiting MMP16 and stimulating p38 MAPK signaling. Oncotarget. 2018;9(31):22047–22057.
- Gao W-L, Liu M, Yang Y, et al. The imprinted H19 gene regulates human placental trophoblast cell proliferation via encoding miR-675 that targets Nodal Modulator 1 (NOMO1). RNA Biol. 2012;9(7):1002–1010.
- Zhang H, Xu C, Yang R, et al. Silencing of nodal modulator 1 inhibits the differentiation of P19 cells into cardiomyocytes. Exp Cell Res. 2015;331(2):369–376.
- Flyer JN, Zuckerman WA, Richmond ME, et al. Prospective study of adenosine on atrioventricular nodal conduction in pediatric and young adult patients after heart transplantation. Circulation. 2017;135(25):2485–2493.
- Shen MM. Nodal signaling: developmental roles and regulation. Development. 2007;134(6):1023–1034.
- Cao L, Li L, Li Y, et al. Loss of the Nodal modulator Nomo results in chondrodysplasia in zebrafish. Curr Mol Med. 2018;18(7):448–458.
- Smits AM, van Vliet P, Metz CH, et al. Human cardiomyocyte progenitor cells differentiate into functional mature cardiomyocytes: an in vitro model for studying human cardiac physiology and pathophysiology. Nat Protoc. 2009;4(2):232–243.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408.
- Cheng Z, Zhang Q, Yin A, et al. The long non-coding RNA uc.4 influences cell differentiation through the TGF-beta signaling pathway. Exp Mol Med. 2018;50(2):e447.
- Goumans M-J, de Boer TP, Smits AM, et al. TGF-beta1 induces efficient differentiation of human cardiomyocyte progenitor cells into functional cardiomyocytes in vitro. Stem Cell Res. 2007;1(2):138–149.
- Ren X-P, Wu J, Wang X, et al. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation. 2009;119(17):2357–2366.
- Tang Y, Zheng J, Sun Y, et al. MicroRNA-1 regulates cardiomyocyte apoptosis by targeting Bcl-2. Int Heart J. 2009;50(3):377–387.
- Liang D, Zhen L, Yuan T, et al. miR-10a regulates proliferation of human cardiomyocyte progenitor cells by targeting GATA6. PLoS ONE. 2014;9(7):e103097.
- Huang J, Cui H, Peng X, et al. The association between splenocyte apoptosis and alterations of Bax, Bcl-2 and caspase-3 mRNA expression, and oxidative stress induced by dietary nickel chloride in broilers. IJERPH. 2013;10(12):7310–7326.
- Newell M, Baker K, Postovit LM, et al. A Critical Review on the Effect of Docosahexaenoic Acid (DHA) on Cancer Cell Cycle Progression. Int J Mol Sci. 2017;18(8):1784.
- Välimäki MJ, Ruskoaho HJ. Targeting GATA4 for cardiac repair. IUBMB Life. 2020;72(1):68–79.
- Zhao R, Watt AJ, Battle MA, et al. Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Dev Biol. 2008;317(2):614–619.
- Sheng J-J, Jin J-P. Gene regulation, alternative splicing, and posttranslational modification of troponin subunits in cardiac development and adaptation: a focused review. Front Physiol. 2014;5:165.
- de Lemos JA. Increasingly sensitive assays for cardiac troponins: a review. JAMA. 2013;309(21):2262–2269.
- Jiang S, Zhang S. Differentiation of cardiomyocytes from amniotic fluid-derived mesenchymal stem cells by combined induction with transforming growth factor β1 and 5-azacytidine. Mol Med Rep. 2017;16(5):5887–5893.
- Qian Q, Qian H, Zhang X, et al. 5-Azacytidine induces cardiac differentiation of human umbilical cord-derived mesenchymal stem cells by activating extracellular regulated kinase. Stem Cells Dev. 2012;21(1):67–75.
- Li J, Wang Z, Ren L, et al. Antagonistic interaction between Nodal and insulin modulates pancreatic β-cell proliferation and survival. Cell Commun Signal. 2018;16(1):79.
- García-Tuñón I, Vuelta E, Lozano L, et al. Establishment of a conditional Nomo1 mouse model by CRISPR/Cas9 technology. Mol Biol Rep. 2020;47(2):1381–1391.
- Schier AF. Nodal signaling in vertebrate development. Annu Rev Cell Dev Biol. 2003;19:589–621.
- Zhang H, Zhou L, Yang R, et al. Identification of differentially expressed genes in human heart with ventricular septal defect using suppression subtractive hybridization. Biochem Biophys Res Commun. 2006;342(1):135–144.