
Abstract
Direct compression (DC) is the simplest and most economical way to produce pharmaceutical tablets. Ideally, it consists of only two steps: dry blending of a drug substance(s) with excipients followed by compressing the powder mixture into tablets. In this study, immediate-release film-coated tablets containing either Sitagliptin phosphate or Sitagliptin hydrochloride were developed using DC technique. After establishing the optimum ratio of ductile and brittle excipients, five formulations were compressed into tablets using a rotary press and finally film coated. Both powders and tablets were examined by standard pharmacopoeial methods. It has been shown that the simultaneous use of excipients with different physical properties, i.e. ductile microcrystalline cellulose and brittle anhydrous dibasic calcium phosphate, produces a synergistic effect, allowing preparation of Sitagliptin DC tablets with good mechanical strength (tensile strength over 2 N/mm2), rapid disintegration (shorter than 2 min), and fast release of the drug substance (85% of the drug is dissolved within 15 min). It was found that the type of calcium phosphate excipient used had a large effect on the properties of the sitagliptin tablets. All formulations developed showed good chemical stability, even when stored under stress conditions (50 °C/80% RH).
Introduction
The oral route of drug administration is the most preferred by patients due to its high comfort and safety of the use (Kumar et al. Citation2012; Viswanathan et al. Citation2017). Despite the development of many modern pharmaceutical oral solid dosage forms (OSDF), tablets are still the most commonly used due to the highest patient compliance and relatively low production costs. Compressed tablets comprise the majority of OSDF, however, their manufacturing can be a complex process requiring lots of formulation expertise (Natoli et al. Citation2017).
There are many different technologies to produce compressed tablets including wet granulation (e.g. high-shear or fluid-bed granulation), dry granulation (roller compaction or slugging), and direct compression (DC) (see ) (Meeus Citation2011). All these processing technologies offer many benefits, but they are not free of certain disadvantages, which must be taken into account with respect to the properties of active substances and excipients used. Wet granulation techniques are probably the most widely used since they can enhance flowability and increase compressibility/compatibility of powder blends, ensure uniform distribution of the drug substance(s) within the powder mixtures as well as obtain controllable drug release. On the other hand, the processes are long-lasting and consist of many different stages during which substances are exposed to heat and moisture which can trigger their physical transformation or/and chemical degradation (Kristensen and Schaefer Citation1987; Shanmugam Citation2015). DC is so far the shortest, the most efficient and the least complex way of pharmaceutical tablets production. Ideally, it consists of only two consecutive unit processes: mixing of one or more drug substances with excipients followed by compression of the powder blend into tablets (). Such technological process allows saving of time, reduction of the energy consumption and consequently cutting down the cost of production (Mirani et al. Citation2011). Moreover, during the process, powders are not exposed to heat or moisture so drug substances sensitive to temperature (e.g. valsartan, proteins, enzymes) or to moisture (e.g. acetylsalicylic acid, vitamins) can be safely processed without the risk of triggering chemical degradation. What should be kept in mind, are challenges connected with poor flowability and/or compressibility of powders, which can lead to poor physical properties of the resulting tablets. Moreover, differences in particle size or density of ingredients can cause segregation during processing, which in turn can lead to inhomogeneity and lack of uniformity of the final dosage units.
Figure 1. Different ways of manufacturing of oral solid dosage forms.
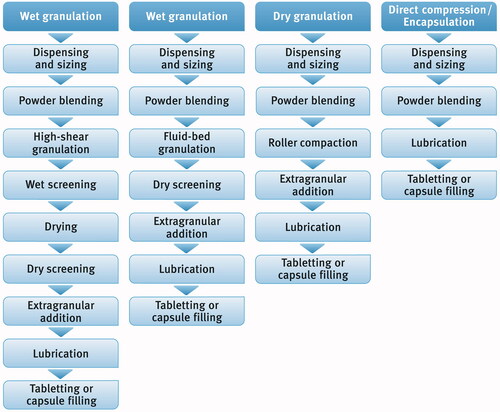
The DC process requires a very critical selection of excipients exhibiting multifunctional properties. Their flowability, compactibility, dilution potential must sufficiently compensate for the potentially adverse properties of drug substances as well as other ingredients present in a formulation (Rojas et al. Citation2012; Mangal et al. Citation2015; Rojas Citation2015). There is a variety of high functional DC excipients available on the market including lactose (Muñoz-Ruiz et al. Citation1993; Ilić et al. Citation2009) microcrystalline cellulose (Albers et al. Citation2006; Thoorens et al. Citation2014), calcium phosphates (Schmidt and Herzog Citation1993; Landín et al. Citation1994), starch (Lawal Citation2019) as well as co-processed materials comprising two or more substances, e.g. lactose and cellulose or microcrystalline cellulose and silicon dioxide (Nachaegari and Bansal Citation2004; Saha and Shahiwala Citation2009).
The present study focuses on the simultaneous use of two common excipients, microcrystalline cellulose (MCC) and anhydrous dibasic calcium phosphate (DCPA), in the development and manufacturing of compressed tablets. MCC is a ductile substance which undergoes plastic deformation during compression. After exceeding the yield stress, its particles change shape irreversibly, which causes an increase in their surface area and a strengthening of the interparticle bonds. Therefore, MCC offers excellent compactibility and thus the possibility of producing tablets with very good mechanical strength. Nevertheless, MCC is known to have a high sensitivity to lubricants and high tabletting speed. Tablets based on it may exhibit extended disintegration time (Ilić et al. Citation2013; Thoorens et al. Citation2014; Chaerunisaa et al. Citation2020). DCPA is a brittle, inorganic compound, which during tabletting undergoes brittle fracture. Under compaction pressure, DCPA particles break up into many small fragments. This increases their specific surface area and the number of potential binding sites, as well as eliminates their lubricant sensitivity. Directly compressible DCPA grades exhibit very good flow and tabletting properties. Moreover, they can measurably facilitate the penetration of liquids into the tablet interior and accelerate the disintegration time. They dissolve in the acid environment of the stomach, which may further facilitate the release of drug substances from immediate-release tablets (Almaya and Aburub Citation2008; Zakowiecki et al. Citation2019; Wagner et al. Citation2022).
A good tabletting mixture should provide, in addition to good flowability, an appropriate balance between plasticity and brittleness, which ensures its effective compression. Of course, the properties of a drug substance itself, such as poor flowability and/or compactibility, may play a large role here. However, the availability of a drug substance, particularly in the early phase of formulation development, may be limited, making it difficult to fully assess and understand its mechanical properties. Therefore, the unfavorable properties of the active component must be compensated for by the right choice of the other inactive ingredients of the pharmaceutical composition. Here, providing such a combination of ductile deformation and brittle fracture through the use of excipients can prove to be extremely advantageous (Sun Citation2017). The benefits of employing ductile and brittle substances simultaneously in pharmaceutical compositions have already been reported. Examples of such combinations include alpha-lactose monohydrate as a brittle component, and maize starch or MCC as a ductile material (Gupta et al. Citation2006; Zheng and Ternik Citation2008; Saha and Shahiwala Citation2009). The benefits of such a solution have also been recognized by manufacturers of pharmaceutical excipients, resulting in the development and commercialization of advanced products that are particularly suitable for use in DC (Schwarz and Fichtner Citation2016; Thoorens et al. Citation2018).
In the present study, DCPA and MCC were used to prepare immediate-release (IR) film-coated tablets containing Sitagliptin by DC method. Sitagliptin is an anti-diabetic drug used to treat type 2 diabetes and was first marketed as Januvia® tablets in 2006 by Merck & Co. Januvia® is available as film-coated tablets containing 25 mg, 50 mg, or 100 mg of Sitagliptin as the phosphate monohydrate salt (Zerilli and Pyon Citation2007; MSD Citation2021). Generic drug products containing other salts, such as Sitagliptin hydrochloride monohydrate or Sitagliptin fumarate, will soon be available on the market (EMA Citation2021; Galenicum Health S.L. Citation2021).
During the studies presented here, formulations for two commercially available salts of Sitagliptin were developed, i.e. phosphate monohydrate and hydrochloride monohydrate. It was intended that the compositions developed should enable a DC and should therefore exhibit suitable functional properties in terms of flowability and compactibility. Hence, a mixture of directly compressible grades of MCC (VIVAPUR® 102) and DCPA was employed as filler/diluent. VIVAPUR® 102 has an average particle size of approximately 130 µm, a bulk density ranging 280–330 g/L and specific surface area of around 0.41 m2/g () (Vehovec et al. Citation2012; JRS Pharma Citation2022). In the case of DCPA, two products were tested:
Figure 2. Example particle of (A) VIVAPUR® 102 (SEM micrograph, magnification of ×1000), (B) DI-CAFOS® A150 (SEM micrograph, magnification of ×1000), (C) DI-CAFOS® A60 (SEM micrograph, magnification of ×1000).

DI-CAFOS® A150 with an average particle size of approximately 150 µm, a porous structure and a large specific surface area of around 23 m2/g, which bulk and tapped density are respectively 720 g/L and 840 g/L ().
very dense DI-CAFOS® A60 with a bulk and tapped density of 1330 g/L and 1500 g/L respectively, average particle size of approximately 60 µm and a very low specific surface area of around 0.25 m2/g () (Zakowiecki et al. Citation2019; Wagner et al. Citation2022).
At the start, the study investigated the effect of the ratio of brittle to ductile material on the flowability and tabletting properties of powder mixtures. The composition considered to be optimal was used for the preparation of tablets. The developed tablets were evaluated for the performance tests as described in the USP/NF monograph for Sitagliptin Tablets as well as in terms of their dissolution in liquids of various pH values. The stability of the developed formulations was tested under stressed conditions (50 °C and 80% RH) according to ICH Q1A(R2) guideline (EMA Citation2003). Characteristics of the developed formulations and their dissolution behavior were compared to the reference product, Januvia® 25 mg, 50 mg, and 100 mg film-coated tablets.
Materials and methods
Drug substances: Sitagliptin phosphate monohydrate (Moehs BCN, S.L., Barcelona, Spain) and Sitagliptin hydrochloride monohydrate (Amino Chemicals, Marsa, Malta).
Fillers/diluents: MCC VIVAPUR® 102 from JRS Pharma (Rosenberg, Germany), DCPA DI-CAFOS® A60 and DI-CAFOS® A150 from Chemische Fabrik Budenheim KG (Budenheim, Germany).
Disintegrant: Croscarmellose sodium Ac-Di-Sol® SD-711 from FMC BioPolymer (Brussels, Belgium).
Lubricants: Magnesium stearate Ligamed® MF-2-V from Peter Greven Fett-Chemi (Venlo, The Netherlands), sodium stearyl fumarate ALUBRA® PG 100 from FMC BioPolymer (Newark, USA).
Film-coating system: AquaPolish® P pink 640.20 PVA produced by Biogrund GmbH (Huenstetten, Germany).
Reference products: Januvia® 25 mg, 50 mg, and 100 mg film-coated tablets (Merck Sharp & Dohme B.V., Haarlem, The Netherlands).
Determining the ratio of brittle to plastic material
Powder mixtures of 1 kg each were prepared as per the compositions given in . The substances were weighed on a SARTORIUS LP3200D balance (Sartorius Lab Instruments GmbH & Co. KG, Göttingen, Germany). DCPA (DI-CAFOS® A150), MCC and croscarmellose sodium were mixed in a Turbula® mixer (Willy A. Bachofen AG, Muttenz, Switzerland) at 32 rpm for 15 min, and after the addition of the lubricant for another 5 min. The obtained mixtures were analyzed in terms of their bulk and tapped density as well as characterized for their flow characteristics using angle of repose and Carr index according to description given in Chapter 2.9.36. “Powder flow” of Ph.Eur. 10.0. Additionally, moisture content was determined with the loss on drying (LoD) using a moisture analyzer MX50 (A&D Company, Ltd. Tokyo, Japan) at the temperature of 105 °C and an endpoint of less than 0.03% mass change in 1 min.
Table 1. Powder mixture compositions with different ratios of brittle and ductile components.
The powder mixtures were compressed into tablets under three compaction forces of 10 kN, 15 kN, and 20 kN (equivalent to 200 MPa, 300 MPa, and 400 MPa respectively), with a Fette 102i rotary tablet press (Fette Compacting GmbH, Schwarzenbek, Germany) run at a 60 rpm press speed, using flat faced punches of 8 mm in diameter (Natoli Engineering Co., Inc., Saint Charles, MO). The target weight of the tablets was 250 mg.
Tablets hardness (expressed as tensile strength), weight and thickness were analyzed using a Semi-Automatic Tablet Testing System SmartTest 50 (Sotax AG, Aesch, Switzerland) and averages calculated based on the analysis of 10 randomly selected tablets.
Friability was tested with a friability tester Friabilator (USP) EF-2 (Electrolab, Mumbai, India). The number of tablets corresponding to 6.5 g were weighed and tested at a speed of 25 rpm for 4 min. The tablets were weighed again, and the mass was compared with their initial weight.
Disintegration test was carried out with an SDx-01 disintegration tester (Secom GmbH, Hamburg, Germany) in 900 ml of purified water at the temperature of 37 °C. Disintegration times of 6 individual tablets were recorded.
Preparation of Sitagliptin film-coated tablets
Five tableting mixtures (formulations) were prepared with the compositions shown in . The mixtures labeled F1–F3 contained sitagliptin phosphate monohydrate, and F4–F5 included sitagliptin hydrochloride monohydrate. Formulation F1 was a reproduction of the reference formulation (Januvia®), and employed two lubricants, magnesium stearate and sodium stearyl fumarate. Formulations F2 and F3 as well as F4 and F5 differed in the type of anhydrous calcium phosphate used. All ingredients were weighed on a SARTORIUS LP3200D balance (Sartorius Lab Instruments GmbH & Co. KG, Göttingen, Germany). First, the drug substance, DCPA, MCC and croscarmellose sodium were mixed in a Turbula® mixer (Willy A. Bachofen AG, Muttenz, Switzerland) at 32 rpm for 10 min, and after the addition of the lubricant(s) for additional 5 min.
Table 2. Compositions of Sitagliptin phosphate and Sitagliptin hydrochloride tablet cores.
Each powder mixture shown in was compressed into tablets with a Fette 102i rotary tablet press (Fette Compacting GmbH, Schwarzenbek, Germany) using concave punches manufactured by Fette Compacting GmbH, Schwarzenbek, Germany. The strength of the tablets was regulated by their weight. Thus, 25 mg tablets had a weight of 100 mg, 50 mg tablets had a weight of 200 mg, and 100 mg tablets had a weight of 400 mg. Detailed information on tableting parameters and setup is given in .
Table 3. Tabletting data.
Tablet cores were coated with an PVA-based film coating, AquaPolish® P pink 640.20 PVA (Biogrund GmbH, Huenstetten, Germany) in a Solidlab 1 drum coater (Hüttlin GmbH, Schopfheim, Germany). The coating system was dispersed in water in a weight ratio of 15%: 85% and agitated gently for 45 min. The tablets were coated to 4% theoretical weight gain using the following coating parameters: inlet air temperature of 60 °C and air flow of 40 m3/h, which allowed the temperature of the tablet bed to be maintained at approximately 46 °C. Atomizing air pressure of 1.0 bar and a spray rate of 2 g/min were used during the process.
Analysis of film-coated tablets
The tablets produced were subjected to performance tests as described in the USP/NF monograph for Sitagliptin tablets. The developed formulations were compared to Januvia® 25 mg, 50 mg, and 100 mg film-coated tablets (Merck Sharp & Dohme B.V., Haarlem, Netherlands).
A disintegration test was performed for 6 individual tablets using an SDx-01 disintegration tester (Secom GmbH, Hamburg, Germany) in 900 ml of purified water at the temperature of 37 °C. The acceptance criterion as per the monograph: disintegration time no longer than 5 min.
Uniformity of dosage unit was assessed with the weight variation (WV) method based on analysis of 10 individual tablets. The content of the drug substance was determined by the UV/Vis method at 267 nm detection wavelength using a T70 UV/VIS Split-Beam Spectrophotometer equipped with flow-through quartz cuvettes with 1 mm path length (PharmaTest AG, Hainburg, Germany). Sample solutions with a nominal concentration of the drug substance of 0.5 mg/mL were prepared by sonicating the suitable amount of the tablets in water for 5 min and filtering through a 0.45 μm Minisart®-RC syringe filter (Sartorius, Goettingen, Germany). The applied analytical method was initially checked for linearity and specificity as per ICH Q2(R2) guideline (EMEA Citation1995) and was found to be sufficient for the intended purpose of the analysis. The maximum acceptance value was adopted in accordance with USP/NF Chapter <905> "Uniformity of Dosage Units" and was (AV) L1 = 15.0.
Tablets hardness (expressed as tensile strength), weight and thickness were analyzed using a Semi-Automatic Tablet Testing System SmartTest 50 (Sotax AG, Aesch, Switzerland) and averages calculated based on the analysis of 10 randomly selected tablets.
Dissolution of the drug substance from the tablets was evaluated using a USP apparatus I (baskets) PTWS 820 D (Pharma Test Apparatebau AG, Hainburg, Germany) by placing 1 tablet in 900 ml of dissolution medium at the temperature of 37 °C and mixing at 100 rpm for 30 min. The effect of the pH of the environment on the dissolution rate was evaluated in media of various pH, i.e. 0.1 M hydrochloric acid (pH 1.0), phosphate buffer pH 4.5, and phosphate buffer pH 7.2 prepared as per Ph.Eur. 10.0, Chapter 5.17.1. “Recommendations on dissolution testing”. The analysis took 0.5 h with sampling points at 5, 10, 20, and 30 min. The samples, after filtering through a 0.45 μm Minisart®-RC syringe filter (Sartorius, Goettingen, Germany), were analyzed using a T70 UV/VIS Split-Beam Spectrophotometer equipped with flow-through quartz cuvettes with 1 mm path length (PharmaTest AG, Hainburg, Germany) at 267 nm detection wavelength. The applied analytical method was initially checked for linearity and specificity as per ICH Q2(R2) guideline (EMEA Citation1995) and was found to be sufficient for the intended purpose of the analysis.
The developed tablets as well as the reference product were placed in low density polyethylene (LDPE) bags and stored in an HPP 750 stability chamber (Memmert, Büchenbach, Germany) under stress conditions (50 °C and 80% RH) for 3 months. Changes in the amount of impurities were examined every month by an HPLC method using a Dionex UltiMateTM 3000 HPLC System equipped with a VWD-3400RS Variable Wavelength Detector (Dionex Softron GmbH, Germering, Germany). Separation was carried out using a SynergiTM 4 u Hydro-RP 80 Å 150 × 4.6 mm LC column (Phenomenex, Aschaffenburg, Germany) whose temperature was maintained at around 25 °C. A linear gradient elution with a flow rate of 1.5 ml/min was used. The gradient program starter from 85% v/v of dilute ortho-phosphoric acid (pH 2.0) and 15% v/v of acetonitrile. Within 7 min this ratio changed to 50:50% v/v. Sample solutions with a nominal concentration of the drug substance of 1 mg/mL were prepared by sonicating the suitable amount of the tablets in water for 5 min and filtering through a 0.45 μm Minisart®-RC syringe filter (Sartorius, Goettingen, Germany). About 5 µL of a sample solution was injected into the chromatographic system and chromatograms were recorded at a detection wavelength of 267 nm. The applied analytical method was initially checked for linearity, specificity as per ICH Q2(R2) guideline (EMEA Citation1995) as well as peak separation ability and found to be sufficient for the intended purpose of the analysis. Additionally, at the beginning and end of the study, tablets were examined in term of hardness, disintegration time and dissolution characteristic using the methods described earlier.
Results
The effect of the ratio of brittle to plastic material
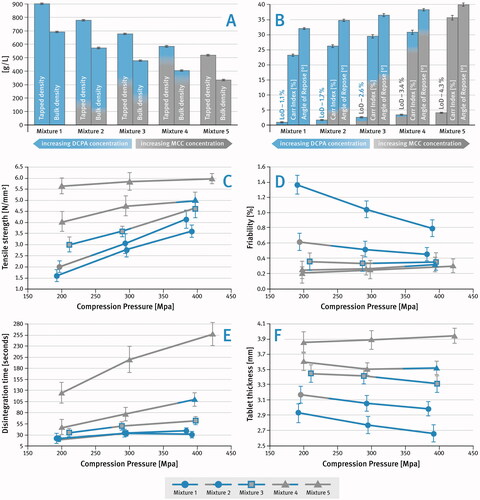
shows how varying the ratio between the content of the ductile component (MCC) and the brittle component (DCPA) affects the physical properties of powder mixtures such as bulk and tapped density, moisture content as well as flow characteristics (expressed here as Carr Index and Angle of Repose). The powder mixtures listed in were evaluated for their tabletability at increasing compaction pressures, and the produced tablets were analyzed for their physical properties. The results are shown in .
Figure 3. Effect of brittle to plastic material ratio on: (A) bulk and tapped density (means of n = 3), (B) moisture content (LoD) and flowability of powder mixtures (means of n = 3), (C) tablet hardness (tensile strength) (means of n = 10), (D) tablet friability (means of n = 3), (E) tablet disintegration time (means of n = 6), (F) tablet thickness (means of n = 10); SD is indicated by the error bars.
Comparison of physical properties of sitagliptin film-coated tablets
Each of the formulations with the compositions given in was compressed into tablets of three strengths, i.e. 25, 50, and 100 mg as per information in . The film-coated tablets were analyzed for their physical properties as described in the Materials and Methods section. A comparison of the results with those obtained for the reference products (labeled as RP) is shown in . The acceptance criteria for each investigated parameter are marked with red lines on the graphs.
Figure 4. (A) Weight variation test – acceptance values (AV), (B) comparison of tablet disintegration time (means of n = 6), (C) comparison of tablet hardness (tensile strengths) (means of n = 10), (D) comparison of tablet thickness (the reference product not included) (means of n = 10); SD is indicated by the error bars.
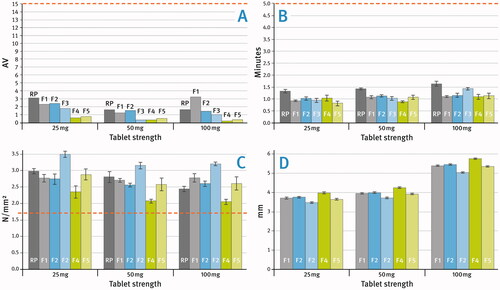
Reference product is not included in due to a different tablet diameter than that of tablets labeled as F1–F5. This graphic primarily shows the effect of different types of DCPA (DI-CAFOS® A150 or DI-CAFOS® A60) on tablet size.
Dissolution study of sitagliptin film-coated tablets in dissolution media of various pH
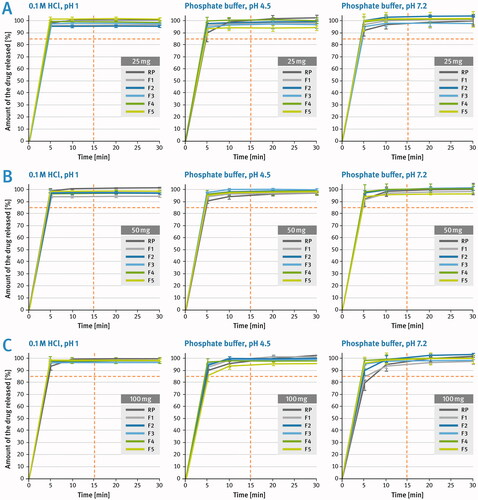
The dissolution rate of the developed film-coated tablets as well as the reference product (labeled as RP) was tested in dissolution media of various pH in the range of 1.0–7.2 as per description given in the Materials and Methods section. A comparison of dissolution profiles is shown in . The acceptance criterion is marked with a red dotted line and was based here on various guidelines that suggest for rapidly dissolving IR tablets more than 85% of the drug substance be dissolved within the first 15 min of testing in media of different pH (FDA Citation1997; EMA Citation2017).
Figure 5. Comparison of the dissolution profiles of Sitagliptin film-coated tablets: (A) 25 mg strength, (B) 50 mg strength, (C) 100 mg strength, in media with pH 1.0, 4.5 and 7.2. Means of n = 3, SD is indicated by the error bars.
Stability study
A comparison of the chemical stability of the developed formulations and the reference product during storage under stress conditions (50 °C/80% RH) is shown in . It is expressed as the sum of all impurities found in the tablets by HPLC analysis at time zero (T0), after 1, 2, and 3 months (subsequent columns) as described in the Materials and Methods section. shows the comparison of selected physical properties of the tablets at time zero (T0) and after 3 months of storage under stress conditions. For the dissolution test, the amount of drug released during the first ten minutes of the test conducted in 0.1 M hydrochloric acid was compared.
Figure 6. Comparison of the changes in total impurities content within 3-months storage under stress condition (50 °C/80% RH) of Sitagliptin film-coated tablets. Means of n = 3, SD is indicated by the error bars.
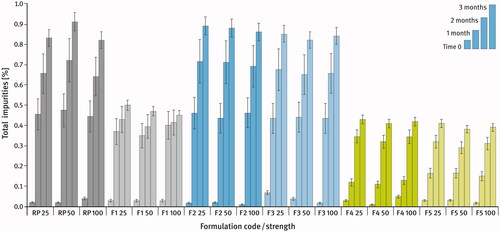
Table 4. Comparison of selected properties of Sitagliptin film-coated tablets after production (T0) and stored under stress condition (50 °C/80% RH) for 3 months.
Discussion
The aim of this study was to develop directly compressible pharmaceutical formulations containing a model drug substance, sitagliptin, using a mixture of excipients commonly used as filler/diluents, i.e. brittle DCPA and ductile MCC. Initially, the study investigated the effect of the ratio of brittle to plastic material on both the flowability and compaction properties of tableting mixtures. Several powder mixtures with different ratios of these two substances were prepared as shown in . In addition to the two fillers/diluents, the mixtures contained a disintegrant (croscarmellose sodium) and a lubricant (magnesium stearate). The results of the analyses shown in indicate that the changes in the properties of the powder mixtures, such as density (both bulk and tapped), flow properties (expressed as Carr Index or angle of repose) as well as moisture content (LoD), were directly proportional to the changes in the ratio of DCPA to MCC. The increase in the concentration of MCC resulted in a decrease in the density of the powder blends, in other words, an increase in the volume occupied by the powders. This is due to the difference in true density of the two fillers/diluents, which is 2.89 g/cm3 for DCPA and 1.51–1.67 g/cm3 for MCC (Rowe Citation2009). In addition, the increase in the concentration of MCC in the powder mixtures caused a deterioration of their powder flowability and an increase in moisture content. All these factors suggest that the ratio of brittle to plastic material can influence the process of tablet manufacturing, as well as the properties of the final product. A larger volume of powders, especially in combination with their poorer flowability, can have a negative effect on powder filling in the dies during tabletting. In addition, it can cause an increase in size/volume of tablets. A higher moisture content can have a negative impact on the stability of tablets containing moisture sensitive drug substances.
The results of the analysis of the mechanical properties of the tablets (hardness and friability) obtained by compressing the powder blends described in shown in , indicate that as the concentration of MCC increased, both the hardness and the friability of the tablets produced improved. On the other hand, it can be observed that as the concentration of DCPA in the powder mixture increased, so did the susceptibility to applied compression pressure, resulting in a substantial improvement of the mechanical properties of the tablets produced. DCPA is a brittle material that undergoes fragmentation during compression and often requires higher compression forces to reach its full potential. Even a small addition of a plastic component, such as MCC, can effectively compensate for this property. As shown in , a higher concentration of MCC in the tabletting mixture measurably increased the disintegration time of the tablets, which to some extent is related to their higher hardness. It can be noticed, however, that even a small addition of DCPA considerably reduced the disintegration time of the tablets, which is due to the increased porosity of the tablets and the easier penetration of water into their interior (Zakowiecki et al. Citation2019). The disintegration time of tablets containing mainly the plastic material was very sensitive to the applied compression pressure. At the same time, the presence of the brittle material mitigated the impact of the compression pressure on the disintegration of tablets.
The results of the tablet thickness measurements show that an increase in DCPA content resulted in a substantial reduction in tablet size (). Furthermore, the thickness of tablets containing high concentration of DCPA decreased measurably with increasing compression pressure. It may even be noted that tablets containing a high concentration of plastic material tended to expand when compressed at higher compression pressures. It should also be mentioned here that DCPA is less sensitive to over-lubrication (Almaya and Aburub Citation2008; Kushner and Moore Citation2010) and therefore its use allows application of a higher concentration of lubricant in the tabletting mixture without adversely affecting its compression properties.
The studies have shown that the use in tabletting mixtures of excipients exhibiting different deformation types i.e. brittle and plastic, produced a synergistic effect which allowed maintaining the advantageous qualities of these substances, while at the same time mitigating their unfavorable properties. In the case of tabletting mixtures containing MCC and DCPA, the tabletting mixtures exhibited very good tabletting properties, allowing the production of tablets of high hardness and low friability, and, at the same time, fast disintegration time and small size. Such mixtures give quite a lot of flexibility during formulation development. Depending on the qualities of a drug substance, the physical properties of the tablets may be customized easily only by adjusting the ratio of plastic to brittle substances.
Based on the study conducted, a mixture containing MCC and DCPA in a weight ratio of 1:1 compressed under a compression pressure of around 300 MPa was selected as optimal for the further development of sitagliptin formulations. Five compositions shown in were developed, each of which was compressed into tablets of three strengths (i.e. 25 mg, 50 mg, and 100 mg) as described in . In the course of the initial trials, it was observed that in the case of sitagliptin, both phosphate monohydrate and hydrochloride monohydrate, it was necessary to use relatively large amounts of lubricant compared to those commonly used. With insufficient lubrication, part of the material was removed from the surface of the tablets by punches during tableting (picking) resulting in the production of damaged tablets (see ).
Figure 7. Tablets containing 1% w/w of magnesium stearate showing picking problem (A) and these with 2% w/w of the lubricant (B).

In the documents relating to the reference product, attention is drawn to the fact that two different lubricants are listed in the tablet composition, i.e. magnesium stearate and sodium stearyl fumarate (EMA Citation2007; MSD Citation2021). The need to use two such excipients may be related to the above-described problem of tablet picking. In such case, this would justify the use of sodium stearyl fumarate, which is less hydrophobic than magnesium stearate, and would reduce adverse effects of a high concentration of lubricant on a drug substance release. On the other hand, this could indicate the use of dry granulation in the production process, which also requires the use of a lubricant.
In the case of the formulations developed in this study, the use of 2% w/w magnesium stearate has successfully eliminated this problem (see ) and enabled effective coating of the tablets produced.
The coated tablets were analyzed as described in the Materials and Methods section and compared with the reference product. The two performance tests as per the relevant pharmacopoeial monograph for Sitagliptin Tablets dealt with disintegration time and uniformity of dosage units. The results shown in demonstrate that all analyzed tablets met the required acceptance criteria (which were marked with red lines). Disintegration times were much shorter than the required 5 min and did not exceed 2 min for all tested tablets. The slightly longer disintegration time of the reference product may be related to differences in tablet geometry. Nevertheless, given such rapid disintegration, minor differences between the tablets analyzed are rather non-essential. Uniformity of dosage units, here demonstrated as Weight Variation was in each case much below the required Acceptance Value (AV) 15. It seems like formulations based on sitagliptin hydrochloride monohydrate (F4 and F5) showed a slightly better performance with this respect.
All tablets produced demonstrated adequate mechanical properties as presented in . It is generally accepted that a tensile strength greater than 1.7 N/mm2 is sufficient for robust tablets (Pitt and Heasley Citation2013; Leane et al. Citation2015). It may be noted that sitagliptin phosphate monohydrate possesses better tabletting properties than sitagliptin hydrochloride monohydrate and allows producing harder tablets (compare respective formulations F2 and F4 versus F3 and F5).
Comparing formulations F2 with F3, and similarly F4 with F5, which had in pairs the same composition, differing only in the type of DCPA used (see ), it may be noted that the use of the dense one (DI-CAFOS® A60) makes it possible to produce tablets of greater hardness. In addition, tablets based on this type of DCPA had a smaller size (see ), which is due to its very low volume (both bulk and tapped) in comparison with other excipients.
Regardless of the differences in tablet size and hardness, all tablets analyzed showed very rapid release rates of the drug substance in dissolution media of various pH from the physiological range (). The dissolution profiles were very fast and repetitive as indicated by relatively small standard deviation values.
Sitagliptin is a BCS Class I compound showing high solubility and complete absorption (EMA Citation2016; Charoo et al. Citation2022). The EMA “Guideline on the Investigation of Bioequivalence” and the FDA guidance “Waiver of in Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System” state that in case of very rapidly dissolving dosage forms, where more than 85% of the drug is dissolved within 15 min in media of different pH, dissolution profiles may be assumed as similar without further mathematical evaluation (EMA Citation2010; FDA Citation2017). In view of this, it can be considered that all the formulations developed in this study showed similar dissolution profiles to the reference products.
The results of the chemical stability studies (see ) clearly indicate that of the two sitagliptin salts under investigation, the hydrochloride one was more stable (formulations F4 and F5), even when stored for 3 months under stress conditions (50 °C/80% RH) in packaging that did not provide sufficient protection against the external conditions, i.e. LDPE bags (compare formulations F2 and F3 with F4 and F5). The chemical stability of different strengths of the same tablet formulation was not substantially different from each other. Furthermore, no measurable differences with respect to the amount of impurities generated were observed between formulations differing only in the type of DCPA used (F2 and F3 or F4 and F5). Interestingly, when comparing the reference product and the F1 formulation, which were analogous in terms of the qualitative and quantitative composition, one can see a difference in the rate of impurities grow. This may be attributed to the different source of the drug substance and/or the different grades and origin of the same excipients used in both preparations. An interesting observation can be made when comparing F1 and F2 formulations, where the difference in composition related mainly to the amount of lubricant used. The rest of the formulation components, i.e. the drug substance, grades and batches of excipients used, were exactly the same. It can be assumed with high probability that due to the hydrophobic nature of lubricants, their twice higher concentration in the first formulation shielded particles of all ingredients more effectively and clearly reduces the impact of external conditions on the drug substance, contributing to a more effective chemical stabilization of the tablets.
summarizes the changes in selected physical properties of the tablets when stored under stress conditions for 3 months. It can be clearly seen that all analyzed tablets softened substantially, losing from about 45% to as much as 65% of their initial hardness (tensile strength). This may be related to the absorption of moisture by the tablets, as indicated by an increase in their weight. Nevertheless, no clear correlation was observed between the amount of moisture absorbed and the decrease in tablet tensile strength. Tablets containing porous grade of DCPA (DI-CAFOS® A150) i.e. formulations F1, F2, and F4 absorbed relatively high amount of moisture (approximately 1.3–1.9% increase in tablet weight) which is comparable to the moisture absorption of the reference product. Tablets containing dense DCPA with lower porosity (DI-CAFOS® A60) i.e., formulations F3 and F5, absorbed relatively less moisture (approximately 0.1–0.8% increase in tablet weight), but even for these formulations the tablets were vastly softer that at the initial ones than at the beginning of the stability studies.
It is important to note here that in this study all the formulations were deliberately exposed to very harsh environmental conditions without sufficient protection from humidity and temperature. The results show that although the tablets are film-coated, they still need to be adequately protected from moisture to retain their physical properties during storage. In this regard, the formulations developed in this study responded to the stress conditions in a manner analogous to the reference product. Nonetheless, in terms of chemical stability, it can be seen that sitagliptin hydrochloride monohydrate offers great advantages over sitagliptin phosphate monohydrate, offering a safer option when developing new generic formulations of this drug substance.
Conclusions
The conducted studies have shown that proper adjustment of the ratio of brittle to ductile fillers/diluent (DCPA and MCC, respectively) in a tablet formulation allowed for successful development of IR film-coated tablets of the model drug substance, sitagliptin by DC technology. The synergistic action of both components made it possible to prepare tablets with appropriate mechanical strengths and required performance for both sitagliptin salts, i.e. phosphate monohydrate and hydrochloride monohydrate, which were comparable with the commercial reference product. It was found that the type of DCPA used had a large effect on the properties of the sitagliptin tablets. The developed formulations stored under stress conditions (50 °C/80% RH) in packaging that does not provide protection from the outside conditions (LDPE) showed stability similar to or better than the reference formulation. In this regard, the hydrochloride salt of sitagliptin appears to be more stable.
Statement of human and animal rights
This article does not contain any studies with human and animal subjects performed by any of the authors.
Acknowledgment
AquaPolish® P pink 640.20 PVA has been developed and kindly donated by Biogrund GmbH (Huenstetten, Germany). Ligamed® MF-2-V has been kindly donated by Peter Greven Fett-Chemi (Venlo, The Netherlands).
Disclosure statement
Daniel Zakowiecki Peter Edinger, Markos Papaioannou and Tobias Hess are employee of Chemische Fabrik Budenheim KG and the company had no role in funding. Bartlomiej Kubiak and Anna Terlecka work for Adamed Pharma S.A. and the company had no role in funding. The companies or any other organization had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results. The authors declare no conflicts of interest.
Data availability statement
All relevant data/results collected in this study are presented in the article.
Additional information
Funding
References
- Albers J, Knop K, Kleinebudde P. 2006. Brand-to-brand and batch-to-batch uniformity of microcrystalline cellulose in direct tableting with a pneumohydraulic tablet press. Pharm Ind. 68(12):1420–1428.
- Almaya A, Aburub A. 2008. Effect of particle size on compaction of materials with different deformation mechanisms with and without lubricants. AAPS PharmSciTech. 9(2):414–418.
- Chaerunisaa AY, Sriwidodo S, Abdassah M. 2020. Microcrystalline cellulose as pharmaceutical excipient. In: Ahmad U, Akhtar J, editors. Pharmaceutical formulation design - recent practices. London: IntechOpen; p. 41–62.
- Charoo NA, Abdallah DB, Bakheit AA, Haque KU, Hassan HA, Abrahamsson B, Cristofoletti R, Langguth P, Mehta M, Parr A, et al. 2022. Biowaiver monograph for immediate-release solid oral dosage forms: sitagliptin phosphate monohydrate. J Pharm Sci. 111(1):2–13.
- [EMA] European Medicines Agency. 1995. ICH Q 2 (R1) validation of analytical procedures: text and methodology. London (UK). [accessed 2022 July 7] https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-2-r1-validation-analytical-procedures-text-methodology-step-5_en.pdf
- [EMA] European Medicines Agency. 2003. ICH Q1A(R2) stability testing of new drug substances and products. London (UK). [accessed 2022 June 7] https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-1-r2-stability-testing-new-drug-substances-products-step-5_en.pdf
- [EMA] European Medicines Agency. 2007. European public assessment report – scientific discussion for Januvia, INN: sitagliptin. [accessed 2022 June 7] https://www.ema.europa.eu/en/documents/scientific-discussion/januvia-epar-scientific-discussion_en.pdf
- [EMA] European Medicines Agency. 2010. Committee for medicinal products for human use (CHMP). Guideline on the investigation of bioequivalence. London (UK). [accessed 2022 June 7] https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf
- [EMA] European Medicines Agency. 2016. Sitagliptin film-coated tablets 25, 50 and 100 mg product-specific bioequivalence guidance. London (UK). [accessed 2022 June 7] https://www.ema.europa.eu/en/documents/scientific-guideline/sitagliptin-film-coated-tablets-25-50-100-mg-product-specific-bioequivalence-guidance_en.pdf
- [EMA] European Medicines Agency. 2017. Reflection paper on the dissolution specification for generic solid oral immediate release products with systemic action. [accessed 2022 June 7] https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-dissolution-specification-generic-solid-oral-immediate-release-products-systemic_en.pdf
- [EMA] European Medicines Agency. 2021. Sitagliptin SUN - Assessment Report. Amsterdam, Netherlands. [accessed 2022 June 7] https://www.ema.europa.eu/en/documents/assessment-report/sitagliptin-sun-epar-public-assessment-report_en.pdf
- [FDA] U.S. Food and Drug Administration. 1997. Guidance for industry. Dissolution testing of immediate release solid oral dosage forms. Rockville (MD, USA). [accessed 2022 June 7] https://www.fda.gov/regulatory-information/search-fda-guidance-documents/dissolution-testing-immediate-release-solid-oral-dosage-forms
- [FDA] U.S. Food and Drug Administration. 2017. Center for Drug Evaluation and Research, Guidance for Industry - Waiver of in vivo bioavailability and bioequivalence studies for immediate release solid oral dosage forms based on a biopharmaceutics classification system. Silver Spring (MD, USA). [accessed 2022 June 7] https://www.gmp-navigator.com/files/guidemgr/UCM070246.pdf. https://www.federalregister.gov/documents/2017/12/26/2017-27786/waiver-of-in-vivo-bioavailability-and-bioequivalence-studies-for-immediate-release-solid-oral-dosage
- Galenicum Health S.L. 2021. Sitagliptin Galenicum Tablet, film coated 25mg – Summary of Product Characteristics. [accessed 2022 June 7] https://myhealthbox.eu/en/sitagliptin-galenicum-tablet-film-coated-25mg/5195140
- Gupta P, Nachaegari SK, Bansal AK. 2006. Improved excipient functionality by coprocessing. In: Katdare A, Chaubal MV, editors. Excipient development for pharmaceutical, biotechnology, and drug delivery systems. New York (NY): Informa Healthcare; p. 109–126.
- Ilić I, Govedarica B, Šibanc R, Dreu R, Srčič S. 2013. Deformation properties of pharmaceutical excipients determined using an in-die and out-die method. Int J Pharm. 446(1–2):6–15.
- Ilić I, Kása P Jr, Dreu R, Pintye-Hódi K, Srcic S. 2009. The compressibility and compactibility of different types of lactose. Drug Dev Ind Pharm. 35(10):1271–1280.
- JRS Pharma. 2022. VIVAPUR® Microcrystalline Cellulose. [accessed 2022 June 7] https://www.jrspharma.com/pharma_en/technical-info/brochures/brochures/mcc-leaflet-vivapur.php
- Kristensen HG, Schaefer T. 1987. Granulation: a review on pharmaceutical wet-granulation. Drug Dev Ind Pharm. 13(4–5):803–872.
- Kumar S, Gupta SK, Sharma PK. 2012. A review on recent trends in oral drug delivery-fast dissolving formulation technology. Adv Biol Res. 6(1):6–13.
- Kushner J, Moore F. 2010. Scale-up model describing the impact of lubrication on tablet tensile strength. Int J Pharm. 399(1–2):19–30.
- Landín M, Martínez-Pacheco R, Gómez-Amoza JL, Souto C, Concheiro A, Rowe RC. 1994. Dicalcium phosphate dihydrate for direct compression: characterization and intermanufacturer variability. Int J Pharm. 109(1):1–8.
- Lawal MV. 2019. Modified starches as direct compression excipients – effect of physical and chemical modifications on tablet properties: a review. Starch. 71(1–2):1800040.
- Leane M, Pitt K, Reynolds G, Manufacturing Classification System (MCS) Working Group. 2015. A proposal for a drug product Manufacturing Classification System (MCS) for oral solid dosage forms. Pharm Dev Technol. 20(1):12–21.
- Mangal S, Meiser F, Morton D, Larson I. 2015. Particle engineering of excipients for direct compression: understanding the role of material properties. Curr Pharm Des. 21(40):5877–5889.
- Meeus L. 2011. Direct compression versus granulation. Pharm Technol Europe. 23(3):21–22. [accessed 2022 June 7]
- Mirani AG, Patankar SP, Borole VS, Pawar AS, Kadam VJ. 2011. Direct compression high functionality excipient using coprocessing technique: a brief review. Curr Drug Deliv. 8(4):426–435.
- [MSD] Merck Sharp & Dohme (UK) Limited. 2021. JANUVIA 25mg, 50mg, 100mg film-coated tablets – Summary of Product Characteristics. [accessed 2022 June 7] https://www.medicines.org.uk/emc/product/321/smpc#gref
- Muñoz-Ruiz A, Perales M, Antequera MV, Payán Villar T, Muñoz-Muñoz N, Jiménez-Castellanos M. 1993. Rheology and compression characteristics of lactose based direct compression excipients. Int J Pharm. 95(1–3):201–207.
- Nachaegari SK, Bansal AK. 2004. Coprocessed excipients for solid dosage forms. Pharm Technol. 28(1):52–64.
- Natoli D, Levin M, Tsygan L, Liu L. 2017. Development, optimization, and scale-up of process parameters: tablet compression. In: Qiu Y, Chen Y, Zhang GGZ, Yu L, Mantrii RV, editors. Developing solid oral dosage forms: pharmaceutical theory and practice. London: Academic Press; p. 917–951.
- Pitt KG, Heasley MG. 2013. Determination of the tensile strength of elongated tablets. Powder Technol. 238:169–175.
- Rojas J, Buckner I, Kumar V. 2012. Co-proccessed excipients with enhanced direct compression functionality for improved tableting performance. Drug Dev Ind Pharm. 38(10):1159–1170.
- Rojas J. 2015. Excipient design by co-processing for direct compression applications. In Narang AS, Boddu SHS, editors. Excipient applications in formulation design and drug delivery. Cham, Switzerland: Springer; p. 589–612.
- Rowe RC, Sheskey PJ, Quinn ME. 2009. Handbook of pharmaceutical excipients. 6th ed. London: Pharmaceutical Press; p. 94–131.
- Saha S, Shahiwala AF. 2009. Multifunctional coprocessed excipients for improved tabletting performance. Expert Opin Drug Deliv. 6(2):197–208.
- Schmidt PC, Herzog R. 1993. Calcium phosphates in pharmaceutical tableting. 1. Physico-pharmaceutical properties. Pharm World Sci. 15(3):105–115.
- Schwarz E, Fichtner V. 2016. Direct compression excipient based on lactose, cellulose and starch (EP 3 002 005 A1). [accessed 2022 June 7] https://patents.google.com/patent/EP3002005A1/en.
- Shanmugam S. 2015. Granulation techniques and technologies: recent progresses. Bioimpacts. 5(1):55–63.
- Sun CC. 2017. Recent advances in tabletting research. Tablets&Capsules. 15(4):30–33.
- Thoorens G, Krier F, Leclercq B, Carlin B, Evrard B. 2014. Microcrystalline cellulose, a direct compression binder in a quality by design environment – a review. Int J Pharm. 473(1–2):64–72.
- Thoorens G, Leclercq B, Ruszkay T. 2018. Microcrystalline cellulose and calcium phosphate compositions useful as pharmaceutical excipients (EP 2 498 818 B1). [accessed 2022 June 7] https://data.epo.org/publication-server/document?iDocId=5535375&iFormat=0.
- Vehovec T, Gartner A, Planinšek O, Obreza A. 2012. Influence of different types of commercially available microcrystalline cellulose on degradation of perindopril erbumine and enalapril maleate in binary mixtures. Acta Pharm. 62(4):515–528.
- Viswanathan P, Muralidaran Y, Ragavan G. 2017. Challenges in oral drug delivery: a nano-based strategy to overcome. In Andronescu E, Grumezescu AM, editors. Micro and nano technologies, Nanostructures for oral medicine. New York (NY): Elsevier; p. 173–201.
- Wagner M, Hess T, Zakowiecki D. 2022. Studies on the pH-dependent solubility of various grades of calcium phosphate-based pharmaceutical excipients. J Pharm Sci. 111(6):1749–1760.
- Zakowiecki D, Lachmann M, Hess T. 2019. Eye on excipients. Tablets Capsules. 7(4):39–43. [accessed 2022 June 7] https://www.researchgate.net/publication/333207856_Eye_on_Excipients_Tablets_Capsules_Magazine.
- Zerilli T, Pyon EY. 2007. Sitagliptin phosphate: a DPP-4 inhibitor for the treatment of type 2 diabetes mellitus. Clin Ther. 29(12):2614–2634.
- Zheng JY, Ternik RL. 2008. Development of low-dose solid oral tablets using direct compression. In: Zheng JY, editor. Formulation and analytical development for low-dose oral drug products. Hobooken (NJ): John Wiley&Sons Inc.; p. 159–204.