
ABSTRACT
This review summarizes the carcinogenic mechanisms for 109 Group 1 human carcinogens identified as causes of human cancer through Volume 106 of the IARC Monographs. The International Agency for Research on Cancer (IARC) evaluates human, experimental and mechanistic evidence on agents suspected of inducing cancer in humans, using a well-established weight of evidence approach. The monographs provide detailed mechanistic information about all carcinogens. Carcinogens with closely similar mechanisms of action (e.g. agents emitting alpha particles) were combined into groups for the review. A narrative synopsis of the mechanistic profiles for the 86 carcinogens or carcinogen groups is presented, based primarily on information in the IARC monographs, supplemented with a non-systematic review. Most carcinogens included a genotoxic mechanism.
Introduction
In the early 1970s, the International Agency for Research on Cancer (IARC) started the IARC Monographs Programme to evaluate the carcinogenic risk of chemicals and other agents to humans. The Programme regularly identifies agents to evaluate, undertaking comprehensive reviews of the available human, animal and laboratory evidence of their carcinogenic potential. The review process involves international committees of experts (Working Groups) and an exhaustive review of the available literature of the human and animal evidence of increased cancer risk occurrence for each agent. As part of the evaluation of potentially carcinogenic agents, the Working Groups also reviewed the available data on mechanistic pathways by which such agents may act as carcinogens. The procedures used by IARC to prepare these reviews is described in each Monograph Volume; a detailed description of the process as currently applied is given in the Preamble to the IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, available at http://monographs.iarc.fr/ENG/Preamble/index.php.
The first IARC Monograph was published in 1972 and examined 19 organic and inorganic substances. Since that initial Volume, a total of 123 Monographs have been published or are in preparation (up to October 2018), addressing a wide range of carcinogenic and potentially carcinogenic agents, including pharmaceuticals, industrial chemicals, radiation, personal habits and biological agents. The present review addresses the Monographs up to and including Volume 106 (published in 2014). These IARC Monographs identified 109 agents that were classified in ‘Group 1, carcinogenic to humans’.
In 2008–2009, six IARC Monograph Working Groups were formed to produce six volumes of Monograph Volume 100, ‘A Review of Human Carcinogens’. The Volume 100 Monographs were intended to provide a review and update of the 107 Group-1 human carcinogens that had been identified in Monograph Volumes 1–99. It was published in six Volumes in 2012. These Volumes examined the Group-1 agents in six broad groups:
Pharmaceuticals, Vol. 100A (IARC Citation2012a)
Biological agents, Vol. 100B (IARC Citation2012b)
Arsenic, Metals, Fibers, and Dusts, Vol. 100C (IARC Citation2012c)
Radiation, Vol. 100D (IARC Citation2012d)
Personal Habits and Indoor Combustions, Vol. 100E (IARC Citation2012e)
Chemical Agents and Related Occupations, Vol. 100F (IARC Citation2012f)
During the period 2012–2014, six additional Monographs were published. These identified two more Group-1 human carcinogens, one each in Volume 105 (IARC Citation2014a) and Volume 106 (IARC Citation2014b).
Volume 100 examined and updated the available scientific evidence for the 107 Group-1 human carcinogens identified in Monograph Volumes 1–99. Since many Group-1 human carcinogens are closely related (e.g. three isotopes of radium are listed separately but have identical carcinogenic mechanisms), Volume 100 grouped the 107 Group-1 human carcinogens into 84 chapters, combining carcinogens that shared common features. The current review examines a total of 86 groupings of Group-1 human carcinogens: the 84 groups from Volume 100 and the two Group-1 human carcinogens from Volumes 105 and 106. The information in this review is presented in 86 sections corresponding to the chapter classification for Group-1 human carcinogens reported in the Monographs.
The purpose of this paper is to provide an overview of the biological mechanisms by which Group-1 human carcinogens exert their carcinogenic effects in humans, focusing specifically on the 86 distinct Group-1 human carcinogens. For simplicity, these will be referred to as ‘agents’ in the review.
Identification of mechanistic information
All agents classified by IARC as Group-1 human carcinogens in Monographs 1 through 106 were reviewed. The narrative summary present in the review for each agent is derived from two sources. The primary source of these narrative summaries is the material presented in the IARC Monographs. The monograph mechanistic descriptions were supplemented with information obtained from non-systematic PubMed literature searches to identify key new mechanistic information that had been published subsequent to the release of the IARC Monographs.
For each agent discussed in Volume 100, mechanistic information presented in Section 4 (‘Mechanistic and other relevant data’) of the relevant IARC Monograph was summarized to identify the major carcinogenic mechanism(s). A similar procedure was followed for the Group-1 agents identified in Volumes 105 and 106, based upon the mechanistic considerations therein. The mechanistic details presented in the Monographs reflect the conclusions of the international expert Working Groups that prepared the Monographs. The Monographs provides a detailed review and update of animal and human evidence on Group-1 human carcinogens at the time they were prepared in 2008–2009. Selected references cited in earlier Monographs were included for some agents in the relevant Volume 100–106 mechanistic summary.
A narrative synopsis was written for each agent, describing the mechanism(s) of carcinogenesis. The summaries of the mechanistic data available in the IARC Monographs presented in this paper were kept relatively short; full descriptions of this information, which for many agents is extensive, may be found in the original Monographs.
The information obtained from the Monographs was supplemented with a (non-systematic) PubMed search to retrieve additional mechanistic information on these 86 agents that had become available after publication of the Monographs. Prior to undertaking the PubMed search, each of the mechanisms identified from the Monographs were classified into one of 24 toxicological endpoints. The endpoints had been identified at a meeting of participants at a workshop on ‘Tumour-site Concordance and Mechanisms of Carcinogenesis’, which convened in Lyon in April/November, 2012. The goal of the PubMed search for each agent was to determine if there was evidence to support inclusion of any new toxicological endpoints to the narrative synopsis. Al-Zoughool and colleagues present a more detailed description of this process (Al-Zoughool et al. Citation2019). For endpoints or mechanisms found to be linked to a carcinogen and not already identified in the synopsis, an addition was made to the narrative summary. Additional mechanistic information was identified for 50 of the 86 agents/agent groups.
The additional information identified through the PubMed search was used for a sensitivity analysis designed to gauge the extent to which additional information not reported in the Monographs might impact the exploratory analysis of key characteristics of human carcinogens.
Narrative reviews of carcinogenic mechanisms for 86 human carcinogens
The review process produced 86 qualitative narrative summaries. These are presented in the order in which they appeared in the IARC Monographs, starting with ‘Pharmaceuticals’ (Volume 100A) and ending with ‘Chemical Agents and Related Occupations’ (Volume 100F). The two Group-1 human carcinogens identified in Volumes 105 and 106 are discussed after the agents in Volume 100F.
As noted above, some Group-1 human carcinogens share common mechanisms. For example, all agents that emit ionizing radiation (Volume 100D) are carcinogenic through a common mechanism. Similarly, many agents in Volume 100F are aromatic amines that possess overlapping mechanistic pathways. In order to avoid repeating the summary of these common mechanisms in multiple narrative summaries, two synopses are presented at the start of the sections for Volume 100D and 100F to discuss these common modalities. Where appropriate, the agent-specific reviews need to be read in conjunction with these two summaries.
Each of the narrative summaries follows a common presentation. The first paragraph is a quotation from the corresponding IARC Monograph, providing the overview of the main carcinogenic mechanism(s) for the agent under review. This is followed by a summary of the exposure routes and cancers that are linked to the agent. The next paragraphs present the narrative synopsis of the mechanistic information provided in the Monograph. No primary references are provided to this material they have been sourced from the well-referenced IARC Monographs. Finally, additional mechanistic data, identified through the literature review, are summarized in a separate section.
Volume 100A: pharmaceuticals
Busulfan
‘Busulfan is a direct-acting alkylating agent that is carcinogenic via a genotoxic mechanism.’ (IARC Citation2012a, 43)
Busulfan (1,4-butanediol dimethane sulfonate) is a direct-acting bifunctional alkylating agent which has been widely used for the treatment of chronic myeloid leukemia prior to the introduction of Imatinib. Busulfan produces acute myeloid leukemia.
The primary mechanism of carcinogenesis is through genotoxicity. Busulfan binds covalently to cellular macromolecules including DNA, RNA, and proteins. Consequently, this drug is capable of producing mono-adducts, intrastrand DNA–DNA cross-links, and DNA–protein cross-links. In vitro studies with human and rodent cells treated with busulfan showed formation of chromosomal aberrations, sister chromatid exchange, and mutations. In vivo treatment of rodents with busulfan induced dominant lethal mutations, and increased the frequency of chromosomal aberrations or micronuclei in bone marrow, intestine, embryonic liver, and germ cells. Patients treated with busulfan for chronic myeloid leukemia were found to exhibit elevated frequencies of sister chromatid exchange and chromosomal aberrations in their peripheral blood lymphocytes. Evidence suggests that busulfan directly induces losses or deletions affecting chromosomes 5 or 7 (e.g. loss of heterozygosity for TP53). Chromosomal alterations and deletions were reported in a variety of experimental models and in lymphocytes of exposed humans.
Chlorambucil
‘Chlorambucil is a direct-acting alkylating agent that is carcinogenic via a genotoxic mechanism.’ (IARC Citation2012a, 53)
Chlorambucil is an antineoplastic agent used primarily to treat leukemias and lymphomas. Chlorambucil induces acute myeloid leukemia.
Chlorambucil forms covalent DNA adducts. The compound contains two chloro-ethyl groups, one of which reacts with the N7 position of guanine or adenine. The second chloro-ethyl group subsequently reacts with cellular proteins, or with a DNA base to form a stable DNA inter-strand cross-link, leading to mitotic delay. Failure to repair these lesions leads to mutations. Chlorambucil has been tested for genotoxicity in several short-term assays in vitro and in vivo. This compound is mutagenic in bacteria after metabolic activation. This drug produces a range of genetic damages including gene conversion in yeast, sex-linked recessive mutations in Drosophila, mutations in Chinese hamster ovary cells, and clastogenic effects in human lymphocytes in vitro, and in animals in vivo. Exposure to chlorambucil increases the frequency of micronuclei and chromosomal aberrations in rat bone-marrow and spleen in vivo.
Additional information
Chlorambucil produces immunosuppression, leading to its use in treatment of conditions such as idiopathic membranous nephropathy (Chen et al. Citation2014b). The relevance to human carcinogenesis remains unclear.
Methyl-CCNU
‘Methyl-CCNU is a direct-acting alkylating agent that is carcinogenic via a genotoxic mechanism.’ (IARC Citation2012a, 60).
Methyl-CCNU (N-(2-chloroethyl)-N’-(4-methylcyclohexyl)-N-nitrosourea) is an anti-neoplastic agent that was used as an investigational drug to treat various cancers but induces acute myeloid leukemia.
Methyl-CCNU is a bifunctional antineoplastic agent that is carcinogenic through a genotoxic mechanism. Genotoxicity arises from electrophilic alkylating metabolites produced by spontaneous chemical decomposition and CYP-P450-mediated metabolism. The metabolites induce alkylation and carbamoylation of cellular macromolecules, including DNA and protein. Alkylation produces G-C crosslinking in DNA. Carbamoylation of certain proteins may contribute to inhibition of DNA-repair.
Genotoxic effects induced by methyl-CCNU were reported in a range of short-term assays. These include the induction of DNA adducts in the bone marrow, spleen and colon of treated mice, and in the kidney, liver and lung of treated rats. Micronuclei are more frequently seen in bone-marrow erythrocytes of treated mice. Chromosomal aberrations, micronuclei, sister chromatid exchange, and DNA strand-breaks were detected in human or rodent cells treated in vitro with methyl-CCNU. Patients treated with this drug demonstrated enhanced frequency of sister chromatid exchange and elevated levels of chromosome aberrations in peripheral blood lymphocytes.
Cyclophosphamide
‘Cyclophosphamide, after its bio-activation to alkylating metabolites, is carcinogenic via a genotoxic mechanism’. (IARC Citation2012a, 82).
Cyclophosphamide is an antineoplastic agent that is widely used in cancer treatment for its immunosuppressive properties. Metabolism of this drug has been extensively studied with respect to carcinogenicity. The parent compound is not carcinogenic by itself and requires hepatic metabolic activation. The two primary metabolites with carcinogenic potential are phosphoramide mustard and acrolein. Cyclophosphamide induces cancer of the bladder and acute myeloid leukemia.
Phosphoramide mustard binds covalently to DNA, producing various types of DNA adducts. Studies with the Comet assay revealed evidence of single-strand DNA breaks and related lesions. Several biomarkers of genotoxicity were detected more frequently in patients treated with cyclophosphamide than controls. In vitro testing noted a wide range of mutagenic effects in both human cells (chromosomal aberrations, sister chromatid exchange, DNA damage) and rodent cells (morphological transformation, chromosomal aberrations, sister chromatid exchange, mutations, unscheduled DNA synthesis (UDS)). These effects were enhanced following incubation in the presence of an exogenous metabolic activation system. In vivo testing in mice found evidence of DNA-adduct formation and, in rodents, dominant lethal mutations, chromosomal aberrations, micronuclei, sister chromatid exchange, mutations, and DNA damage.
Acrolein, in addition to displaying DNA binding, was linked to development of cystitis, which contributes to carcinogenesis through a promotion effect.
Additional information
Alterations in DNA-methylation patterns are associated with cyclophosphamide-induced chronic cystitis in mice (Choi et al. Citation2013) and during cyclophosphamide-induced teratogenesis (Gueta et al. Citation2010).
Etoposide in combination with cisplatin and bleomycin; cisplatin and bleomycin; etoposide
‘Etoposide in combination with cisplatin and bleomycin is carcinogenic via a genotoxic mechanism.’ (IARC Citation2012a., 101).
Etoposide, cisplatin and bleomycin are frequently administered to patients as combined chemotherapy. The three agents exert differing mechanisms of action and carcinogenic potential. Etoposide has been most extensively studied and the most notable carcinogenic profile. Etoposide in combination with cisplatin and bleomycin initiates acute myeloid leukemia. As a separate agent, etoposide was also classified as a Group-1 human carcinogen.
All three drugs interfere with the ability of DNA polymerase to synthesize DNA. Etoposide binds to topoisomerase IIα and affects DNA replication. Topoisomerase IIα reduces DNA tangles and supercoils by producing double-strand DNA breaks that are subsequently re-ligated. The etoposide-topoisomerase IIα complex interferes with DNA re-ligation, enhancing the persistence of DNA double-strand breaks. These complexes also directly block advancing DNA-replication fork. Cisplatin and bleomycin also enhance double-strand breaks. Each of these individual drugs induces sister chromatid exchange and aneuploidy.
Etoposide was also shown to initiate chromosomal breakages, rearrangements, and translocations within the MLL (mixed lineage leukemia) gene in experimental systems, e.g. in mouse embryonic stem cells and in CD34-positive hematopoietic in culture, including human long-term repopulating hematopoietic stem cells.
Melphalan
‘Melphalan is a direct-acting alkylating agent that is carcinogenic via a genotoxic mechanism’ (IARC Citation2012a, 113).
Melphalan is used in the treatment of several neoplasms, including multiple myeloma. Melphalan induces acute myeloid leukemia.
Melphalan (4-[bis(chloroethyl)amino]phenylalanine) is a direct-acting, bifunctional, alkylating agent that binds to cellular macromolecules including DNA, RNA and proteins. This compound was found to produce various types of DNA adducts and induce DNA interstrand cross-links. Adducts and cross-links were observed in in vitro studies with rodent cells and in patients treated with melphalan. These effects were noted particularly in the cancer-related genes TP53 and N-RAS. In vitro and in vivo studies found an increased frequency of dominant lethal mutations, chromosomal aberrations, micronuclei, and DNA strand-breaks in rodents and in rodent cells treated with melphalan. Similar effects were detected in human cells treated in vitro. Mutations in the HPRT gene were also noted. In human patients treated with melphalan, chromosomal aberrations and sister chromatid exchange were found in peripheral lymphocytes.
MOPP
‘The MOPP combination as well as individual components, except for prednisone, are genotoxic, and induce cancer via a genotoxic mechanism.’ (IARC Citation2012a, 126).
MOPP refers to a four-drug chemotherapeutic regimen composed of: mechlorethamine, oncovin, procarbazine, and prednisone. The individual agents were subjected to separate IARC reviews. MOPP was superseded by more recent therapeutic alternatives. MOPP initiates cancer of the lung and acute myeloid leukemia.
Mechlorethamine (chlormethine, nitrogen mustard) is a bifunctional alkylating agent that binds to DNA and produces mono-adducts and cross–links. In vitro studies show that this drug induces chromosomal aberrations, sister chromatid exchange, and unscheduled DNA synthesis in both rodent and human cells. In vivo studies in mice produced dominant lethal mutations and micronuclei in bone marrow. Chromosomal aberrations were noted in one study of treated patients.
Oncovin (vincristine sulfate) is a vinca alkaloid that interferes with microtubule assembly and spindle formation, thereby blocking cell replication. Vincristine sulfate induces micronuclei in the bone marrow of mice and hamsters treated in vivo, and aneuploidy and transformation in hamster embryo cells, but does not induce sister chromatid exchange or structural chromosomal aberrations.
Procarbazine is a methyl-hydrazine derivative. Carcinogenicity requires metabolism to a reactive intermediate (a methyl-diazonium cation) that methylates DNA. In vivo studies demonstrated that procarbazine induced micronuclei and structural chromosomal aberrations in mice, sister chromatid exchange in mice and Chinese hamsters, and DNA damage in rodents.
Prednisone is a synthetic glucocorticoid with multiple modes of action. This compound produces a range of anti-inflammatory and immunosuppressive effects. There is no evidence that it causes mutagenicity or direct DNA damage.
Several studies on the carcinogenicity of the MOPP combination therapy confirm the findings described above for the individual components.
Additional information
A small study in workers involved in the production of vincristine sulfate (n = 15) found evidence for increased frequencies of micronuclei, DNA strand breaks and higher mutation frequency in the HPRT gene (Hongping et al. Citation2006). This was confirmed an in vitro experiment that exposed cells from two non-occupationally exposed subjects to vincristine sulfate (Jiang et al. Citation2008).
Tamoxifen
‘There is strong evidence that in rat liver, tamoxifen is a genotoxic carcinogen through a pathway involving α-hydroxylation to produce 4-hydroxytamoxifen, sulfation of the α-hydroxy metabolite, and subsequent DNA-adduct formation. Evidence for the role of this pathway in induction of human endometrial tumours is less compelling; rather, the data suggest that the carcinogenicity of tamoxifen is associated with an oestrogen-receptor-dependent pathway.’ (IARC Citation2012a, 155).
Tamoxifen is a first-line drug for the treatment of metastatic breast cancer in post-menopausal women. In addition to use in chemotherapy, it was suggested for therapy as a chemo-preventive agent for women at high risk of breast cancer. Tamoxifen produces endometrial cancer.
‘The available evidence indicates that tamoxifen is both a genotoxic carcinogen and a tumour promoter in rat liver, and that humans are likely to be less susceptible to the genotoxicity of the drug.’ (IARC Citation2012a, 148).
Evidence for a genotoxic mechanism of tamoxifen-induced carcinogenesis in humans is not compelling. There are conflicting reports on formation of tamoxifen-DNA adducts in humans, with some investigators finding adducts in endometrial, colon and white blood cells. Studies in rats and mice have been more consistent in reporting adducts in liver. The suggestion was made that the production of adducts is linked to a minor phase-I metabolic pathway, i.e. sulfotransferase-mediated sulfation, specifically by the STA2 isoform of the enzyme SULT2A1, which may explain the discrepant results. Most investigators have not detected DNA adducts in uterus and other extra-hepatic tissues from rats.
Tamoxifen induces micronuclei in metabolically proficient human cells and initiates aneuploidy and chromosomal aberrations. Moreover, both tamoxifen and 4-hydroxy-tamoxifen induce mutations in the lacI reporter gene and the cII gene in the livers of Big Blue® transgenic rats.
There is supporting evidence for a non-genotoxic pathway for tamoxifen-induced carcinogenesis in humans. Tamoxifen is a selective estrogen-receptor modulator. In the endometrial endothelium, this drug acts as an agonist, stimulating cellular proliferation. Evidence suggests that the genes targeted by tamoxifen activation of the estrogen receptor differ from those stimulated by estrogen itself. This mechanism may be responsible for the differential action of tamoxifen in distinct tissues, and may contribute to carcinogenicity.
Additional information
Tamoxifen was found to induce changes in DNA-methylation patterns in vivo in humans and animals (Liggett et al. Citation2011; Pathak et al. Citation2009; Tryndyak et al. Citation2006) and induce changes in the expression patterns of microRNAs in the liver of treated rats (Pogribny et al. Citation2007; Tryndyak et al. Citation2006). Tamoxifen was also reported to induce changes in cell proliferation as well as expression of telomerase activity in human carcinoma cell lines (Aldous et al. Citation1999). This compound also produced a decrease in histone methylation (Tryndyak et al. Citation2006). In more recent studies, tamoxifen exhibited epigenetic effects in mouse liver and DNA-adduct formation in reproductive organs of mice and humans (de Conti et al. Citation2014; Hernandez-Ramon et al. Citation2014). Tamoxifen induced a significant reduction in fat mass in adipose mice and transiently stimulated the production of reactive oxygen species (ROS) in these mice in vivo, and in murine adipocytes exposed in vitro (Liu et al. Citation2015). The growth-inhibitory effects of tamoxifen on MCF7 human breast cancer cells were associated with enhanced levels of ROS production and lipid peroxidation (Sajadimajd, Yazdanparast, and Roshanzamir Citation2016).
Thiotepa
‘Thiotepa is an alkylating agent that is carcinogenic via a genotoxic mechanism.’ (IARC Citation2012a., 168).
Thiotepa (N,N’,N”-triethylenethiophosphoramide) was used for chemotherapy but has been largely replaced by newer agents. Thiotepa causes leukemia.
Thiotepa is rapidly metabolized to triethylenephosphoramide (TEPA). Thiotepa and TEPA are alkylating agents that both appear to contribute to the carcinogenic effects. There is strong evidence that both thiotepa and TEPA form DNA adducts and DNA cross-links. Thiotepa is cytotoxic and produces mutations. The rate of adduct formation is elevated if DNA-repair mechanisms are blocked. Absence of p53 activity also exacerbates the mutagenic effect. Thiotepa induced micronuclei and chromosomal aberrations in bone marrow of mice and rats, and chromosomal aberrations and sister chromatid exchange in Rhesus monkeys. Patients receiving therapy with thiotepa displayed higher levels of chromosomal aberrations in peripheral lymphocytes than controls.
Treosulfan
‘treosulfan is carcinogenic via a genotoxic mechanism.’ (IARC Citation2012A, 173)
Treosulfan [(2S,3S)-2,3-dihydroxy-4-methylsulfonyloxybutyl]methanesulfonate] is used in the treatment of ovarian cancer. Treosulfan causes acute myeloid leukemia.
Treosulfan is a pro-drug converted non-enzymatically to a mono-epoxide and a diepoxide. Evidence suggests that the carcinogenic properties of treosulfan are derived from these metabolites. Treosulfan is a bifunctional alkylating agent that alkylates DNA and creates inter-strand cross–links. This compound is mutagenic in Salmonella typhimurium strains TA100 and TA1535 in the absence of metabolic activation, and in Chinese hamster cells. Among the few in vivo studies available there are two reports that show treosulfan-induced micronucleus formation in mouse bone-marrow.
Diethylstilbestrol
‘Neonatal exposure to diethylstilbestrol causes persistent changes in gene expression and DNA methylation patterns in diethylstilbestrol target tissues (prostate and uterus), and there is some evidence that hormone responsiveness is permanently altered in the mammary and prostate tissue of exposed mice. It is likely that two or more of these factors (see below) in combination are responsible for the carcinogenic effects of diethylstilbestrol, with estrogen receptor-mediated effects and genotoxicity conceivably both being involved, while other factors may be contributory. The early developmental changes in the female and male genital tract caused by exposure to diethylstilbestrol in utero or – in rodents – neonatally, may result in epigenetic events that create a tissue and cellular environment conducive for the mechanisms responsible for the transplacental carcinogenic effects of diethylstilbestrol in humans and animals.’ (IARC Citation2012a, 206).
Diethylstilbestrol (DES) was used in the past to prevent miscarriage, to treat prostate cancer and as a livestock growth-stimulant. DES is no longer commercially available in the USA. Diethylstilbestrol produces cancer of the breast as well as clear-cell adenocarcinoma of the cervix and vagina in women who were exposed to the drug in utero.
Understanding the mechanism of carcinogenesis for DES is challenging. While this drug produces cancer in women exposed as adults, it is remarkable that exposure in utero induces cancer in the offspring of the exposed woman. There is some evidence that such exposure might affect even their grandchildren. This complicates consideration of carcinogenic mechanisms.
IARC Volume 100A provides an extensive discussion of DES. The summary of the mechanisms of action suggests that multiple pathways are likely to be involved. Five mechanistic categories were explored.
Direct genotoxicity
In vivo studies in animals demonstrated some evidence of DNA-adduct formation after exposure to DES. However, it appears that these adducts may be lipid hydroperoxide- and malondialdehyde-DNA adducts induced by oxidative stress. Aneuploidy, sister chromatid exchange, chromosomal aberrations and micronuclei were reported in some species and tissues but the evidence is conflicting. In vitro studies found evidence for the production of aneuploidy. Diethylstilbestrol inhibited the polymerization of microtubules in human fibroblasts and in prostate cancer cells. This may underlie the development of aneuploidy. There is little evidence that DES is mutagenic. There are data suggesting that mitochondria might be a target for the drug: adducts in mitochondrial DNA were detected and there is evidence of oxidative metabolism of DES in mitochondria. In addition, mitochondria are known to express functional estrogen receptors α and β.
Cell proliferation and apoptosis
Treatment with DES increases the mitotic rate and stimulates cell proliferation in selected tissues. Diethylstilbestrol might immortalize primary animal embryo cells in vitro and transform human breast cell lines.
Modulation of the immune system
Several studies found evidence for modulation of the immune-system by DES. This effect is dose-dependent, appears to be mediated by the thymus, and is different in males and females.
Estrogen receptor-mediated effects
In-utero exposure to DES produces long-term alterations in hormonal response of reproductive tissues in both male and female offspring. These effects were modified in animal models where estrogen-receptor levels could be modulated.
Effects on gene expression (hormonal imprinting)
In-utero exposure to DES produced persistently elevated expression of several genes, including proto-oncogenes such as c-Fos and c-Myc. There was evidence of hypo-methylation of the promoter regions of these genes.
Additional information
Diethylstilbestrol may exert epigenetic effects as evidenced by (1) induced changes in DNA-methylation patterns (Bromer et al. Citation2009; Tang et al. Citation2008a), (2) histone deacetylation (Warita et al. Citation2010) and (3) down-regulation of expression of micro-RNAs (Hsu et al. Citation2009). Numerous long-term effects have been described in breast and reproductive tissues of DES-exposed humans (Newbold Citation2012).
Estrogen-only menopausal therapy
‘Receptor-mediated responses to hormones are a plausible and probably necessary mechanism for estrogen-induced carcinogenesis. In addition, there is support for a genotoxic effect of estrogenic hormones or their associated by-products such as reactive oxygen species. The types of DNA damage found in cells and tissues exposed to estrogens are consistent with such genotoxic effects. Current knowledge does not allow a conclusion as to whether either of these mechanisms is the major determinant of estrogen-induced cancer. It is entirely possible that both mechanisms contribute to and are necessary for estrogen carcinogenesis.’ (IARC Citation2012a, 241).
The term conjugated estrogens refers to a group of at least 8 related compounds, of which estradiol is the most potent. These are most commonly used for hormone replacement in women who have entered menopause mainly as a result of having a hysterectomy. Epidemiological evidence has established estrogen as a cause of endometrial and ovarian cancer.
There is little evidence that estrogens directly affect DNA. However, the major metabolic pathway for estrogens is mediated by the enzymes CYP1A1 and 1B1. This pathway produces o-quinone compounds that form several DNA adducts with various effects on DNA replication. Further metabolism of estrogens leads to redox cycling and the generation of reactive oxygen species that are known to damage DNA. This mechanism is invoked to underpin estrogen-induced tumor initiation/promotion.
There is some evidence that estradiol induces DNA strand-breaks, sister chromatid exchange and chromosomal aberrations in animal and human cells. Aneuploidy was also noted in exposed animals. However, mutations are not consistently seen in in vitro studies.
Estrogen therapy increases cellular proliferation through stimulation of nuclear estrogen receptor-mediated signaling pathways. Estrogen receptors (ERα and ERβ) were recently identified in the mitochondria and may be involved in deregulation of mitochondrial bioenergetics and creation of oxidative stress.
Additional information
Estrogen-only replacement therapy was found to elevate DNA methylation (Friso et al. Citation2007; Wu et al. Citation2010a), and increase telomerase activity (Bayne et al. Citation2007; Kyo et al. Citation1999). A more recent review highlights the involvement of both genetic and epigenetic changes in carcinogenic mechanisms of endometrial cancer (Banno et al. Citation2014).
Combined estrogen–progestogen menopausal therapy
‘Current knowledge indicates that hormone receptor-mediated responses are a plausible and probably necessary mechanism for hormonal carcinogenesis by combined estrogen-progestogen menopausal therapy. There is also support for the potential involvement of genotoxic effects of the estrogenic hormones used in combined estrogen–progestogen menopausal therapy, or their associated metabolic by-products including the formation of DNA adducts, and reactive oxygen species that damage DNA.’ (IARC Citation2012a, 277).
Combined estrogen–progestogen therapy has replaced estrogen-only therapy in an attempt to avoid the increased risk of endometrial cancer associated with estrogens. However, the epidemiological evidence indicates that the combination therapy also produces both breast and endometrial cancer.
The potential mechanisms of action for estrogens are discussed above. Progestogens, including those used in combined estrogen–progestogen menopausal therapy, appear to display the capacity to stimulate cell proliferation in the breast while inhibiting cell proliferation in the uterus. The magnitude of these effects varies for different synthetic progestogens, with a suggestion that medroxyprogesterone acetate is active.
There is no evidence linking progestogen to direct DNA damage. An in vitro study with human breast cancer cells in growth medium containing progesterone showed that the cells released paracrine factors that stimulated vascular endothelial growth-factor (VEGF) receptors and induced proliferation of endothelial cells and breast cancer cells.
Data suggested that progestogen, when taken in combination with estrogens, reduced the carcinogenic potential of the estrogen. The mechanism for this effect is unclear, although there is evidence indicates that binding of progesterone to androgen receptors may be part of the mechanism. This is based upon the hypothesis that signaling pathways are linked, leading to suppression of estrogen-induced functions.
Combined estrogen–progestogen oral contraceptives
‘Hormone-receptor-mediated responses are probably a necessary mechanism for hormonal carcinogenesis by combined estrogen–progestogen oral contraceptives. Because estrogen mediates the expression of progesterone receptors, the presence of estrogen in these combined estrogen–progestogen oral contraceptives may be essential for progestogen-mediated cell proliferation. There is also support for the involvement of genotoxic effects of the metabolic by-products of estrogenic hormones in combined estrogen–progestogen oral contraceptives, or of the reactive oxygen species generated in response to them.’ (IARC Citation2012a, 310).
Combined estrogen–progestogen oral contraceptives produces cancer of the breast, in situ and invasive cancer of the uterine cervix, and cancer of the liver.
The use of an estrogen-progestogen combination as an oral contraceptive differs from the use of these same drugs for post-menopausal treatment, mainly because of differences in dosage, patterns of administration, and the age of the patients. The active chemical agents are the same. Therefore, potential carcinogenic mechanisms are likely to also be similar.
Additional information
Estrogen was shown to increase DNA methylation (Friso et al. Citation2007; Wu et al. Citation2010a), and enhance telomerase activity (Bayne et al. Citation2007; Kyo et al. Citation1999).
Azathioprine
‘Azathioprine is carcinogenic via two mechanisms: (a) as an immunosuppressant it is associated with post-transplant lymphoproliferative disorders that generally have a viral etiology, and (b) because it causes 6-thioguanine to accumulate in patient’s DNA, it also contributes to cancer development via DNA damage’ (IARC Citation2012a, 328-9).
Azathioprine (6-[(1-methyl-4-nitro-1H-imidazol-5-yl)sulfanyl]-7H-purine) is used as an adjunct immuno-suppressant for patients undergoing renal transplantation. It is a pro-drug that is converted to 6-mercaptopurine, which is itself used in treatment of acute lymphocytic leukemia. This compound produces squamous cell carcinoma of the skin and non-Hodgkin lymphoma.
Metabolism of azathioprine leads to its conversion to 6-thioguanine (6-TG), which may become incorporated into replicating DNA. The thiol group is subject to chemical methylation, which results in the formation of a modified base (S-methyl-thioguanine) in the DNA strand. While insertion of 6-TG is not frequent, it is highly miscoding during replication. This mechanism may underlie the carcinogenic effect of azathioprine.
There are conflicting reports on the formation of chromosomal aberrations in azathioprine-treated patients. There is, however, evidence for induction of chromosomal aberrations, but not sister chromatid exchange, in human lymphocytes treated in vitro.
Azathioprine has a distinct mechanism of action in transplant patients who develop lymphoproliferative cancer. This mechanism is linked to the presence of Epstein-Barr virus (EBV). The immunosuppressive effect of azathioprine increases the risk of activating EBV infection. The carcinogenic mechanism of EBV is discussed below.
Chlornaphazine
‘Chlornaphazine is a bifunctional alkylating agent with genotoxic/mutagenic activity. In addition, the presence of sulfate esters of 2-naphthylamine as intermediates in the metabolism of chlornaphazine in rats is consistent with the production of 2-naphthylamine and the increased incidence of bladder tumours in humans.’ (IARC Citation2012a, 333).
Chlornaphazine (N,N-bis(2-chloroethyl)-2-naphthylamine) is an antineoplastic agent that has been utilized to treat Hodgkin’s lymphoma but is no longer in used clinically. This drug has been subject to limited research. In one small epidemiological study there was a link to bladder cancer. The IARC Working Group concluded that chlornaphazine produces cancer of the urinary bladder.
As a bifunctional alkylating agent, chlornaphazine likely shares mechanisms with other members of this class. The limited data available show evidence of genotoxic effects. In vitro studies noted induction of chromosomal aberrations in Chinese hamster cells; micronuclei in bone-marrow cells of mice and rats; production of mutations in mouse lymphoma cells; and unscheduled DNA synthesis in rat hepatocytes.
Ciclosporin (also: cyclosporine)
‘Ciclosporin is an immunosuppressant; long-term immunosuppression is linked to an increased risk of cancer. There are at least two facets to this. First, immunosuppression per se is associated with cancer, for example in individuals positive for the human immunodeficiency virus (HIV). Pharmacological immunosuppression is associated with an increased incidence of a similar spectrum of malignancies. These generally have a viral etiology. Examples include the EBV-related post-transplant lymphoproliferative disorders, and HPV-related cervical carcinoma. In addition to these malignancies that usually arise early after immunosuppression is initiated, there are late effects – such as the development of skin cancer – that may have a different aetiology that could reflect direct or indirect effects of ciclosporin on DNA.’ (IARC Citation2012a, 343).
Ciclosporin is a potent immunosuppressive drug that is used in patients undergoing organ transplantation which has causally been associated with several cancers, most of which display a viral etiology such as Kaposi sarcoma, cervical cancer, and non-Hodgkin lymphoma.
Ciclosporin enhances the synthesis of transforming growth factor β (TGF-β) and consequent activation of its dependent transcriptional activators. Studies in cultured human pulmonary adenocarcinoma cells treated with ciclosporin exhibited evidence of a metaplastic phenotype. It is not clear if this effect contributes to the carcinogenicity of ciclosporin.
There is little evidence that ciclosporin directly initiates DNA damage. One study reported an increased frequency of chromosomal aberrations in lymphocytes of kidney-transplant patients treated with ciclosporin. A second study noted elevated number of sister chromatid exchange in peripheral blood lymphocytes of exposed individuals. There is evidence that ciclosporin may produce a rise in the level of double-strand DNA breaks. Ciclosporin induces oxidative stress, which gives rise to an increase in reactive oxygen species (ROS) levels and higher rates of single-strand DNA breaks, which may be converted to double-strand breaks during DNA replication. This might lead to a chronic excess of double-strand breaks, which may result in cancer development. Several reports showed that ciclosporin inhibits the repair of UV-induced DNA damage.
Additional information
Significant alterations in microRNAs were induced in human proximal tubular epithelial cells after exposure to cyclosporine A (Chen et al. Citation2015).
Aristolochic acids
‘Key steps in the mechanism by which aristolochic acid causes tumours in experimental animals have been identified, and are consistent with events occurring in patients with urothelial cancers associated with aristolochic acid nephropathy and Balkan endemic nephropathy. The same DNA adducts identified in humans are also found in experimental animals’ (IARC Citation2012a, 359).
The term ‘aristolochic acids’ refers to components in the plant species Aristolochia, which contains a mixture of aristolochic acid I and its demethylated derivative, aristolochic acid II. These plants are commonly used in traditional Chinese medicine. Plants containing aristolochic acid induce cancer of the renal pelvis and ureter.
The cancers associated with a weight-loss regimen of herbal ingredients that contained aristolochic acids directly relate to the mechanism of action of these acids. The toxicity of aristolochic acids I and II has been inferred from effects seen in patients diagnosed with kidney nephropathy after the use of herbal mixtures containing Aristolochia species, which led to rapidly progressive fibrosing interstitial nephritis. The same aristolochic acid-specific DNA adducts identified in experimental animals exposed to aristolochic acid or herbal products containing aristolochic acid were also found in urothelial tissue of aristolochic-acid nephropathy patients, in renal tissue from Balkan endemic nephropathy patients, and in tumor tissue from residents of endemic villages.
High doses of aristolochic acids produce severe necrosis of renal tubules, splenic and thymic atrophy and ulceration of the forestomach in animals. Aristolochic acids are consistently active in in vivo and in vitro genotoxicity tests. Metabolism of aristolochic acids leads to production of electrophilic cyclic N-acylnitrenium ions, which react with DNA to produce adducts. These adducts were identified and detected in exposed animals and in urothelial tissues from patients with nephropathy subsequent to intake of aristolochic acid. The DNA adducts may lead to mutations that activate oncogenes or inactivate tumor suppressor genes (e.g. TP53 or RAS). In rodent tumors, activation of RAS oncogenes were discovered through a specific CAA→CTA transversion mutation in codon 61. In one nephropathy patient, a similar mutation was found in codon 139, exon 5 of the RAS gene.
Additional information
Aristolochic acid can induce apoptosis of endothelial cells of the human umbilical vein in vitro (Shi and Feng Citation2011).
Methoxsalen plus ultraviolet-A (UVA) radiation
‘Methoxsalen in combination with UVA is carcinogenic via a genotoxic mechanism that involves photo-activation’ (IARC Citation2012a, 372).
Methoxsalen (8-Methoxypsoralen) is a drug derived from plants. In psoralen-UVA (PUVA) therapy, this chemical is used in combination with ultraviolet light as a photosensitizing agent for the treatment of psoriasis and other skin lesions. Treatment requires activation of the psoralen with high-intensity long-wavelength ultraviolet light (UVA). Carcinogenic effects also require activation by UVA. Hence, mechanistic information need to pertain to the combination of the drug and exposure to UVA. Methoxsalen, in combination with UVA radiation, produces squamous cell carcinoma of the skin.
PUVA was found to produce DNA adducts and other forms of DNA damage in a range of prokaryotic and eukaryotic cells. Methoxsalen preferentially intercalates into DNA at 5ʹ-TpA sites. Exposure to UVA induces photo-activation leading to DNA alkylation. In PUVA-exposed Chinese hamster ovary cells, bi-adducts were observed that might be major PUVA-induced pre-mutagenic lesions in mammalian cells.
In vitro studies with human cells treated with PUVA demonstrated induction of chromosomal aberrations, sister chromatid exchange, mutations, DNA damage, and DNA crosslinks. Similar effects, in addition to unscheduled DNA synthesis, were found in cultured rodent cells. PUVA transforms mouse C3H10T1/2 cells. Mitotic recombination and mutation were found in fungi, and mutation and DNA damage in bacteria exposed to PUVA. Treatment with PUVA also produces reactive oxygen species (including singlet oxygen and superoxide) that may play a role in PUVA-induced cytotoxicity.
Phenacetin
‘While there is evidence of genetic damage caused by phenacetin in various experimental systems, similar data are not available in humans’. (IARC Citation2012a, 395)
Phenacetin (N-(4-ethoxyphenyl)acetamide), first released in 1887, was an analgesic and fever-reducing drug used in both human and veterinary medicine, until the drug was implicated in kidney disease (nephropathy) in patients who ingested chronic excessive doses. Analgesic mixtures containing phenacetin induce cancer of the renal pelvis and ureter. The carcinogenicity of these mixtures is related to the phenacetin component.
Evidence for a carcinogenic mechanism for phenacetin is conflicting. There are no apparent studies showing genetic or other effects in humans.
Phenacetin induced chromosomal alterations and DNA damage in target and non-target tissues, micronuclei in bone-marrow erythrocytes in mice and rats, DNA damage in the kidney of mice and in the urinary bladder of rats, cell proliferation in the urothelium of the kidney, the bladder, and the renal pelvis in rats, and DNA synthesis in the nasal respiratory and olfactory mucosa of rats. Phenacetin also induced chromosomal aberrations in Chinese hamster cells in vitro and DNA strand-breaks in rat and human cells from the urinary bladder in vitro, but not in rat hepatocytes after exposure in vivo. In rat kidney cells, the metabolite N-hydroxy-phenacetin, but not parent compound phenacetin, induced micronucleus formation. Phenacetin exhibited mutagenicity when tested in bacterial systems in the presence of a metabolic system derived from hamster or rat liver. Mutagenicity was not detected when a metabolic system from mice was employed. Oral feeding of phenacetin to mice with a deficiency in nucleotide-excision repair produced an increased mutation frequency in a LacZ reporter gene in the kidney.
Volume 100B: biological agents
There is an extensive literature concerning the molecular modes of action of biological agents discussed in Volume 100B. Many of the molecular processes underlying infection by a biological agent and subsequent propagation of the agent in the body are relevant to potential carcinogenic mechanisms. It is not feasible to provide details on all the relevant material in the brief summaries presented here. A synopsis of the key processes will thus be presented. The reader is referred to Volume 100B and the literature cited therein for more detail.
Epstein-barr virus
‘Mechanistic data that strongly support an oncogenic role of EBV in human cancer can be summarized as follows: (a) EBV immortalizes normal B cells in culture, (b) one or several EBV gene products are expressed in all EBV-associated cancers, (c) at the molecular level, these EBV-encoded gene products associated with latent viral infection induce cell proliferation, block apoptosis, induce genomic instability or modulate cell migration. These events occur before or during tumour initiation. Several of these gene products are also involved in mechanisms contributing to continued tumour maintenance, cell growth, and progression.’ (IARC Citation2012b, 80).
The Epstein-Barr virus (EBV) is a ubiquitous virus that infects up to 95% of the world population by the time subjects reach adulthood. Once the initial infection is controlled by the immune system, EBV persists in a latent state inside B-cells of the immune system. Evidence suggests that re-activation by an external agent is required to trigger carcinogenicity of EBV. Activating agents include the following: infections (especially malaria in connection with Burkitt lymphoma), immunosuppressive drugs (azathioprine, ciclosporin), immunodeficiency and possibly exposure to food products such as salted fish in China; and certain chemicals. EBV initiates several types of lymphoma and cancer of the nasopharynx.
EBV expresses six latent nuclear proteins and three latent membrane proteins. All of these are multi-functional and affect cell-signaling pathways that may contribute to tumorigenesis. In addition, EBV expresses two non-coding RNAs that contribute to B-cell transformation and more than 22 microRNAs (miRNAs) that target genes relevant to carcinogenesis.
In Monograph Volume 100B the mechanistic evidence for EBV-associated oncogenesis – apart from the immortalization of B-cells – is summarized. EBV infection of human cells in vitro affects their phenotype. EBV infection of human cells is linked to cell proliferation, apoptosis, and cell migration induced by single EBV proteins or combinations thereof, primarily by enhanced expression or ‘knock-down’ of single proteins. Induction of EBV-positive lymphoproliferative diseases or lymphomas was observed after infection of animals (New World monkeys) with EBV, or upon transplantation of EBV-infected human B lymphocytes to immunosuppressed mice (SCID mice, nude mice). At the molecular level, EBV-encoded gene products are associated with genomic instability or modulation of cell migration. The EBV-specific nuclear antigens EBNA-1 and EBNA-3C and latent membrane protein LMP-1 independently promote genomic instability, as detected by non-clonal chromosomal aberrations, DNA breaks and phosphorylation of histone H2AX.
Additional information
EBV induced oxidative stress in human B lymphocytes, epithelial, and lymphoblastoid cell lines in vitro (Gargouri et al. Citation2009; Kamranvar and Masucci Citation2011; Lassoued et al. Citation2008), and elevated levels of micronuclei in human cells in vitro (Gualandi et al. Citation2001; Wu et al. Citation2010b). Altered DNA methylation (Kusano et al. Citation2006; Matsusaka et al. Citation2011) and down-regulation of micro-RNAs (Shinozaki et al. Citation2010) were reported in gastric carcinomas associated with EBV infection. EBV altered DNA-methylation patterns in vitro (Grafodatskaya et al. Citation2010; Niller et al. Citation2016; Skalska et al. Citation2010) and induced histone modifications (Anderton et al. Citation2011; Maruo et al. Citation2011). Further, EBV-encoded LMP-1 induced microRNA-10b (Li et al. Citation2010) and downregulated miRNA-203 (Yu et al. Citation2012).
Immune effects attributed to EBV comprise induction of release of chemotactic cytokines or chemokines in human neutrophils through accumulation of mRNA for interleukin-8 (IL-8) and macrophage inflammatory protein-1 α (MIP-1 α) (Chiu et al. Citation2013). EBV utilizes epigenetic gene regulation in the cellular host to establish latent infection (Hammerschmidt Citation2015). Host miRNA expression during primary B-cell infection by EBV initiated dynamic changes in several miRNAs: oncogenic miRNAs were induced, and tumor suppressor miRNAs were predominantly repressed (Forte et al. Citation2012). EBV-immortalized B-lymphoblastoid cell lines are characterized by genome-wide demethylation and rearrangement of heterochromatic histone marks (Niller et al. Citation2014).
Chronic infection with hepatitis B virus
‘There is strong evidence to support an indirect role for HBV in hepatocarcinogenesis resulting from chronic necro-inflammatory hepatic disease (cirrhosis), as well as moderate evidence for a direct role largely associated with the protein HBx (transcribed from the HBV genome)’. (IARC Citation2012b, 123)
‘At the molecular level, the genesis of HBV-induced hepatocellular carcinoma (HCC) is a complex, multifaceted, and multistep process, an essential component being a series of genetic or epigenetic changes in the genes that govern cell proliferation and cell death.’ (IARC Citation2012b, 113).
The Hepatitis B virus (HBV) is a common DNA virus that displays strong geographic variability. This virus produces acute and chronic hepatitis and liver cirrhosis, and may be present in an inactive carrier state. HBV is established as a causal agent for liver cirrhosis and hepatocellular carcinoma (HCC).
A large proportion of HCC arises in the presence of chronic hepatitis or cirrhosis, suggesting that a chronic necro-inflammatory process contributes to cancer development (see Figure 4.1, p116 in Monograph 100B). Oxidative stress and upregulation of inducible nitric oxide synthase (iNOS) were demonstrated in chronic viral hepatitis. This process leads to the generation of reactive oxygen species (ROS) or reactive nitrogen species (RNS) that produce DNA adducts and lead to mutations and genomic instability. Telomerase activation may also occur. HBV might integrate into the cellular genome. While not required for virus production, integration was found in over 85% of HBV-related HCC’s. Increased cellular proliferation from an inflammatory response might produce double-strand DNA breaks and facilitate viral integration. Integration occurs at random throughout the DNA, but integration sites near a variety of genes associated with cellular division and genomic stability were noted. Integration might lead to a variety of chromosomal aberrations, including deletions, translocations, duplications or amplifications.
The HBV-encoded protein HBx functions through protein–protein interactions. This virus activates transcription of a wide variety of proteins involved in regulation of cellular function, including p53. A wide variety of cis-elements are responsive to HBx, including many transcription factors. HBx directly interferes with DNA repair by forming a complex with the DNA-repair protein XAP-1. Methylation and epigenetic gene silencing are important mechanisms for HCC in the early stages of tumor development. HBx may contribute to this process by deregulating expression of DNA methyltransferases.
Additional information
Several studies documented effects of HBx on factors that may affect cancer risk, including c-myc, p53, p21 and miRNAs (Hou et al. Citation2009; Ura et al. Citation2009; Yano et al. Citation2013; Zhang et al. Citation2011). The impact on histone functioning through the induction of metastasis-associated protein-1 (MTA1) and histone deacetylase (HDAC) was confirmed (Yoo et al. Citation2008). Differential expression of 188 miRNAs was noted both in vitro (Ura et al. Citation2009) and in vivo (Zhang et al. Citation2011).
HBV proteins induced apoptosis in humans and animals in vivo and in vitro (Clippinger, Gearhart, and Bouchard Citation2009; Terradillos et al. Citation1998), and promoted cellular proliferation and differentiation in human cells (Guo et al. Citation2009; Peng et al. Citation2005). MicroRNA deregulation is an early event and becomes stronger throughout the various steps of HBV-associated hepatocarcinogenesis; miRNA-145 is a candidate tumor-suppressive miRNA which plays an important role in HCC development (Gao et al. Citation2011). Specific microRNAs, reported to be associated with various aspects of hepatitis B biology were classified and their role in multiple aspects related to HBV was defined (Sarkar and Chakravarty Citation2015). Epigenetic changes induced by the HBx protein include aberrations in DNA methylation, post-translational modification of histones, microRNA expression, and epigenetic control of HBV covalently closed circular DNA (cccDNA) (Koumbi and Karayiannis Citation2015; Tian and Ou Citation2015; Tian et al. Citation2013). Single-nucleotide polymorphisms (SNPs) in the genes encoding miR-146a and miR-196a-2 influence susceptibility to HCC from HBV infection (Xu et al. Citation2013).
Chronic infection with hepatitis C virus
‘Although there is strong evidence that HCV is one of the leading causes of HCC (hepatocellular carcinoma), there is still much to understand regarding the mechanism of HCV-induced transformation. While liver fibrosis resulting from long-lasting chronic inflammation and liver regeneration after immune-mediated cell death are likely factors contributing to the development of HCC, the direct role of HCV proteins remains to be determined. Many in vitro studies have shown that HCV expression may interfere with cellular functions that are important for cell differentiation and cell growth.’ (IARC Citation2012b, 158)
Hepatitis C virus (HCV) is an RNA virus that is endemic globally. This virus is predominantly transmitted via blood with a high prevalence among intravenous drug users, especially those who share needles. Acute infection develops into persistent infection in up to 90% of cases. HCV induces hepatocellular carcinoma and non-Hodgkin lymphoma.
HCV replicates exclusively within the cellular cytoplasm. Therefore, HCV does not directly interact with DNA, and potentially pro-carcinogenic events are restricted to the cytoplasm. HCV replication is directly linked to the endoplasmic reticulum and lipid metabolism. The proteins expressed by HCV interact with cellular components. Chronic inflammation and endoplasmic reticulum stress lead to oxidative stress and disruption of the intracellular redox state that gives rise to genomic damage. Several HCV proteins interact directly with cellular signaling cascades and affect cell metabolism and replication. The HCV core protein may interact with cyclin/CDK complexes and affect cell-cycle control. HCV produces steatosis by impairing lipid excretion and metabolism and by enhancing lipid genesis in the liver. The carcinogenic effect of HCV is enhanced through a positive feedback loop involving steatosis, insulin resistance and endoplasmic reticulum stress.
Additional information
HCV affects the epigenetic apparatus of the cell by changing DNA methylation in humans (Feng et al. Citation2010; Lim, Park, and Jang Citation2012; RipoRipoli et al. Citation2011), and by inducing alterations in miRNA levels in human cells (CermelCermelli et al. Citation2011; Ishida et al. Citation2011; Peveling-Oberhag et al. Citation2012; Ura et al. Citation2009). HCV interferes with the pathway controlling apoptosis (Pavio et al. Citation2005) but the impact on cellular proliferation is unclear (Pavio et al. Citation2005; Zhou et al. Citation2012). HCV interferes with DNA-damage repair (Lai et al. Citation2008; Machida et al. Citation2010; Pal et al. Citation2010). Aberrant methylation of the promoter region of the tumor suppressor gene SPINT2/HAI-2 was proposed as an epigenetic mechanism in HCV-induced hepatocarcinogenesis (Ramadan et al. Citation2015). Disease progression from chronic hepatitis C to cirrhosis and HCC is associated with increasing DNA promoter methylation (Zekri Ael et al. Citation2014). Telomere length was reduced in patients with chronic active HCV and in patients in remission, compared with healthy controls (Biron-Shental et al. Citation2013), and mRNAs of histone-modifying genes were more than 8-fold overexpressed in HCC tissues; among these, the expression level of the histone-lysine N-methyltransferase gene SUV39H2 was associated with HCV infection (Hung et al. Citation2014).
Kaposi sarcoma herpes virus
‘At the molecular level, KSHV-encoded gene products associated with latent viral infection induce cell proliferation, block apoptosis, induce genomic instability or modulate cell migration and tumour progression.’ (IARC Citation2012b, 195).
Kaposi sarcoma herpes virus (KSHV) is a DNA virus and a member of the herpes virus family. This virus is able to produce life-long latent infections with CD19-positive B-cells being a major reservoir. KSHV is mainly transmitted via saliva with an infection peak between ages 6 and 10 years in high-prevalence countries. The virus induces Kaposi sarcoma.
Infection with KSHV converts primary endothelial cells into spindle cells. KSHV proteins were shown to interfere with apoptosis. There is little evidence to suggest that KSHV produces DNA damage or genetic instability. Five KSHV proteins were found to possess cell-transforming properties in vitro. Three other proteins affect cell-cell regulation and tumor cell survival. KSHV produces many alterations to cellular gene expression and transcription. These changes tend to stimulate cell proliferation and modulate differentiation potential.
Additional information
Natural killer cells from subjects infected with KSHV (both asymptomatic and those who developed Kaposi sarcoma) exhibited changes in expression of several genes including those encoding CD161 receptors (Dupuy et al. Citation2012). KSHV-encoded microRNAs induce B-cell proliferation (Boss et al. Citation2011). KSHV produced cell proliferation and differentiation in vivo and in vitro (Dupuy et al. Citation2012; Morris et al. Citation2012).
Infection with human immunodeficiency virus-1
‘HIV-1 increases human cancer risk indirectly, primarily by immunosuppression. Suggested mechanisms include HIV-1-mediated immune dysregulation, in particular B-cell hyper-activation, and perhaps the effects of the secreted HIV-1 Tat protein. However, unlike what is known about other cancer-associated viruses, there is no evidence that HIV-1 infection by itself leads to cell transformation or immortalization.’ (IARC Citation2012b, 240).
Human immunodeficiency virus-1 (HIV-1) is the causative agent of acquired immune deficiency syndrome (AIDS). HIV-1 is a RNA virus that transcribes its RNA core into DNA through the action of reverse transcriptase, which subsequently is integrated in the host cell DNA. This virus primarily infects CD4-positive T-cells, macrophages and dendritic cells. There is no evidence that HIV-1 causes cancer directly. However HIV-1 increases cancer risk because it gives rise to a severe immunodeficiency, leading to an enhanced risk from secondary carcinogens. For example, the risk of non-Hodgkin’s lymphoma is enhanced as a result of ‘the profound depletion of CD4-positive T lymphocytes that is caused by HIV-1 and allows the dysregulation of B-cell control, and the expression of the effects of lymphotrophic viruses’ (IARC Citation2012b, 223). Despite the integration of the cDNA transcript of the viral RNA in the genome, there is no apparent evidence that HIV induces chromosomal or genetic damage.
Additional information
HIV induces telomerase activity in monocyte-derived macrophages (Reynoso et al. Citation2012).
Human papillomavirus types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58 and 59
The characterization of the mechanisms of action of the HPV oncoproteins in in vitro and in vivo assays provides compelling evidence for a direct role of high-risk mucosotropic HPVs in the development of cervical cancer. The mechanisms involve immortalization, transformation, inhibition of apoptosis, induction of genomic instability, and deregulation of the immune response. A common feature of mucosotropic HPV-associated cancers is the expression of the viral genes E6 and E7. The E6 and E7 oncogenes of HPV16 and HPV18 have been the most extensively studied and were found to confer a similar set of biological phenotypes (e.g. immortalization, inhibition of DNA-damage response, genomic instability, and inhibition of differentiation) on epithelial cells from multiple human tissues in which HPV-associated cancers are found. The E6 and E7 proteins of the same HPVs (16 and 18) share similar sets of biochemical properties (e.g., for E6: inactivation of p53, induction of hTERT, binding to PDZ; for E7: inactivation of pRb and related pocket proteins, activation of E2Fs). Suppression of HPV16/18 E6 and E7 gene expression in cell lines derived from human cervical cancers leads to senescence or apoptosis.’ (IARC Citation2012b, 294).
The HPV family consists of DNA viruses of over 100 types with 4 main genus groups. The HPV family most commonly infect mucosal tissue (the α genus) or cutaneous tissue (the β genus). HPV displays long persistence without overt signs of infection. The carcinogenic potency varies by type, with HPV16 and HPV18 exhibiting the strongest connection to cervical cancer etiology. Transcription of the viral genome produces 7 proteins of which two (E6 and E7) are most strongly associated with carcinogenesis. HPV initiates cervical cancer.
The E6 and E7 proteins of the α-genus HPV types target cellular proteins for degradation through the ubiquitin proteasome pathway. Multiple target proteins were identified. Of particular relevance for E6 is the p53 protein, while E7 targets pRb, p107, p130 and related pocket proteins. E6 also enhances telomerase activity through an unknown mechanism. E7 alters transcriptional regulations through AP1 transcription factors, histone deacetylases and MPP2. HPV may compromise normal DNA-repair processes and cellular response to DNA damage. This leads to genetic instability and chromosomal abnormalities. E7 stimulates cellular proliferation while E6 inhibits apoptosis. Persistence of viral infection and expression of E6/7 is required for carcinogenesis, which suggests that the virus alters the immune response.
Additional information
HPV infection induces epigenetic effects as follows: changes in DNA methylation (Jiang et al. Citation2012; Sartor et al. Citation2011; Weiss et al. Citation2011), enhanced histone modifications (Hsu et al. Citation2012; McLaughlin-Drubin, Crum, and Munger Citation2011), modulated expression of microRNAs (Dreher et al. Citation2011; Greco et al. Citation2011), and impaired DNA repair (Bajpai et al. Citation2013; Rey, Lee, and Park Citation1999). Studies indicate that HPV-induced immortalization of keratinocytes is associated with a sequential and progressive rise in promoter methylation of a subset of genes (Schutze et al. Citation2015), and that epigenetic changes contribute to maintaining a malignant phenotype in HPV-positive oropharyngeal squamous cell carcinomas (Anayannis, Schlecht, and Belbin Citation2015).
Human T-cell lymphotropic virus type-1
‘There is strong mechanistic evidence supporting a role of HTLV-1 in human carcinogenesis. The viral protein Tax has the ability to immortalize and transform human T cells. At the leukaemic stage, the expression of Tax is often not maintained, but the viral protein HBZ continues to be expressed and supports the sustained growth of the leukaemic cells.’ (IARC Citation2012b, 332).
Human T-cell lymphotropic virus type-1 (HTLV-1) is a complex retrovirus that contains two copies of its genomic RNA which infect multiple cell types but only induce cell transformation in T-lymphocytes. HTLV-1produces adult T-cell lymphoma/leukemia.
HTLV-1 expresses 6 proteins of which the Tax protein is essential for carcinogenesis. Tax was found to activate and repress multiple genes, modulate the cell-cycle and repress apoptosis. Transcription pathways activated by HTLV-1 include those of NF-κB, CREB, SRF, myc and AP-1. HTLV-1 inhibits the function of p53. There is associated genomic instability and development of aneuploidy. Epigenetic silencing was noted around a common breakpoint region in chromosome 10p11.2. Long-term viral persistence is required for carcinogenesis, suggesting that immune system abnormalities may be important.
Additional information
HTLV-1 increases the frequency of micronuclei (Majone and Jeang Citation2000), sister chromatid exchange (Chieco-Bianchi et al. Citation1988) and induces chromosomal aberrations in humans (Maruyama et al. Citation1992; Whang-Peng et al. Citation1993). HTLV-1 induces angiogenesis by establishing gap-junction intercellular communication with endothelial cells (Bazarbachi et al. Citation2004; El-Sabban et al. Citation2002). HTLV-1 is implicated as a causal agent for several inflammatory diseases including spastic paraparesis, dermatitis, and inflammatory lung diseases suggesting that production of chronic inflammation may play an important role for carcinogenesis (Satou et al. Citation2011; Yamamoto-Taguchi et al. Citation2013). Alterations in cellular microRNA expression by HTLV-1 in infected cell lines affect a subset of miRNAs associated with deregulation of host gene expression and signal transduction (Ruggero et al. Citation2010). HTLV-1 inhibits proteins involved in biogenesis and maturation of cellular miRNAs, resulting in a perturbation of the expression profile of host miRNAs (Moles and Nicot Citation2015). Adult T-cell leukemia/lymphoma (ATL) genomes are characterized by prominent CpG island DNA hyper-methylation. In ATL a markedly higher number of genes were significantly hyper-methylated than hypo-methylated in comparison to healthy controls which was associated with transcriptional silencing (Kataoka et al. Citation2015). The HTLV-specific oncoproteins Tax and HBZ might both activate and silence distinct cellular promoters by interacting with cellular enzymes involved in histone modification (Minarovits et al. Citation2016).
Chronic infection with Opisthorchis viverrini; chronic infection with Clonorchis sinensis
‘Liver-fluke-induced cholangiocarcinoma is likely the result of chronic inflammation that involves the activation of oxidative stress pathways. Metabolic products excreted from liver flukes are highly immunogenic and may stimulate cell proliferation and anti-apoptosis directly.’ (IARC Citation2012b, 365).
Opisthorchis viverrini and Clonorchis sinensis are liver flukes that are endemic in Asia, particularly in China, Thailand, Viet Nam, the Lao People’s Democratic Republic and the Republic of Korea. Their life-cycles involve snails and fish as intermediary hosts. Human infection is predominantly arises from eating raw or undercooked fish. These two liver flukes produce cholangiocarcinoma.
Liver flukes produce histopathological changes characterized by inflammation, hyperplasia and metaplasia with a high frequency of changes in the bile duct. Liver flukes excrete various metabolic products that are highly immunogenic. These organisms elevate cell proliferation through stimulation of the retinoblastoma protein Rb and cyclin D1. The immune response leads to endogenous production of NDMA and nitric oxide (NO) and to nitrosation of amines. Liver-fluke infection is linked to diffuse nitrosative and oxidative DNA damage and adduct formation. There is little evidence to support any direct genetic or epigenetic effects.
Additional information
Expression of c-Ski, TGF-β and Smad4, was significantly up-regulated (Boonmars et al. Citation2011) by Opisthorchis viverrini in a hamster model and humans. There was increased expression of proteins related to stress response, DNA replication and repair, and cell structure (Khoontawad et al. Citation2010). Elevated expression of DNA-repair enzymes was also reported (Khoontawad et al. Citation2010; Loilome et al. Citation2012). Aberrant hyper-methylation of certain loci is a common event in liver fluke-related cholangiocarcinoma and may potentially contribute to cholangiocarcinogenesis (Sriraksa et al. Citation2011).
Chronic infection with Schistosoma haematobium
‘S. haematobium with egg deposition in the tissue leads to severe inflammation of the urinary bladder wall resulting in increased oxidative stress. This increase points towards a relationship between oxidative stress induced by continuous and chronic inflammation due to schistosome infection, and possibly nitric-oxide-mediated DNA genotoxicity and alkylation of DNA by N-nitroso compounds.’ (Schistosoma haematobium, IARC Volume 100B, p 382). ‘Several studies indicate that the carcinogenicity of S. haematobium is a multifactorial and multistage process in which several mechanisms are involved. S. haematobium eggs induce chronic inflammation and irritation in the urinary bladder. The inflammatory response around the eggs gives rise to genotoxic factors and products that may cause genomic instabilities of host cells, leading to modifications in the regulation of tumour-suppressor genes and oncogenes as well as stimulation of a proliferative response of the host cells to repair tissue damage caused by the inflammation.’ (IARC Citation2012b, 378)
Schistosoma haematobium is a parasite that is endemic to tropical regions in Africa and the Middle East. Schistosoma haematobium produces squamous cell carcinoma of the bladder.
Schistosoma haematobium reproduces in a human host where eggs are excreted in urine, pass through a fresh-water snail and infect the human host who comes in contact with the water where the snails live. The adult worm attaches to venous blood vessels surrounding the bladder. Subsequently eggs are then released through the bladder wall. Approximately 50% of the eggs remain in the bladder wall and initiate a local inflammatory response. There is evidence for DNA-adduct formation and a rise in gene methylation. Oxidative stress markers are also elevated.
Additional information
Chinese hamster ovary cells treated in culture with S. haematobium total antigen showed enhanced proliferation, an increased proportion of cells in S-phase, reduction of apoptosis, down-regulation of tumor suppressor p27, and up-regulation of the anti-apoptotic protein Bcl-2 (Botelho et al. Citation2009). The parasite also down-regulates the transcriptional activity of the estrogen receptor in mouse and HCV29 human urothelial cells (Botelho et al. Citation2012).
Chronic infection with Helicobacter pylori
‘Multiple lines of evidence point to a central role for a chronic gastric inflammatory response and resulting oxidative stress in H. pylori-associated gastric carcinogenesis. This leads to altered cellular turnover accompanied by changes in gene expression, methylation, and mutation’ (IARC Citation2012b, 422).
Helicobacter pylori is a gram-negative bacterium that exists mainly in mucous-secreting gastric cells. This bacterium produces non-cardia gastric carcinoma and mucosa-associated lymphoid tissue (MALT) lymphoma. There is evidence that chronic H. pylori infection reduces the risk of adenocarcinoma of the esophagus perhaps through production of gastric atrophy.
Non-cardia gastric carcinoma normally arises in areas of chronic inflammation. H. pylori is the primary cause of gastritis. An especially intense inflammatory response to H. pylori is postulated to induce more serious damage in gastric epithelial cells, more rapid cell turnover, and eventual emergence of epithelial cells carrying cancer-prone mutations. H. pylori is not directly genotoxic in vitro although there is limited evidence in vivo for an increased frequency of DNA strand-breaks, micronuclei, and DNA adducts. Several reports showed that H. pylori alters the expression of specific oncogenes and tumor suppressor genes implicated in gastric carcinogenesis. Hyper-methylation of E-cadherin and p14 was reported likely as a consequence of the inflammatory process induced by H. pylori. Infection with H. pylori promotes the nuclear translocation of β-catenin, thereby activating downstream β-catenin-responsive genes including cyclin D. H. pylori upregulates the p53 homologue p73 in gastric cells, which leads to promotion of apoptosis, and decreases expression of the cell-cycle inhibitory protein p27, which is known to be lost in aggressive gastric cancers.
Additional information
H. pylori interferes with intercellular gap-junctional communication (Tao et al. Citation2007; Xu et al. Citation2011). Epigenetic mechanisms in gastric cancer have been reviewed (Chiariotti et al. Citation2013; Gigek et al. Citation2012). H. pylori induced epigenetic dysregulation (hyper-methylation) of the transcriptional regulator, forkhead box (Fox) protein FOXD3 to promote gastric carcinogenesis (Cheng et al. Citation2013). In addition to H. pylori-associated virulence factors, epigenetic changes in infected host cells, such as DNA methylation and miRNAs, may play a significant part in gastric cancer development and progression of the precancerous cascade (Valenzuela et al. Citation2015).
Volume 100C: arsenic, metals, fibers, dust
Arsenic and arsenic compounds
‘For inorganic arsenic and its metabolites, the evidence points to weak or non-existent direct mutagenesis, which is seen only at highly cytotoxic concentrations. On the other hand, long-term, low-dose exposure to inorganic arsenic likely causes increased mutagenesis as a secondary effect of genomic instability, perhaps mediated by higher levels of reactive oxygen species, as well as co-mutagenesis with other agents. The major underlying mechanisms observed at low concentrations include the rapid induction of oxidative DNA damage and inhibition of DNA repair, followed by changes in DNA-methylation patterns, aneuploidy, and gene amplification. Gene amplification, altered DNA methylation, and aneuploidy lead to altered gene expression, and genomic instability.’ (IARC Citation2012c, 84).
Arsenic is a common element that is widely present in the environment. Arsenical compounds are used in the manufacture of pigments, glass products, semi-conductors, leather preservatives, catalysts, pyrotechnics, anti-fouling agents in paints, pharmaceutical substances, dyes and soaps, ceramics, and alloys (automotive solder and radiators). Arsenic is also used in electrophotography manufacturing and in synthesis of a wide range of chemicals. The main route of human exposure is via ingestion with high levels of arsenic in drinking-water providing a major route of intake in some regions. Chronic exposure to arsenic induces cancers of lung, bladder and skin.
Arsenicals do not react directly with DNA. The major underlying carcinogenic mechanisms observed at low concentrations are rapid induction of oxidative DNA damage and inhibition of DNA repair. Some forms of arsenic induce chromosomal aberrations in vitro, but this is significant only at toxic doses. Chronic low-dose exposure in animal models produces genomic instability and leads to chromosomal aberrations and micronucleus formation. AsIII interferes with spindle function during mitosis. Increased mutagenesis is observed as a consequence of enhanced genomic instability.
Global hypo-methylation of DNA, combined with local hyper-methylation of selected genes, was noted in animal models and humans exposed to arsenic. Gene amplification was reported in some animal experiments. An adverse effect on DNA repair and p53 function was detected, although other studies reported increased DNA-repair activity, perhaps related to antioxidant effects in response to arsenic-induced oxidative damage. Arsenic stimulates an inflammatory response and interferes with apoptosis during chronic exposure. Arsenic was found to induce androgen independence in human prostate epithelial cells, which led to a 6-fold rise in expression of the K-RAS oncogene. Low levels of arsenic stimulate angiogenesis through an inflammatory process, leading to blood-vessel remodeling in the liver.
Additional information
Arsenic initiates alterations in gene expression. In human cell lines, arsenic upregulated genes associated with cytotoxicity and cell proliferation (Dodmane et al. Citation2013). In SC/PC rat kidney cells, chronic exposure to low doses of arsenite elevated secretion of matrix metalloproteinase (MMP), enhanced Cox-2 expression, and increased the rate of cell proliferation (Tokar et al. Citation2013). Mammalian cell transformation and immortalization were detected following exposure to inorganic arsenic compounds in cell culture (Takahashi, Barrett, and Tsutsui Citation2002). There is increasing evidence from human studies suggesting that arsenic exposure is associated with epigenetic alterations (Argos Citation2015; Roy et al. Citation2015). Arsenic was shown to (1) alter methylation levels of both global DNA and gene promoters, (2) induce histone acetylation, methylation, and phosphorylation and (3) affect microRNA expression (Bustaffa et al. Citation2014; Ren et al. Citation2011). Analysis of CpG methylation in newborn cord blood of arsenic-exposed mothers identified a number of genes in which methylation levels were associated with differences in birth outcomes such as gestational age and placental weight (Rojas et al. Citation2015).
Beryllium and beryllium compounds
‘Several molecular mechanisms operate in beryllium-induced carcinogenesis. Whereas mutagenicity tests with beryllium have shown only weakly positive or negative results, chromosomal aberrations and aneuploidy were observed in vivo in mice, at nontoxic concentrations. Beryllium is capable of producing oxidative stress, which can lead to cell injury in the form of DNA damage, activation of proto-oncogenes, and apoptotic mechanisms. The toxicity of beryllium in the lung may lead to cell killing and compensatory cell proliferation. Furthermore, the beryllium-induced chronic inflammatory response with attendant release of cytokines from beryllium-reactive CD4+ T-cells could also play a part in the development of a carcinogenic response in lung tissue. Inflammatory processes induced by beryllium may also cause an increase in reactive oxygen species, mediate cell turnover, and alter cell-signaling pathways. Furthermore, down-regulation of genes involved in DNA synthesis, repair and recombination has also been reported. Thus, the processes underlying beryllium-induced carcinogenesis are clearly complex, with several possible interactive mechanisms.’ (IARC Citation2012c, 116).
Beryllium is an uncommon metal that is used primarily in alloys or in beryllium oxide ceramics. Beryllium produces lung cancer in occupational settings. Inhalation seems to be required for carcinogenesis. There is no apparent evidence of a cancer risk from ingestion of beryllium.
There is little evidence for direct genotoxicity of beryllium, although down-regulation of genes involved in DNA synthesis and DNA repair was observed. The mechanisms associated with the carcinogenicity of beryllium have not been extensively studied. Beryllium is known to produce a chronic allergic-type lung response and disease called berylliosis. The inflammatory response to this condition leads to enhanced cell proliferation, oxidative stress and altered cell-signaling pathways. Studies in mice showed formation of reactive oxygen species with marked increases in apoptosis and activation of caspase 8.
Additional information
Exposure to beryllium induced expression of the cell-adhesion molecule I-CAM1 on the surface of small-airway epithelial cells and enhanced the release of soluble I-CAM1 into the extracellular medium (Rodriguez et al. Citation2008). In microorganisms and in mammalian cells, soluble beryllium compounds may give rise to replication infidelity during in vitro DNA synthesis, and produce forward gene mutations (Leonard and Lauwerys Citation1987). Beryllium also induced morphological cell transformation in vitro (Pienta, Poiley, and Lebherz Citation1977). An in vivo study in mice suggested that beryllium-induced oxidative stress and unrepaired DNA damage which may be due to down-regulation of DNA-repair genes (Attia et al. Citation2013).
Cadmium and cadmium compounds
‘Several mechanisms have been identified that potentially contribute to cadmium-induced carcinogenesis. Direct binding to DNA appears to be of minor importance, and mutagenic responses are weak. Convincing evidence exists on disturbances of DNA-repair and tumour-suppressor proteins, which lead to chromosomal damage and genomic instability. Further reported effects include changes in DNA-methylation patterns as well as interactions with signal-transduction processes, which may contribute to the deregulation of cell growth.’ (IARC Citation2012c, 140).
Cadmium is used widely as a key component in Ni-Cd batteries. Exposure of the general public is predominantly through ingestion of contaminated food and drinking-water. Smokers receive exposure from cigarets. Occupational exposure is mainly through inhalation. Cadmium causes lung cancer.
Cadmium does not directly damage DNA when studied in cell extracts. However, in mammalian cells in vitro, and in rodents in vivo, this metal induces double-strand DNA breaks, chromosomal aberrations and micronuclei. Cadmium is not mutagenic in bacteria, but enhances the mutagenicity of UVR, alkylation, and oxidation in mammalian cells by inhibiting several types of DNA repair. The latter activity may be due to displacement of zinc by cadmium from zinc-finger structures. Cadmium induces oxidative stress both in vitro and in vivo. Since cadmium is not redox-active by itself, this effect may be attributed to inhibition of antioxidant enzymes.
Cadmium interacts with a multitude of cellular signal-transduction pathways. In vitro studies in several cell types demonstrated that this metal (1) induced the receptor-mediated release of inositol-1,4,5-trisphosphate and calcium, (2) activated various mitogenic protein kinases, transcription and translation factors, and (3) enhanced expression of cellular proto-oncogenes such as c-fos, c-myc, and c-jun. Cadmium also inhibits the negative controls of cell proliferation. This metal (1) inactivates the tumor suppressor protein p53, (2) inhibits the p53 response to damaged DNA, and (2) reduces DNA methylation.
Additional information
Cadmium affects genes involved in growth regulation of initiated cells during the promotion stage of in vitro cell transformation (Fang, Mar, and Cho Citation2002). Induction of DNA hypo-methylation was reported in humans in vivo and in human cells in vitro after exposure to cadmium (Hossain et al. Citation2012; Huang et al. Citation2008). Studies of the effect of cadmium on rates of apoptosis are conflicting with one group finding no marked effect in the rat testis (Zhu et al. Citation2011) while others reported higher rates of apoptosis in rat liver (Yu, He, and Chen Citation2007). Several studies indicated that cadmium is able to induce various epigenetic changes in plant and mammalian cells in vitro and in vivo (Wang et al. Citation2012a). There is also evidence that prenatal exposure to cadmium alters epigenetic signatures in the DNA of the placenta and of the newborn, with distinct exposure-related patterns of DNA methylation in fetal and maternal DNA (Sanders et al. Citation2014; Vilahur, Vahter, and Broberg Citation2015).
Chromium (VI) compounds
‘Several mechanisms are involved in the carcinogenesis induced by chromium (VI) including induction of DNA damage, generation of oxidative stress and induction of aneuploidy leading to cell transformation. With respect to DNA damage, the spectrum of lesions induced by chromium (VI) appears to depend strongly on the cellular reductant involved. Thus, under physiological conditions with ascorbate as the major reductant, the generation of pre-mutagenic ternary chromium–ascorbate–DNA adducts appears to be of major relevance, which may be linked to the increased number of mismatch-repair-resistant cells observed in chromate-induced lung tumours.’ (IARC Citation2012c, 163).
Chromium (VI) compounds contain chromium in an oxidation state that occurs predominantly in manufactured products. Chromium (VI) is reduced to chromium (III) in the presence of reducing agents such as iron or oxidizable organic matter. The general population is exposed to these compounds via inhalation from anthropogenic sources or from drinking-water. Occupational exposure is through inhalation or dermal contact. Most studies on human cancer risk were conducted in occupational settings. Chromium (VI) compounds produce lung cancer.
Chromium (VI) compounds are genotoxic, both in vitro and in vivo. Workers exposed to chromium (VI)-containing dust exhibited elevated levels of DNA strand-breaks, sister chromatid exchange and micronuclei in lymphocytes. Dominant lethal mutations were observed in male mice exposed to chromium (VI).
Chromium (VI) may be reduced to Chromium (III) in the body. When the reduction of chromium (VI) occurs extracellularly, the resulting chromium (III) cannot traverse the cell membrane. However, chromium (III) may also be formed through a series of intracellular processes that involve ascorbate and cysteine as key reductants. This process leads to the formation of DNA adducts (mainly ternary adducts), DNA–DNA and DNA–protein cross-links, DNA strand-breaks and oxidative DNA base-pair modifications. Chronic exposure to chromium (VI) may lead to the outgrowth of mismatch repair-deficient clones. The reduction of chromium (VI) might release potentially toxic intermediates such as hydroxyl radicals, superoxide and nitric oxide, which may give rise to oxidative stress.
Additional information
Epigenetic effects of Chromium (VI) were reported. In human bronchial epithelial cells, treatment with Cr(VI) reduced the acetylation level of histone H4K16, whereas Cr(VI) at low doses elevated the concentration of histone biotinylation (Chen et al. Citation2016; Xia et al. Citation2014). In the same cell type, hyper-methylation of the p16 protein was noted at the CpG islands, CpG1, CpG31 and CpG32 after treatment with Cr(VI). There was a positive correlation between the extent of CpG1 methylation in p16 and cell damage, and a negative correlation with the p16 expression level. The CpG1 methylation level of p16 is an epigenetic biomarker of the effect of Cr(VI) treatment (Hu et al. Citation2016).
Nickel compounds
‘The ultimate carcinogenic species in nickel carcinogenesis is the nickel ion Ni(II). Nickel compounds are not mutagenic in bacteria, and only weakly mutagenic in mammalian cells under standard test procedures, but they may induce DNA damage, chromosomal aberrations, and micronuclei in vitro and in vivo. Nickel compounds act as co-mutagens with a variety of DNA-damaging agents. Thus, disturbances of DNA repair appear to be important. A further important mechanism is the occurrence of epigenetic changes, mediated by altered DNA methylation patterns and histone modification. Inflammation may also contribute to nickel-induced carcinogenesis.’ (IARC Citation2012c, 210).
Nickel is a metal that is widely used in manufacturing and industrial processes. This metal is a component of a wide range of alloys and compounds such as oxides, sulfides and salts. The general population is predominantly exposed through ingestion of contaminated food. Occupational exposure is mainly via inhalation. Nickel induces lung cancer and cancers of the nasal cavity and paranasal sinuses.
The ultimate carcinogenic/genotoxic species is the Ni2+ ion, although this ion does not react directly with DNA. However, genotoxic effects were consistently detected in exposed humans, experimental animals, and cultured cells. Oxidative DNA damage, chromosomal damage, and weak mutagenicity were found in mammalian cells. Other genotoxic effects including sister chromatid exchange, chromosomal aberrations, and micronuclei were found only at toxic levels of nickel. These genotoxic effects may be mainly secondary to inflammation and oxidative stress. Nickel (a redox-active metal) was shown to increase the level of reactive oxygen species in many cell types. This may involve catalysis of Fenton-type reactions. Nickel inhibits nucleotide-excision and base-excision repair of DNA damage. Specific inhibition of XPA and 3-methyladenine-DNA glycosylase II was reported. Nickel compounds are able to initiate silencing of genes specifically located near heterochromatin. Nickel also induces ubiquination and phosphorylation of histones.
Additional information
Crystalline nickel sulfide was found to induce genomic instability in transformed human broncho-epithelial cells (Chen et al. Citation2004). Nickel induced post-translational histone modifications and affected the associated enzymes (Chervona, Arita, and Costa Citation2012). Nickel also altered the global levels of post-translational histone modifications in exposed nickel-refinery workers (Arita et al. Citation2012). Alterations in gene expression profiles were also reported in these workers (Arita et al. Citation2013). Nickel was also noted to induce microRNA expression (Chiou et al. Citation2015; Humphries, Wang, and Yang Citation2016).
Asbestos (chrysotile, amosite, crocidolite, tremolite, actinolite, and anthophyllite)
‘The mechanistic basis for asbestos-induced carcinogenicity is a complex interaction between crystalline mineral fibres and target cells in vivo. The following general mechanisms have been proposed for the carcinogenicity of asbestos fibre: asbestos and erionite fibres have been shown to generate free radicals that directly induce genotoxicity as assessed by breaks and oxidized bases in DNA. Asbestos fibres have also been shown to interfere with the mitotic apparatus by direct physical interaction resulting in aneuploidy and polyploidy. In addition, asbestos induces macrophage activation and persistent inflammation, which generate reactive oxygen and nitrogen species contributing to tissue injury, genotoxicity, and epigenetic alterations. These effects are associated with the activation of intracellular signaling pathways, resistance to apoptosis, and stimulation of cell proliferation.’ (IARC Citation2012c, 294)
Asbestos is a generic name for a range of naturally occurring mineral silicate fibers with 6 main varieties. This material has been extensively studied both epidemiologically and mechanistically. The primary route of exposure is inhalation. Ingestion from drinking-water is a secondary source. Workers might expose family members through fibers attached to their clothing. All forms of asbestos produce mesothelioma and cancers of the lung, larynx, and ovary.
The mechanism by which asbestos is carcinogenic is complex and involves interactions between the fibers and target cells (see Figure 4.2, p.289 in IARC Citation2012c). The major carcinogenic mechanism involves an inflammatory response, which gives rise to chronic lung fibrosis or asbestosis. Alveolar macrophages phagocytose the fibers, resulting in the release of cytokines, growth factors, and oxidants ((see Figure 4.1, p.284 in IARC Citation2012c). Phagocytosis may be incomplete or fail, which may lead to apoptosis of the macrophage. This response might generate an excess of ROS and RNS, which initiates a wide range of down-stream genetic and epigenetic events leading to DNA damage and carcinogenesis. Inhalation of asbestos fibers is thus associated with excess generation of ROS and RNS metabolites, cell injury, apoptosis, and persistent lung inflammation. As mentioned above, asbestos fibers also physically interfere with the mitotic machinery of the cell.
Additional information
Epigenetic effects after exposure to asbestos have been described. Differential expression of microRNAs between pleural mesothelioma and mesothelial cells was reported, suggesting the potential role of miRNAs as oncogenes or tumor suppressor genes in mesothelioma oncogenesis. Dysregulated miRNAs are potential diagnostic or prognostic biomarkers for malignant mesothelioma, which might facilitate the surveillance procedure of asbestos-exposed subjects (Minoia et al. Citation2011; Sturchio et al. Citation2012). One of three genes hyper-methylated in pleural mesothelioma was also highly methylated in sarcomatoid-type peritoneal mesothelioma (Hama et al. Citation2012). The role of epigenetics in malignant pleural mesothelioma was reviewed where increasing evidence indicates that the lack of response of this disease to chemotherapy is due to epigenetic effects such as histone acetylation or DNA methylation (Vandermeers et al. Citation2013).
Erionite
Erionite is a naturally occurring fibrous mineral that belongs to a group of hydrated aluminosilicates called zeolites. Natural zeolites have many commercial uses, most of which are based upon the ability of these minerals to selectively absorb molecules from air or liquids. Erionite was used as a noble-metal-doping catalyst in a hydrocarbon cracking process. Although erionite displays a morphology similar to that of amphibole asbestos, it has different chemical and physical properties. Erionite initiates mesothelioma. The potency of erionite to induce mesothelioma seems higher than for any type of asbestos.
The carcinogenic mechanism is similar to that of asbestos (see above).
Leather dust
The term ‘leather dust’ is used to indicate a range of exposures found in leather goods manufacturing, leather tanning and processing, and in leather footwear industries. Leather is produced from the skin or hide of animals through a tanning process using either vegetable tannins or chromium (III) sulfate. Leather is frequently treated with chemicals such as benzidine-based dyes and chemical solvents (e.g. toluene, benzene, acrylic resins, polyurethane). Leather dust is created from the handling and processing of leather throughout the tanning and manufacturing process, leading to complex and variable exposures. Leather dust produces nasal cancers and paranasal sinus cancers, most commonly of the adenocarcinoma type.
The mechanisms by which leather dust is carcinogenic are discussed under wood dust (see below).
Crystalline silica dust in the form of quartz or cristobalite
‘Three mechanisms have been proposed for the carcinogenicity of crystalline silica in rats. First, exposure to crystalline silica impairs alveolar macrophage-mediated particle clearance, which results in macrophage activation, and the sustained release of chemokines and cytokines, which induce genotoxicity, injury, and proliferation of lung epithelial cells leading to the development of lung cancer. Second, extracellular generation of free radicals by crystalline silica depletes antioxidants in the lung-lining fluid, and induces epithelial cell injury followed by epithelial cell proliferation. Third, crystalline silica particles are taken up by epithelial cells followed by intracellular generation of free radicals that directly induce genotoxicity. The Working Group considered the first mechanism as the most prominent based on the current experimental data on inhalation or intra-tracheal instillation of silica in rats, although the other mechanisms cannot be excluded. It is unknown which of these mechanisms occur in humans exposed to crystalline silica dust.’ (IARC Citation2012c, 396).
The predominant commercial categories of silica dust are sand/gravel, quartz crystals and diatomite, which is used in filtration and as an abrasive. The main route of human exposure is inhalation. Chronic occupational exposure to silica produces a fibrotic lung disease termed silicosis. Silica dust produces lung cancer.
Silica dust does not directly damage DNA. However, silica provokes a strong inflammatory response with macrophage activation and release of cytokines and chemokines. Reactive oxygen species are created by macrophages and through direct chemical interaction with the silicates. Silica particles may be directly phagocytosed into lung epithelial cells and then generate ROS. This leads to DNA damage including strand breaks. Quartz crystals have been shown to chemically deplete antioxidants in lung tissue.
Additional information
Crystalline silica induces apoptosis in rat macrophages in vitro (Persson Citation2005). A rise in Hprt mutation frequency was detected in alveolar epithelial cells obtained from rats exposed to α-quartz (Driscoll et al. Citation1995). Quartz-treated, transformed and tumorigenic cells exhibited an elevation in the expression of TGF-beta1/beta2 mRNA transcripts (Williams et al. Citation1996). There is some evidence of epigenetic effects after exposure to silica dust. Analysis of miRNA expression patterns in the lungs of silica-treated rats showed some miRNAs up-regulated and others down-regulated. These altered miRNAs may be involved in lung fibrosis of these exposed rats (Faxuan et al. Citation2012). Alterations in global DNA methylation were found in lung tissues from human silicosis patients (Zhang et al. Citation2016).
Wood dust
‘Potential mechanisms responsible for the carcinogenicity of wood dust include tissue injury induced by the deposition of wood dust particles in the sinonasal region, impaired ciliary clearance, direct genotoxicity, and indirect genotoxicity secondary to chronic inflammation. Wood or leather dusts may also act as carrier for other genotoxic agents (e.g. chromate). There is weak evidence for these mechanisms in cellular assays, short-term animal assays, or assays for genotoxicity with peripheral blood cells or buccal epithelial cells obtained from workers exposed to wood dust’. (IARC Citation2012c, 459).
Dust from wood products is a complex substance that varies considerably depending upon the tree species from which the wood is derived. Most human exposure arises from woodworking activities in construction or furniture building. ‘Furniture and cabinet making’ was examined in previous IARC reviews, and identified as a cause of nasal adenocarcinoma. Wood dust induces cancer of the nasal cavity, paranasal sinuses and nasopharynx. Evidence suggests that exposure to dust from hardwood trees is associated with a higher risk of cancer.
The mechanism underlying carcinogenesis for wood dust is not known. The mechanism with greatest support is based upon a combination of impaired mucociliary clearance of wood-dust particles from the nose and sinus area leading to mechanical irritation, chronic inflammation and increased cellular proliferation. Direct experimental evidence to support this model is lacking. Extracts from some species of wood (oak and beech) detected mutagenicity in bacteria and rat hepatocytes. Wood dust is also known to act as a carrier for other carcinogens. There is strong evidence that oak and beech wood contain chromium compounds. These mechanistic considerations also pertain to leather dust.
Additional information
Exposure to wood dust induced cytotoxicity, gave rise to formation of ROS and activated the apoptotic caspase-3 enzyme in animal and human cell lines in vitro (Naarala et al. Citation2003; Pylkkanen et al. Citation2009). Solvent extracts of natural woods induced chromosome aberrations in respiratory cells in culture (Zhou, Norpoth, and Nelson Citation1995). Exposure to wood dust modulated macrophage-derived cytokines and chemokines (Maatta et al. Citation2005).
Volume 100D: radiation
IARC Monograph Volume 100D (IARC Citation2012d) includes separate discussions of carcinogenicity of agents based on the type of radiation they release. A broad distinction can be made between ionizing and ultraviolet radiation. Four chapters in IARC Monograph Volume 100D discuss ionizing radiation. A discussion on mechanistic aspects, which applies to all types of ionizing radiation is given in the Monograph on X- and γ-radiation.
The first section below provides discussion of the general mechanism by which ionizing radiation is carcinogenic. In subsequent sections, relevant agent-specific issues are discussed.
General mechanisms related to ionizing radiation
‘All types of ionizing radiation transfer their energy to biological material in clusters of ionization and excitation events, primarily through a free-electron-mediated mechanism. In cells, energy deposition from all types of ionizing radiation results in a wide variety of molecular damage: in DNA, this includes base damage and single- and double-strand breaks, some of which may be clustered and form complex lesions. Subsequent processing of these lesions may lead to chromosomal aberrations and mutations. There is much evidence pointing to DNA damage being of primary importance in the biological outcome of exposure to ionizing radiation. Genome-wide sequencing of tumours has shown wide heterogeneity in constituent mutations, indicating there may be multiple pathways to tumour formation. In addition, there is emerging consensus that epigenetic factors are important in tumorigenic processes associated with radiation exposure. Notably, ionizing radiation induces effects such as genomic instability and bystander effectsFootnote1, which are epigenetic in origin. Also important are the interactions at the tissue level between radiation-damaged cells and normal cells.’ (IARC Citation2012d, p.209-210).
High-energy radiation is capable of ionizing molecules. Several types of ionizing radiation may be distinguished as follows: X-rays, γ-rays, neutron radiation, radiation from radionuclides that emit α-particles, and radiation from radionuclides that emit β-particles. The source, energy levels, and depth of penetration into tissue vary across different radiation types. However, the mechanisms by which these sources interact with tissue and by which carcinogenic is initiated are essentially similar.
Everyone is exposed to background ionizing radiation from soil, building materials, cosmic rays and radon gas. Exposure to external radiation accounts for approximately 40% of the average worldwide natural radiation dose, the remainder being due to internal exposure, mainly from radon. The mean exposure levels due to medical uses of radiation have been increasing, in particular due to widespread use of computed tomography (CT), angiography, and interventional irradiation procedures in developed countries. Approximately half of the external radiation exposure is now related to medical procedures. Studies on populations exposed to intense ionizing radiation levels from nuclear bomb explosions have contributed substantially to our knowledge of cancer risk and our understanding of radiation carcinogenesis. Internal radiation exposure arises from radionuclides (e.g. radon-222 or iodine-131) deposited in the body upon absorption of products from sources such as natural decay of radioactive building materials, by-products of nuclear explosion testing, accidental release of radiation (e.g. Chernobyl), or from radiotherapy and diagnostic procedures. Internal exposure originates predominantly from α- and β-particles.
Two main mechanistic models underlying the carcinogenicity of ionizing radiation have been proposed: the mutation theory (changes in DNA that result in miscoding) and a non-genetic effect theory (epigenetic factors). Bystander effects have also been proposed. The mutation theory assumes a mechanistic model for carcinogenesis that is purely genetic: ionizing radiation produces damage to DNA, repair processes fail to correct (all) the damage, which leads to mutations in dividing cells. Carcinogenesis results from the accumulation of such damage through clonal expansion of the mutated cell. The non-genetic model is focused on epigenetic factors. These factors affect the dynamic functioning of the cell rather than initiating changes in DNA. A wide range of epigenetic alterations were reported and contribute to carcinogenesis. Ionizing radiation might induce epigenetic changes. Both these mechanisms, as well as bystander effects, are likely to contribute to carcinogenesis from ionizing radiation. These mechanisms interact in complex ways with interactions also occurring with remote and nearby normal cells and other host factors.
Ionizing radiation deposits energy in cellular macromolecules, leading to a wide range of damage. DNA damage results either from direct ionization of its constituent atoms or indirectly through reactions with free radicals – e.g. the hydroxyl radical – produced by interaction of the radiation with water molecules. There is strong evidence that ionizing radiation is capable of producing a wide-range of DNA damage and mutations, leading to large-scale gene deletion, gross chromosomal damage and genetic instability. DNA damage includes: base-pair damage, single-strand breaks, double-strand breaks, DNA–protein cross-links, sister chromatid exchange and combinations of these. Observed events include: chromosomal aberrations, gene-sequence and mini-satellite mutations, and apoptosis. Ionizing radiation produces damage to bone marrow which may lead to immunosuppression.
Additional information
There is strong evidence that ionizing radiation induces mutations in the TP53 gene and in other loci (Coates et al. Citation2003; Knoops et al. Citation2007; Sankaranarayanan Citation1991; Tamminga and Kovalchuk Citation2011). Epigenetic effects that are induced by ionizing radiation include alterations in DNA methylation in humans and animals in vivo (Giotopoulos et al. Citation2006; Koturbash et al. Citation2007; Krakowczyk et al. Citation2010), and changes in micro-RNA expression (Koturbash et al. Citation2007; Templin et al. Citation2011). Ionizing radiation induces telomere shortening and dysfunction in radiation workers (Ilyenko, Lyaskivska, and Bazyka Citation2011). Similar effects were found in human and animal cell lines in vitro (Berardinelli et al. Citation2014; Li et al. Citation2012; Sgura et al. Citation2006; Slijepcevic, Natarajan, and Bryant Citation1998). There is consistent evidence that germline mutations in DNA repair genes were associated with increased sensitivity to carcinogenesis from ionizing radiation (Chokkalingam and Buffler Citation2008; Garcia-Quispes et al. Citation2011; Rajaraman et al. Citation2008; Santos et al. Citation2012).
Solar and ultraviolet radiation
Following exposure to the individual components of ultraviolet radiation, i.e. UVA, UVB or UVC, there is an overlapping profile of DNA damage, in particular for cyclobutane-pyrimidine dimers. However, the proportion of different base-pair changes shows variation depending on the radiation wavelength and the cell type or species used. The mechanisms leading to the formation of these photoproducts may also be different. Cyclobutane-pyrimidine dimers at cytosine-containing DNA sequences are formed following exposure to both UVA and UVB individually in human skin ex vivo. Human cells have repair systems that eliminate DNA photoproducts: the absence of these enzymes, as seen in Xeroderma Pigmentosum (XP) patients, leads to an increased risk of developing squamous cell carcinomas and melanomas, which supports a major role of DNA photoproducts in photo-carcinogenesis. Mutations have been detected in human cells exposed to UVA, UVB and UVC’ (IARC Citation2012d, 89-90).
Human exposure to ultraviolet radiation (UVR) originates primarily from sunlight. Important secondary exposure sources are tanning beds, UVB phototherapy of psoriasis, and welding. The carcinogenicity of solar radiation is related to the UVR component. Approximately 5% of solar terrestrial radiation is UVR, and solar radiation is the major source of human exposure to UVR. The UV component of terrestrial radiation from the midday sun comprises approximately 95% UVA and 5% UVB; UVC and most of UVB are removed from extraterrestrial radiation by stratospheric ozone. Solar radiation induces cutaneous malignant melanoma, squamous cell carcinoma of the skin and basal cell carcinoma of the skin. The use of UV-emitting tanning devices produces cutaneous malignant melanoma and ocular melanoma. Evidence is unclear as to the relative carcinogenicity of different wavelengths of UVR (i.e. UVA, UVB and UVC).
High-energy UVR (e.g. UVB/C) interacts directly with DNA to produce damage. UVA interacts mainly with endogenous or exogenous photosensitizers, which release ROS and RNS that lead to DNA damage. A common effect is production of cyclobutane dimers between adjacent pyrimidine bases. UVR is mutagenic in in vitro and in vivo tests across a wide range of species. Mutations usually are targeted at two adjacent pyrimidines. The most common mutations are tandem CC→VR which is mutagenic in in vitro and in vivo tests such as rare genetic condition Xeroderma pigmentosum, which is defective in the NER pathway. Tandem CC→VR mutations are associated with more extensive DNA damage and an increased cancer risk. This DNA damage leads to genomic instability, including chromatid breakage and elevated levels of sister chromatid exchange. Alterations of gene expression profiles were noted with use of cDNA micro-arrays. Solar-simulated UV (but not UVA and UVB) produced significant immunosuppression in humans. A bystander effect was also described.
Additional information
In cultured Chinese hamster ovary cells, 254-nm low-intensity continuous wave UV light markedly enhanced the level of sister chromatic exchange (SCE) (Rasmussen, Hammer-Wilson, and Berns Citation1989). In in vitro and in vivo tests, UVC and natural sunlight were found to induce single-strand DNA breaks in human cells (Rodrigues-Junior et al. Citation2012; Squires et al. Citation2004). In mouse embryonic fibroblasts harboring a humanized p53 gene, UV produced mutations in this gene (Liu et al. Citation2004). UVB initiated aberrant DNA methylation and histone modifications in mouse skin and human squamous cell carcinoma (Nandakumar et al. Citation2011). Histone modifications were detected in human fibroblasts and hairless mouse skin exposed to UVB/UVA (Kim et al. Citation2013, Citation2009; Nandakumar et al. Citation2011). In vitro investigations with keratinocytes demonstrated that UVB induced apoptosis (Cai et al. Citation2009; Jantschitsch et al. Citation2009). One study (Whiteman, Whiteman, and Green Citation2001) suggested that sun exposure in childhood carries a higher risk of cancer than a similar exposure later in life, indicating a higher susceptibility in childhood. Acute UV exposure was found to produce angiogenesis while chronic exposure reduced angiogenesis and contributed to photo-aging (Chung and Eun Citation2007); the relevance of these observations to carcinogenesis is unclear. Compared with the relatively moderate mutagenic effects, acute UVA/UVB exposure of human keratinocytes induced significant epigenomic and transcriptomic alterations (Shen et al. Citation2017).
X-radiation and γ-radiation
A mechanistic summary is given above in the section ‘General mechanisms related to ionizing radiation’.
X- and γ-radiation have been established as causal agents for a wide range of cancers including cancers of salivary gland, esophagus, stomach, colon, lung, bone, basal cells of the skin, female breast, kidney, urinary bladder, brain and CNS, and thyroid. X- and γ-radiation also induce leukemia (except chronic lymphocytic leukemia). The general mechanisms discussed above are applicable. X- and γ-radiation penetrate matter to a greater depth than α- and β-particles, and exert different radiation tracks in tissues. The higher penetrating power also increases the range of tissues affected.
Additional information
Semen abnormalities, sperm DNA damage and global hyper-methylation were detected in health workers occupationally exposed to ionizing (X, β, gamma) radiation (Kumar et al. Citation2013). Exposure of pregnant mice to low-dose ionizing radiation (X-rays) produced epigenetic alterations specifically in male offspring which were reduced with antioxidants, indicating that these responses are mediated in part by oxidative stress (Bernal et al. Citation2013). Alterations in global DNA methylation patterns and hypo-methylation at specific cancer-related genes were observed in human colorectal carcinoma cells exposed to ionizing (gamma) radiation (Bae et al. Citation2015). Expression levels of some microRNAs vary significantly after exposure to ionizing radiation. These miRNAs are related to modulation of the expression of several post-transcriptional targets in DNA damage-response pathways, and to regulation of DNA-damage repair (Marta et al. Citation2015).
Neutron radiation
A mechanistic summary is given above in the section ‘General mechanisms related to ionizing radiation’.
The general mechanisms by which neutron radiation is carcinogenic are similar to those described above. Neutrons are electrically neutral and interact with the body predominantly through interactions with atomic nuclei. This leads to molecular damage being clustered in space, which was suggested to decrease the effectiveness of DNA repair. According to the Monograph there is inadequate evidence that neutron radiation is carcinogenic to humans, due to lack of data on human exposure to ‘pure’ neutrons, which is extremely rare. The classification of neutron radiation as a Group-1 human carcinogen was based predominantly upon data from animal investigations (sufficient evidence in animals) and on similarities with X- and γ-radiation with respect to biological effects and physical properties.
Internalized radionuclides that emit α-particles
A mechanistic summary is given above in the section ‘General mechanisms related to ionizing radiation’.
Radionuclides emitting α-particles were reviewed separately when adequate data were available, and evaluated as follows. There is sufficient evidence in humans for the carcinogenicity of radon-222, its decay products, and of underground hematite mining with exposure to radon (all of which produce cancer of the lung). Similarly, there is sufficient evidence in humans for the carcinogenicity of radium-224 (bone sarcomas), radium-226 (bone sarcomas and carcinomas of the paranasal sinuses and mastoid process) and radium-228 (bone sarcomas). In addition, there is sufficient evidence in humans for the carcinogenicity of thorium-232 as stabilized thorium-232 dioxide in colloidal form (Thorotrast). Diagnostic injection of Thorotrast initiates primary liver cancer, leukemia (except chronic lymphocytic leukemia), cancer of the extrahepatic bile ducts, and of gallbladder. There is also sufficient evidence in humans for the carcinogenicity of the α-emitter plutonium-239 (cancer of the lung, liver and bone).
The general carcinogenic mechanisms are as outlined above. Alpha-particles possess a limited ability to penetrate tissue. Internal exposure is necessary for carcinogenesis. Alpha-particles emitted by radionuclides induce chromosomal aberrations in circulating lymphocytes and gene mutations in humans in vivo. This Group-1 classification was considered to be applicable to all α-particle-emitting radionuclides, based mainly upon the following considerations: (1) all radionuclides that emit α-particles and that have been adequately studied were found to induce cancer in humans and experimental animals and (2) α-particles emitted by radionuclides, irrespective of their source, produce the same pattern of secondary ionizations, and the same pattern of localized damage to biological molecules, including DNA. These effects, observed in vitro, include DNA double-strand breaks, chromosomal aberrations, gene mutations, and cell transformation.
Internalized radionuclides that emit β-particles
A mechanistic summary is given above in the section ‘General mechanisms related to ionizing radiation’.
Radionuclides emitting β-particles were reviewed separately when adequate data were available, and evaluated as follows. There is sufficient evidence in humans for the carcinogenicity of exposures during childhood and adolescence to short-lived radioisotopes of iodine, including iodine-131. These exposures produce cancer of the thyroid. Similarly, there is sufficient evidence in humans for the carcinogenicity of therapeutic administration of phosphorus-32, as phosphate, which induces acute leukemia in patients with polycythemia vera. In addition, there is sufficient evidence in humans for the carcinogenicity of external and internal exposures to fission products, including strontium-90, which initiates solid cancers and leukemia. This Group-1 classification was considered to be applicable to all β-particle-emitting radionuclides, based predominantly upon the following considerations: (1) all radionuclides that emit β-particles and that have been adequately studied, were noted to induce cancer in humans and experimental animals and (2) β-particles emitted by radionuclides, irrespective of their source, produce similar patterns of secondary ionizations and the same type of localized damage to biological molecules including to DNA. These effects include DNA double-strand breaks, chromosomal aberrations in circulating lymphocytes and gene mutations in humans in vivo, and cell transformation.
Volume 100E: personal habits and indoor combustions
Tobacco smoking
‘The pathways by which tobacco products cause cancer essentially recapitulate established mechanisms of carcinogenesis by individual compounds of tobacco smoke. Most carcinogens, either directly or after metabolism catalyzed by multiple cytochrome P450 enzymes, react with nucleophilic sites in DNA to form adducts, which, if left unrepaired by cellular DNA-repair enzymes, persist and cause mistakes during DNA replication, giving give rise to permanent mutations. If these occur in important regions of critical growth-controlling genes such as the oncogene KRAS or the tumour suppressor gene TP53, cell growth can become severely unregulated and cancer can result. Multiple studies of mutations in KRAS, TP53, and other growth-controlling genes in lung tumours from smokers, some of which report thousands of mutations, are fully consistent with this overall concept. There are over 70 established carcinogens in cigarette smoke, and multiple DNA adducts are present in the lungs and other tissues of smokers, and sister chromatid exchange as well as other genetic effects are consistently observed. However, only relatively few DNA adducts in smokers’ lungs have been structurally characterized and the relationship between specific adducts and the consequent mutations in critical genes is still unclear. Other processes that contribute to cancer induction by tobacco products include inflammation, tumour promotion, oxidative damage, co-carcinogenesis, and direct activation of cellular growth pathways by constituents of smoke. However, the details of the interaction of these processes with the DNA-damage pathways and their role in specific cancers caused by tobacco products are still not fully understood’. (IARC Citation2012e, 165-6).
Tobacco smoking is a potent initiating source of cancers at multiple sites, including lung, oral cavity, naso-, oro- and hypopharynx, nasal cavity and accessory sinuses, larynx, esophagus, stomach, pancreas, colorectum, liver, kidney (body and pelvis), ureter, urinary bladder, uterine cervix and ovary (mucinous) as well as myeloid leukemia.
Over 5300 substances were identified in tobacco smoke including more than 70 carcinogens. Of these, 16 are IARC Group-1 carcinogens, including nitrosamines (e.g. NNK and NNN), polycyclic aromatic hydrocarbons (e.g. benzo[a]pyrene), 1,3-butadiene and cadmium. The carcinogenic mechanisms associated with tobacco smoking reflect those of the constituent carcinogens. It is not feasible to review all these agents here.
A conceptual framework for smoking-induced carcinogenesis is presented in IARC Monograph Volume 100E (see Figure 4.1, p.133 in IARC Citation2012e). The major pathway involves (1) binding of carcinogens or their metabolites to DNA, (2) formation of adducts, and (3) induction of mutations in key genes such as TP53 and KRAS. Indeed, a study that sequenced 623 cancer-related genes in 188 human lung adenocarcinomas noted multiple somatic mutations in critical growth-controlling genes, consistent with the chronic attack of cellular DNA by tobacco-smoke carcinogens and their metabolically activated forms. Mutagenicity of urine excreted by smokers, sister chromatid exchange and micronuclei in buccal cells. Other genetic effects have been consistently observed. Other cancer-related effects were also reported including (1) direct activation of cellular signaling pathways (see Figure 4.5, p.146 in IARC Citation2012e), (2) induction of chronic inflammation and (3) epigenetic mechanisms including enzymatic methylation of gene promoters, histone modification and RNA-mediated gene silencing.
There is evidence that the carcinogenic mechanisms may be different for some cancer sites affected by tobacco smoke. Interaction effects with alcohol intake were also documented where synergistic effects are perhaps due to alcohol enhancing the solubility of tobacco-derived carcinogens, or to interactions with metabolic enzymes of the cytochrome-P450 family.
Additional information
Exposure to cigaret smoke produces protein adducts both in vivo and in vitro (Rainey et al. Citation2009; von Stedingk et al. Citation2011). Components of cigaret smoke interact with cell receptors and cell-signaling pathways to mediate a variety of effects, such as stimulation of DNA synthesis in lung adenocarcinoma cells (Schuller et al. Citation1999), and immunosuppression (Geng et al. Citation1995). Other established responses attributed to cigaret smoke such as induction of gene expression and genotoxicity may be mediated by the aryl hydrocarbon receptor (Dertinger, Silverstone, and Gasiewicz Citation1998; Meek and Finch Citation1999; Rochette et al. Citation2009). In normal human respiratory epithelia and lung cancer cells cultured in the presence of cigaret smoke condensate, cigaret smoke mediated epigenetic repression of miR-487b during pulmonary carcinogenesis and miR-217 during esophageal adenocarcinogenesis (Xi et al. Citation2015, Citation2013). Prenatal exposure to tobacco smoke was associated with reproducible epigenetic changes, such as increased CpG methylation, that persisted into childhood (Breton et al. Citation2014; Markunas et al. Citation2014).
Second-hand tobacco smoke
‘Second-hand tobacco smoke’ is classified in IARC Monograph Volume 100E (IARC Citation2012e) as a distinct Group-1 carcinogen which induces lung cancer and strongly associated with cancers of larynx and pharynx. In addition, there is sufficient evidence in humans for carcinogenicity of parental smoking – with an emphasis on prenatal exposures –, which induces hepatoblastoma in children.
Second-hand tobacco smoke is a mixture of exhaled mainstream smoke and side-stream smoke, which is the smoke originating from the tip of the cigaret/cigar when no inhalation is occurring. The chemical composition of second-hand smoke is qualitatively similar to mainstream smoke inhaled directly by the smoker. There are quantitative differences in the concentrations of carcinogens. In addition, human exposure to second-hand tobacco smoke is affected by dispersion in the air in the environment, leading to lower doses of exposure.
The conceptual model for understanding mechanisms by which tobacco smoke causes cancer in active smokers is discussed above. This model also applies to second-hand tobacco smoke since it contains all of the same carcinogens and toxicants as mainstream cigaret smoke, although at lower concentrations. Thus the mechanisms by which second-hand smoke is carcinogenic are similar as for tobacco smoking (see above).
Smokeless tobacco
‘Smokeless tobacco products have much lower levels of carcinogens and toxicants that result from combustion, so the effects of these agents are not seen to a significant extent. The most prevalent strong carcinogens in smokeless tobacco are the tobacco-specific nitrosamines; other nitrosamines, PAHs, aldehydes and metals are also present, and there are large amounts of some inorganic salts that may contribute to inflammation … … Multiple studies demonstrate that tobacco specific nitrosamines are absorbed and metabolised in smokeless tobacco users … … There is evidence for DNA-adduct formation in oral tissues of smokeless tobacco users, and sister chromatid exchange, chromosomal aberrations, and micronuclei have been observed. Many studies have demonstrated RAS and TP53 mutations in smokeless tobacco users … … Oxidative stress and reactive oxygen species could play a significant part in cancer induction by smokeless tobacco, particularly at high pH. Chronic local inflammation and irritation can also be induced by smokeless tobacco’. (IARC Citation2012e, 132).
The term smokeless tobacco implies use of unburned tobacco in the finished product. Smokeless tobacco products may be inhaled or applied orally (chewed, sucked, gargled or applied directly to the gums). The chemical composition depends upon the method of manufacture employed. Smokeless tobacco produces cancer of the oral cavity, esophagus and pancreas. Multiple carcinogens were identified in smokeless tobacco products and the mechanisms by which smokeless tobacco is carcinogenic reflect these carcinogens. DNA-adduct formation was documented in oral tissues of smokeless tobacco users, and there is evidence of chromosomal damage such as sister chromatid exchange and micronuclei and mutations in RAS and TP53. Localized inflammation and oxidative stress were also reported along with production of reactive oxygen species (ROS).
N ‘ -Nitrosonornicotine (NNN) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)
‘NNK and NNN are the most abundant strong carcinogens in smokeless tobacco; their uptake and metabolic activation have been clearly documented in users of smokeless tobacco. Combined application of NNN and NNK to the oral mucosa of rats induced oral tumours, consistent with their induction by smokeless tobacco. One of the mechanisms of carcinogenicity is cytochrome-P450-mediated α-hydroxylation, which leads to the formation of DNA and haemoglobin adducts that have been detected in users of tobacco.’ (IARC Citation2012e, 328).
Virtually all commercial tobacco products contain NNN and NNK, which always occur together. These compounds are mainly formed during the curing of tobacco. There is a large variation in levels of these compounds in mainstream smoke and side-stream smoke of cigarets and in smokeless tobacco products. This is predominantly attributed to differences in tobacco types utilized for various products such as in agricultural practices, curing methods, and in manufacturing. There is not marked direct evidence of carcinogenicity of NNN or NNK in humans, mainly due to the difficulty in distinguishing the direct effect of these two nitrosamines from that of other carcinogens present in tobacco smoke. There is strong evidence of carcinogenicity of these two substances in animals.
Some mechanistic discussions specifically linked to NNN/NNK are presented here. Pyridyloxobutyl-DNA adducts of NNN and NNK are formed at the N7- and O6-positions of deoxyguanosine (dG) and the O2-positions of thymidine and deoxycytidine. Metabolic activation of NNK also leads to N7-methyl-dG and O6-methyl-dG, identical to the DNA adducts formed from nitrosodimethylamine and other DNA-methylating agents.
Nicotine, NNN and NNK bind to nicotinic and other cellular receptors, resulting in activation of protein kinases A and B, and other changes. Nicotine and NNK enhance the expression of survivin, an inhibitor of apoptosis, in normal human bronchial epithelial cells. This results in decreased apoptosis, enhanced angiogenesis, and increased cell transformation.
Additional information
Overexpression of DNA methyltransferase 1 (DNMT1) is correlated with smoking status and low expression of retinoic acid receptor β (RARβ) in esophageal tissue from SCC patients. In esophageal squamous epithelial cells in vitro, NNK induced hypermethylation of the RARβ promoter through upregulation of DNMT1which finally led to enhancement of cell proliferation and inhibition of apoptosis (Wang et al. Citation2012b). NNK also induced circulating microRNA deregulation in early lung carcinogenesis (Wu et al. Citation2014). Long non-coding RNAs (lncRNA) are postulated to be important epigenetic regulators of a variety of cancers. The long non-coding RNA NR 026689 was found associated with NNK-induced lung carcinogenesis (Wu et al. Citation2016).
Betel quid and areca nut
‘Betel quid and areca-nut ingredients and extracts show a variety of genetic and related effects. Continuous local irritation of buccal epithelial cells caused by betel quid and its ingredients, particularly areca nut and slaked lime, can give rise to chronic inflammation, oxidative stress and cytokine production. Reactive oxygen species generated during chewing of betel quid and other genotoxic reactants formed from arecoline and areca nut-derived nitrosamines, can lead to DNA damage in exposed oral keratinocytes. Persistent oxidative stress can drive affected cells to uncontrolled proliferation and hyperplastic/dysplastic lesions. Chronic occurrence of these toxic insults in the oral cavity of chewers could drive these pre-neoplastic cells towards full malignancy’ (IARC Citation2012e, 363).
Betel-quid chewing is an ancient practice on the Indian subcontinent and many parts of Asia, which is still prevalent today. The term ‘quid’ denotes a mixture of substances that is placed and retained in the mouth, and often swallowed. Betel-quid is made of a folded betel leaf containing various products such as areca nut, tobacco, slaked lime, and catechu. Areca nut, the major constituent of a betel quid, is the fruit of the Areca catechu, a palm tree that grows in South and South-East Asia and the Pacific islands. Chewing betel quid with tobacco products produces cancer of the oral cavity, pharynx and esophagus. Betel quid without added tobacco induces cancers of the oral cavity and esophagus.
Betel quids that contain tobacco products would pose the same carcinogenic hazards as described for smokeless tobacco. In addition, other components of betel quid, such as arecoline, areca nut, betel leaf, slaked lime, and catechu, were examined for carcinogenesis. Secondary and tertiary amines in betel quids may be converted to nitrosamines in the saliva and stomach. There is no evidence that the non-tobacco components of betel quid directly induce DNA damage. However, irritation of oral tissues from the quid, combined with the generation of reactive oxygen species, produce indirect DNA damage. Slaked lime produces a high-pH environment that exacerbates these effects. Increased frequencies of micronuclei were detected in oral exfoliated cells in chewers of betel quid without tobacco. Micronucleus formation was also observed in precancerous lesions in the oral cavity of chewers of betel quid alone, and of betel quid with tobacco. Elevated levels of sister chromatid exchange and micronucleus formation were observed in cultured peripheral lymphocytes collected from chewers of areca nut without tobacco and slaked lime, and with tobacco. DNA strand breaks were induced in both animal and human cell cultures. TP53 is downregulated and the function of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) altered.
Areca nut contains several alkaloids and tannins. Arecoline is the most common alkaloid in areca nut, which also contains a variety of amines that are nitrosated in saliva during quid chewing. All these substances contribute to carcinogenicity. Aqueous extracts of areca nut produce a variety of cellular effects including: DNA strand-breaks, sister chromatid exchange, micronucleus formation, gene mutations, chromosomal aberrations and increased cellular proliferation. These were found in both in vivo animal tests and in vitro experiments with mammalian cells. Similar effects were noted in tests with areca-nut alkaloids. Arecoline inhibited TP53 expression in human epithelial cells. Irritation of oral tissues from the quid, combined with the generation of ROS, produce indirect DNA damage.
Additional information
Areca quid chewing and arecoline elevate the expression of calcium-binding proteins, which might be involved in oral submucous fibrosis, a pre-cancerous lesion (Yu et al. Citation2013). Areca-nut extract up-regulate cyclooxygenase inflammatory signaling in the oral cavity (Chiang et al. Citation2008). and induce the TGF-β pathway during the progression of oral submucous fibrosis (Khan et al. Citation2012). Extracts of lime-based betel quid were shown to induce cell transformation (Lin et al. Citation2003). Arecoline produced alterations in expression of several genes catalyzing histone methylation, acetylation, and demethylation in human K-562 myelogenous leukemia cells (Lin et al. Citation2011). Silencing of the Rarb (retinoic acid receptor β) gene, by hyper-methylation, was reported in areca nut-induced oral squamous cell carcinoma in mice (Lai et al. Citation2014).
Alcohol consumption, ethanol in alcoholic beverages, acetaldehyde associated with the consumption of alcoholic beverages
‘Although alcoholic beverages may contain several potentially carcinogenic compounds, this synthesis focuses on the role of ethanol and acetaldehyde in the carcinogenesis associated with alcoholic beverages … … . The role of ethanol metabolism in tumour initiation is implied by the associations observed between different forms of cancer and polymorphisms in genes involved in the oxidation of ethanol. Whether, or to what degree, these associations are explained by redox changes, formation of radicals, effects on intermediary metabolism and/or effects on other pro-carcinogens cannot be established from current findings … . Acetaldehyde is a cytotoxic, genotoxic, mutagenic and clastogenic compound. It is carcinogenic in experimental animals’ (IARC Citation2012e, 470-1).
The term ‘Alcoholic beverages’ refers to a wide group of drinks that are produced by fermentation. Alcohol consumption produces cancers of the oral cavity, pharynx, larynx, esophagus, colorectum, liver (hepatocellular carcinoma), and female breast.
There is an extensive literature concerning the carcinogenic mechanisms underlying the carcinogenicity of alcoholic beverages. While alcoholic beverages contain many ingredients, not all of which are present in all types of alcoholic drinks, all forms of alcoholic beverage share carcinogenic potential. Hence, the main focus for cancer risk has been on two factors 1) ethanol as the primary non-water constituent of alcoholic drinks, and 2) acetaldehyde, which is the initial metabolic product of ethanol in humans. In addition to being directly carcinogenic, alcohol may act as an agent to facilitate the transport of other carcinogens into the body. This mechanism is not considered further here.
Ethanol
The induction of the metabolic enzyme CYP2E1 by ethanol produces various reactive oxygen species, which lead to the formation of lipid peroxidation products such as 4-hydroxy-nonenal and to the condition of oxidative stress. The increase in reactive oxygen species and oxidative stress, which damage the DNA and affect its repair, has been associated with ethanol-induced carcinogenesis in many organs, such as the breast, liver and pancreas.
Ethanol mediates hepatocellular injury, fibrotic changes and cirrhosis. Ethanol increases levels of sex hormones such as estrogens and androgens, perhaps through inhibition of liver catabolism of sex steroids due to ADH-mediated alcohol oxidation and an increase in the hepatic redox state. Reduced folate levels are associated with alcohol intake but may reflect general life-style factors rather than a biological mechanism. In animals in vivo, ethanol induced DNA adducts, DNA strand-breaks, sister chromatid exchange and dominant lethal mutations.
Acetaldehyde
Ethanol is metabolized to acetaldehyde by three major pathways: the alcohol dehydrogenase (ADH) pathway, the microsomal ethanol oxidizing cytochrome P450 (CYP) pathway, and the catalase-H2O2 system. Acetaldehyde, to which many deleterious effects of ethanol can be attributed, is oxidized to acetate primarily by aldehyde dehydrogenases (ALDHs). Over the past decade, epidemiological evidence of enhanced cancer risks among heterozygous carriers of the inactive ALDH enzyme has become much stronger, in particular for esophageal cancer: practically all studies conducted in East-Asian populations who consumed alcoholic beverages show significantly increased odds ratios for carriers of the inactive ALDH allele. In addition, several studies have demonstrated associations between the polymorphism of ADH1B and upper aero-digestive tract cancers, which have been explained either by more active ADH producing more acetaldehyde or by less active ALDH causing prolonged exposure to lower levels of ethanol-derived acetaldehyde. These data imply that acetaldehyde is the key compound in the development of cancers of the esophagus and other upper aero-digestive tract cancers associated with alcoholic beverage consumption
Acetaldehyde induces DNA-adducts, DNA–protein crosslinks, DNA strand-breaks, sister chromatid exchange, chromosomal aberrations, and micronuclei in eukaryotic cells in vitro. There is evidence for gene-environment interaction in cancer risk with regard to the ALDH2 and ADH1B genes. Alcoholics have been found to exhibit significantly higher levels of chromosomal aberrations and cells with micronuclei than either non-drinking controls or abstinent alcoholics (formerly heavy drinkers who had been abstinent for at least a year).
Additional information
Ethanol induced histone modifications (Agudelo et al. Citation2011; D’Addario et al. Citation2011; Guo et al. Citation2011; Zhao et al. Citation2011b) and other epigenetic changes (D’Addario et al. Citation2011; Figueiredo et al. Citation2009; Hicks, Middleton, and Miller Citation2010; Hu et al. Citation2009; Schernhammer et al. Citation2010). There is also evidence of increased expression of miRNAs in humans who consume alcohol (Avissar et al. Citation2009; Tang et al. Citation2008b). Alcohol intake was found to produce inflammation in some target tissues such as pancreas (Vonlaufen et al. Citation2007) and skin (Farkas and Kemeny Citation2013).
Chinese-style salted fish
‘Possible mechanisms for the association of consumption of Cantonese-style salted fish with an increased risk of nasopharyngeal carcinoma are the endogenous formation of N-nitroso compounds and/or their formation due to the processing of the fish – i.e. a reaction between secondary and tertiary amines in the fish and nitrate/nitrite in the crude salt used – and activation of the oncogenic Epstein-Barr virus. These two mechanisms are not mutually exclusive.’ (IARC Citation2012e, 510).
Chinese-style salted fish is popular in Chinese populations along the south China coast and South-eastern Asian countries. Various methods of preparation are used, including: salting, brining, dry-salting, and pickle curing. Weather conditions can lead to insect infestation and growth of bacteria such as Staphylococci. Consumption of Chinese-style salted fish produces nasopharyngeal carcinoma.
The carcinogenic mechanisms are not well established. One study found that DMSO-extracts of Chinese-style salted fish were mutagenic in S. typhimurium TA 100 and TA 98 in the presence of metabolic activation, but two studies that employed n-hexane and ethyl acetate extracts found no marked mutagenesis in these systems either in the absence or presence of a metabolic activation system from rat liver. Nevertheless, these samples contained high levels of precursors that upon nitrosation in vitro with sodium nitrite under acidic conditions would yield direct-acting genotoxic (probably N-nitroso) compounds. In one study, urine samples collected from rats fed Chinese-style salted fish showed mutagenic activity in TA 100 and TA 98.
There is also evidence that Chinese-style salted fish might re-activate EBV, which is known to induce nasopharyngeal carcinoma (see EBV above). Active EBV affects many different cellular processes that may lead to carcinogenesis, including induction of genomic instability, activation of NADPH oxidase, and increased expression of inducible nitric oxide synthase. These enzymes produce ROS and RNS-mediated DNA damage. These findings indicate that reactivation by Chinese-style salted fish of latent EBV in infected cells may play a part in nasopharyngeal cancer, by promoting genomic instability via induction of oxidative and nitrative DNA damage.
Indoor emissions from household combustion of coal
‘Chemical analyses and bioassay-directed fractionation of smoky coal emissions have identified polycyclic aromatic hydrocarbons (PAHs) as an important chemical class that accounts for much of their mutagenicity and carcinogenicity. The epidemiological link between exposure to smoky coal emissions and an increased risk for lung cancer is strengthened mechanistically by the fact that the mutation spectra of the TP53 tumour-suppressor gene and the KRAS oncogene in the lung tumours from non-smokers exposed to smoky coal emissions reflect an exposure to PAHs and differs from the mutation spectra found in these genes in lung tumours from cigarette smokers. Thus, the mutation spectra in lung tumours from non-smokers whose cancers are linked to smoky coal emissions reflect the primary DNA damage induced by the most prominent class of mutagens/carcinogens in these emissions’ (IARC Citation2012e, 532).
Coal is a major fuel used for cooking and heating in homes through much of Asia, particularly in China. Coal is a highly variable fuel depending on the source and method of processing. Combustion of coal indoors, in household appliances such as cooking and heating stoves, may lead to incomplete combustion that produces by-products including polycyclic aromatic hydrocarbons (PAHs) and aldehydes. In addition, combustion releases contaminants such as silicates, sulfur, and mercury. Emissions from household combustion of coal induce lung cancer.
Exposure to smoky coal emissions results in the excretion of several PAHs and their metabolites, e.g. methyl- and hydroxyl-PAHs, and exposed individuals display elevated levels of DNA adducts and accumulation of the p53 protein. It was also reported that exposure to coal emissions is associated with increased levels of DNA–protein crosslinks, unscheduled DNA synthesis, sister chromatid exchange, chromosomal aberrations, micronucleus formation, and hyper-methylation or deletion of the p16 gene in peripheral blood lymphocytes. In many studies, extracts or condensates of coal emissions were found to be mutagenic in S. typhimurium with or without metabolic activation.
Polycyclic aromatic hydrocarbons are the predominant carcinogenic agents originating from coal emissions. PAHs are metabolized rapidly to more soluble metabolites and reactive species, such as phenols, dihydrodiols, epoxides, quinones and tetrols, some of which interact with DNA to produce adducts. Metabolism to PAH-epoxides is a major component of the carcinogenic mechanism. Other potential mechanisms include production of ROS, interruption of gap-junctional intercellular communication, cell-cycle dysregulation, induction of apoptosis and immuno-suppression. Some of these effects may be mediated by activation of the aryl-hydrocarbon receptor.
Volume 100F: chemical agents and related occupations
Overview of carcinogenic mechanisms for aromatic amines
In IARC Monograph Volume 100F (IARC Citation2012f), a number of aromatic amines are reviewed. A general overview of carcinogenic mechanisms for aromatic amines is presented in Monograph Volume 99. Extracts from the overview are presented here.
Exposures to aromatic amines have long been known to produce cancer of the urinary bladder in humans. These substances also induce neoplasms at many different organ sites in lab animals. Multiple metabolic pathways are involved in the activation of aromatic amines to DNA-reactive intermediates.
Metabolism is initiated in the liver with either N-oxidation by cytochrome P450-associated enzymes, or N-acetylation by N-acetyl-transferase 2 (NAT2). The latter reaction provides a detoxification pathway for aromatic amines since it reduces the amount of parent compound that may undergo N-oxidation and N-hydroxylation. Oxidation to N-hydroxyarylamine is mainly mediated by the enzyme CYP1A2, but the iso-enzymes CYP1A1 and CYP4B1 may also be involved. The N-hydroxy metabolite is highly electrophilic and forms adducts with liver DNA at the C8 positions of deoxyguanosine and deoxyadenosine. N-hydroxyarylamines may be transported in free form to the blood or be conjugated with glucuronide. The acid-labile glucuronidated intermediate is excreted via the kidney and hydrolyzed in the bladder lumen where it eventually forms the N-hydroxy metabolite again. The acidic pH of urine enhances the hydrolysis reaction and thus represents an additional risk factor for aromatic amine-related bladder cancer. NAT1-mediated O-acetylation may represent the final activation step of N-hydroxyarylamines which occurs in the bladder epithelium and forms N-acetoxyarylamine. Breakdown of this unstable aromatic ester produces the highly reactive aryl-nitrenium ion that may serve as electrophilic intermediate leading to DNA adducts and tumor initiation.
Other activation pathways of aromatic amines to DNA-reactive intermediates include (1) sulfotransferase-mediated activation of N-hydroxyarylamine to an N-sulfate ester, (2) myeloperoxidase- and lactoperoxidase-mediated pathways that catalyze activation in the mammary gland, (3) peroxidative activation by prostaglandin H synthase, and (4) non-enzymatic protonation of the N-hydroxylamine nitrogen. Genotoxic aromatic amines may induce tumor formation at different sites depending upon substrate specificity and different bio-activation pathways.
The genotoxic effects of aromatic amines are well established on the basis of mutagenicity and clastogenicity observed in numerous in vitro and in vivo assays that demonstrated the capability of these compounds to form DNA adducts after metabolic activation to electrophilic intermediates. The predominant site for covalent binding of aromatic amines to DNA is the C8 position of guanine, but adducts at other sites, including C8 of adenine and N2 of guanine, were also identified. As DNA adducts may lead to somatic point mutations, it is reasonable to assume that activated aromatic amines may lead to bladder-tumor development by inducing mutations in key genes such as the TP53 tumor-suppressor gene and the HRAS oncogene, both involved in bladder carcinogenesis. Organ specificity and inter- and intra-species differences in cancer susceptibility to aromatic amines may be related to polymorphisms in genes that regulate DNA-repair, since deficient DNA-repair capacity is associated with enhanced bladder-cancer risk, and to polymorphisms in genes that encode enzymes involved in activation or detoxification pathways.
4-Aminobiphenyl
‘There is strong mechanistic evidence indicating that the carcinogenicity of 4-aminobiphenyl in humans operates by a genotoxic mechanism of action that involves metabolic activation, formation of DNA adducts, and induction of mutagenic and clastogenic effects. Metabolic activation to DNA-reactive intermediates occurs by multiple pathways including N-oxidation in the liver, O-acetylation in the bladder, and peroxidative activation in the mammary gland and other organs.’ (IARC Citation2012f, 50).
4-Aminobiphenyl was used mainly in industrial settings as an antioxidant in rubber, as a dye intermediate, and in the detection of sulfates. This compound use in industry was phased out in the mid-1950s. Non-industrial exposure occurs primarily through smoking. Exposure to 4-aminobiphenyl initiates bladder cancer.
4-Aminobiphenyl is a member of the aromatic amine family. that shares carcinogenic mechanisms with other members of this family (see overview, above). The genotoxic effects of aromatic amines (including 4-aminobiphenyl) are ‘well established on the basis of mutagenicity and clastogenicity observed in numerous in vitro and in vivo assays that show the capability of these compounds to form DNA adducts after metabolic activation to electrophilic intermediates’ (IARC Citation2012f, 48). Micronuclei were observed in several studies. 4-Aminobiphenyl is activated through N-hydroxylation in the liver. The resulting metabolite is highly electrophilic and forms DNA adducts. The N-hydroxy metabolite is glucuronidated and excreted through the kidney. However, in the acidic environment in the bladder lumen the N-hydroxy metabolite is formed again. NAT1-mediated O-acetylation may then occur, which leads to the production of the highly reactive aryl nitrenium ion, and formation of DNA adducts. Mutations were induced when 4-aminobiphenyl was tested on S. typhimurium in the presence of S9-mediated metabolic activation. 4-Aminobiphenyl induced mutations at the HPRT locus and chromosomal instability in human bladder epithelial cells. 4-Aminobiphenyl-mediated mutations were found in h-Ras (mouse) and TP53 (human bladder cells).
Additional information
In human mammary epithelial cells exposed to 4-aminobiphenyl, a concentration-dependent reduction was observed in methylation of lysine 4 of histone H3 (Bradley et al. Citation2007). In 4-ABP-treated HepG2 cells, the expression of 27 miRNAs was at least 3-fold higher than controls. In addition, the expression of 6 genes involved in different DNA-repair pathways was reduced (Huan et al. Citation2014).
Benzidine
‘There is strong mechanistic evidence indicating that the carcinogenicity of benzidine in humans operates by a genotoxic mechanism of action that involves metabolic activation, formation of DNA adducts, and induction of mutagenic and clastogenic effects. Metabolic activation to DNA-reactive intermediates occurs by multiple pathways including N-oxidation in the liver, O-acetylation in the bladder, and peroxidative activation in the mammary gland and in other organs.’ (IARC Citation2012f, 61).
Benzidine is an aromatic amine with a long history of use in the production of dyes, particularly azo dyes used for wool, cotton and leather. Industrial production was phased out in the 1960s, with bans on its use in many countries. Most exposure is from occupational contact. The general public may have low exposure through contact with consumer goods that contain azo-based dyes. Benzidine produces bladder cancer.
General metabolism and carcinogenic mechanisms are similar to those discussed in the section on 4-aminobiphenyl (see also the overview, above). Benzidine and its two active metabolites, N-acetylbenzidine and N,N ‘-diacetylbenzidine, were detected in the urine of workers exposed to benzidine. The predominant DNA adduct identified in exfoliated bladder cells is N ‘-(deoxyguanosin-8-yl)-N-acetyl-benzidine.
Significant increases in the frequency of chromosomal aberrations were noted in peripheral blood lymphocytes of workers exposed to benzidine or benzidine-based dyes. In workers exposed to benzidine, the amount of mutant p53 protein rose with higher exposure. Similarly, benzidine induced DNA lesions in p53 in the bladder, liver, and lung of exposed rats, increased the frequency of micronucleated mouse bone-marrow cells, induced unscheduled DNA synthesis in mice, and elevated number of DNA strand-breaks in liver of exposed rats.
Additional information
In early studies, some epigenetic effects of benzidine were described. In benzidine-induced hepatomas from B6C3F1 mice, the Ha-Ras and Ki-Ras oncogenes were hypo-methylated in tumor as compared to adjacent non-tumor tissue, and elevated oncogene expression was detected (Vorce and Goodman Citation1989). In vivo studies showed that benzidine induces apoptosis, cell proliferation and differentiation (Chen et al. Citation2014a; Lentz et al. Citation2010).
Dyes metabolized to benzidine
‘There is strong mechanistic evidence indicating that benzidine-based dyes are converted by azoreduction to benzidine in humans and in experimental animals and, consequently, produce DNA adducts and genotoxic effects similar to those of benzidine.’ (IARC Citation2012f, 71).
Three benzidine-based dyes were reviewed in Monograph Volume 100F: Direct Black 38, Direct Blue 6 and Direct Brown 95. Clear and consistent epidemiological evidence to establish that these dyes induce cancer in humans is lacking. However, animal studies demonstrated that these agents are carcinogenic and there is strong evidence that these dyes are metabolized to benzidine, a known carcinogen in humans. The carcinogenic mechanisms are similar to the active metabolite benzidine.
4,4 ‘ -Methylenebis(2-chlorobenzenamine)
‘There is strong mechanistic evidence indicating that the carcinogenicity of 4,4 ‘ -methylene-bis(2-chlorobenzenamine) involves a genotoxic mechanism of action that includes metabolic activation, formation of DNA adducts, and induction of mutagenic and clastogenic effects in humans. Metabolic activation to DNA-reactive intermediates occurs by multiple pathways including N-oxidation in the liver, O-acetylation in the bladder, and peroxidative activation in the mammary gland and other organs. The genotoxicity of 4,4 ‘ -methylene-bis(2-chlorobenzenamine) is well documented and its toxicological profile is similar to that of o-toluidine (see below), thus indicating a common mode of action. It has also been shown to cause the formation of sister chromatid exchange and micronuclei in urothelial cells and lymphocytes of exposed workers.’ (IARC Citation2012f, 80).
4,4ʹ-Methylenebis(2-chlorobenzenamine), commonly known as methylenebis(ortho-chloroaniline) (MOCA or MBOCA) is employed as a curing agent in production of some types of polyurethane. Industrial exposure is the most common route of contact with MOCA, although environmental contamination has created some exposure risk for the general public. There is a lack of direct epidemiological evidence that MOCA induces cancer in humans. However, it is a multi-organ carcinogen in experimental animals. Overall, studies on the metabolism, genotoxicity, and animal carcinogenicity of MOCA indicate that this substance acts similarly to other aromatic amines that are known to initiate cancer of the urinary bladder in humans.
MOCA is a member of the aromatic amine family and shares carcinogenic mechanisms with other such amines (see overview, above). N-oxidation of MOCA leads to production of DNA adducts and mutagenesis. Elevated levels of micronuclei were noted in exfoliated bladder epithelial cells and peripheral lymphocytes of exposed workers. MOCA (1) induced mutations at the HPRT locus in human lymphoblastoid cells, (2) stimulated prophage induction in E. coli, (3) produced aneuploidy in S. cerevisiae, (4) unscheduled DNA synthesis in cultured mouse hepatocytes, (5) transformation in several mammalian cell cultures, and (6) sister chromatid exchange in lymphocytes of rats treated in vivo and in Chinese hamster ovary cells in vitro. MOCA was shown to induce immortalization of cells in culture.
2-Naphthylamine
‘There is strong mechanistic evidence indicating that the carcinogenicity of 2-naphthylamine operates by a genotoxic mechanism of action that involves metabolic activation, formation of DNA adducts, and induction of mutagenic and clastogenic effects. Metabolic activation to DNA-reactive intermediates occurs by multiple pathways including N-oxidation in the liver, O-acetylation in the bladder, and peroxidative activation in the mammary gland and other organs.’ (IARC Citation2012f, 90).
2-Naphthylamine was utilized commercially as an intermediate in the manufacture of dyes, as an antioxidant in the rubber industry, and in the production of 2-chloro-naphthalene. Because of carcinogenicity occurrence, the manufacture and use of 2-naphthylamine was prohibited in the European Union since 1998. In the USA, it was a strictly regulated substance. 2-Naphthylamine is also produced during burning of tobacco (smoking), cooking oils and similar substances. This compound induces cancer of the urinary bladder.
2-Naphthylamine is a member of aromatic amine family and shares carcinogenic mechanisms with other members (see overview, above). 2-Naphthylamine was mutagenic in S. typhimurium strains TA98 and TA100, and in Chinese hamster ovary cells in the presence or absence of an exogenous activating system. 2-Naphthylamine produced DNA strand breaks and induced sister chromatid exchange.
Ortho-Toluidine
‘The N-oxidized metabolite of o-toluidine, N-hydroxy-o-toluidine, was mutagenic in S. typhimurium strain TA100. Other reported effects of o-toluidine include the induction of sister chromatid exchange, aneuploidy, unscheduled DNA synthesis, DNA strand-breaks, and cell transformation in vitro, and the induction of micronuclei in peripheral blood of rats treated in vivo … … … There is moderate mechanistic evidence indicating that the carcinogenicity of ortho-toluidine involves metabolic activation, formation of DNA adducts, and induction of DNA-damaging effects.’ (IARC Citation2012f, 98).
ortho-Toluidine is used in a variety of industrial processes including the production of herbicides and dyes. Exposure is highest in occupational settings, with non-occupational exposure predominantly through tobacco smoking and use of hair dyes. ortho-Toluidine has consistently been associated with an elevated risk of bladder cancer in humans.
ortho-Toluidine is a member of aromatic amine family and shares general carcinogenic mechanisms with other members (see the overview, above). However, the metabolism of o-toluidine is still under investigation, and available data indicate a preferential ring-oxidation or N-acetylation rather than N-oxidation. It was suggested that carcinogenesis may involve peroxidative activation of the chemical, catalyzed by prostaglandin H synthase in the epithelium of the urinary bladder.
Auramine and auramine production
‘There are insufficient mechanistic data relevant to the carcinogenicity of auramine in humans. Auramine induces DNA strand-breaks in experimental animals.’ (IARC Citation2012f, 104).
Auramine and its salts were employed mainly in production of dyes, in particular those used for dyeing paper. Workers involved in manufacturing this dye are the major group exposed, but utilization of this substance was banned in many countries. Auramine induces hepatoma and lymphoma in experimental animals. Auramine production produces bladder cancer in humans.
Auramine is a member of aromatic amine family and shares general carcinogenic mechanisms with other members (see the overview, above). However, the metabolism of auramine has not been studied in detail. The finding that auramine-induced intra-chromosomal recombination in S. cerevisiae was less in the presence of a free-radical scavenger (N-acetylcysteine) suggests that auramine may induce genotoxic effects in yeast by generating free radicals. Commercial preparations of auramine were mutagenic in several strains of S. typhimurium, when tested with metabolic activation systems. In vitro investigations demonstrated DNA strand-breaks, unscheduled DNA synthesis, micronucleus formation and induction of deletions and aneuploidy in S. cerevisiae. Formation of DNA strand-breaks was confirmed in in vivo rat studies.
Magenta and magenta production
‘There are insufficient mechanistic data relevant to the carcinogenicity of magenta in humans or experimental animals.’ (IARC Citation2012f, 109) .
Magenta refers to a group of 4 dyes: Magenta I, II III and 0 (Basic Red 09). Their chemical structures are similar. Workers involved in manufacturing these dyes are the major group exposed. Magenta production produces bladder cancer in humans. A component of commercial magenta, CI Basic Red 9, induced liver tumors in rats and mice.
Magenta dyes are members of the aromatic amine family and share general carcinogenic mechanisms with other members (see overview, above). However, the metabolism of magenta dyes has not been directly studied and no apparent information on specific carcinogenic mechanisms of magenta was published. Magenta was mutagenic in S. typhimurium strains TA98, TA100, and TA1535 when tested in the presence of metabolic activation.
Benzo[a]pyrene
‘Benzo[a]pyrene is metabolically activated to a series of reactive intermediates by CYP450 and related enzymes under control of the aryl-hydrocarbon receptor … … All … … pathways reflect genotoxic mechanisms, as they involve alterations to DNA. Benzo[a]pyrene is pleotropic and has the ability to affect many cell- and organ-based systems. … … (T)here are probably many modes of carcinogenic action operating to different extents in vivo. These include mechanisms that involve AhR, oxidative stress, immunotoxicity and epigenetic events … … The most commonly encountered … … mechanistically relevant DNA lesion is the anti-benzo[a]pyrene-7,8-diol-9,10-oxide-DNA adduct. The formation of this adduct is consistent with anti-benzo[a]pyrene-7,8-diol-9,10-epoxide-associated genotoxic effects in surrogate tissues and with the mutation pattern in the TP53 gene in lung tumours from humans exposed to PAH mixtures that contain benzo[a]pyrene. The fact that … … benzo[a]pyrene itself induce genotoxic effects like sister chromatid exchange, chromosomal aberrations, micronuclei, DNA damage (detected by use of the comet assay) and 8-deoxyguanosine, supports the notion that benzo[a]pyrene contributes to human cancer’ (IARC Citation2012f, 137).
Benzo[a]pyrene is a member of the polycyclic aromatic hydrocarbon (PAH) family which is a widespread environmental contaminant produced through incomplete combustion of organic material. High levels are found in tobacco smoke. The IARC review of benzo[a]pyrene does not report epidemiological data on cancer risk in humans specifically, because human exposure to benzo[a]pyrene normally involve exposure to complex agents (IARC Citation2012f). However, exposure to benzo[a]pyrene-containing mixtures was linked to human tumors of the lung, bladder, esophagus and lymphatic system. There is strong evidence of multi-site carcinogenesis in animals exposed to benzo[a]pyrene.
A number of potential carcinogenic mechanisms were identified for benzo[a]pyrene (listed above). Most involve the initial metabolic transformation of benzo[a]pyrene into reactive intermediates and ultimate carcinogens such as benzo[a]pyrene-7,8-diol-9,10-epoxides. Key mechanisms include (1) creation of stable and depurinating DNA adducts, (2) repetitive redox cycling which generates ROS, (3) interaction with the Aryl hydrocarbon receptor (AhR), (4) immunosuppression and (5) epigenetic changes. Binding to the AhR initiates perturbations in cell-signaling. Benzo[a]pyrene produces single-strand DNA breaks and mutations in p53 in vitro. Benzo[a]pyrene and/or its metabolites were shown to enhance cell proliferation in several human cell lines.
‘The current understanding of mechanisms underlying benzo[a]pyrene-induced carcinogenesis in experimental animals is almost solely based on two complementary pathways: those of the diol-epoxides and the radical cations. Each provides a different explanation for the effects observed in experimental animals in specific tissues.’ (IARC Citation2012f, 132). A short description of these two major pathways and of several other mechanisms proposed for benzo[a]pyrene carcinogenesis is presented here.
The diolepoxide mechanism
The diolepoxide mechanism features a sequence of metabolic transformations: oxidation of benzo[a]pyrene to yield benzo[a]pyrene-7,8-oxide (by CYP1A1 and CYP1B1), reduction to benzo[a]pyrene-7,8-diol (by epoxide hydrolase), and further oxidation to benzo[a]pyrene-7,8-diol-9,10-epoxide (by CYP1A1 and CYP1B1). Each class of metabolic intermediate was shown to be genotoxic and carcinogenic.
The radical-cation mechanism
The radical-cation mechanism has been studied exclusively in connection with mouse-skin tumorigenesis. One-electron oxidation of benzo[a]pyrene by CYPs or peroxidases creates a radical cation with a free electron localized on carbon 6, as a consequence of ionization potential and geometric configuration. In mouse skin, this radical cation gives rise to the formation of covalent adducts with guanine (at the C8 carbon and the N7 nitrogen) and adenine (at the N7 nitrogen). These adducts are unstable and are thought to generate apurinic sites in mouse skin. However, only low levels of apurinic sites were measured in the epidermis of mice treated with benzo[a]pyrene, and no apparent studies to date reported an increase in the number of apurinic sites in lung tissues treated with benzo[a]pyrene.
Meso-region methylation and benzylic oxidation
The mechanism of meso-region methylation and benzylic oxidation features methylation of benzo[a]pyrene to 6-methylbenzo[a]pyrene, with S-adenosylmethione as the carbon donor. This process was noted to occur in vitro, and in vivo in rat liver. 6-Methylbenzo[a]pyrene is further metabolized by CYPs to 6-hydroxymethylbenzo[a]pyrene and then conjugated to sulfate by 3ʹ-phosphoadenosine-5ʹ-phosphosulfate sulfotransferase to 6-[(sulfooxy)methyl]-benzo[a]pyrene. This reactive sulfate ester forms DNA adducts in vivo. These benzo[a]pyrene-DNA adducts have only been measured in rat liver, which is not a target for benzo[a]pyrene-induced carcinogenesis. There is no apparent evidence to date that this mechanism operates in lung.
Formation of orthoquinone/reactive oxygen species
This mechanism features enzymatic oxidation of benzo[a]pyrene-7, 8-diol to the ortho-quinone, benzo[a]pyrene-7, 8-quinone, by aldo-keto reductases. Benzo[a]pyrene-7, 8-quinone reacts with DNA to yield both stable and depurinating DNA adducts in vitro and also undergoes repetitive redox cycling which generates ROS that damage DNA.
Aryl hydrocarbon-receptor (AhR) mechanism
The AhR regulates the transcription of a series of genes including Cyp1A1, Cyp1A2, Nqo1, Aldh3a1 (encoding aldehyde dehydrogenase 3A1), UGT1a6 (uridine 5ʹ-diphosphateglucuronosyl transferase), and Gsta1 (glutathione S-transferase A1). All these genes are activated by AhR-ligands, including benzo[a]pyrene, via the AhR-mediated aromatic hydrocarbon response element. The AhR plays a part in the response to oxidative stress in cell-cycle regulation and apoptosis.
Immunosuppression mechanism
Benzo[a]pyrene induces immunosuppression in adult mice by altering the cell-mediated responses. Immune development in offspring is also altered following in utero exposure to benzo[a]pyrene. It is postulated that PAHs, including benzo[a]pyrene, act predominantly through their AhR-mediated CYP-derived metabolites (diolepoxides, quinones) to activate oxidative and electrophilic signaling pathways in lymphoid and non-lymphoid cells, including myeloid cells, epithelial cells, and other cell types. Further, there is evidence that PAHs suppress immunity by p53-dependent pathways, by modulating signaling pathways in lymphocytes via non-genotoxic mechanisms, and oxidative stress.
Epigenetic mechanisms
Benzo[a]pyrene and/or its metabolites were found to increase cell proliferation in several human cell lines, affect apoptosis, and alter DNA methylation.
Additional information
Genome-wide H3K9 histone acetylation profiles were altered in benzo[a]pyrene-treated MCF7 breast cancer cells (Sadikovic et al. Citation2008). Benzo[a]pyrene induced hyper-methylation in humans and animals (Damiani et al. Citation2008; Fang et al. Citation2013; Subach et al. Citation2006; Tang et al. Citation2012; Yauk et al. Citation2008). Benzo[a]pyrene increased the expression of miRNAs in human cells in vitro (Li et al. Citation2012; Zhao et al. Citation2011a) and decreased global and gene-specific DNA methylation during zebrafish development (Fang et al. Citation2013).
Coal gasification
‘There is strong evidence from experimental studies for a genotoxic mode of action for coal gasification samples. Although there are no human studies, it is highly likely that genotoxicity is the mechanism relevant to the carcinogenic effects of coal-gasification emissions, predominantly due to the presence of mutagenic PAHs.’ (IARC Citation2012f, 150).
Coal gasification is the process during which coal is converted into a gaseous mixture that might be burned with lower environmental impact. Coal is reacted with oxygen, steam and carbon dioxide to form a gas containing hydrogen and carbon monoxide. During this process, which is essentially incomplete combustion, the heat evolved is consumed and sulfur and nitrogen in the coal are converted to hydrogen sulfide (rather than sulfur dioxide) and ammonia (rather than nitrogen oxides), respectively. These reduced forms of sulfur and nitrogen are easily isolated, captured and used, making gasification a clean-coal technology with a better environmental performance than coal combustion. Workers in coal gasification may be exposed to many compounds, including asbestos, silica, amines, arsenic, cadmium, lead, nickel, vanadium, PAH and other hydrocarbons, sulfur dioxide, sulfuric acid, and aldehydes. Occupational exposure during coal gasification produces lung cancer.
Carcinogenic mechanisms reflect those of the substances released. The PAH and alkylated by-product fractions were mutagenic in S. typhimurium in the presence of an exogenous metabolic activation system. Mice fed a diet containing coal tar from a gas-plant residue showed a complex pattern of aromatic adducts in multiple tissues. Topical application of coal tar also produced DNA adducts. Several of the PAHs found in ambient air in gas works are mutagenic (i.e. benz[a]anthracene, benzo[a]pyrene, benzo[ghi]perylene) and carcinogenic (i.e. benz[a]anthracene, benzo[a]pyrene). Despite the absence of human studies, a genotoxic mechanism of carcinogenesis was established.
Occupational exposures during coal-tar distillation
‘Studies in experimental systems and in tissues of humans provide strong evidence for a genotoxic mechanism underlying the effects of occupational exposures during coal-tar distillation in humans. The detection of anti-benzo[a]pyrene-7,8-diol-9,10-epoxide-DNA adducts in the peripheral blood lymphocytes of exposed workers suggests the participation of benzo[a]pyrene in the genotoxic mechanism of this exposure in humans.’ (IARC Citation2012f, 158).
Coal tar is obtained by cooling the gas that is formed during the destructive distillation of coal to approximately ambient temperature. Workers involved in coal-tar distillation are exposed to a wide range of PAHs. Exposures from the coal-tar distillation process have been causally linked to skin cancer with a particular relationship to scrotal cancer in men.
Emissions from coal-tar pitch and roofing-tar were mutagenic in S. typhimurium in the presence of an exogenous metabolic activation system, and in two mammalian cell systems in the presence and absence of an exogenous metabolic activation system. These emissions induced sister chromatid exchange in Chinese hamster ovary cells and enhanced viral transformation in Syrian hamster embryo cells both in absence and presence of an exogenous metabolic activation system. Coal tar applied topically to the skin of male mice produced a complex pattern of DNA adducts and markedly elevated the mutation frequency in lambda-lacZ transgenic mice (MutaMouse). DNA strand-breaks were detected in exposed mice. The urine from psoriasis patients undergoing coal-tar treatments was mutagenic in bacteria. Peripheral blood lymphocytes of workers occupationally exposed to coal tars exhibited increased chromosomal damage.
Coal-tar pitch
‘There is strong evidence from experimental data that coal-tar pitch has a genotoxic mechanism of action. There is moderate evidence in humans for a genotoxic mechanism underlying the effects of exposures during roofing and paving with coal-tar pitch, based on one study.’ (IARC Citation2012f, 165).
Coal-tar pitch is the residue from the distillation of coal-tar. Coal-tar pitch is utilized in the roofing and paving industries. Exposure during roofing occurs both when the old roofing material is removed and the new is applied. Coal-tar application for paving was phased out in most of Europe between 1963 and 1995. Coal-tar is occasionally employed for treatment of psoriasis. Emissions from coal-tar pitch induce lung cancer.
Carcinogenic mechanisms reflect those of the constituent agents. Coal-tar pitch and roofing-tar emissions were mutagenic in bacteria in presence of an exogenous metabolic activation system, and in mammalian cells with and without metabolic activation. These agents also induced sister chromatid exchange in Chinese hamster ovary cells. Coal-tar pitches contain several PAHs that are genotoxic and carcinogenic in experimental studies. In human studies, increasing exposure was associated with higher levels of DNA strand-breaks.
Additional information
A coal tar mixture was found to activate phospholipase A2 in human coronary artery endothelial cells producing histone fragmentation and poly(ADP)ribose polymerase cleavage and eventually leading to elevated rates of apoptosis (Tithof et al. Citation2011). The application of coal tar and UV light to treat psoriasis produced increased apoptosis and genotoxicity (Borska et al. Citation2010). Occupational exposure to coal-tar pitch induced telomere shortening in DNA of leukocytes in peripheral blood of exposed workers (Wang et al. Citation2015).
Coke production
‘Overall, these data strongly indicate a mutagenic/genotoxic mode of action for occupational exposures during coke production, based on experimental and human studies. […] There is ample mechanistic support for the respiratory carcinogenic effects of occupational exposures during coke production in humans, in part through analysis of exposure to benzo[a]pyrene. This is based on direct measurement of anti-benzo[a]pyrene-7,8-diol-9,10-oxide-DNA adducts in peripheral blood lymphocytes (surrogate tissue) and on the identification of genotoxic effects consistent with those induced by anti-benzo[a]pyrene-7,8-diol- 9,10-oxide or benzo[a]pyrene. It is also consistent with the known carcinogenic activity of this epoxide in lung tissues in experimental animals. Moreover, the influence of GST polymorphisms on levels of anti-benzo[a]pyrene-7,8-diol-9,10-oxide-DNA adducts is suggestive of the presence of reactive electrophilic intermediates, such as anti-benzo[a]pyrene-7,8-diol-9,10-oxide. Since coke-oven emissions are complex mixtures, these exposures could have more than one underlying mechanism of action. The fact that chronic exposure to PAH in non-smoking coke-oven workers induced both gene-specific (e.g. in the TP53 gene) and global methylation changes in peripheral blood lymphocytes, suggests an epigenetic mechanism.’ (IARC Citation2012f, 175).
Coke is produced from coal and used as a fuel and reducing agent in industrial processes used in iron foundries and similar sites. Production creates exposures to coal dust, various gaseous by-products such as coke-oven gas and various minerals and other contaminants. Emissions from coke production induce lung cancer.
Coke-oven emissions are complex mixtures and there may be more than one underlying carcinogenic mechanism. These emissions are mutagenic in bacteria and mammalian cells inducing DNA damage, sister chromatid exchange and morphological cell transformation. Exposure to coke-oven emissions produced benzo[a]pyrene-diolepoxide-DNA adducts in mice. In humans, peripheral blood lymphocytes of coke-oven workers displayed increased frequencies of sister chromatid exchange. The urine of coke-oven workers was mutagenic in S. typhimurium in the presence of an exogenous metabolic system. Both gene-specific (e.g. in the TP53 gene) and global methylation changes were detected in peripheral blood lymphocytes of non-smoking coke-oven workers, which suggests that an epigenetic mechanism may be involved in carcinogenicity development.
Additional information
Aberrant gene promoter methylation was detected in sputum from individuals exposed to smoky coal (Liu et al. Citation2008). Comparative micro-RNA analyses of non-small cell lung cancers (NSCLCs) from areas in China where smoky coal was used/not used showed strong down-regulation of miR-144 associated with smoky-coal burning (Pan et al. Citation2015). Methylation of CpG islands was elevated in the genes p14 (ARK), p15 (INK4b) and p16 (INK4a) in coke-oven workers (Zhang et al. Citation2015).
Untreated or mildly treated mineral oils
‘There is weak evidence on the mechanism underlying the effects in humans of exposures to mineral oils. This evidence is based on genotoxic (mutagenic) activity of mineral oils in bacteria and a single cytogenetic study of glassworkers exposed to aerosols of mineral oils.’ (IARC Citation2012f, 193).
Mineral oils are produced from crude petroleum oils by use of a wide range of complex chemical processes. The final products are a heterogeneous group of complex chemical mixtures. PAHs are common components, unless the mineral oil is highly refined. Untreated mineral oils (i.e. those that are not highly refined) induce cancer of the skin, specifically of the scrotum. Highly refined mineral oils have not been linked to human cancer.
The carcinogenic mechanism for mineral oils has not been extensively examined. The presence of PAHs in untreated mineral oil may provide the basis for a genotoxic carcinogenic mechanism. In a cytogenetic study, chromosomal damage – chromatid breaks, chromosome breaks, and chromosome exchanges – was increased in glassmakers (smokers and non-smokers) compared with controls. In lab studies, mutagenic activities of mineral-oil samples were significantly correlated with the amount of 3–7-ring polycyclic aromatic compounds for a subgroup of oil samples.
Shale oils
‘Shale oils are genotoxic in experimental systems. There are only few data to determine an underlying mechanism for the carcinogenicity of shale oils.’ (IARC Citation2012f, 204).
Oil shale is sedimentary rock that contains mainly mineral components and organic matter (kerogen). To recover the oil from the shale, the kerogen is decomposed thermally, and the resulting liquid fraction is recovered as shale oil, a complex mixture of hydrocarbons and polar components. Shale oils have been causally linked to skin cancer, particularly in human scrotum . Epidemiological data pertaining to other cancer sites, including lung – a clear target organ in rats and mice – remain inadequate to draw conclusions.
Human studies showed conflicting results on the genotoxicity of shale oil. A small group of workers exhibited increased levels of chromosomal breakage, but the workers were also involved with coke production. Elevated levels of DNA adducts were not observed, but the study groups were small. Shale oils are genotoxic in experimental systems. Crude shale oil and oil-shale retort waters were highly mutagenic in S. typhimurium in absence of an exogenous metabolic activation system and demonstrated mutagenic activity in bacteria, fungi, and mammalian cells in culture. Unfractionated shale oil, and the basic and PAH-containing fractions derived from it, produced a positive response in the morphological cell-transformation assay with Syrian hamster embryo cells. Shale oil induced a significant rise in sister-chromatid exchange and chromosomal abnormalities in human lymphocytes in vitro.
Soot, as found in occupational exposure of chimney sweeps
‘Extracts of soots contain carcinogenic polycyclic aromatic hydrocarbons and are genotoxic. Based on a small number of genotoxicity studies in exposed humans, there is moderate evidence of a genotoxic mode of action underlying the carcinogenic hazards associated with occupational exposures as a chimney sweep. The detection of anti-benzo[a]pyrene-7,8-diol-9,10-epoxide-DNA adducts in the peripheral blood lymphocytes of chimney sweeps suggests involvement of benzo[a]pyrene in the genotoxic effect of this exposure in humans.’ (IARC Citation2012f, 213).
Soot is the by-product of combustion of various organic (carbon-containing) materials. The chemical composition is highly variable, depending upon the material being burned and the conditions of combustion. Most soots contain a substantial quantity of PAHs. The carcinogenicity of soots was first noted among chimney sweeps by Percival Pott in 1775. Soot produces skin cancer (primarily of the scrotum) and lung cancer.
The PAH components of soot are believed to contribute toward carcinogenicity through genotoxic mechanisms that have been discussed elsewhere. Experimental studies on soots showed mutagenicity. Extracts of soot samples from domestic sources were mutagenic in S. typhimurium, both in presence and absence of an exogenous metabolic system. Further, extracts of particulate emissions from wood-combustion induced sister chromatid exchange in Chinese hamster ovary cells, transformation of Syrian hamster embryo cells, and mutation in S. typhimurium. In several human investigations, occupational exposure of chimney sweeps was linked to increased levels of DNA adducts and higher frequencies of micronuclei in stimulated lymphocytes.
Occupational exposure during aluminum production
‘Air-emission samples from aluminum smelters were mutagenic in bacteria. There were mixed reports on the mutagenicity of urine from exposed workers. DNA-adduct studies of blood samples from aluminum-smelter workers also gave mixed results. Based on both experimental and human studies, there is weak-to-moderate evidence for a genotoxic mechanism underlying the effects of occupational exposures during aluminium production.’ (IARC Citation2012f, 221).
Aluminum production as defined in IARC Monograph Volume 100F (IARC Citation2012f, 215) encompasses the electrolytic reduction of alumina (Al2O3) to aluminum, and the casting of aluminum into ingots. Bauxite mining, alumina production from bauxite, alloying and fabrication of aluminum products (sheets, wires, foil, etc.) were not considered.
Workers in aluminum production are primarily exposed to PAHs: in the electrolytic reduction process anodes are formed continuously from a paste of petroleum coke and coal-tar pitch, which bakes to form carbon, replacing the anode that is being consumed. Occupational exposures in this industry and the related carbon electrode-manufacturing industry were monitored most intensively with respect to PAHs. Exposures have decreased steadily since the 1950s but PAHs are still the main carcinogens to which workers are exposed. Aluminum production induces cancers of the bladder and lung.
Carcinogenic mechanisms related to coke and coal-tar pitch would apply to aluminum production according to the process where anode consumption occurs.
Air-emission samples from aluminum smelters and samples from air-filters in anode-processing rooms were mutagenic in S. typhimurium strains TA98 and TA100, in the presence of an exogenous metabolic activation system. Mixed results were reported for the mutagenicity of urine from exposed workers and for the presence of DNA-adducts in blood lymphocytes of workers in aluminum production.
Aflatoxins
‘There is strong evidence that the carcinogenicity of aflatoxins operates by a genotoxic mechanism of action that involves metabolic activation to a genotoxic epoxide metabolite, formation of DNA adducts, and modification of the TP53 gene. In human hepatocellular carcinoma from areas where exposure to aflatoxins is high, up to 50% of tumours have been shown to harbour a specific point mutation in the TP53 tumour-suppressor gene.’ (IARC Citation2012f, 244).
The largest proportion of aflatoxins found in foodstuffs globally is produced by the common fungus Aspergillus flavus which produces only B aflatoxins and the closely related species A. parasiticus which generates both B and G aflatoxins. Aflatoxin M1 is a metabolite of aflatoxin B1 that occurs in milk from animals consuming feed contaminated with B aflatoxins. The fungi grow as a contaminant on foodstuffs, primarily maize, peanuts and cottonseed. Aflatoxins produce hepatocellular carcinoma. Concurrent infection with HBV increases the cancer risk.
Aflatoxin B1 (AFB1) – the most common and most potent of the aflatoxins – induced mutations in S. typhimurium strains TA98 and TA100, and initiated unscheduled DNA synthesis, chromosomal aberrations, sister chromatid exchange, micronucleus formation and cell transformation in various in vivo and in vitro mammalian systems. Aflatoxin B1 also produced immunosuppression. In the case of mutagenicity, this is strongly dependent on metabolic activation. Aflatoxin B1 ‘is metabolized predominantly in the liver to an AFB1–8,9-exoepoxide, which forms a pro-mutagenic AFB1-N7-guanine adduct in DNA, giving rise to G→T transversion mutations. In human hepatocellular cancers in areas where aflatoxin exposure is high, up to 50% of tumors have been shown to harbor a specific AGG→AGT point mutation in codon 249 of the TP53 tumor-suppressor gene (codon 249Ser mutation)’ (IARC Citation2012f, 244).
Exposure to aflatoxin induced adducts to DNA and albumin, gene mutations and chromosomal alterations, including micronuclei and sister chromatid exchange, and mitotic recombination. DNA and protein adducts of aflatoxin were detected in many studies of human liver and body fluids. Aflatoxin induced mutations in S. typhimurium strains TA98 and TA100 in the presence of an exogenous bio-activation system (rat-liver S9).
Additional information
A reverse association was found in cancer-free subjects between AFB1 exposure (measured in urine) and global DNA methylation (measured in leukocytes). These alterations in DNA methylation may represent an epigenetic biomarker of dietary AFB1 exposure (Wu et al. Citation2013). Expression levels of miRNA-429, miRNA-24, and miRNA-1268a were tested in aflatoxin B1-related hepatocellular carcinoma samples and in adjacent tissue. Overexpression of these miRNAs in the tumor samples modulated prognosis, recurrence-free survival, and overall survival (Huang et al. Citation2013; Liu et al. Citation2014; Long et al. Citation2016). The epigenetic footprint associated with early-onset aflatoxin-induced HCC was studied in primary human hepatocytes exposed to AFB1. A range of persistent hyper/hypo-methylated genes was identified. A number of the hypo-methylated and up-regulated genes played a clear role in carcinogenesis (Rieswijk et al. Citation2016).
Benzene
‘Benzene may act by causing chromosomal damage (aneuploidy, deletions and translocations) through inhibition of topoisomerase II, disruption of microtubules and other mechanisms; by generating oxygen radicals that lead to point mutations, strand breaks and oxidative stress; by causing immune system dysfunction that leads to decreased immune-surveillance; by altering stem-cell pool sizes through haematotoxic effects; by inhibiting gap-junction intercellular communication; and by altering DNA methylation and perhaps specific microRNAs … … There is strong evidence that benzene metabolites, acting alone or in concert, produce multiple genotoxic effects at the level of the pluripotent haematopoietic stem cell resulting in chromosomal changes in humans consistent with those seen in haematopoietic cancer. A variety of genotoxic changes, including chromosomal abnormalities, have been found in the lymphocytes of workers exposed to benzene.’ (IARC Citation2012f, p.283 & p.285).
Benzene is an intermediate in the manufacture of a wide range of organic chemicals. This chemical occurs naturally in petroleum products including gasoline and was used as an additive in non-leaded gasoline to raise the octane rating. Most exposure to benzene occurs through industrial contact. Occupational exposure occurs via inhalation or dermal absorption of solvents in the rubber, paint and parts-manufacturing industries. This compound also occurs during crude-oil refining and chemical manufacturing, a large component of which entails exposure to gasoline. Workers involved in the transport of crude oil and gasoline and in the dispensing of gasoline at service stations, as well as street workers, taxi drivers and others employed at workplaces with exposure to exhaust gases from motor vehicles also experience exposure to benzene. The primary sources of exposure to benzene for the general population are ambient air that contains tobacco smoke, air contaminated with benzene (for example, in areas with heavy traffic, around gasoline filling-stations), consumption of contaminated water, or eating contaminated food. Benzene induces acute myeloid leukemia (AML) and acute non-lymphocytic leukemia (ANLL).
Benzene requires metabolism to active metabolites – in particular, benzoquinones – to display its carcinogenic potential. The initial metabolic step involves cytochrome P450 (CYP)-dependent oxidation to benzene oxide, most of which spontaneously rearranges to phenol, which is either excreted or further metabolized to hydroquinone and 1,4-benzo-quinone. Benzoquinone formation from hydroquinone via myeloperoxidase in the bone marrow was suggested as a key step in the carcinogenicity attributed to benzene. There is considerable evidence for an important role of this metabolic pathway to benzoquinone.
In multiple studies in different occupational populations in many countries, a variety of genotoxic changes, including chromosomal abnormalities were detected in lymphocytes of workers exposed to benzene. Exposure to benzene is associated with typical chromosomal changes seen in AML (e.g. 5q-/-5 and t(8,21)) in peripheral blood cells of heavily exposed workers. Benzene induced chromosomal aberrations, micronuclei and sister chromatid exchange in bone-marrow cells of mice, chromosomal aberrations in bone-marrow cells of rats and Chinese hamsters, and sperm-head anomalies in mice treated in vivo. Benzene induced chromosomal aberrations and mutations in human cells in vitro. Benzene was reported to induce cell transformation, aneuploidy, and – in Drosophila – somatic mutation and crossing-over in spermatogonia. Benzene inhibited the DNA-related enzyme topoisomerase II, which is essential for maintenance of proper chromosome structure and segregation.
Benzene-induced leukemia might commence as a mutagenic event in stem or progenitor cells; subsequent genomic instability allows for sufficient mutations to be acquired in a relatively short time. Eight genetic pathways were suggested to lead to development of AML (see Figure 4.2, p.280. IARC Citation2012f). Benzene has been linked to five of these genetic pathways.
Additional information
Several investigators showed that exposure to benzene may be associated with altered DNA methylation both in humans and animals (Gao et al. Citation2010; Xing et al. Citation2013; Yang et al. Citation2014). Benzene metabolites such as hydroquinone and p-benzoquinone generated phosphorylated histones in human promyelocytic leukemia cells (Ishihama, Toyooka, and Ibuki Citation2008) and disrupted global DNA methylation via inhibition of DNA methyltransferase activity, which is a potential epigenetic mechanism (Hu et al. Citation2014).
Bis(chloromethyl)ether and chloromethyl methyl ether (technical grade)
‘There is moderate to strong evidence that bis(chloromethyl)ether and chloromethyl methyl ether, which are powerful alkylating agents, operate by a genotoxic mechanism. This mechanism is likely to be similar to that of other strong alkylating agents, involving modification of DNA and resultant mutations.’ (IARC Citation2012f, 305).
Bis(chloromethyl)ether (BCME) and chloromethyl methyl ether (CMME) are used primarily as chemical intermediates and alkylating agents. BCME is used as a lab reagent in manufacture of plastics, ion-exchange resins, and polymers. Historical uses of BCME include crosslinking of cellulose, synthesis of styrene and polymers, surface treatment of vulcanized rubber to increase adhesion, and manufacture of flame-retardant fabrics. CMME is used as an alkylating agent and as an industrial solvent to manufacture dodecylbenzyl chloride, water repellants, ion-exchange resins, and polymers, and as a chloromethylating reagent. Exposure to these chemicals is strictly regulated worldwide. Small quantities are currently produced to be used only in enclosed systems for the synthesis of other chemicals. BCME and CMME produce lung cancer.
BCME and CMME belong to the group of chloroalkyl ethers which are rapidly hydrolyzed to form hydrochloric acid, methanol and formaldehyde. Our understanding of the carcinogenic mechanisms of BCME and CMME is limited, with only few studies of genotoxicity and cytotoxicity available. However, both compounds are known to be potent alkylating agents which suggests that these compounds might operate by a genotoxic mode of action. In vitro, CMME enhanced virus-induced transformation of Syrian hamster embryo cells and elicited unscheduled DNA synthesis. As one of the primary metabolites of BCME, formaldehyde may contribute to the carcinogenic mechanism. However, since BCME is more potent as a carcinogen, formaldehyde may not be the predominant agent.
1,3-Butadiene
‘There is strong evidence that the carcinogenicity of 1,3-butadiene in humans operates by a genotoxic mechanism that involves formation of reactive epoxides, interaction of these direct-acting mutagenic epoxides with DNA, and resultant mutagenicity. The metabolic pathways for butadiene in experimental animals have also been demonstrated in humans.’ (IARC Citation2012f, 333).
Butadiene is employed primarily in the production of synthetic rubbers and polymers, which are used in a wide variety of industrial and consumer products. Butadiene is also utilized as an intermediate in the production of chloroprene and other chemicals. The highest exposures to butadiene occur in occupational settings in several industrial activities, such as petroleum refining and related operations, production of C4 fractions containing butadiene, production and distribution of gasoline, production of purified butadiene monomer, and manufacture of rubber and plastic products, such as tires, hoses and a variety of molded objects. There is some low-level airborne exposure of the general public. 1,3-Butadiene induces cancer of the hematolymphatic system.
The first step in the metabolism of butadiene involves cytochrome P450 (CYP)-mediated oxidation to epoxybutene, which may be metabolized by conjugation with glutathione (GSH) mediated by glutathione S-transferase (GST), by hydrolysis catalyzed by epoxide hydrolase (EH), or by oxidation to multiple diastereomers of diepoxybutane. Dihydroxy-butene formed by hydrolysis of epoxybutene may be oxidized to epoxybutanediol. The latter epoxides are also detoxified by GST or EH. Each of the epoxide intermediates may contribute to the mutagenicity and carcinogenicity attributed to butadiene. Factors that influence their relative contributions include concentration in tissues, reactivity with DNA, and repair of the ensuing DNA adducts. Variability in the expression of key enzymes involved in the biotransformation of butadiene may exert an effect on metabolite concentrations in tissues, and on subsequent mutagenic response.
There is strong evidence that the carcinogenicity of butadiene involves a genotoxic mechanism of action mediated by reactive epoxide metabolites. 1,3-Butadiene and its metabolites produce DNA adducts in animals and humans, with the N7 position of guanine being the major target site. The metabolites are mutagenic and genotoxic at multiple sites in mice and rats, and in a variety of other test systems. An AT-TA transversion mutation was consistently found across all biological systems. Mutations in K-Ras, H-Ras, p53, p16/p15 and β-catenin were detected in mice treated with 1,3-butadiene. Lab-controlled modulation of key determinants of 1,3-butadiene metabolism was shown to affect genotoxicity. Micronucleus formation and sister chromatid exchange were reported.
Additional information
Butadiene induces altered DNA methylation and histone modifications in animals (Koturbash et al. Citation2011a, Citation2011b). Demethylation of repetitive DNA sequences and alterations in histone-lysine acetylation were seen in liver and lung tissues of butadiene-exposed mice, whereas DNA methylation alterations were not significant in kidneys of these mice. (Chappell et al. Citation2014).
2,3,7,8-Tetrachlorodibenzo-para-dioxin, 2,3,4,7,8-pentachlorodibenzofuran, 3,3ʹ,4,4ʹ,5-pentachlorobiphenyl
‘There is strong evidence to support a receptor-mediated mechanism that operates in humans for carcinogenesis associated with 2,3,7,8-tetrachlorodibenzo-para-dioxin (TCDD), where the primary mechanism is the promotion of tumour development through modification of cell replication and apoptosis, with a secondary mechanism related to increases of oxidative stress causing DNA damage. The conservation of the aryl hydrocarbon receptor and the related signaling pathways and responses across species, including humans, add additional strength to the notion that this mechanism is active in humans. There is strong evidence to support a receptor-mediated mechanism for 2,3,4,7,8-pentachlorodibenzofuran- and 3,3ʹ,4,4ʹ,5-pentachlorobiphenyl-associated carcinogenesis based upon extensive evidence showing activity identical to 2,3,7,8-tetrachlorodibenzo-para-dioxin (TCDD) for every step of the mechanism described for TCDD-associated carcinogenesis in humans, including receptor binding, gene expression, protein-activity changes, cellular replication, oxidative stress, promotion in initiation-promotion studies and complete carcinogenesis in laboratory animals.’ (IARC Citation2012f, 371).
2,3,7,8-Tetrachlorodibenzo-para-dioxin (TCDD) has no known commercial applications. This compound occurred as a contaminant in chlorophenoxy herbicides, including 2,4,5-trichlorophenoxy-acetic acid, which were widely used in the 1960s and 1970s to control weeds. TCDD is also generated during thermal processes such as incineration, in metal-processing, and in the bleaching of paper pulp with free chlorine. The strongest evidence in humans for the carcinogenicity of TCDD is for all cancers combined.
Polychlorinated dibenzofurans (PCDFs) are not manufactured commercially other than for scientific research purposes. Release of PCDFs, including 2,3,4,7,8-pentachlorodibenzofuran (PeCDF) into the environment is mainly from combustion and incineration. The latter substance is the major congener emitted from cement kilns burning hazardous waste (approximately 20% of the total emission). Other major sources of PeCDF are metal smelting, refining, and processing; chemical manufacturing/processing such as production of chlorophenols, PCBs and vinyl chloride as well as pulp bleaching.
Mixtures of polychlorobiphenyls (PCBs), including 3,3ʹ,4,4ʹ,5-pentachlorobiphenyl (PCB 126), were produced for commercial purposes during half a century until 1977, to be used as dielectric insulating fluids for transformers and capacitors. PCB 126 and other PCBs were also employed in hydraulic fluids, plastics, and paints. The manufacture and use of PCBs in the USA were discontinued in 1977, but PCBs are still released into the environment through utilization and disposal of products containing PCBs, as by-products during the synthesis of certain organic chemicals, and during combustion of some waste materials.
When IARC Monograph 100F was published, causal links with human cancer had not been established for PeCDF and PCB 126Footnote2. Mechanistic evidence was used to support their classification as Group-1 carcinogens.
Most, if not all of the effects of TCDD are related to compound binding to and activation of the aryl hydrocarbon receptor (AhR). This receptor is expressed in most mammalian tissues. In addition to TCDD, many other halogenated aromatic compounds bind to this receptor, including the coplanar PCBs and PCDFs and dibenzofurans. It is generally proposed that the toxic and carcinogenic effects of dioxin and other halogenated compounds are due to high affinity to AhR, and to sustained pleiotropic response from a large number of genes – many of which encode drug-metabolizing enzymes –, subsequent to formation of the receptor-ligand complex. TCDD induces sustained AhR activation due to its long half-life in the body and continuing environmental exposure. Binding to the AhR leads to the modification of the activity of a wide range of Phase-I and -II genes. Through cross-talk, activation of the AhR also affects other receptor-mediated pathways such as those associated with the estrogen receptor and the retinoic-acid receptor β. This leads to up-regulation of enzymes and enhanced production of reactive intermediates in the xenobiotic metabolism, which increases oxidative stress. TCDD was shown to increase cellular proliferation both in vivo and in vitro in several tissues and cells, possibly via interaction with protein-kinase C signaling, inhibition of senescence, or activation of growth-signaling factors.
The receptor-mediated mechanism of action for TCDD-associated carcinogenesis in humans is suggested to be also the mechanism underlying the carcinogenicity of 2,3,4,7,8-PeCDF and PCB 126. The primary events are promotion of carcinogenesis through the activation of cellular replication and alterations in cellular senescence and apoptosis through AhR. These congeners, through activation of an array of metabolic enzymes, enhance the risk for oxidative stress as an indirect initiator of carcinogenesis, which makes these congeners complete carcinogens. The conservation of the AhR and related signaling pathways across species support this mechanism of action in humans.
Additional information
TCDD is immunotoxic (Kerkvliet Citation2002) and immunosuppressive (Vineis and Zahm Citation1988). The capacity of T-cells isolated from TCDD-exposed industrial workers to proliferate upon interleukin-2 stimulation was significantly diminished. TCDD exerted a long-term immunosuppressive-effect on T-helper cell function (Tonn et al. Citation1996). TCDD initiated alterations in DNA methylation, which altered gene expression (McClure et al. Citation2011; Singh et al. Citation2011), and produced genomic instability (Korkalainen et al. Citation2012). Acetylation of histones H3 and H4 and tri-methylation of histone H3 were detected at the promoter regions of CYP1A1 and CYP1B1 in human MCF-7 breast cancer cells and in human HepG2 hepatic cancer cells exposed to TCDD (Beedanagari et al. Citation2010). TCDD dysregulated expression of microRNAs, miR101a and miR122, in mice in vivo. The gene COX-2, a target of miR101a, may play a significant role in induction of liver damage in these exposed mice (Yoshioka, Higashiyama, and Tohyama Citation2011). Prenatal exposure of mice to TCDD triggered a significant modulation of the microRNA expression profile in thymocytes (Singh et al. Citation2012).
Ethylene oxide
‘There is strong evidence that the carcinogenicity of ethylene oxide, a direct-acting alkylating agent, operates by a genotoxic mechanism. A dose-related increase in the frequency of ethylene oxide-derived haemoglobin adducts has been observed in exposed humans and rodents, and a dose-related increase in the frequency of ethylene oxide-derived DNA adducts has been demonstrated in exposed rodents. Ethylene oxide consistently acts as a mutagen and a clastogen at all phylogenetic levels, it induces heritable translocations in the germ cells of exposed rodents, and a dose-related increase in the frequency of sister chromatid exchange, chromosomal aberrations and micronucleus formation in the lymphocytes of exposed workers.’ (IARC Citation2012f, 395-6).
Ethylene oxide is an important raw material used in the manufacture of chemical derivatives that are the basis for major consumer goods in virtually all industrialized countries. More than half of the ethylene oxide produced worldwide is employed in the manufacture of mono-ethylene glycol. Other important derivatives of ethylene oxide include di-ethylene glycol, tri-ethylene glycol, poly(ethylene) glycols, ethylene glycol ethers, ethanol-amines, and ethoxylation products of fatty alcohols, fatty amines, alkyl phenols, cellulose and poly(propylene) glycol. Only a small proportion (0.05%) of the annual production of ethylene oxide is utilized directly in the gaseous form as a sterilizing agent, fumigant and insecticide. This agent is well known as a sterilant of drugs, hospital equipment, disposable and reusable medical items. Most human exposure is occupational, although some exposure occurs from air pollution and tobacco smoking.
There has been no conclusive causal association between ethylene oxide and any human cancer although there are suggestive links (i.e. limited evidence) with lymphatic and hematopoietic cancers (specifically lymphoid tumors, i.e. non-Hodgkin’s lymphoma, multiple myeloma and chronic lymphocytic leukemia), and breast cancer. The classification as a Group-1 carcinogen – mechanistic upgrade – is based upon evidence of carcinogenesis in animals and on strong mechanistic evidence.
There is an extensive literature on the metabolism of and carcinogenic mechanisms for ethylene oxide. This agent was noted to be genotoxic and mutagenic in numerous assays and cell types. Increases in gene mutations and chromosomal alterations were observed in multiple systems. In vitro and in vivo studies showed that ethylene oxide binds to cellular macromolecules, which results in a variety of adducts to DNA, RNA and protein. Dose-dependent elevation in sister-chromatid exchange and chromosomal aberrations were observed in exposed workers. In vivo studies in mice and rats using mutagenicity assays with reporter genes such as Hprt or the LacI-transgene demonstrated a significantly increased mutation frequencies after exposure to ethylene oxide.
Formaldehyde
‘The current data strongly indicate that genotoxicity plays an important role in the carcinogenicity of formaldehyde in nasal tissues in humans, and that cellular replication in response to formaldehyde-induced cytotoxicity promotes the carcinogenic response. Three possible mechanisms, all focused around genotoxicity, are moderately supported as the underlying mechanism for induction of haematological malignancies in humans. Further research is needed to decide which of the mechanisms is the most important.’ (IARC Citation2012f, 430).
Formaldehyde is produced worldwide on a large scale by catalytic, vapor-phase oxidation of methanol, and mainly used in the production of various types of resins, as an intermediate in the manufacture of industrial chemicals, and directly – in aqueous solution known as formalin – as a disinfectant and preservative. Formaldehyde is formed endogenously in mammals, including humans, as a consequence of oxidative metabolism. This compound is also found in natural products (e.g. certain foods), cigarets, and automobile exhausts. Formaldehyde induces cancer of the nasopharynx and leukemia. Formaldehyde exhibits a genotoxic mechanism of action in the production of nasopharyngeal cancer.
Formaldehyde has a genotoxic mechanism of action for the production of nasopharyngeal cancer. Micronucleus formation was found in cells of the nasal and oral mucosa of formaldehyde-exposed humans. DNA–protein crosslinks were noted in circulating white blood cells of exposed workers. Sister chromatid exchange, micronucleus formation and chromosomal aberrations were reported in several investigations in blood lymphocytes of chronically exposed workers.
Studies in laboratory animals that inhaled formaldehyde demonstrated genotoxic effects in nasal tissues. DNA strand-breaks were induced by formaldehyde in vivo in mouse liver and rat lung cells. There is consistent in vitro evidence of formaldehyde-induced mutations, encompassing both clastogenic effects and direct DNA mutation. Formaldehyde displayed mutagenic potential in several bacterial systems, both with and without bio-activation. Formaldehyde induced deletions, point mutations, insertions, and cell transformation in in vitro assays with mammalian cells. Chromosomal aberrations, micronuclei and sister chromatid exchange were all increased in vitro in numerous rodent and human primary cells and cell lines treated with formaldehyde.
The current data indicate that genotoxicity plays an important role in the carcinogenicity of formaldehyde in nasal tissues in humans, and that cellular replication in response to formaldehyde-induced cytotoxicity promotes the carcinogenic response. There is less understanding of the mechanism underlying carcinogenesis for leukemia, and concerns have been raised regarding whether inhaled formaldehyde can reach the bone marrow, which would be necessary for a direct carcinogenic mechanism. Three possible mechanisms, all focused around genotoxicity, are supported by the available data as the underlying mechanism for induction of hematological malignancies in humans: formaldehyde (1) may damage stem cells in the bone marrow directly, as most other leukemogens do; (2) may damage hematopoietic stem cells or progenitor cells circulating in the peripheral blood; and (3) may damage the primitive pluripotent stem cells present in the nasal turbinates and/or the olfactory mucosa. Further research is needed to decide which of the mechanisms is the most important.
Additional information
Formaldehyde produced global genomic hypomethylation in 16HBE cells (Liu et al. Citation2011) and induced changes in microRNA expression profiles in humans (Rager et al. Citation2013, Citation2011). At low dose, formaldehyde initiated cell proliferation and reduced apoptosis, while higher levels led to increased apoptosis and excessively high concentrations produced necrosis (Szende and Tyihak Citation2010). Formaldehyde was found to produce DNA adducts (Lu et al. Citation2010, Citation2012; Wang et al. Citation2007). A chromosome-wide aneuploidy study revealed significant elevation in the frequencies of monosomy, trisomy, tetrasomy, and structural aberrations of multiple chromosomes in formaldehyde-exposed workers compared with controls. The detection of increased levels of monosomy 7 and structural aberrations of chromosome 5 is particularly relevant, as these changes are frequently observed in acute myeloid leukemia (Lan et al. Citation2015).
Sulfur mustard
‘Data from a variety of sources all strongly support a genotoxic mechanism underlying the carcinogenic action of mustard gas/sulfur mustard, mainly based on the observation that this chemical is a bi-functional alkylating agent. The direct reaction of this agent with DNA likely initiates a cascade of genetic events that lead to cancer. There is evidence to support DNA-alkylation leading to crosslink formation, inhibition of DNA synthesis and repair, point mutation, and induction of chromosome-type and chromatid-type aberrations. In addition, the production of reactive oxygen species and cytotoxicity – other reported contributors to the mechanism of action – could act complementary to DNA alkylation’ (IARC Citation2012f, 446).
Sulfur mustard (also known as mustard gas) was used as a chemical warfare agent in various military conflicts since World War I. Sulfur mustard is acutely toxic and produces death within three days of exposure. At lower persistent exposure levels, it induces lung cancer.
Sulfur mustard (1,1ʹ-thiobis (2-chloroethane) is an alkylating agent that produces DNA inter-strand cross-links. In vitro studies found that sulfur mustard induced DNA adducts in human cells and various animal models. Alkylation by sulfur mustard affects transcriptional processes and may lead to truncated transcripts by impairing RNA polymerase via an alkylated promoter. Cells in late G1- or S-phase are at highest risk. Cells engaged in proliferation following tissue injury are particular targets. In humans exposed to sulfur mustard from leaking munitions, elevated rates of sister chromatid exchange were observed. Animal studies demonstrated chromosomal aberrations, micronuclei and mutations. Sulfur mustard produces free-radical-mediated oxidative stress. Sulfur mustard also inhibits anti-oxidant enzyme activity. There is evidence that carcinogenicity attributed to sulfur mustard is based upon a genotoxic mechanism of action that involves DNA alkylation leading to cross-link formation, inhibition of DNA synthesis and repair, induction of point mutations, and chromosome-type and chromatid-type aberrations.
Additional information
A significant rise of global DNA methylation was detected in early endothelial cells in vitro, and in human skin in vivo after exposure to sulfur mustard (Steinritz et al. Citation2016). In addition, alterations in miRNA expression were observed in early endothelial cells exposed to sub-lethal sulfur mustard concentrations (Schmidt et al. Citation2016).
Vinyl chloride
‘Numerous studies on the toxicokinetics, metabolism, genotoxicity, and molecular biology of vinyl chloride provide strong evidence that the carcinogenicity of this chemical involves a genotoxic mechanism of action, mediated by reactive metabolites. Key events in the pathway of vinyl chloride-induced liver carcinogenesis include metabolic activation to reactive metabolites, binding of the metabolites to DNA, pro-mutagenic action of these adducts leading to G→A and A→T transitions, and the effects of such mutations on the functioning of proto-oncogenes and tumour suppressor genes at the gene and protein levels.’ (IARC Citation2012f, 472).
Vinyl chloride is employed primarily (> 95%) in manufacture of polyvinyl chloride (PVC) – which comprises approximately 12% of the total use of plastic worldwide – and mainly in the production of plastic piping. Minor uses of vinyl chloride monomer (VCM) include synthesis of chlorinated solvents such as 1,1,1-trichloroethane, and the production of ethylene diamine for the manufacture of resins.
The main route of occupational exposure to vinyl chloride is by inhalation, which occurs primarily in vinyl chloride/PVC plants and in PVC-processing plants. VCM is present in mainstream smoke of cigarets and cigars. Non-occupational exposure levels for the general population are low. Vinyl chloride induces angiosarcoma of the liver and hepatocellular carcinoma.
Vinyl chloride is readily absorbed upon inhalation and rapidly metabolized in the liver. The primary metabolites are highly reactive chloroethylene oxide, and its rearrangement product chloroacetaldehyde. Both bind to proteins, DNA and RNA and form ethenoadducts. Chloroethylene oxide is the most reactive with nucleotides. Vinyl chloride is mutagenic, usually in the presence of metabolic activation, in various assays with bacteria, yeast or mammalian cells. This compound is also clastogenic in vivo and in vitro. Vinyl chloride induces unscheduled DNA synthesis, increases the frequency of sister chromatid exchange in rat and human cells, and elevates the frequency of chromosomal aberrations, DNA strand breaks and micronucleus formation in mice, rats, and hamsters in vivo. Polymorphic variations in metabolic genes (e.g. those of the CYP450 family) or DNA repair genes may alter carcinogenicity but do not affect the underlying mechanisms.
Additional information
In workers exposed to vinyl chloride, hypermethylation of the 5ʹCpG island in the p16 gene was observed in 72% of the hepatocellular carcinomas examined (Weihrauch et al. Citation2001).
Isopropyl alcohol manufacture by the strong-acid process
‘Little information on possible mechanisms of carcinogenicity of inorganic acid mists is available. The increased incidence of cancer of the paranasal sinuses in workers involved in the strong-acid process of isopropyl alcohol manufacture may be due to exposure to the strong acid mists and/or the presence of diisopropyl sulphate, an intermediate that shows sufficient evidence of carcinogenicity in experimental animals. Available data suggest that localized low pH from inhalation of inorganic acid mists could damage DNA and lead to neoplasia. There is no evidence that would support the occurrence of DNA damage by any other mechanism of carcinogenesis.’ (IARC Citation2012f, 483).
Isopropyl alcohol can be prepared via indirect or direct hydration of propylene, and through catalytic hydrogenation of acetone. Indirect hydration of propylene involves reaction of propylene with sulfuric acid to produce mono- and di-isopropyl sulfates, which are then hydrolyzed to isopropanol. Epidemiological studies showed that exposure of humans during the manufacture of isopropyl alcohol by the strong-acid process produced cancer of the nasal cavity (paranasal sinuses). However, neither sulfuric acid nor isopropyl alcohol are classified as human carcinogens.
Workers exposed to sulfuric acid mists exhibited a rise in incidence of symptoms and macroscopic and microscopic changes of nasal mucosa, including squamous metaplasia and atypia in an exposure-response relationship. Significant increases in incidence of sister chromatid exchange, micronucleus formation and chromosomal aberrations in peripheral lymphocytes were detected in one study of workers at a sulfuric acid plant (40 exposed, 42 controls).
There is little direct evidence concerning a carcinogenic mechanism. It is hypothesized that local alterations in pH due to inhalation of the low-pH acid mist might lead to DNA damage through depurination of DNA and deamination of cytidine. Low pH may also alter the integrity of enzymes. However, direct experimental support for this mechanism is currently lacking. In addition, estimation of the impact of exposure to acid mists on pH levels in tissues is complex due to variation in droplet size and inhalation patterns.
Additional information
Isopropyl alcohol may induce immunosuppression in human cells in vitro (Carignan, Desy, and de Campos-Lima Citation2012; Desy et al. Citation2008) and interfere with inflammatory responses in rats (Kasuga et al. Citation1992).
Mists from strong inorganic acids
‘While it is plausible that conditions of localized low pH from inhalation of inorganic acid mists could damage DNA and increase cancer risks, the evidence supporting DNA-damage induction or any other mechanism as the cause of the observed cancers due to the inorganic acid mists is weak.’ (IARC Citation2012f, 493).
The mechanistic information related to ‘Mists from strong inorganic acids’ was presented in the previous section on ‘Isopropyl alcohol manufacture by the strong-acid process’. Major industries with exposure to strong inorganic acid mists include those that manufacture phosphate fertilizer, isopropanol (isopropyl alcohol), synthetic ethanol (ethyl alcohol), sulfuric acid, nitric acid, and lead batteries. Exposure also occurs during copper smelting, and pickling and other acid treatments of metals. Exposure to strong inorganic acid mists containing sulfuric acid may occur by inhalation, ingestion, and dermal contact and the effects depend upon many factors including droplet size, proximity to the source, and control measures such as ventilation and containment. Mists from strong inorganic acids induce cancer of the larynx.
It is hypothesized that the same carcinogenic mechanisms are likely in operation for mists from strong organic acids as described for isopropyl alcohol manufacture by the strong-acid process (see above). As noted in that section, there is a lack of a clear mechanism but it is hypothesized to be related to exposure of tissue to low-pH conditions.
Occupational exposures during iron and steel founding
‘There is moderate evidence that extracts of particles collected from a steel foundry act via a genotoxic mechanism, based on bacterial mutation studies. There is weak evidence for a genotoxic mechanism of action for exposures during iron and steel founding, based on DNA-adduct studies.’ (IARC Citation2012f, 505).
The processes in iron and steel founding generally comprise pattern-making, molding and core-making, melting, pouring and shake-out, and fettling. Substantial exposures to silica dust (respirable quartz) and carbon monoxide continue to occur in many foundries. There is exposure to airborne PAHs resulting mainly from the thermal decomposition of carbonaceous ingredients commonly added to foundry sand. In addition, some steel-foundry workers (e.g. fettlers) are exposed to airborne chromium and nickel compounds. Workers may also be exposed to other chemicals, including phenol, formaldehyde, isocyanates and various amines. Foundry work induces lung cancer.
Cancer risk and carcinogenic mechanisms reflect those of the specific substances to which workers are exposed. In several studies extracts of particulates from samples collected at a steel foundry, or filter extracts, were mutagenic in S. typhimurium in the presence or absence of an exogenous metabolic activation system. Workers in an iron foundry were examined for the presence of aromatic DNA adducts in lymphocytes using of [32P]-postlabelling. There was a significant correlation between the estimated exposures and DNA-adduct levels. In another population of foundry workers, immunochemical analysis of aromatic DNA-adducts showed a positive trend with exposure. When exposure to PAHs decreased over a period of 5 years, the levels of DNA adducts also fell.
Occupational exposure as a painter
‘The multiple genetic and cytogenetic effects observed among workers employed as painters or in the paint industry provide strong evidence in support of genotoxicity as one mechanism underlying the observed increase in cancer risk. However, due to the complexity and changing nature of the exposure mixtures and the potential interactions between exposures as a painter, other mechanisms are also likely.’ (IARC Citation2012f, 531).
Painting involves exposure to a diverse range of chemicals, including components of the paint (pigments, dyes, binders, solvents and additives) and from preparation of the surface being painted. Painting is estimated to involve over a thousand different substances, many of which are potentially carcinogenic. Azo pigments based upon 3,3ʹ-dichlorobenzidine are commonly present in paint; asbestos was used as a filler until the early 1990s; the main organic solvents utilized in paints are toluene, xylene, aliphatic compounds, ketones, alcohols, esters, and glycol ethers, although modern solvent-based paints contain markedly less solvent and less hazardous solvents than a decade ago. Occupational exposure as a painter induces mesothelioma, and cancers of the urinary bladder and lung. A positive association was observed between maternal exposure to painting (including pre-conception and during pregnancy) and childhood leukemia in the offspring.
Carcinogenic mechanisms reflect those of the specific chemical exposures. Many of these have been described previously.
Six studies among painters showed consistently and significantly elevated frequencies of chromosomal aberrations, and three of these reported an association with years of employment. Five of 6investigations reported significant elevation in frequency of micronuclei among painters. Chromosomal aberrations and micronuclei were found both in cultured lymphocytes and in buccal cells. Significantly elevated frequencies of sister chromatid exchange were detected in 4 of 7 studies among painters. Increased levels of DNA strand-breaks and DNA adducts were also reported.
Occupational exposures in the rubber-manufacturing industry
‘The multiple genetic and cytogenetic effects observed among workers employed in the rubber-manufacturing industry provide strong evidence to support genotoxicity as one mechanism for the observed increase in cancer risks. However, due to the complexity and changing nature of the exposure mixture and the potential interactions between exposures in the rubber-manufacturing industry, other mechanisms are also likely to play a part.’ (IARC Citation2012f, 559).
Workers in the rubber-manufacturing industry are exposed to a wide variety of chemicals, the composition of which varies across industrial sites and sectors. A major exposure arises from dusts and fumes created during the rubber-making and vulcanization processes. Potential exposures include N-nitrosamines, PAHs, solvents, and phthalates. Inhalation is the main route of exposure, although workers may also be exposed dermally to cyclohexane-soluble compounds. Occupational exposure in the rubber-manufacturing industry produces leukemia, lymphoma, and cancers of the urinary bladder, lung, and stomach.
Carcinogenic mechanisms reflect those of the specific chemical exposures. Several studies among workers in the rubber-manufacturing industry provided evidence of chromosomal aberrations, sister-chromatid exchange, micronucleus formation, premature chromosome condensation, DNA breakage, DNA-adduct formation, mutagenicity in urine, and mutation in the HPRT gene. For each of these endpoints, in most studies a positive response was observed in exposed workers compared with non-exposed controls.
Post-Volume 100 Group-1 human carcinogens
Diesel-engine exhaust
‘ … … there is strong mechanistic evidence that diesel-engine exhaust, as well as many of its components, can induce lung cancer in humans through genotoxic mechanisms that include DNA damage, gene and chromosomal mutation, changes in relevant gene expression, the production of reactive oxygen species and inflammatory responses. In addition, the co-carcinogenic, cell-proliferative and/or tumour-promoting effects of other known and suspected human carcinogens present in diesel engine exhaust probably contribute to its carcinogenicity in the human lung.’ (IARC Citation2014a, 464).
The diesel engine was invented in 1898 and has undergone extensive development leading to higher efficiency and emission control. Diesel engines power a wide range of automotive vehicles (including passenger cars, buses, ships and commercial vehicles) and also provide power for sites where electricity is generated. Occupational exposures to diesel-engine exhausts occur for workers using these vehicles and machines. The general public is also exposed to exhaust fumes from vehicles. Inhalation is the main route of exposure. Diesel-engine exhaust produces lung cancer.
Diesel exhaust is a complex chemical mixture, containing gaseous and particulate components. The carcinogenic potential of diesel-engine exhaust arises from these component chemicals such as nitrogen oxides, sulfur, ozone, acrolein, benzene, formaldehyde, naphthalene, PAHs, lead, arsenic, and chromium. Several of these substances have individually been classified as Group-1 or Group-2 agents. There is evidence that the carcinogenic effect of diesel-engine exhaust is predominantly related to the particulate components.
Carcinogenic mechanisms for diesel-engine exhaust are complex, with component chemicals displaying strong evidence of a range of genotoxic effects including: sister chromatid exchange, mutation in bacterial and animal models, chromosomal aberrations, DNA adducts, DNA strand-breaks and unscheduled DNA synthesis. PAHs (including benzo[a]pyrene) and nitro-PAHs are components with strong carcinogenic potential. These compounds require activation by phase-I metabolic enzymes and lead to DNA-adduct formation. Mechanistic aspects for benzo[a]pyrene were previously described. Exposure to whole diesel-engine exhaust produced increased levels of sister chromatid exchange, micronuclei, and bulky DNA adducts. In lung tissue, exposure to particulates from diesel-engine exhaust induced a marked immunological responses, characterized by production of ROS and oxidative DNA damage. Levels of pro-inflammatory mediators were elevated with evidence for enhanced gene expression. Humans exposed to diesel-engine exhaust developed airway inflammation and also excreted high levels of 1-hydroxypyrene, a marker of PAH exposure.
‘Genes involved in phase I and II metabolism, oxidative stress, antioxidant response, immune/inflammatory response and cell cycle/apoptosis, and those that respond to cell damage were upregulated in cultured rat alveolar epithelial cells exposed to fractionated organic solvent extracts of diesel engine exhaust particles. Exposure to whole diesel-engine exhaust induced sister chromatid exchange in lung cells and lung tissues, increased the levels of bulky DNA adducts and enhanced oxidative DNA damage in rodents, caused mutations in transgenic rats and induced angiogenesis and vasculogenesis in mice’. (IARC Citation2014a, 462–3).
Additional information
Diesel-engine exhaust stimulated various cell receptors, an effect that may be mediated through benzo[a]pyrene (Chaloupka et al. Citation1993; Fertuck, Matthews, and Zacharewski Citation2001; Li et al. Citation2011; Owens et al. Citation2006; Vrabie, Jonker, and Murk Citation2009). Acute exposure of human asthmatic volunteers to diesel exhaust led to modifications in the expression of various microRNAs (Yamamoto et al. Citation2013). Diesel-exhaust particles enhanced the expression of microRNA-21 (miR-21) and activated the PTEN/PI3K/AKT pathway in human bronchial epithelial cells (Zhou et al. Citation2015). Prenatal exposure of mice to diesel-exhaust particles produced genome-wide disruption of the DNA-methylation profile in brains of offspring (Tachibana et al. Citation2015). In human volunteers, expression of specific microRNAs and genes associated with bronchial immune responses were significantly modulated upon exposure to diesel exhaust (Rider et al. Citation2016).
Trichloroethylene
‘In experimental animals and humans, oxidative metabolism of trichloroethylene is catalyzed by cytochrome P450 enzymes, whereas GSH conjugation of trichloroethylene is catalyzed by GST enzymes. The formation of reactive metabolites of trichloroethylene in the kidney from processing of GSH-conjugation metabolites in situ has been observed in experimental animals and in human kidney cells. The reactive GSH-conjugation metabolites of trichloroethylene are genotoxic on the basis of consistent results in several available test systems.’ (IARC Citation2014b, 189).
Trichloroethylene is a chlorinated solvent that has been produced commercially since the 1920s by chlorination of ethylene or acetylene. In the 1930s, it was introduced in the dry-cleaning industry, but was replaced by tetrachloroethylene in the 1950s. Trichloroethylene has industrial applications in the cleaning and degreasing of metal parts, as an anesthetic, a heat-transfer medium, an extraction agent for fats and oils, and as an intermediate in production of other chemicals. Currently, trichloroethylene is still employed as a spot remover in dry cleaning. Trichloroethylene usage has declined dramatically since 1970 due to environmental and health concerns and government regulations. Exposure is mainly through inhalation and largely confined to occupational sites. Trichloroethylene induces renal cancer.
Trichloroethylene is rapidly absorbed following inhalation and distributed throughout the body, predominantly in tissues with a high lipid content. All effects attributed to trichloroethylene are due to its metabolites. Of the two main metabolic pathways (CYP- and GSH-mediated), the GSH-conjugated metabolites are highly reactive, while CYP metabolites are chemically stable. The initial GSH-conjugation occurs in the liver and produces S-(1,2-dichlorovinyl) glutathione (DCVG) which is subsequently metabolized in the kidney.
The carcinogenic mechanism underlying trichloroethylene exposure is not known comprehensively. Trichloroethylene is not genotoxic and not a direct-acting mutagen. In contrast, the products of GSH-mediated metabolism (e.g. DCVG, dichloroacetic acid, chloral hydrate) are genotoxic, particularly in the kidney. This genotoxicity was observed in experimental animals and in human kidney cells. Trichloroethylene and its metabolites also alter the function of the immune system.
Limited studies in humans suggest that trichloroethylene-exposed subjects may develop chromosomal aberrations and sister chromatid exchange, although these effects may be due to GSH-metabolites or to co-exposure to stabilizing agents that are known mutagens. In bacteria, yeast and mammalian systems, trichloroethylene induces mutations after metabolic activation. In vitro and in vivo studies demonstrated some evidence that exposure to trichloroethylene leads to chromosomal aberrations, micronucleus formation and sister chromatid exchange. Trichloroethylene is nephrotoxic in animals and humans, but experimental evidence linking this effect to development of renal cell carcinoma is lacking. Animal and human evidence suggests that trichloroethylene possesses immunotoxic properties, increasing the risk of autoimmune disease. The precise role of this effect in carcinogenesis is unclear.
Additional information
Trichloroethylene induced changes in DNA methylation which have been linked to teratogenesis (Palbykin et al. Citation2011) and decreased global DNA methylation in human lymphoblastoid cells (Tabish et al. Citation2012) and human hepatic cells (Zhang et al. Citation2014). Trichloroethylene was shown to increase CYP2H1 expression in embryonic chick hearts (Makwana et al. Citation2013), and to up- and down-regulate a large number of genes in Sprague-Dawley rat embryo hearts (Collier et al. Citation2003). Trichloroethylene induced alterations in gene expression and DNA methylation in the livers of mice treated in vivo (Jiang et al. Citation2014). This compound also induced alterations in the methylation of histone H3K79 in human liver cells (Deng et al. Citation2017).
Discussion
The overview of mechanisms underlying human cancer development presented in this review indicates that important mechanisms may be identified for most of the Group-1 human carcinogens reviewed by the IARC Monographs. For some agents (e.g. 2,3,4,7,8-pentachlorodibenzofuran, PeCDF), the strong human relevance of the available mechanistic data served as a basis for the classification of the chemical as a Group-1 human carcinogen in the absence of sufficient epidemiological evidence in humans. However, in other cases (e.g. mineral and shale oils, and trichloroethylene) the carcinogenic mechanism in humans is not clear. A more quantitative analysis of the mechanisms underlying human cancer occurrence (Krewski et al. Citation2019) noted that 10 of the 109 individual agents considered here were placed in Group-1 on the basis of mechanistic upgrades in the absence of convincing human epidemiological data.
This review is based upon the expert opinion of IARC Monographs Working Groups concerning the available primary data. There was no attempt to conduct a full systematic review of the evidence, as the sheer volume of the literature supporting mechanistic data for these 86 agents precludes inclusion in this review of all the citations to support the narrative overviews. Supporting references, as well as a more exhaustive discussion of the mechanistic summaries may be found in the primary IARC Monographs in which the agents were reviewed. A reference to the appropriate IARC Monograph Volume is provided in the agent-specific sections.
The most common carcinogenic mechanisms identified through this review was genotoxicity. Other common carcinogenic mechanisms included oxidative stress, immunosuppression, chronic inflammation, interaction with cellular receptors, and epigenetic effects such as modifications of histones and other structural cellular components.
It is apparent from the narrative summaries that genotoxic mechanisms feature prominently. Interpretation of this pattern needs to consider the potential for selection effects initiated by the absence of research on non-genotoxic mechanisms for some agents. Consideration also needs to be given to the process by which agents are selected for review in IARC Monographs. It is possible that genotoxic agents may have been more commonly selected for review, especially in the earlier years of the IARC review programme. Future evaluations may reveal a higher proportion of agents without direct genotoxicity, particularly if reviews consider agents with different modes of action.
Many Group-1 carcinogens with established carcinogenic mechanisms displayed multiple modes of action. While genotoxicity is common, many agents may act according to additional mechanistic pathways. For example, beryllium is genotoxic but also induces oxidative stress, increases cell proliferation, alters cell-signaling pathways and modifies DNA repair. Diethylstilbestrol has genotoxic, receptor-mediated and epigenetic effects that contribute to carcinogenesis. It is not always clear which mechanism is the predominant or driving mechanism for a carcinogen. For some agents, multiple modes of action may be required for carcinogenesis.
The summaries in this review focused on classes of carcinogenic mechanisms associated with an agent, rather than on specific genetic mutations that play a role in the progression to cancer. For example, the IARC Monographs commonly list mutations in specific oncogenes (e.g. TP53) as a mechanism. While mutations in these specific genes are important because they are directly linked to modifications in cellular function that lead to cancer, the action of the agent is to induce mutations, which may be randomly distributed throughout the genome. The narrative mechanistic summaries emphasized the general type of action of the agent (e.g. production of mutations) rather than the specific mutation. It would be useful to extend the exploration of mutations to identify the genetic drivers involved, along with the associated signaling pathways (Vogelstein et al. Citation2013).
A number of the Group-1 human carcinogens represent composite exposures or conditions associated with human behavior (e.g. tobacco smoking or painters). Tobacco smoking does not expose subjects to a single carcinogenic agent as there are over 100 known or suspected carcinogens contained within tobacco smoke. Many of the occupational exposures during painting involve individuals being exposed to multiple potential or known carcinogens. In these cases, the carcinogenic risk and mechanisms would be expected to reflect those of the component exposures. Interactions among multiple exposures are possible and might result in carcinogenic mechanisms not associated with the individual agents; these potential interactive effects have not been widely studied.
The composite nature of many human carcinogens presents challenges in summarizing the associated carcinogenic mechanisms. It is not feasible to report on all potential mechanisms related to each known carcinogen contained in the composite exposure. The summaries presented above attempt to capture the core mechanisms without providing details for each component agent. In some cases, reference was made to other summaries. For example, the discussion of ‘Betel quid with added tobacco’ was cross-referenced to the section on ‘Tobacco smoking’.
The scientific literature cited in the Monograph Volumes 100A–100F was published before 2010, with many of the citations were published prior to 2000. The volume 100 summaries would thus contain no information regarding mechanisms that were identified more recently and that may not have been widely studied for many agents. One major area of under-representation would be epigenetic effects.
The data collection process used as the foundation for the narrative reviews provided an opportunity to examine the potential impact of research conducted subsequent to the publication of the IARC Monographs. This was done through classification the narrative information into broad mechanisms of carcinogenesis.
Attempts have been made to categorize carcinogenic mechanisms into groups that reflect different broad mechanisms (Smith Citation2019). In 2012, participants at IARC workshops on ‘Tumour-site Concordance and Mechanisms of Carcinogenesis’ developed an initial classification scheme using 24 toxicological endpoints. Discussion following the meeting led to a re-design of the classification system with the development of 10 key characteristics of human carcinogens (Smith Citation2019; Smith et al. Citation2016). These 24 toxicological endpoints have been mapped to the 10 key characteristics of human carcinogens using an algorithm described by Al-Zoughool and colleagues (Al-Zoughool et al. Citation2019). We applied this algorithm to the results of the narrative reviews (Krewski et al. Citation2019), examining the impact of the supplemental PubMed search on the number of agents classified as displaying each of the 10 key characteristics.
illustrates the classification across the 10 key characteristics of the identified carcinogenic mechanisms of the 86 Group-1 agents discussed in this article. The figure indicates the number of carcinogens that have a mechanism for each of the 10 key characteristics. The figure shows the counts based on the mechanistic information abstracted from the IARC Monographs and also displays the additional mechanisms linked to an agent based on the PubMed search. For each key characteristic except genotoxicity, the supplemental PubMed search produced an increase in the number of agents demonstrating a particular key characteristic. The most notable rise in the number of additional agents identified was reported for epigenetic effects, which may be ascribed to the increased interest in epigenetics as a carcinogenic mechanism since 2010. In relative terms, receptor mediated effects demonstrated the greatest increase in proportion of additional agents identified.
Figure 1. Distribution of key characteristics of 86 group-1 agents (data abstracted from the monographs and the supplementary PubMed search shown in grey and black, respectively).
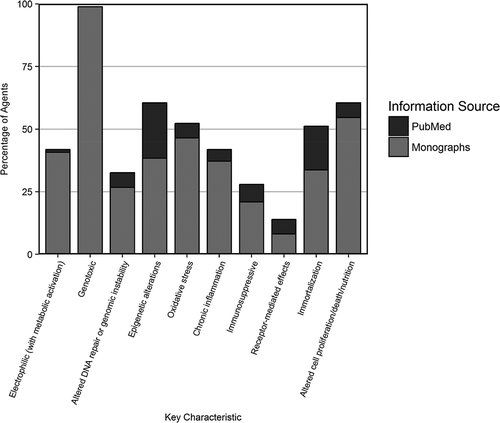
It should be noted that the PubMed search used to complement the information in the Monographs was not a systematic review but rather it was a targeted search, which was designed to identify newly described potential carcinogenic mechanisms for each agent. While the conduct of a full systematic review was beyond the scope of this project, the results of the sensitivity analysis suggest that the IARC Monographs provide an account of the mechanisms by which Group-1 agents induce cancer in humans.
A fuller discussion of the results of an exploratory analysis of the key characteristics associated with the 86 Group-1 agents considered here is available (Krewski et al. Citation2019).
The development of the key characteristics of human carcinogens (Smith et al. Citation2016) provides a useful framework around which to organize summaries of the available mechanistic information on mechanisms by which agents may produce cancer in humans. Recent IARC Monographs have incorporated consideration of the 10 key characteristics into the mechanistic discussion in Sections 4 of the Monographs. In identifying relevant data on these key characteristics, IARC has started to employ formal methods of systematic review in developing mechanistic summaries. Future evaluations of the mechanisms underlying human cancer occurrence will benefit from this new approach to developing mechanistic summaries reported in Sections 4 of the IARC Monographs.
Acknowledgments
We are grateful to Abdallah Alami for assistance in the final formatting of this article. D. Krewski is the Natural Sciences and Engineering Research Council of Canada Chair in Risk Science at the University of Ottawa.
Notes
1. A bystander effect occurs when non-irradiated cells exhibit effects caused by radiation as a result of chemical signals (messengers) received from nearby irradiated cells. Similar effects can be seen in response to chemical exposures (Asur et al. Citation2010; Asur, Thomas, and Tucker Citation2009).
2. PCBs as a group were reviewed in Monograph Volume 107. They were classified as Group 1 human carcinogens. Volume 107 was outside the scope of the current overview.
Related Research Data
References
- Agudelo, M., N. Gandhi, Z. Saiyed, V. Pichili, S. Thangavel, P. Khatavkar, A. Yndart-Arias, and M. Nair. 2011. Effects of alcohol on histone deacetylase 2 (HDAC2) and the neuroprotective role of trichostatin A (TSA). Alcohol. Clin. Exp. Res. 35 (8):1550–56. doi:10.1111/j.1530-0277.2011.01492.x.
- Aldous, W. K., A. J. Marean, M. J. DeHart, L. A. Matej, and K. H. Moore. 1999. Effects of tamoxifen on telomerase activity in breast carcinoma cell lines. Cancer 85 (7):1523–29.
- Al-Zoughool, M., M. Bird, J. Rice, R. A. Baan, M. Billard, N. Birkett, D. Krewski, and J. M. Zielinski. 2019. Development of a database on key characteristics of human carcinogens. J. Toxicol. Environ. Health B. In Press. doi:10.1080/10937404.2019.1642593.
- Anayannis, N. V., N. F. Schlecht, and T. J. Belbin. 2015. Epigenetic mechanisms of human papillomavirus-associated head and neck cancer. Arch. Pathol. Lab. Med. 139 (11):1373–78. doi:10.5858/arpa.2014-0554-RA.
- Anderton, J. A., S. Bose, M. Vockerodt, K. Vrzalikova, W. Wei, M. Kuo, K. Helin, J. Christensen, M. Rowe, P. G. Murray, et al. 2011. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin’s Lymphoma. Oncogene 30 (17):2037–43. doi:10.1038/onc.2010.579.
- Argos, M. 2015. Arsenic exposure and epigenetic alterations: recent findings based on the illumina 450K DNA methylation array. Curr. Environ. Health Rep. 2 (2):137–44. doi:10.1007/s40572-015-0052-1.
- Arita, A., A. Munoz, Y. Chervona, J. Niu, Q. Qu, N. Zhao, Y. Ruan, K. Kiok, T. Kluz, H. Sun, et al. 2013. Gene expression profiles in peripheral blood mononuclear cells of Chinese nickel refinery workers with high exposures to nickel and control subjects. Cancer Epidemiol. Biomarkers Prev. 22 (2):261–69. doi:10.1158/1055-9965.EPI-12-1011.
- Arita, A., J. Niu, Q. Qu, N. Zhao, Y. Ruan, A. Nadas, Y. Chervona, F. Wu, H. Sun, R. B. Hayes, et al. 2012. Global levels of histone modifications in peripheral blood mononuclear cells of subjects with exposure to nickel. Environ. Health Perspect. 120 (2):198–203. doi:10.1289/ehp.1104140.
- Asur, R., M. Balasubramaniam, B. Marples, R. A. Thomas, and J. D. Tucker. 2010. Involvement of MAPK proteins in bystander effects induced by chemicals and ionizing radiation. Mutat. Res. 686 (1–2):15–29. doi:10.1016/j.mrfmmm.2009.12.007.
- Asur, R. S., R. A. Thomas, and J. D. Tucker. 2009. Chemical induction of the bystander effect in normal human lymphoblastoid cells. Mutat. Res. 676 (1–2):11–16. doi:10.1016/j.mrgentox.2009.02.012.
- Attia, S. M., G. I. Harisa, M. H. Hassan, and S. A. Bakheet. 2013. Beryllium chloride-induced oxidative DNA damage and alteration in the expression patterns of DNA repair-related genes. Mutagenesis 28 (5):555–59. doi:10.1093/mutage/get032.
- Avissar, M., M. D. McClean, K. T. Kelsey, and C. J. Marsit. 2009. MicroRNA expression in head and neck cancer associates with alcohol consumption and survival. Carcinogenesis 30 (12):2059–63. doi:10.1093/carcin/bgp277.
- Bae, J. H., J. G. Kim, K. Heo, K. Yang, T. O. Kim, and J. M. Yi. 2015. Identification of radiation-induced aberrant hypomethylation in colon cancer. BMC Genomics 16:56. doi:10.1186/s12864-015-1229-6.
- Bajpai, D., A. Banerjee, S. Pathak, S. K. Jain, and N. Singh. 2013. Decreased expression of DNA repair genes (XRCC1, ERCC1, ERCC2, and ERCC4) in squamous intraepithelial lesion and invasive squamous cell carcinoma of the cervix. Mol. Cell. Biochem. 377 (1–2):45–53. doi:10.1007/s11010-013-1569-y.
- Banno, K., M. Yanokura, M. Iida, K. Masuda, and D. Aoki. 2014. Carcinogenic mechanisms of endometrial cancer: involvement of genetics and epigenetics. J. Obstet. Gynaecol. Res. 40 (8):1957–67. doi:10.1111/jog.12442.
- Bayne, S., M. E. Jones, H. Li, and J. P. Liu. 2007. Potential roles for estrogen regulation of telomerase activity in aging. Ann. N. Y. Acad. Sci. 1114:48–55. doi:10.1196/annals.1396.023.
- Bazarbachi, A., R. Abou Merhi, A. Gessain, R. Talhouk, H. El-Khoury, R. Nasr, O. Gout, R. Sulahian, F. Homaidan, H. de The, et al. 2004. Human T-cell lymphotropic virus type I-infected cells extravasate through the endothelial barrier by a local angiogenesis-like mechanism. Cancer Res. 64 (6):2039–46.
- Beedanagari, S. R., R. T. Taylor, P. Bui, F. Wang, D. W. Nickerson, and O. Hankinson. 2010. Role of epigenetic mechanisms in differential regulation of the dioxin-inducible human CYP1A1 and CYP1B1 genes. Mol. Pharmacol. 78 (4):608–16. doi:10.1124/mol.110.064899.
- Berardinelli, F., A. Sgura, A. Di Masi, S. Leone, G. A. Cirrone, F. Romano, C. Tanzarella, and A. Antoccia. 2014. Radiation-induced telomere length variations in normal and in Nijmegen Breakage Syndrome cells. Int. J. Radiat. Biol. 90 (1):45–52. doi:10.3109/09553002.2014.859400.
- Bernal, A. J., D. C. Dolinoy, D. Huang, D. A. Skaar, C. Weinhouse, and R. L. Jirtle. 2013. Adaptive radiation-induced epigenetic alterations mitigated by antioxidants. Faseb J. 27 (2):665–71. doi:10.1096/fj.12-220350.
- Biron-Shental, T., A. Amiel, R. Anchidin, R. Sharony, R. Hadary, and Y. Kitay-Cohen. 2013. Telomere length and telomerase reverse transcriptase mRNA expression in patients with hepatitis C. Hepatogastroenterology 60 (127):1713–16.
- Boonmars, T., Z. Wu, S. Boonjaruspinyo, A. Puapairoj, B. Kaewsamut, I. Nagano, S. Pinlaor, P. Yongvanit, O. Wonkchalee, A. Juasook, et al. 2011. Involvement of c-Ski oncoprotein in carcinogenesis of cholangiocacinoma induced by Opisthorchis viverrini and N-nitrosodimethylamine. Pathol. Oncol. Res. 17 (2):219–27. doi:10.1007/s12253-010-9300-8.
- Borska, L., C. Andrys, J. Krejsek, K. Hamakova, J. Kremlacek, V. Palicka, D. Ranna, and Z. Fiala. 2010. Genotoxic and apoptotic effects of Goeckerman therapy for psoriasis. Int. J. Dermatol. 49 (3):289–94. doi:10.1111/j.1365-4632.2009.04258.x.
- Boss, I. W., P. E. Nadeau, J. R. Abbott, Y. Yang, A. Mergia, and R. Renne. 2011. A Kaposi’s sarcoma-associated herpesvirus-encoded ortholog of microRNA miR-155 induces human splenic B-cell expansion in NOD/LtSz-scid IL2Rgammanull mice. J. Virol. 85 (19):9877–86. doi:10.1128/JVI.05558-11.
- Botelho, M., A. C. Ferreira, M. J. Oliveira, A. Domingues, J. C. Machado, and J. M. Da Costa. 2009. Schistosoma haematobium total antigen induces increased proliferation, migration and invasion, and decreases apoptosis of normal epithelial cells. Int. J. Parasitol. 39 (10):1083–91. doi:10.1016/j.ijpara.2009.02.016.
- Botelho, M. C., R. Ribeiro, N. Vale, P. Oliveira, R. Medeiros, C. Lopes, J. C. Machado, and J. M. Correia Da Costa. 2012. Inactivation of estrogen receptor by Schistosoma haematobium total antigen in bladder urothelial cells. Oncol. Rep. 27 (2):356–62. doi:10.3892/or.2011.1552.
- Bradley, C., R. van der Meer, N. Roodi, H. Yan, M. B. Chandrasekharan, Z. W. Sun, R. L. Mernaugh, and F. F. Parl. 2007. Carcinogen-induced histone alteration in normal human mammary epithelial cells. Carcinogenesis 28 (10):2184–92. doi:10.1093/carcin/bgm100.
- Breton, C. V., K. D. Siegmund, B. R. Joubert, X. Wang, W. Qui, V. Carey, W. Nystad, S. E. Haberg, C. Ober, D. Nicolae, et al.; Bridge consortium Asthma. 2014. Prenatal tobacco smoke exposure is associated with childhood DNA CpG methylation. PLoS ONE. 9 (6):e99716. doi:10.1371/journal.pone.0099716.
- Bromer, J. G., J. Wu, Y. Zhou, and H. S. Taylor. 2009. Hypermethylation of homeobox A10 by in utero diethylstilbestrol exposure: an epigenetic mechanism for altered developmental programming. Endocrinology 150 (7):3376–82. doi:10.1210/en.2009-0071.
- Bustaffa, E., A. Stoccoro, F. Bianchi, and L. Migliore. 2014. Genotoxic and epigenetic mechanisms in arsenic carcinogenicity. Arch. Toxicol. 88 (5):1043–67. doi:10.1007/s00204-014-1233-7.
- Cai, B. X., S. L. Jin, D. Luo, X. F. Lin, and J. Gao. 2009. Ginsenoside Rb1 suppresses ultraviolet radiation-induced apoptosis by inducing DNA repair. Biol. Pharm. Bull. 32 (5):837–41. doi:10.1248/bpb.32.837.
- Carignan, D., O. Desy, and P. O. de Campos-Lima. 2012. The dysregulation of the monocyte/macrophage effector function induced by isopropanol is mediated by the defective activation of distinct members of the AP-1 family of transcription factors. Toxicol. Sci. 125 (1):144–56. doi:10.1093/toxsci/kfr283.
- Cermelli, S., A. Ruggieri, J. A. Marrero, G. N. Ioannou, and L. Beretta. 2011. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 6 (8):e23937. doi:10.1371/journal.pone.0023937.
- Chaloupka, K., N. Harper, V. Krishnan, M. Santostefano, L. V. Rodriguez, and S. Safe. 1993. Synergistic activity of polynuclear aromatic hydrocarbon mixtures as aryl hydrocarbon (Ah) receptor agonists. Chem. Biol. Interact. 89 (2–3):141–58.
- Chappell, G., T. Kobets, B. O’Brien, N. Tretyakova, D. Sangaraju, O. Kosyk, K. G. Sexton, W. Bodnar, I. P. Pogribny, and I. Rusyn. 2014. Epigenetic events determine tissue-specific toxicity of inhalational exposure to the genotoxic chemical 1,3-butadiene in male C57BL/6J mice. Toxicol. Sci. 142 (2):375–84. doi:10.1093/toxsci/kfu191.
- Chen, C. D., Z. L. Wu, J. K. Chen, and W. D. Ji. 2004. Crystalline nickel sulfide-induced genomic instability in transformed human broncho-epithelial cells. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 22 (1):57–59.
- Chen, D., T. Kluz, L. Fang, X. Zhang, H. Sun, C. Jin, and M. Costa. 2016. Hexavalent chromium (Cr(VI)) down-regulates acetylation of histone H4 at Lysine 16 through induction of stressor protein Nupr1. PLoS ONE 11 (6):e0157317. doi:10.1371/journal.pone.0157317.
- Chen, J., A. Zmijewska, D. Zhi, and R. B. Mannon. 2015. Cyclosporine-mediated allograft fibrosis is associated with micro-RNA-21 through AKT signaling. Transpl. Int. 28 (2):232–45. doi:10.1111/tri.12471.
- Chen, M. H., L. C. Hsu, J. L. Wu, C. W. Yeh, J. N. Tsai, Y. C. Hseu, and L. S. Hsu. 2014a. Exposure to benzidine caused apoptosis and malformation of telencephalon region in zebrafish. Environ. Toxicol. 29 (12):1428–36. doi:10.1002/tox.21873.
- Chen, Y., A. Schieppati, X. Chen, G. Cai, J. Zamora, G. A. Giuliano, N. Braun, and A. Perna. 2014b. Immunosuppressive treatment for idiopathic membranous nephropathy in adults with nephrotic syndrome. Cochrane Database Syst Rev 10:Cd004293. doi:10.1002/14651858.CD004293.pub3.
- Cheng, A. S., M. S. Li, W. Kang, V. Y. Cheng, J. L. Chou, S. S. Lau, M. Y. Go, C. C. Lee, T. K. Ling, E. K. Ng, et al. 2013. Helicobacter pylori causes epigenetic dysregulation of FOXD3 to promote gastric carcinogenesis. Gastroenterology 144 (1):122–133 e9. doi:10.1053/j.gastro.2012.10.002.
- Chervona, Y., A. Arita, and M. Costa. 2012. Carcinogenic metals and the epigenome: understanding the effect of nickel, arsenic, and chromium. Metallomics 4 (7):619–27. doi:10.1039/c2mt20033c.
- Chiang, S. L., P. H. Chen, C. H. Lee, A. M. Ko, K. W. Lee, Y. C. Lin, P. S. Ho, H. P. Tu, D. C. Wu, T. Y. Shieh, et al. 2008. Up-regulation of inflammatory signalings by areca nut extract and role of cyclooxygenase-2-1195G>a polymorphism reveal risk of oral cancer. Cancer Res. 68 (20):8489–98. doi:10.1158/0008-5472.CAN-08-0823.
- Chiariotti, L., T. Angrisano, S. Keller, E. Florio, O. Affinito, P. Pallante, C. Perrino, R. Pero, and F. Lembo. 2013. Epigenetic modifications induced by helicobacter pylori infection through a direct microbe-gastric epithelial cells cross-talk. Med. Microbiol. Immunol. 202 (5):327–37. doi:10.1007/s00430-013-0301-6.
- Chieco-Bianchi, L., D. Saggioro, A. Del Mistro, A. Montaldo, F. Majone, and A. G. Levis. 1988. Chromosome damage induced in cord blood T-lymphocytes infected in vitro by HTLV-I. Leukemia 2 (12 Suppl):223S–232S.
- Chiou, Y. H., S. H. Liou, R. H. Wong, C. Y. Chen, and H. Lee. 2015. Nickel may contribute to EGFR mutation and synergistically promotes tumor invasion in EGFR-mutated lung cancer via nickel-induced microRNA-21 expression. Toxicol. Lett. 237 (1):46–54. doi:10.1016/j.toxlet.2015.05.019.
- Chiu, W. C., C. M. Chen, T. T. Cheng, H. L. You, S. F. Yu, L. H. Weng, H. Y. Huang, C. C. Huang, and C. J. Chen. 2013. EBV-encoded small RNA1 and nonresolving inflammation in rheumatoid arthritis. Kaohsiung J. Med. Sci. 29 (11):606–10. doi:10.1016/j.kjms.2013.04.002.
- Choi, I. S., K. Yu, J. Kim, E. De Guzman, D. J. Weisenberger, S. Oghamian, H. J. Kim, K. H. Lee, C. Carroll, B. N. Trinh, et al. 2013. Alterations in deoxyribonucleic acid (DNA) methylation patterns of Calca, Timp3, Mmp2, and Igf2r are associated with chronic cystitis in a cyclophosphamide-induced mouse model. Urology 82 (1):253.e9-15. doi:10.1016/j.urology.2013.04.010.
- Chokkalingam, A. P., and P. A. Buffler. 2008. Genetic susceptibility to childhood leukaemia. Radiat Prot Dosimetry 132 (2):119–29. doi:10.1093/rpd/ncn255.
- Chung, J. H., and H. C. Eun. 2007. Angiogenesis in skin aging and photoaging. J. Dermatol. 34 (9):593–600. doi:10.1111/j.1346-8138.2007.00341.x.
- Clippinger, A. J., T. L. Gearhart, and M. J. Bouchard. 2009. Hepatitis B virus X protein modulates apoptosis in primary rat hepatocytes by regulating both NF-kappaB and the mitochondrial permeability transition pore. J. Virol. 83 (10):4718–31. doi:10.1128/JVI.02590-08.
- Coates, P. J., S. A. Lorimore, K. J. Lindsay, and E. G. Wright. 2003. Tissue-specific p53 responses to ionizing radiation and their genetic modification: the key to tissue-specific tumour susceptibility? J. Pathol. 201 (3):377–88. doi:10.1002/path.1456.
- Collier, J. M., O. Selmin, P. D. Johnson, and R. B. Runyan. 2003. Trichloroethylene effects on gene expression during cardiac development. Birth Defects Res. Part A Clin. Mol. Teratol. 67 (7):488–95. doi:10.1002/bdra.10073.
- D’Addario, C., S. Johansson, S. Candeletti, P. Romualdi, S. O. Ogren, L. Terenius, and T. J. Ekstrom. 2011. Ethanol and acetaldehyde exposure induces specific epigenetic modifications in the prodynorphin gene promoter in a human neuroblastoma cell line. Faseb J. 25 (3):1069–75. doi:10.1096/fj.10-168534.
- Damiani, L. A., C. M. Yingling, S. Leng, P. E. Romo, J. Nakamura, and S. A. Belinsky. 2008. Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Res. 68 (21):9005–14. doi:10.1158/0008-5472.CAN-08-1276.
- de Conti, A., V. Tryndyak, M. I. Churchwell, S. Melnyk, J. R. Latendresse, L. Muskhelishvili, F. A. Beland, and I. P. Pogribny. 2014. Genotoxic, epigenetic, and transcriptomic effects of tamoxifen in mouse liver. Toxicology 325:12–20. doi:10.1016/j.tox.2014.08.004.
- Deng, R. X., X. H. Ren, J. W. Ruan, J. Zheng, J. C. Zhong, W. X. Lu, X. Q. Zou, and J. J. Liu. 2017. An investigation of trichloroethylene-induced effects on histone methylation in L-02 hepatic cells. Zhonghua Yu Fang Yi Xue Za Zhi 51 (4):347–52. doi:10.3760/cma.j.issn.0253-9624.2017.04.013.
- Dertinger, S. D., A. E. Silverstone, and T. A. Gasiewicz. 1998. Influence of aromatic hydrocarbon receptor-mediated events on the genotoxicity of cigarette smoke condensate. Carcinogenesis 19 (11):2037–42. doi:10.1093/carcin/19.11.2037.
- Desy, O., D. Carignan, M. Caruso, and P. O. de Campos-Lima. 2008. Immunosuppressive effect of isopropanol: down-regulation of cytokine production results from the alteration of discrete transcriptional pathways in activated lymphocytes. J. Immunol. 181 (4):2348–55. doi:10.4049/jimmunol.181.4.2348.
- Dodmane, P. R., L. L. Arnold, S. Kakiuchi-Kiyota, F. Qiu, X. Liu, S. I. Rennard, and S. M. Cohen. 2013. Cytotoxicity and gene expression changes induced by inorganic and organic trivalent arsenicals in human cells. Toxicology 312:18–29. doi:10.1016/j.tox.2013.07.008.
- Dreher, A., M. Rossing, B. Kaczkowski, D. K. Andersen, T. J. Larsen, M. K. Christophersen, F. C. Nielsen, and B. Norrild. 2011. Differential expression of cellular microRNAs in HPV 11, −16, and −45 transfected cells. Biochem. Biophys. Res. Commun. 412 (1):20–25. doi:10.1016/j.bbrc.2011.07.011.
- Driscoll, K. E., L. C. Deyo, B. W. Howard, J. Poynter, and J. M. Carter. 1995. Characterizing mutagenesis in the hprt gene of rat alveolar epithelial cells. Exp. Lung Res. 21 (6):941–56.
- Dupuy, S., M. Lambert, D. Zucman, S. P. Choukem, S. Tognarelli, C. Pages, C. Lebbe, and S. Caillat-Zucman. 2012. Human Herpesvirus 8 (HHV8) sequentially shapes the NK cell repertoire during the course of asymptomatic infection and Kaposi sarcoma. PLoS Pathog. 8 (1):e1002486. doi:10.1371/journal.ppat.1002486.
- El-Sabban, M. E., R. A. Merhi, H. A. Haidar, B. Arnulf, H. Khoury, J. Basbous, J. Nijmeh, H. de The, O. Hermine, and A. Bazarbachi. 2002. Human T-cell lymphotropic virus type 1-transformed cells induce angiogenesis and establish functional gap junctions with endothelial cells. Blood 99 (9):3383–89. doi:10.1182/blood.v99.9.3383.
- Fang, M. Z., W. Mar, and M. H. Cho. 2002. Cadmium affects genes involved in growth regulation during two-stage transformation of Balb/3T3 cells. Toxicology 177 (2–3):253–65.
- Fang, X., C. Thornton, B. E. Scheffler, and K. L. Willett. 2013. Benzo[a]pyrene decreases global and gene specific DNA methylation during zebrafish development. Environ. Toxicol. Pharmacol. 36 (1):40–50. doi:10.1016/j.etap.2013.02.014.
- Farkas, A., and L. Kemeny. 2013. Alcohol, liver, systemic inflammation and skin: a focus on patients with psoriasis. Skin Pharmacol Physiol 26 (3):119–26. doi:10.1159/000348865.
- Faxuan, W., Z. Qin, Z. Dinglun, Z. Tao, R. Xiaohui, Z. Liqiang, and L. Yajia. 2012. Altered microRNAs expression profiling in experimental silicosis rats. J Toxicol Sci 37 (6):1207–15.
- Feng, Q., J. E. Stern, S. E. Hawes, H. Lu, M. Jiang, and N. B. Kiviat. 2010. DNA methylation changes in normal liver tissues and hepatocellular carcinoma with different viral infection. Exp. Mol. Pathol. 88 (2):287–92. doi:10.1016/j.yexmp.2010.01.002.
- Fertuck, K. C., J. B. Matthews, and T. R. Zacharewski. 2001. Hydroxylated benzo[a]pyrene metabolites are responsible for in vitro estrogen receptor-mediated gene expression induced by benzo[a]pyrene, but do not elicit uterotrophic effects in vivo. Toxicol. Sci. 59 (2):231–40. doi:10.1093/toxsci/59.2.231.
- Figueiredo, J. C., M. V. Grau, K. Wallace, A. J. Levine, L. Shen, R. Hamdan, X. Chen, R. S. Bresalier, G. McKeown-Eyssen, R. W. Haile, et al. 2009. Global DNA hypomethylation (LINE-1) in the normal colon and lifestyle characteristics and dietary and genetic factors. Cancer Epidemiol. Biomarkers Prev. 18 (4):1041–49. doi:10.1158/1055-9965.EPI-08-0926.
- Forte, E., R. E. Salinas, C. Chang, T. Zhou, S. D. Linnstaedt, E. Gottwein, C. Jacobs, D. Jima, Q. J. Li, S. S. Dave, et al. 2012. The Epstein-Barr virus (EBV)-induced tumor suppressor microRNA MiR-34a is growth promoting in EBV-infected B cells. J. Virol. 86 (12):6889–98. doi:10.1128/jvi.07056-11.
- Friso, S., S. Lamon-Fava, H. Jang, E. J. Schaefer, R. Corrocher, and S. W. Choi. 2007. Oestrogen replacement therapy reduces total plasma homocysteine and enhances genomic DNA methylation in postmenopausal women. Br. J. Nutr. 97 (4):617–21. doi:10.1017/S0007114507433013.
- Gao, A., X. Zuo, Q. Liu, X. Lu, W. Guo, and L. Tian. 2010. Methylation of PARP-1 promoter involved in the regulation of benzene-induced decrease of PARP-1 mRNA expression. Toxicol. Lett. 195 (2–3):114–18. doi:10.1016/j.toxlet.2010.03.005.
- Gao, P., C. C. Wong, E. K. Tung, J. M. Lee, C. M. Wong, and I. O. Ng. 2011. Deregulation of microRNA expression occurs early and accumulates in early stages of HBV-associated multistep hepatocarcinogenesis. J. Hepatol. 54 (6):1177–84. doi:10.1016/j.jhep.2010.09.023.
- Garcia-Quispes, W. A., G. Perez-Machado, A. Akdi, S. Pastor, P. Galofre, F. Biarnes, J. Castell, A. Velazquez, and R. Marcos. 2011. Association studies of OGG1, XRCC1, XRCC2 and XRCC3 polymorphisms with differentiated thyroid cancer. Mutat. Res. 709-710:67–72. doi:10.1016/j.mrfmmm.2011.03.003.
- Gargouri, B., J. Van Pelt, F. El Feki Ael, H. Attia, and S. Lassoued. 2009. Induction of Epstein-Barr virus (EBV) lytic cycle in vitro causes oxidative stress in lymphoblastoid B cell lines. Mol. Cell. Biochem. 324 (1–2):55–63. doi:10.1007/s11010-008-9984-1.
- Geng, Y., S. M. Savage, L. J. Johnson, J. Seagrave, and M. L. Sopori. 1995. Effects of nicotine on the immune response. I. Chronic exposure to nicotine impairs antigen receptor-mediated signal transduction in lymphocytes. Toxicol. Appl. Pharmacol. 135 (2):268–78. doi:10.1006/taap.1995.1233.
- Gigek, C. O., E. S. Chen, D. Q. Calcagno, F. Wisnieski, R. R. Burbano, and M. A. Smith. 2012. Epigenetic mechanisms in gastric cancer. Epigenomics 4 (3):279–94. doi:10.2217/epi.12.22.
- Giotopoulos, G., C. McCormick, C. Cole, A. Zanker, M. Jawad, R. Brown, and M. Plumb. 2006. DNA methylation during mouse hemopoietic differentiation and radiation-induced leukemia. Exp. Hematol. 34 (11):1462–70. doi:10.1016/j.exphem.2006.06.008.
- Grafodatskaya, D., S. Choufani, J. C. Ferreira, D. T. Butcher, Y. Lou, C. Zhao, S. W. Scherer, and R. Weksberg. 2010. EBV transformation and cell culturing destabilizes DNA methylation in human lymphoblastoid cell lines. Genomics 95 (2):73–83. doi:10.1016/j.ygeno.2009.12.001.
- Greco, D., N. Kivi, K. Qian, S. K. Leivonen, P. Auvinen, and E. Auvinen. 2011. Human papillomavirus 16 E5 modulates the expression of host microRNAs. PLoS ONE 6 (7):e21646. doi:10.1371/journal.pone.0021646.
- Gualandi, G., L. Giselico, M. Carloni, F. Palitti, P. Mosesso, and A. M. Alfonsi. 2001. Enhancement of genetic instability in human B cells by Epstein-Barr virus latent infection. Mutagenesis 16 (3):203–08. doi:10.1093/mutage/16.3.203.
- Gueta, K., N. Molotski, N. Gerchikov, E. Mor, S. Savion, A. Fein, V. Toder, N. Shomron, and A. Torchinsky. 2010. Teratogen-induced alterations in microRNA-34, microRNA-125b and microRNA-155 expression: correlation with embryonic p53 genotype and limb phenotype. BMC Dev. Biol. 10:20. doi:10.1186/1471-213X-10-20.
- Guo, G. H., D. M. Tan, P. A. Zhu, and F. Liu. 2009. Hepatitis B virus X protein promotes proliferation and upregulates TGF-beta1 and CTGF in human hepatic stellate cell line, LX-2. Hbpd Int 8 (1):59–64.
- Guo, W., E. L. Crossey, L. Zhang, S. Zucca, O. L. George, C. F. Valenzuela, and X. Zhao. 2011. Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS ONE 6 (5):e19351. doi:10.1371/journal.pone.0019351.
- Hama, R., Y. Watanabe, K. Shinada, Y. Yamada, Y. Ogata, Y. Yoshida, T. Tamura, T. Hiraishi, R. Oikawa, J. Sakurai, et al. 2012. Characterization of DNA hypermethylation in two cases of peritoneal mesothelioma. Tumour Biol. 33 (6):2031–40. doi:10.1007/s13277-012-0462-8.
- Hammerschmidt, W. 2015. The epigenetic life cycle of epstein-barr virus. Curr. Top. Microbiol. Immunol. 390 (Pt 1):103–17. doi:10.1007/978-3-319-22822-8_6.
- Hernandez-Ramon, E. E., N. A. Sandoval, K. John, J. M. Cline, C. E. Wood, R. A. Woodward, and M. C. Poirier. 2014. Tamoxifen-DNA adduct formation in monkey and human reproductive organs. Carcinogenesis 35 (5):1172–76. doi:10.1093/carcin/bgu029.
- Hicks, S. D., F. A. Middleton, and M. W. Miller. 2010. Ethanol-induced methylation of cell cycle genes in neural stem cells. J. Neurochem. 114 (6):1767–80. doi:10.1111/j.1471-4159.2010.06886.x.
- Hongping, D., L. Jianlin, Z. Meibian, W. Wei, J. Lifen, C. Shijie, Z. Wei, W. Baohong, and H. Jiliang. 2006. Detecting the cytogenetic effects in workers occupationally exposed to vincristine with four genetic tests. Mutat. Res. 599 (1–2):152–59. doi:10.1016/j.mrfmmm.2006.02.003.
- Hossain, M. B., M. Vahter, G. Concha, and K. Broberg. 2012. Low-level environmental cadmium exposure is associated with DNA hypomethylation in Argentinean women. Environ. Health Perspect. 120 (6):879–84. doi:10.1289/ehp.1104600.
- Hou, Z., G. Liu, F. Zheng, and D. Tan. 2009. HBx gene inducing hepatocellular carcinoma in vivo and its mechanism. Zhong Nan Da Xue Xue Bao Yi Xue Ban 34 (4):282–88.
- Hsu, C. H., K. L. Peng, H. C. Jhang, C. H. Lin, S. Y. Wu, C. M. Chiang, S. C. Lee, W. C. Yu, and L. J. Juan. 2012. The HPV E6 oncoprotein targets histone methyltransferases for modulating specific gene transcription. Oncogene 31 (18):2335–49. doi:10.1038/onc.2011.415.
- Hsu, P. Y., D. E. Deatherage, B. A. Rodriguez, S. Liyanarachchi, Y. I. Weng, T. Zuo, J. Liu, A. S. Cheng, and T. H. Huang. 2009. Xenoestrogen-induced epigenetic repression of microRNA-9-3 in breast epithelial cells. Cancer Res. 69 (14):5936–45. doi:10.1158/0008-5472.CAN-08-4914.
- Hu, G., P. Li, Y. Li, T. Wang, X. Gao, W. Zhang, and G. Jia. 2016. Methylation levels of P16 and TP53 that are involved in DNA strand breakage of 16HBE cells treated by hexavalent chromium. Toxicol. Lett. 249:15–21. doi:10.1016/j.toxlet.2016.03.003.
- Hu, J., H. Ma, W. Zhang, Z. Yu, G. Sheng, and J. Fu. 2014. Effects of benzene and its metabolites on global DNA methylation in human normal hepatic L02 cells. Environ. Toxicol. 29 (1):108–16. doi:10.1002/tox.20777.
- Hu, Y., K. L. Willett, I. A. Khan, B. E. Scheffler, and A. K. Dasmahapatra. 2009. Ethanol disrupts chondrification of the neurocranial cartilages in medaka embryos without affecting aldehyde dehydrogenase 1A2 (Aldh1A2) promoter methylation. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 150 (4):495–502. doi:10.1016/j.cbpc.2009.07.007.
- Huan, L. C., J. C. Wu, B. H. Chiou, C. H. Chen, N. Ma, C. Y. Chang, Y. K. Tsen, and S. C. Chen. 2014. MicroRNA regulation of DNA repair gene expression in 4-aminobiphenyl-treated HepG2 cells. Toxicology 322:69–77. doi:10.1016/j.tox.2014.05.003.
- Huang, D., Y. Zhang, Y. Qi, C. Chen, and W. Ji. 2008. Global DNA hypomethylation, rather than reactive oxygen species (ROS), a potential facilitator of cadmium-stimulated K562 cell proliferation. Toxicol. Lett. 179 (1):43–47. doi:10.1016/j.toxlet.2008.03.018.
- Huang, X. Y., J. G. Yao, H. D. Huang, C. Wang, Y. Ma, Q. Xia, and X. D. Long. 2013. MicroRNA-429 Modulates Hepatocellular Carcinoma Prognosis and Tumorigenesis. Gastroenterol Res Pract 2013:804128. doi:10.1155/2013/804128.
- Humphries, B., Z. Wang, and C. Yang. 2016. The role of microRNAs in metal carcinogen-induced cell malignant transformation and tumorigenesis. Food Chem. Toxicol. 98 (Pt A):58–65. doi:10.1016/j.fct.2016.02.012.
- Hung, S. Y., H. H. Lin, K. T. Yeh, and J. G. Chang. 2014. Histone-modifying genes as biomarkers in hepatocellular carcinoma. Int J Clin Exp Pathol 7 (5):2496–507.
- IARC. 2012a. Pharmaceuticals. IARC Monogr. Eval. Carcinogenic Risks Hum. 100A: 1–437.
- IARC. 2012b. Biological agents. IARC Monogr. Eval. Carcinogenic Risks Hum. 100B: 1–441.
- IARC. 2012c. Arsenic, metals, fibres, and dusts. IARC Monogr. Eval. Carcinogenic Risks Hum. 100C: 11–465.
- IARC. 2012d. Radiation. IARC Monogr. Eval. Carcinogenic Risks Hum. 100D: 1–437.
- IARC. 2012e. Personal habits and indoor combustions vol 100E. IARC Monogr. Eval. Carcinogenic Risks Hum. 100E: 1–575.
- IARC. 2012f. Chemical agents and related occupations. IARC Monogr. Eval. Carcinogenic Risks Hum. 100F: 1–599.
- IARC. 2014a. Diesel and gasoline engine exhausts and some nitroarenes. IARC Monogr. Eval. Carcinogenic Risks Hum. 105: 9–699.
- IARC. 2014b. Trichloroethylene, tetrachloroethylene, and some other chlorinated agents. IARC Monogr. Eval. Carcinogenic Risks Hum. 106: 1–514.
- Ilyenko, I., O. Lyaskivska, and D. Bazyka. 2011. Analysis of relative telomere length and apoptosis in humans exposed to ionising radiation. Exp. Oncol. 33 (4):235–38.
- Ishida, H., T. Tatsumi, A. Hosui, T. Nawa, T. Kodama, S. Shimizu, H. Hikita, N. Hiramatsu, T. Kanto, N. Hayashi, et al. 2011. Alterations in microRNA expression profile in HCV-infected hepatoma cells: involvement of miR-491 in regulation of HCV replication via the PI3 kinase/Akt pathway. Biochem. Biophys. Res. Commun. 412 (1):92–97. doi:10.1016/j.bbrc.2011.07.049.
- Ishihama, M., T. Toyooka, and Y. Ibuki. 2008. Generation of phosphorylated histone H2AX by benzene metabolites. Toxicol In Vitro 22 (8):1861–68. doi:10.1016/j.tiv.2008.09.005.
- Jantschitsch, C., S. Majewski, A. Maeda, T. Schwarz, and A. Schwarz. 2009. Infrared radiation confers resistance to UV-induced apoptosis via reduction of DNA damage and upregulation of antiapoptotic proteins. J. Invest. Dermatol. 129 (5):1271–79. doi:10.1038/jid.2008.362.
- Jiang, J., L. J. Zhao, C. Zhao, G. Zhang, Y. Zhao, J. R. Li, X. P. Li, and L. H. Wei. 2012. Hypomethylated CpG around the transcription start site enables TERT expression and HPV16 E6 regulates TERT methylation in cervical cancer cells. Gynecol. Oncol. 124 (3):534–41. doi:10.1016/j.ygyno.2011.11.023.
- Jiang, W., Y. Lu, Z. Chen, S. Chen, M. Zhang, L. Jin, J. Lou, and J. He. 2008. Studying the genotoxicity of vincristine on human lymphocytes using comet assay, micronucleus assay and TCR gene mutation test in vitro. Toxicology 252 (1–3):113–17. doi:10.1016/j.tox.2008.07.057.
- Jiang, Y., J. Chen, J. Tong, and T. Chen. 2014. Trichloroethylene-induced gene expression and DNA methylation changes in B6C3F1 mouse liver. PLoS ONE 9 (12):e116179. doi:10.1371/journal.pone.0116179.
- Kamranvar, S. A., and M. G. Masucci. 2011. The Epstein-Barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia 25 (6):1017–25. doi:10.1038/leu.2011.35.
- Kasuga, F., S. Inoue, T. Asano, and S. Kumagai. 1992. Effects of isopropanol on the development of inflammatory reactions in rats. Food Chem. Toxicol. 30 (7):631–34.
- Kataoka, K., Y. Nagata, A. Kitanaka, Y. Shiraishi, T. Shimamura, J. Yasunaga, Y. Totoki, K. Chiba, A. Sato-Otsubo, G. Nagae, et al. 2015. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 47 (11):1304–15. doi:10.1038/ng.3415.
- Kerkvliet, N. I. 2002. Recent advances in understanding the mechanisms of TCDD immunotoxicity. Int. Immunopharmacol. 2 (2–3):277–91.
- Khan, I., N. Kumar, I. Pant, S. Narra, and P. Kondaiah. 2012. Activation of TGF-beta pathway by areca nut constituents: a possible cause of oral submucous fibrosis. PLoS ONE 7 (12):e51806. doi:10.1371/journal.pone.0051806.
- Khoontawad, J., C. Wongkham, Y. Hiraku, P. Yongvanit, S. Prakobwong, T. Boonmars, P. Pinlaor, and S. Pinlaor. 2010. Proteomic identification of peroxiredoxin 6 for host defence against Opisthorchis viverrini infection. Parasite Immunol. 32 (5):314–23. doi:10.1111/j.1365-3024.2009.01189.x.
- Kim, M. K., S. Lee, E. J. Kim, K. H. Kong, D. H. Lee, and J. H. Chung. 2013. Topical application of anacardic acid (6-nonadecyl salicylic acid) reduces UV-induced histone modification, MMP-13, MMP-9, COX-2 and TNF-alpha expressions in hairless mice skin. J. Dermatol. Sci. 70 (1):64–67. doi:10.1016/j.jdermsci.2012.11.008.
- Kim, M. K., J. M. Shin, H. C. Eun, and J. H. Chung. 2009. The role of p300 histone acetyltransferase in UV-induced histone modifications and MMP-1 gene transcription. PLoS ONE 4 (3):e4864. doi:10.1371/journal.pone.0004864.
- Knoops, L., R. Haas, S. de Kemp, D. Majoor, A. Broeks, E. Eldering, J. P. de Boer, M. Verheij, C. van Ostrom, A. de Vries, et al. 2007. In vivo p53 response and immune reaction underlie highly effective low-dose radiotherapy in follicular lymphoma. Blood 110 (4):1116–22. doi:10.1182/blood-2007-01-067579.
- Korkalainen, M., K. Huumonen, J. Naarala, M. Viluksela, and J. Juutilainen. 2012. Dioxin induces genomic instability in mouse embryonic fibroblasts. PLoS ONE 7 (5):e37895. doi:10.1371/journal.pone.0037895.
- Koturbash, I., A. Boyko, R. Rodriguez-Juarez, R. J. McDonald, V. P. Tryndyak, I. Kovalchuk, I. P. Pogribny, and O. Kovalchuk. 2007. Role of epigenetic effectors in maintenance of the long-term persistent bystander effect in spleen in vivo. Carcinogenesis 28 (8):1831–38. doi:10.1093/carcin/bgm053.
- Koturbash, I., A. Scherhag, J. Sorrentino, K. Sexton, W. Bodnar, J. A. Swenberg, F. A. Beland, F. Pardo-Manuel Devillena, I. Rusyn, and I. P. Pogribny. 2011a. Epigenetic mechanisms of mouse interstrain variability in genotoxicity of the environmental toxicant 1,3-butadiene. Toxicol. Sci. 122 (2):448–56. doi:10.1093/toxsci/kfr133.
- Koturbash, I., A. Scherhag, J. Sorrentino, K. Sexton, W. Bodnar, V. Tryndyak, J. R. Latendresse, J. A. Swenberg, F. A. Beland, I. P. Pogribny, et al. 2011b. Epigenetic alterations in liver of C57BL/6J mice after short-term inhalational exposure to 1,3-butadiene. Environ. Health Perspect. 119 (5):635–40. doi:10.1289/ehp.1002910.
- Koumbi, L., and P. Karayiannis. 2015. The epigenetic control of hepatitis B virus modulates the outcome of infection. Front Microbiol 6:1491. doi:10.3389/fmicb.2015.01491.
- Krakowczyk, L., S. Blamek, J. K. Strzelczyk, A. Plachetka, A. Maciejewski, S. Poltorak, and A. Wiczkowski. 2010. Effects of X-ray irradiation on methylation levels of p16, MGMT and APC genes in sporadic colorectal carcinoma and corresponding normal colonic mucosa. Med. Sci. Monit. 16 (10):CR469–74.
- Krewski, D., M. Al-Zoughool, M. Bird, N. Birkett, M. Billard, and B. Milton. 2019. Analysis of key characteristics of human carcinogens. In Tumour site concordance and mechanisms of carcinogenesis, ed. R. A. Baan, B. W. Stewart, and K. Straif. Lyon: IARC Press.
- Kumar, D., S. R. Salian, G. Kalthur, S. Uppangala, S. Kumari, S. Challapalli, S. G. Chandraguthi, H. Krishnamurthy, N. Jain, P. Kumar, et al. 2013. Semen abnormalities, sperm DNA damage and global hypermethylation in health workers occupationally exposed to ionizing radiation. PLoS ONE 8 (7):e69927. doi:10.1371/journal.pone.0069927.
- Kusano, M., M. Toyota, H. Suzuki, K. Akino, F. Aoki, M. Fujita, M. Hosokawa, Y. Shinomura, K. Imai, and T. Tokino. 2006. Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein-Barr virus. Cancer 106 (7):1467–79. doi:10.1002/cncr.21789.
- Kyo, S., M. Takakura, T. Kanaya, W. Zhuo, K. Fujimoto, Y. Nishio, A. Orimo, and M. Inoue. 1999. Estrogen activates telomerase. Cancer Res. 59 (23):5917–21.
- Lai, C. K., K. S. Jeng, K. Machida, Y. S. Cheng, and M. M. Lai. 2008. Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology 370 (2):295–309. doi:10.1016/j.virol.2007.08.037.
- Lai, Z. L., Y. A. Tsou, S. R. Fan, M. H. Tsai, H. L. Chen, N. W. Chang, J. C. Cheng, and C. M. Chen. 2014. Methylation-associated gene silencing of RARB in areca carcinogens induced mouse oral squamous cell carcinoma. Biomed Res Int 2014:378358. doi:10.1155/2014/378358.
- Lan, Q., M. T. Smith, X. Tang, W. Guo, R. Vermeulen, Z. Ji, W. Hu, A. E. Hubbard, M. Shen, C. M. McHale, et al. 2015. Chromosome-wide aneuploidy study of cultured circulating myeloid progenitor cells from workers occupationally exposed to formaldehyde. Carcinogenesis 36 (1):160–67. doi:10.1093/carcin/bgu229.
- Lassoued, S., R. Ben Ameur, W. Ayadi, B. Gargouri, R. Ben Mansour, and H. Attia. 2008. Epstein-Barr virus induces an oxidative stress during the early stages of infection in B lymphocytes, epithelial, and lymphoblastoid cell lines. Mol. Cell. Biochem. 313 (1–2):179–86. doi:10.1007/s11010-008-9755-z.
- Lentz, S., R. Eversole, J. M. Law, and J. C. Means. 2010. Cellular proliferation, cell death, and liver histology in Gambusia affinis after dietary exposure to benzidine and 2-aminofluorene. Int. J. Toxicol. 29 (3):247–58. doi:10.1177/1091581810363745.
- Leonard, A., and R. Lauwerys. 1987. Mutagenicity, carcinogenicity and teratogenicity of beryllium. Mutat. Res. 186 (1):35–42. doi:10.1016/0165-1110(87)90013-3.
- Li, D., Q. Wang, C. Liu, H. Duan, X. Zeng, B. Zhang, X. Li, J. Zhao, S. Tang, Z. Li, et al. 2012. Aberrant expression of miR-638 contributes to benzo(a)pyrene-induced human cell transformation. Toxicol. Sci. 125 (2):382–91. doi:10.1093/toxsci/kfr299.
- Li, G., Z. Wu, Y. Peng, X. Liu, J. Lu, L. Wang, Q. Pan, M. L. He, and X. P. Li. 2010. MicroRNA-10b induced by Epstein-Barr virus-encoded latent membrane protein-1 promotes the metastasis of human nasopharyngeal carcinoma cells. Cancer Lett. 299 (1):29–36. doi:10.1016/j.canlet.2010.07.021.
- Li, J., P. Kanju, M. Patterson, W. L. Chew, S. H. Cho, I. Gilmour, T. Oliver, R. Yasuda, A. Ghio, S. A. Simon, et al. 2011. TRPV4-mediated calcium influx into human bronchial epithelia upon exposure to diesel exhaust particles. Environ. Health Perspect. 119 (6):784–93. doi:10.1289/ehp.1002807.
- Liggett, T. E., A. A. Melnikov, J. R. Marks, and V. V. Levenson. 2011. Methylation patterns in cell-free plasma DNA reflect removal of the primary tumor and drug treatment of breast cancer patients. Int. J. Cancer 128 (2):492–99. doi:10.1002/ijc.25363.
- Lim, J. S., S. H. Park, and K. L. Jang. 2012. Hepatitis C virus Core protein overcomes stress-induced premature senescence by down-regulating p16 expression via DNA methylation. Cancer Lett. 321 (2):154–61. doi:10.1016/j.canlet.2012.01.044.
- Lin, M. H., F. P. Chou, H. P. Huang, J. D. Hsu, M. Y. Chou, and C. J. Wang. 2003. The tumor promoting effect of lime-piper betel quid in JB6 cells. Food Chem. Toxicol. 41 (11):1463–71.
- Lin, P. C., W. H. Chang, Y. H. Chen, C. C. Lee, Y. H. Lin, and J. G. Chang. 2011. Cytotoxic effects produced by arecoline correlated to epigenetic regulation in human K-562 cells. J. Toxicol. Environ. Health Part A 74 (11):737–45. doi:10.1080/15287394.2011.539123.
- Liu, L., P. Zou, L. Zheng, L. E. Linarelli, S. Amarell, A. Passaro, D. Liu, and Z. Cheng. 2015. Tamoxifen reduces fat mass by boosting reactive oxygen species. Cell Death Dis 6:e1586. doi:10.1038/cddis.2014.553.
- Liu, Q., L. Yang, C. Gong, G. Tao, H. Huang, J. Liu, H. Zhang, D. Wu, B. Xia, G. Hu, et al. 2011. Effects of long-term low-dose formaldehyde exposure on global genomic hypomethylation in 16HBE cells. Toxicol. Lett. 205 (3):235–40. doi:10.1016/j.toxlet.2011.05.1039.
- Liu, Y., Q. Lan, M. Shen, J. Jin, J. Mumford, D. Ren, and P. Keohavong. 2008. Aberrant gene promoter methylation in sputum from individuals exposed to smoky coal emissions. Anticancer Res. 28 (4B):2061–66.
- Liu, Y. X., X. D. Long, Z. F. Xi, Y. Ma, X. Y. Huang, J. G. Yao, C. Wang, T. Y. Xing, and Q. Xia. 2014. MicroRNA-24 modulates aflatoxin B1-related hepatocellular carcinoma prognosis and tumorigenesis. Biomed Res Int 2014:482926. doi:10.1155/2014/482926.
- Liu, Z., M. Hergenhahn, H. H. Schmeiser, G. N. Wogan, A. Hong, and M. Hollstein. 2004. Human tumor p53 mutations are selected for in mouse embryonic fibroblasts harboring a humanized p53 gene. Proc. Natl. Acad. Sci. U.S.A. 101 (9):2963–68. doi:10.1073/pnas.0308607101.
- Loilome, W., S. Kadsanit, N. Namwat, A. Techasen, A. Puapairoj, A. Dechakhamphu, C. Pinitsoontorn, and P. Yongvanit. 2012. Impaired antioxidant enzyme activity and increased DNA repair enzyme expression in hamster liver tissues related to cholangiocarcinoma development. Asian Pac. J. Cancer Prev. 13:Suppl:59–64.
- Long, X. D., X. Y. Huang, J. G. Yao, P. Liao, Y. J. Tang, Y. Ma, and Q. Xia. 2016. Polymorphisms in the precursor microRNAs and aflatoxin B1-related hepatocellular carcinoma. Mol. Carcinog. 55 (6):1060–72. doi:10.1002/mc.22350.
- Lu, K., L. B. Collins, H. Ru, E. Bermudez, and J. A. Swenberg. 2010. Distribution of DNA adducts caused by inhaled formaldehyde is consistent with induction of nasal carcinoma but not leukemia. Toxicol. Sci. 116 (2):441–51. doi:10.1093/toxsci/kfq061.
- Lu, K., H. Gul, P. B. Upton, B. C. Moeller, and J. A. Swenberg. 2012. Formation of hydroxymethyl DNA adducts in rats orally exposed to stable isotope labeled methanol. Toxicol. Sci. 126 (1):28–38. doi:10.1093/toxsci/kfr328.
- Maatta, J., M. L. Majuri, R. Luukkonen, A. Lauerma, K. Husgafvel-Pursiainen, H. Alenius, and K. Savolainen. 2005. Characterization of oak and birch dust-induced expression of cytokines and chemokines in mouse macrophage RAW 264.7 cells. Toxicology 215 (1–2):25–36. doi:10.1016/j.tox.2005.06.021.
- Machida, K., G. McNamara, K. T. Cheng, J. Huang, C. H. Wang, L. Comai, J. H. Ou, and M. M. Lai. 2010. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/Mre11/Rad50 DNA repair pathway in monocytes and hepatocytes. J. Immunol. 185 (11):6985–98. doi:10.4049/jimmunol.1000618.
- Majone, F., and K. T. Jeang. 2000. Clastogenic effect of the human T-cell leukemia virus type I Tax oncoprotein correlates with unstabilized DNA breaks. J. Biol. Chem. 275 (42):32906–10. doi:10.1074/jbc.C000538200.
- Makwana, O., L. Ahles, A. Lencinas, O. I. Selmin, and R. B. Runyan. 2013. Low-dose trichloroethylene alters cytochrome P450-2C subfamily expression in the developing chick heart. Cardiovasc. Toxicol. 13 (1):77–84. doi:10.1007/s12012-012-9180-0.
- Markunas, C. A., Z. Xu, S. Harlid, P. A. Wade, R. T. Lie, J. A. Taylor, and A. J. Wilcox. 2014. Identification of DNA methylation changes in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 122 (10):1147–53. doi:10.1289/ehp.1307892.
- Marta, G. N., B. Garicochea, A. L. Carvalho, J. M. Real, and L. P. Kowalski. 2015. MicroRNAs, cancer and ionizing radiation: Where are we?. Rev. Assoc. Med. Bras. (1992) 61 (3):275–81. doi:10.1590/1806-9282.61.03.275.
- Maruo, S., B. Zhao, E. Johannsen, E. Kieff, J. Zou, and K. Takada. 2011. Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proc. Natl. Acad. Sci. U.S.A. 108 (5):1919–24. doi:10.1073/pnas.1019599108.
- Maruyama, K., T. Fukushima, S. Mochizuki, K. Kawamura, and M. Miyauchi. 1992. Human T-cell leukemia virus type I as agent inducing genetic changes in infected human lymphocytes. Leukemia 6 (Suppl 3):60S–63S.
- Matsusaka, K., A. Kaneda, G. Nagae, T. Ushiku, Y. Kikuchi, R. Hino, H. Uozaki, Y. Seto, K. Takada, H. Aburatani, et al. 2011. Classification of Epstein-Barr virus-positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 71 (23):7187–97. doi:10.1158/0008-5472.CAN-11-1349.
- McClure, E. A., C. M. North, N. E. Kaminski, and J. I. Goodman. 2011. Changes in DNA methylation and gene expression during 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced suppression of the lipopolysaccharide-stimulated IgM response in splenocytes. Toxicol. Sci. 120 (2):339–48. doi:10.1093/toxsci/kfq396.
- McLaughlin-Drubin, M. E., C. P. Crum, and K. Munger. 2011. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. U.S.A. 108 (5):2130–35. doi:10.1073/pnas.1009933108.
- Meek, M. D., and G. L. Finch. 1999. Diluted mainstream cigarette smoke condensates activate estrogen receptor and aryl hydrocarbon receptor-mediated gene transcription. Environ. Res. 80 (1):9–17. doi:10.1006/enrs.1998.3872.
- Minarovits, J., A. Demcsak, F. Banati, and H. H. Niller. 2016. Epigenetic Dysregulation in Virus-Associated Neoplasms. Adv. Exp. Med. Biol. 879:71–90. doi:10.1007/978-3-319-24738-0_4.
- Minoia, C., E. Sturchio, B. Porro, B. Ficociello, A. Zambelli, and M. Imbriani. 2011. microRNAs as biological indicators of environmental and occupational exposure to asbestos. G Ital Med Lav Ergon 33 (4):420–34.
- Moles, R., and C. Nicot. 2015. The emerging role of miRNAs in HTLV-1 infection and ATLL pathogenesis. Viruses 7 (7):4047–74. doi:10.3390/v7072805.
- Morris, V. A., A. S. Punjabi, R. C. Wells, C. J. Wittkopp, R. Vart, and M. Lagunoff. 2012. The KSHV viral IL-6 homolog is sufficient to induce blood to lymphatic endothelial cell differentiation. Virology 428 (2):112–20. doi:10.1016/j.virol.2012.03.013.
- Naarala, J., J. P. Kasanen, P. Pasanen, A. L. Pasanen, A. Liimatainen, S. Pennanen, and J. Liesivuori. 2003. The effects of wood dusts on the redox status and cell death in mouse macrophages (RAW 264.7) and human leukocytes in vitro. J. Toxicol. Environ. Health Part A 66 (13):1221–35. doi:10.1080/15287390306406.
- Nandakumar, V., M. Vaid, T. O. Tollefsbol, and S. K. Katiyar. 2011. Aberrant DNA hypermethylation patterns lead to transcriptional silencing of tumor suppressor genes in UVB-exposed skin and UVB-induced skin tumors of mice. Carcinogenesis 32 (4):597–604. doi:10.1093/carcin/bgq282.
- Newbold, R. R. 2012. Prenatal exposure to diethylstilbestrol and long-term impact on the breast and reproductive tract in humans and mice. J Dev Orig Health Dis 3 (2):73–82. doi:10.1017/s2040174411000754.
- Niller, H. H., F. Banati, D. Salamon, and J. Minarovits. 2016. Epigenetic Alterations in Epstein-Barr Virus-Associated Diseases. Adv. Exp. Med. Biol. 879:39–69. doi:10.1007/978-3-319-24738-0_3.
- Niller, H. H., Z. Tarnai, G. Decsi, A. Zsedenyi, F. Banati, and J. Minarovits. 2014. Role of epigenetics in EBV regulation and pathogenesis. Future Microbiol 9 (6):747–56. doi:10.2217/fmb.14.41.
- Owens, C. V., Jr., C. Lambright, M. Cardon, L. E. Gray Jr., B. K. Gullett, and V. S. Wilson. 2006. Detection of androgenic activity in emissions from diesel fuel and biomass combustion. Environ. Toxicol. Chem. 25 (8):2123–31.
- Pal, S., S. J. Polyak, N. Bano, W. C. Qiu, R. L. Carithers, M. Shuhart, D. R. Gretch, and A. Das. 2010. Hepatitis C virus induces oxidative stress, DNA damage and modulates the DNA repair enzyme NEIL1. J. Gastroenterol. Hepatol. 25 (3):627–34. doi:10.1111/j.1440-1746.2009.06128.x.
- Palbykin, B., J. Borg, P. T. Caldwell, J. Rowles, A. J. Papoutsis, D. F. Romagnolo, and O. I. Selmin. 2011. Trichloroethylene induces methylation of the Serca2 promoter in H9c2 cells and embryonic heart. Cardiovasc. Toxicol. 11 (3):204–14. doi:10.1007/s12012-011-9113-3.
- Pan, H. L., Z. S. Wen, Y. C. Huang, X. Cheng, G. Z. Wang, Y. C. Zhou, Z. Y. Wang, Y. Q. Guo, Y. Cao, and G. B. Zhou. 2015. Down-regulation of microRNA-144 in air pollution-related lung cancer. Sci Rep 5:14331. doi:10.1038/srep14331.
- Pathak, S., N. Kedia-Mokashi, M. Saxena, R. D’Souza, A. Maitra, P. Parte, M. Gill-Sharma, and N. Balasinor. 2009. Effect of tamoxifen treatment on global and insulin-like growth factor 2-H19 locus-specific DNA methylation in rat spermatozoa and its association with embryo loss. Fertil. Steril. 91 (5 Suppl):2253–63. doi:10.1016/j.fertnstert.2008.07.1709.
- Pavio, N., S. Battaglia, D. Boucreux, B. Arnulf, R. Sobesky, O. Hermine, and C. Brechot. 2005. Hepatitis C virus core variants isolated from liver tumor but not from adjacent non-tumor tissue interact with Smad3 and inhibit the TGF-beta pathway. Oncogene 24 (40):6119–32. doi:10.1038/sj.onc.1208749.
- Peng, Z., Y. Zhang, W. Gu, Z. Wang, D. Li, F. Zhang, G. Qiu, and K. Xie. 2005. Integration of the hepatitis B virus X fragment in hepatocellular carcinoma and its effects on the expression of multiple molecules: a key to the cell cycle and apoptosis. Int. J. Oncol. 26 (2):467–73.
- Persson, H. L. 2005. Iron-dependent lysosomal destabilization initiates silica-induced apoptosis in murine macrophages. Toxicol. Lett. 159 (2):124–33. doi:10.1016/j.toxlet.2005.05.002.
- Peveling-Oberhag, J., G. Crisman, A. Schmidt, C. Doring, M. Lucioni, L. Arcaini, S. Rattotti, S. Hartmann, A. Piiper, W. P. Hofmann, et al. 2012. Dysregulation of global microRNA expression in splenic marginal zone lymphoma and influence of chronic hepatitis C virus infection. Leukemia 26 (7):1654–62. doi:10.1038/leu.2012.29.
- Pienta, R. J., J. A. Poiley, and W. B. Lebherz 3rd. 1977. Morphological transformation of early passage golden Syrian hamster embryo cells derived from cryopreserved primary cultures as a reliable in vitro bioassay for identifying diverse carcinogens. Int. J. Cancer 19 (5):642–55. doi:10.1002/ijc.2910190508.
- Pogribny, I. P., V. P. Tryndyak, A. Boyko, R. Rodriguez-Juarez, F. A. Beland, and O. Kovalchuk. 2007. Induction of microRNAome deregulation in rat liver by long-term tamoxifen exposure. Mutat. Res. 619 (1–2):30–37. doi:10.1016/j.mrfmmm.2006.12.006.
- Pylkkanen, L., H. Stockmann-Juvala, H. Alenius, K. Husgafvel-Pursiainen, and K. Savolainen. 2009. Wood dusts induce the production of reactive oxygen species and caspase-3 activity in human bronchial epithelial cells. Toxicology 262 (3):265–70. doi:10.1016/j.tox.2009.06.019.
- Rager, J. E., B. C. Moeller, M. Doyle-Eisele, D. Kracko, J. A. Swenberg, and R. C. Fry. 2013. Formaldehyde and epigenetic alterations: microRNA changes in the nasal epithelium of nonhuman primates. Environ. Health Perspect. 121 (3):339–44. doi:10.1289/ehp.1205582.
- Rager, J. E., L. Smeester, I. Jaspers, K. G. Sexton, and R. C. Fry. 2011. Epigenetic changes induced by air toxics: formaldehyde exposure alters miRNA expression profiles in human lung cells. Environ. Health Perspect. 119 (4):494–500. doi:10.1289/ehp.1002614.
- Rainey, R. P., I. G. Gillman, X. Shi, T. Cheng, A. Stinson, D. Gietl, and A. P. Albino. 2009. Fluorescent detection of lipid peroxidation derived protein adducts upon in-vitro cigarette smoke exposure. Toxicol. Mech. Methods 19 (6–7):401–09. doi:10.1080/15376510903104224.
- Rajaraman, P., P. Bhatti, M. M. Doody, S. L. Simon, R. M. Weinstock, M. S. Linet, M. Rosenstein, M. Stovall, B. H. Alexander, D. L. Preston, et al. 2008. Nucleotide excision repair polymorphisms may modify ionizing radiation-related breast cancer risk in US radiologic technologists. Int. J. Cancer 123 (11):2713–16. doi:10.1002/ijc.23779.
- Ramadan, R. A., M. A. Zaki, A. M. Awad, and L. A. El-Ghalid. 2015. Aberrant methylation of promoter region of SPINT2/HAI-2 gene: an epigenetic mechanism in hepatitis C virus-induced hepatocarcinogenesis. Genet Test Mol Biomarkers 19 (7):399–404. doi:10.1089/gtmb.2015.0025.
- Rasmussen, R. E., M. Hammer-Wilson, and M. W. Berns. 1989. Mutation and sister chromatid exchange induction in Chinese hamster ovary (CHO) cells by pulsed excimer laser radiation at 193 nm and 308 nm and continuous UV radiation at 254 nm. Photochem. Photobiol. 49 (4):413–18.
- Ren, X., C. M. McHale, C. F. Skibola, A. H. Smith, M. T. Smith, and L. Zhang. 2011. An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Environ. Health Perspect. 119 (1):11–19. doi:10.1289/ehp.1002114.
- Rey, O., S. Lee, and N. H. Park. 1999. Impaired nucleotide excision repair in UV-irradiated human oral keratinocytes immortalized with type 16 human papillomavirus genome. Oncogene 18 (50):6997–7001. doi:10.1038/sj.onc.1203180.
- Reynoso, R., M. Wieser, D. Ojeda, M. Bonisch, H. Kuhnel, F. Bolcic, H. Quendler, J. Grillari, R. Grillari-Voglauer, and J. Quarleri. 2012. HIV-1 induces telomerase activity in monocyte-derived macrophages, possibly safeguarding one of its reservoirs. J. Virol. 86 (19):10327–37. doi:10.1128/jvi.01495-12.
- Rider, C. F., M. Yamamoto, O. P. Gunther, J. A. Hirota, A. Singh, S. J. Tebbutt, and C. Carlsten. 2016. Controlled diesel exhaust and allergen coexposure modulates microRNA and gene expression in humans: Effects on inflammatory lung markers. J. Allergy Clin. Immunol. 138 (6):1690–700. doi:10.1016/j.jaci.2016.02.038.
- Rieswijk, L., S. M. Claessen, O. Bekers, M. van Herwijnen, D. H. Theunissen, D. G. Jennen, T. M. de Kok, J. C. Kleinjans, and S. G. van Breda. 2016. Aflatoxin B1 induces persistent epigenomic effects in primary human hepatocytes associated with hepatocellular carcinoma. Toxicology 350-352:31–39. doi:10.1016/j.tox.2016.05.002.
- Ripoli, M., R. Barbano, T. Balsamo, C. Piccoli, V. Brunetti, M. Coco, G. Mazzoccoli, M. Vinciguerra, and V. Pazienza. 2011. Hypermethylated levels of E-cadherin promoter in Huh-7 cells expressing the HCV core protein. Virus Res. 160 (1–2):74–81. doi:10.1016/j.virusres.2011.05.014.
- Rochette, P. J., S. Lacoste, J. P. Therrien, N. Bastien, D. E. Brash, and R. Drouin. 2009. Influence of cytosine methylation on ultraviolet-induced cyclobutane pyrimidine dimer formation in genomic DNA. Mutat. Res. 665 (1–2):7–13. doi:10.1016/j.mrfmmm.2009.02.008.
- Rodrigues-Junior, D. M., A. A. Melo, B. B. Da Silva, and P. V. Lopes-Costa. 2012. Formation of DNA strand breaks in peripheral lymphocytes of rats after exposure to natural sunlight. Biomed. Environ. Sci. 25 (2):245–49. doi:10.3967/0895-3988.2012.02.018.
- Rodriguez, S., Y. A. Kunde, T. M. McCleskey, and E. Hong-Geller. 2008. Upregulation of I-CAM1 in response to beryllium exposure in small airway epithelial cells. Toxicol. Lett. 179 (3):140–47. doi:10.1016/j.toxlet.2008.04.014.
- Rojas, D., J. E. Rager, L. Smeester, K. A. Bailey, Z. Drobna, M. Rubio-Andrade, M. Styblo, G. Garcia-Vargas, and R. C. Fry. 2015. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol. Sci. 143 (1):97–106. doi:10.1093/toxsci/kfu210.
- Roy, R. V., Y. O. Son, P. Pratheeshkumar, L. Wang, J. A. Hitron, S. P. Divya, D. R, D. Kim, Y. Yin, Z. Zhang, et al. 2015. Epigenetic targets of arsenic: emphasis on epigenetic modifications during carcinogenesis. J. Environ. Pathol. Toxicol. Oncol. 34 (1):63–84.
- Ruggero, K., A. Corradin, P. Zanovello, A. Amadori, V. Bronte, V. Ciminale, and D. M. D’Agostino. 2010. Role of microRNAs in HTLV-1 infection and transformation. Mol. Aspects Med. 31 (5):367–82. doi:10.1016/j.mam.2010.05.001.
- Sadikovic, B., J. Andrews, D. Carter, J. Robinson, and D. I. Rodenhiser. 2008. Genome-wide H3K9 histone acetylation profiles are altered in benzopyrene-treated MCF7 breast cancer cells. J. Biol. Chem. 283 (7):4051–60. doi:10.1074/jbc.M707506200.
- Sajadimajd, S., R. Yazdanparast, and F. Roshanzamir. 2016. Augmentation of oxidative stress-induced apoptosis in MCF7 cells by ascorbate-tamoxifen and/or ascorbate-juglone treatments. In Vitro Cell. Dev. Biol. Anim. 52 (2):193–203. doi:10.1007/s11626-015-9961-4.
- Sanders, A. P., L. Smeester, D. Rojas, T. DeBussycher, M. C. Wu, F. A. Wright, Y. H. Zhou, J. E. Laine, J. E. Rager, G. K. Swamy, et al. 2014. Cadmium exposure and the epigenome: Exposure-associated patterns of DNA methylation in leukocytes from mother-baby pairs. Epigenetics 9 (2):212–21. doi:10.4161/epi.26798.
- Sankaranarayanan, K. 1991. Ionizing radiation and genetic risks. III. Nature of spontaneous and radiation-induced mutations in mammalian in vitro systems and mechanisms of induction of mutations by radiation. Mutat. Res. 258 (1):75–97. doi:10.1016/0165-1110(91)90029-u.
- Santos, L. S., S. C. Branco, S. N. Silva, A. P. Azevedo, O. M. Gil, I. Manita, T. C. Ferreira, E. Limbert, J. Rueff, and J. F. Gaspar. 2012. Polymorphisms in base excision repair genes and thyroid cancer risk. Oncol. Rep. 28 (5):1859–68. doi:10.3892/or.2012.1975.
- Sarkar, N., and R. Chakravarty. 2015. Hepatitis B virus infection, MicroRNAs and liver disease. Int J Mol Sci 16 (8):17746–62. doi:10.3390/ijms160817746.
- Sartor, M. A., D. C. Dolinoy, T. R. Jones, J. A. Colacino, M. E. Prince, T. E. Carey, and L. S. Rozek. 2011. Genome-wide methylation and expression differences in HPV(+) and HPV(-) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics 6 (6):777–87. doi:10.4161/epi.6.6.16216.
- Satou, Y., J. Yasunaga, T. Zhao, M. Yoshida, P. Miyazato, K. Takai, K. Shimizu, K. Ohshima, P. L. Green, N. Ohkura, et al. 2011. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 7 (2):e1001274. doi:10.1371/journal.ppat.1001274.
- Schernhammer, E. S., E. Giovannucci, T. Kawasaki, B. Rosner, C. S. Fuchs, and S. Ogino. 2010. Dietary folate, alcohol and B vitamins in relation to LINE-1 hypomethylation in colon cancer. Gut 59 (6):794–99. doi:10.1136/gut.2009.183707.
- Schmidt, A., D. Steinritz, H. Thiermann, V. Meineke, and M. Abend. 2016. Alteration of miRNA expression in early endothelial cells after exposure with sub-lethal sulfur mustard concentrations. Toxicol. Lett. 244:88–94. doi:10.1016/j.toxlet.2015.10.002.
- Schuller, H. M., P. K. Tithof, M. Williams, and H. Plummer 3rd. 1999. The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 59 (18):4510–15.
- Schutze, D. M., J. M. Kooter, S. M. Wilting, C. J. Meijer, W. Quint, P. J. Snijders, and R. D. Steenbergen. 2015. Longitudinal assessment of DNA methylation changes during HPVE6E7-induced immortalization of primary keratinocytes. Epigenetics 10 (1):73–81. doi:10.4161/15592294.2014.990787.
- Sgura, A., A. Antoccia, F. Berardinelli, R. Cherubini, S. Gerardi, C. Zilio, and C. Tanzarella. 2006. Telomere length in mammalian cells exposed to low- and high-LET radiations. Radiat Prot Dosimetry 122 (1–4):176–79. doi:10.1093/rpd/ncl478.
- Shen, Y., M. Stanislauskas, G. Li, D. Zheng, and L. Liu. 2017. Epigenetic and genetic dissections of UV-induced global gene dysregulation in skin cells through multi-omics analyses. Sci Rep 7:42646. doi:10.1038/srep42646.
- Shi, H., and J. M. Feng. 2011. Aristolochic acid induces apoptosis of human umbilical vein endothelial cells in vitro by suppressing PI3K/Akt signaling pathway. Acta Pharmacol. Sin. 32 (8):1025–30. doi:10.1038/aps.2011.74.
- Shinozaki, A., T. Sakatani, T. Ushiku, R. Hino, M. Isogai, S. Ishikawa, H. Uozaki, K. Takada, and M. Fukayama. 2010. Downregulation of microRNA-200 in EBV-associated gastric carcinoma. Cancer Res. 70 (11):4719–27. doi:10.1158/0008-5472.CAN-09-4620.
- Singh, N. P., U. P. Singh, H. Guan, P. Nagarkatti, and M. Nagarkatti. 2012. Prenatal exposure to TCDD triggers significant modulation of microRNA expression profile in the thymus that affects consequent gene expression. PLoS ONE 7 (9):e45054. doi:10.1371/journal.pone.0045054.
- Singh, N. P., U. P. Singh, B. Singh, R. L. Price, M. Nagarkatti, and P. S. Nagarkatti. 2011. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS ONE 6 (8):e23522. doi:10.1371/journal.pone.0023522.
- Skalska, L., R. E. White, M. Franz, M. Ruhmann, and M. J. Allday. 2010. Epigenetic repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP. PLoS Pathog. 6 (6):e1000951. doi:10.1371/journal.ppat.1000951.
- Slijepcevic, P., A. T. Natarajan, and P. E. Bryant. 1998. Telomeres and radiation-induced chromosome breakage. Mutagenesis 13 (1):45–49. doi:10.1093/mutage/13.1.45.
- Smith, M. T. 2019. Key characteristics of carcinogens. In Tumour site concordance and mechanisms of carcinogenesis, ed. R. A. Baan, B. W. Stewart, and K. Straif. Lyon: IARC Press.
- Smith, M. T., K. Z. Guyton, C. F. Gibbons, J. M. Fritz, C. J. Portier, I. Rusyn, D. M. DeMarini, J. C. Caldwell, R. J. Kavlock, P. F. Lambert, et al. 2016. Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environ. Health Perspect. 124 (6):713–21. doi:10.1289/ehp.1509912.
- Squires, S., J. A. Coates, M. Goldberg, L. H. Toji, S. P. Jackson, D. J. Clarke, and R. T. Johnson. 2004. p53 prevents the accumulation of double-strand DNA breaks at stalled-replication forks induced by UV in human cells. Cell Cycle 3 (12):1543–57. doi:10.4161/cc.3.12.1272.
- Sriraksa, R., C. Zeller, M. A. El-Bahrawy, W. Dai, J. Daduang, P. Jearanaikoon, S. Chau-In, R. Brown, and T. Limpaiboon. 2011. CpG-island methylation study of liver fluke-related cholangiocarcinoma. Br. J. Cancer 104 (8):1313–18. doi:10.1038/bjc.2011.102.
- Steinritz, D., A. Schmidt, F. Balszuweit, H. Thiermann, T. Simons, E. Striepling, B. Bolck, and W. Bloch. 2016. Epigenetic modulations in early endothelial cells and DNA hypermethylation in human skin after sulfur mustard exposure. Toxicol. Lett. 244:95–102. doi:10.1016/j.toxlet.2015.09.016.
- Sturchio, E., A. Amadori, J. Businaro, B. Ficociello, P. Ferrazza, C. Colosio, and C. Minoia. 2012. Possible use of microRNAs as biomarkers for monitoring of workers exposed to asbestos. G Ital Med Lav Ergon 34 (3 Suppl):571–73.
- Subach, O. M., V. B. Baskunov, M. V. Darii, D. V. Maltseva, D. A. Alexandrov, O. V. Kirsanova, A. Kolbanovskiy, M. Kolbanovskiy, F. Johnson, R. Bonala, et al. 2006. Impact of benzo[a]pyrene-2ʹ-deoxyguanosine lesions on methylation of DNA by SssI and HhaI DNA methyltransferases. Biochemistry 45 (19):6142–59. doi:10.1021/bi0511639.
- Szende, B., and E. Tyihak. 2010. Effect of formaldehyde on cell proliferation and death. Cell Biol. Int. 34 (12):1273–82. doi:10.1042/CBI20100532.
- Tabish, A. M., K. Poels, P. Hoet, and L. Godderis. 2012. Epigenetic factors in cancer risk: effect of chemical carcinogens on global DNA methylation pattern in human TK6 cells. PLoS ONE 7 (4):e34674. doi:10.1371/journal.pone.0034674.
- Tachibana, K., K. Takayanagi, A. Akimoto, K. Ueda, Y. Shinkai, M. Umezawa, and K. Takeda. 2015. Prenatal diesel exhaust exposure disrupts the DNA methylation profile in the brain of mouse offspring. J Toxicol Sci 40 (1):1–11. doi:10.2131/jts.40.1.
- Takahashi, M., J. C. Barrett, and T. Tsutsui. 2002. Transformation by inorganic arsenic compounds of normal Syrian hamster embryo cells into a neoplastic state in which they become anchorage-independent and cause tumors in newborn hamsters. Int. J. Cancer 99 (5):629–34. doi:10.1002/ijc.10407.
- Tamminga, J., and O. Kovalchuk. 2011. Role of DNA damage and epigenetic DNA methylation changes in radiation-induced genomic instability and bystander effects in germline in vivo. Curr Mol Pharmacol 4 (2):115–25.
- Tang, W. Y., L. Levin, G. Talaska, Y. Y. Cheung, J. Herbstman, D. Tang, R. L. Miller, F. Perera, and S. M. Ho. 2012. Maternal exposure to polycyclic aromatic hydrocarbons and 5ʹ-CpG methylation of interferon-gamma in cord white blood cells. Environ. Health Perspect. 120 (8):1195–200. doi:10.1289/ehp.1103744.
- Tang, W. Y., R. Newbold, K. Mardilovich, W. Jefferson, R. Y. Cheng, M. Medvedovic, and S. M. Ho. 2008a. Persistent hypomethylation in the promoter of nucleosomal binding protein 1 (Nsbp1) correlates with overexpression of Nsbp1 in mouse uteri neonatally exposed to diethylstilbestrol or genistein. Endocrinology 149 (12):5922–31. doi:10.1210/en.2008-0682.
- Tang, Y., A. Banan, C. B. Forsyth, J. Z. Fields, C. K. Lau, L. J. Zhang, and A. Keshavarzian. 2008b. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol. Clin. Exp. Res. 32 (2):355–64. doi:10.1111/j.1530-0277.2007.00584.x.
- Tao, R., M. F. Hu, J. T. Lou, and Y. L. Lei. 2007. Effects of H pylori infection on gap-junctional intercellular communication and proliferation of gastric epithelial cells in vitro. World J. Gastroenterol. 13 (41):5497–500. doi:10.3748/wjg.v13.i41.5497.
- Templin, T., S. Paul, S. A. Amundson, E. F. Young, C. A. Barker, S. L. Wolden, and L. B. Smilenov. 2011. Radiation-induced micro-RNA expression changes in peripheral blood cells of radiotherapy patients. Int. J. Radiat. Oncol. Biol. Phys. 80 (2):549–57. doi:10.1016/j.ijrobp.2010.12.061.
- Terradillos, O., T. Pollicino, H. Lecoeur, M. Tripodi, M. L. Gougeon, P. Tiollais, and M. A. Buendia. 1998. p53-independent apoptotic effects of the hepatitis B virus HBx protein in vivo and in vitro. Oncogene 17 (16):2115–23. doi:10.1038/sj.onc.1202432.
- Tian, Y., and J. H. Ou. 2015. Genetic and epigenetic alterations in hepatitis B virus-associated hepatocellular carcinoma. Virol Sin 30 (2):85–91. doi:10.1007/s12250-015-3582-7.
- Tian, Y., W. Yang, J. Song, Y. Wu, and B. Ni. 2013. Hepatitis B virus X protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis. Mol. Cell. Biol. 33 (15):2810–16. doi:10.1128/mcb.00205-13.
- Tithof, P. K., S. M. Richards, M. A. Elgayyar, F. M. Menn, V. M. Vulava, L. McKay, J. Sanseverino, G. Sayler, D. E. Tucker, C. C. Leslie, et al. 2011. Activation of group IVC phospholipase A(2) by polycyclic aromatic hydrocarbons induces apoptosis of human coronary artery endothelial cells. Arch. Toxicol. 85 (6):623–34. doi:10.1007/s00204-010-0614-9.
- Tokar, E. J., R. J. Person, Y. Sun, A. O. Perantoni, and M. P. Waalkes. 2013. Chronic exposure of renal stem cells to inorganic arsenic induces a cancer phenotype. Chem. Res. Toxicol. 26 (1):96–105. doi:10.1021/tx3004054.
- Tonn, T., C. Esser, E. M. Schneider, W. Steinmann-Steiner-Haldenstatt, and E. Gleichmann. 1996. Persistence of decreased T-helper cell function in industrial workers 20 years after exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Environ. Health Perspect. 104 (4):422–26. doi:10.1289/ehp.96104422.
- Tryndyak, V. P., L. Muskhelishvili, O. Kovalchuk, R. Rodriguez-Juarez, B. Montgomery, M. I. Churchwell, S. A. Ross, F. A. Beland, and I. P. Pogribny. 2006. Effect of long-term tamoxifen exposure on genotoxic and epigenetic changes in rat liver: implications for tamoxifen-induced hepatocarcinogenesis. Carcinogenesis 27 (8):1713–20. doi:10.1093/carcin/bgl050.
- Ura, S., M. Honda, T. Yamashita, T. Ueda, H. Takatori, R. Nishino, H. Sunakozaka, Y. Sakai, K. Horimoto, and S. Kaneko. 2009. Differential microRNA expression between hepatitis B and hepatitis C leading disease progression to hepatocellular carcinoma. Hepatology 49 (4):1098–112. doi:10.1002/hep.22749.
- Valenzuela, M. A., J. Canales, A. H. Corvalan, and A. F. Quest. 2015. Helicobacter pylori-induced inflammation and epigenetic changes during gastric carcinogenesis. World J. Gastroenterol. 21 (45):12742–56. doi:10.3748/wjg.v21.i45.12742.
- Vandermeers, F., S. Neelature Sriramareddy, C. Costa, R. Hubaux, J. P. Cosse, and L. Willems. 2013. The role of epigenetics in malignant pleural mesothelioma. Lung Cancer 81 (3):311–18. doi:10.1016/j.lungcan.2013.05.014.
- Vilahur, N., M. Vahter, and K. Broberg. 2015. The epigenetic effects of prenatal cadmium exposure. Curr. Environ. Health Rep. 2 (2):195–203. doi:10.1007/s40572-015-0049-9.
- Vineis, P., and S. H. Zahm. 1988. Immunosuppressive effects of dioxin in the development of Kaposi’s sarcoma and non-Hodgkin’s lymphoma. Lancet 1 (8575–6):55. doi:10.1016/S0140-6736(88)91032-X.
- Vogelstein, B., N. Papadopoulos, V. E. Velculescu, S. Zhou, L. A. Diaz Jr., and K. W. Kinzler. 2013. Cancer genome landscapes. Science 339 (6127):1546–58. doi:10.1126/science.1235122.
- von Stedingk, H., A. C. Vikstrom, P. Rydberg, M. Pedersen, J. K. Nielsen, D. Segerback, L. E. Knudsen, and M. Tornqvist. 2011. Analysis of hemoglobin adducts from acrylamide, glycidamide, and ethylene oxide in paired mother/cord blood samples from Denmark. Chem. Res. Toxicol. 24 (11):1957–65. doi:10.1021/tx200284u.
- Vonlaufen, A., J. S. Wilson, R. C. Pirola, and M. V. Apte. 2007. Role of alcohol metabolism in chronic pancreatitis. Alcohol Res Health 30 (1):48–54.
- Vorce, R. L., and J. I. Goodman. 1989. Altered methylation of ras oncogenes in benzidine-induced B6C3F1 mouse liver tumors. Toxicol. Appl. Pharmacol. 100 (3):398–410. doi:10.1016/0041-008x(89)90288-3.
- Vrabie, C. M., M. T. Jonker, and A. J. Murk. 2009. Specific in vitro toxicity of crude and refined petroleum products. 1. Aryl hydrocarbon receptor-mediated responses. Environ. Toxicol. Chem. 28 (9):1995–2003. doi:10.1897/08-624.1.
- Wang, B., Y. Li, C. Shao, Y. Tan, and L. Cai. 2012a. Cadmium and its epigenetic effects. Curr. Med. Chem. 19 (16):2611–20. doi:10.2174/092986712800492913.
- Wang, J., S. L. Zhao, Y. Li, M. Meng, and C. Y. Qin. 2012b. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone induces retinoic acid receptor beta hypermethylation through DNA methyltransferase 1 accumulation in esophageal squamous epithelial cells. Asian Pac. J. Cancer Prev. 13 (5):2207–12. doi:10.7314/apjcp.2012.13.5.2207.
- Wang, M., G. Cheng, P. W. Villalta, and S. S. Hecht. 2007. Development of liquid chromatography electrospray ionization tandem mass spectrometry methods for analysis of DNA adducts of formaldehyde and their application to rats treated with N-nitrosodimethylamine or 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol. 20 (8):1141–48. doi:10.1021/tx700189c.
- Wang, Y., X. Duan, Y. Zhang, S. Wang, W. Yao, S. Wang, W. Wang, and Y. Wu. 2015. DNA methylation and telomere damage in occupational people exposed to coal tar pitch. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 33 (7):507–11.
- Warita, K., T. Mitsuhashi, T. Sugawara, Y. Tabuchi, T. Tanida, Z. Y. Wang, Y. Matsumoto, T. Yokoyama, H. Kitagawa, T. Miki, et al. 2010. Direct effects of diethylstilbestrol on the gene expression of the cholesterol side-chain cleavage enzyme (P450scc) in testicular Leydig cells. Life Sci. 87 (9–10):281–85. doi:10.1016/j.lfs.2010.06.020.
- Weihrauch, M., M. Benicke, G. Lehnert, C. Wittekind, R. Wrbitzky, and A. Tannapfel. 2001. Frequent k- ras −2 mutations and p16(INK4A)methylation in hepatocellular carcinomas in workers exposed to vinyl chloride. Br. J. Cancer 84 (7):982–89. doi:10.1054/bjoc.2000.1675.
- Weiss, D., T. Basel, F. Sachse, A. Braeuninger, and C. Rudack. 2011. Promoter methylation of cyclin A1 is associated with human papillomavirus 16 induced head and neck squamous cell carcinoma independently of p53 mutation. Mol. Carcinog. 50 (9):680–88. doi:10.1002/mc.20798.
- Whang-Peng, J., Y. M. Chen, T. Knutsen, W. P. Zhao, and S. Tsai. 1993. Chromosome studies in HTLV-I, -II, and HIV-1, −2 cell lines infected in vivo and in vitro. J. Acquir. Immune Defic. Syndr. 6 (8):930–40.
- Whiteman, D. C., C. A. Whiteman, and A. C. Green. 2001. Childhood sun exposure as a risk factor for melanoma: a systematic review of epidemiologic studies. Cancer Causes Control 12 (1):69–82.
- Williams, A. O., A. D. Knapton, E. T. Ifon, and U. Saffiotti. 1996. Transforming growth factor beta expression and transformation of rat lung epithelial cells by crystalline silica (quartz). Int. J. Cancer 65 (5):639–49. doi:10.1002/(SICI)1097-0215(19960301)65:5<639::AID-IJC14>3.0.CO;2-2.
- Wu, A. H., K. D. Siegmund, T. I. Long, W. Cozen, P. Wan, C. C. Tseng, D. Shibata, and P. W. Laird. 2010a. Hormone therapy, DNA methylation and colon cancer. Carcinogenesis 31 (6):1060–67. doi:10.1093/carcin/bgq009.
- Wu, C. C., M. T. Liu, Y. T. Chang, C. Y. Fang, S. P. Chou, H. W. Liao, K. L. Kuo, S. L. Hsu, Y. R. Chen, P. W. Wang, et al. 2010b. Epstein-Barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 38 (6):1932–49. doi:10.1093/nar/gkp1169.
- Wu, H. C., Q. Wang, H. I. Yang, W. Y. Tsai, C. J. Chen, and R. M. Santella. 2013. Global DNA methylation in a population with aflatoxin B1 exposure. Epigenetics 8 (9):962–69. doi:10.4161/epi.25696.
- Wu, J., X. Li, Y. Xu, T. Yang, Q. Yang, C. Yang, and Y. Jiang. 2016. Identification of a long non-coding RNA NR_026689 associated with lung carcinogenesis induced by NNK. Oncotarget 7 (12):14486–98. doi:10.18632/oncotarget.7475.
- Wu, J. J., T. Yang, X. Li, Y. Xia, Y. Zhao, F. Zou, and Y. G. Jiang. 2014. 4-(Methylnitrosamino)-1-(3-pyridyl) −1-butanone induces circulating microRNA deregulation in early lung carcinogenesis. Biomed. Environ. Sci. 27 (1):10–16. doi:10.3967/bes2014.011.
- Xi, S., S. Inchauste, H. Guo, J. Shan, Z. Xiao, H. Xu, M. Miettenen, M. R. Zhang, J. A. Hong, M. T. Raiji, et al. 2015. Cigarette smoke mediates epigenetic repression of miR-217 during esophageal adenocarcinogenesis. Oncogene 34 (44):5548–59. doi:10.1038/onc.2015.10.
- Xi, S., H. Xu, J. Shan, Y. Tao, J. A. Hong, S. Inchauste, M. Zhang, T. F. Kunst, L. Mercedes, and D. S. Schrump. 2013. Cigarette smoke mediates epigenetic repression of miR-487b during pulmonary carcinogenesis. J. Clin. Invest. 123 (3):1241–61. doi:10.1172/JCI61271.
- Xia, B., X. H. Ren, Z. X. Zhuang, L. Q. Yang, H. Y. Huang, L. Pang, D. S. Wu, J. Luo, Y. L. Tan, J. J. Liu, et al. 2014. Effect of hexavalent chromium on histone biotinylation in human bronchial epithelial cells. Toxicol. Lett. 228 (3):241–47. doi:10.1016/j.toxlet.2014.05.010.
- Xing, C., Q. Chen, G. Li, L. Zhang, M. Zheng, Z. Zou, L. Hou, Q. F. Wang, X. Liu, and X. Guo. 2013. Microsomal epoxide hydrolase (EPHX1) polymorphisms are associated with aberrant promoter methylation of ERCC3 and hematotoxicity in benzene-exposed workers. Environ. Mol. Mutagen. 54 (6):397–405. doi:10.1002/em.21786.
- Xu, C., Y. Chen, X. Chen, and F. Wang. 2011. Effects of different types of Helicobacter pylori on the gap junction intercellular communication in GES-1 cells. Zhong Nan Da Xue Xue Bao Yi Xue Ban 36 (4):294–300. doi:10.3969/j.issn.1672-7347.2011.04.003.
- Xu, Y., L. Li, X. Xiang, H. Wang, W. Cai, J. Xie, Y. Han, S. Bao, and Q. Xie. 2013. Three common functional polymorphisms in microRNA encoding genes in the susceptibility to hepatocellular carcinoma: a systematic review and meta-analysis. Gene 527 (2):584–93. doi:10.1016/j.gene.2013.05.085.
- Yamamoto, M., A. Singh, F. Sava, M. Pui, S. J. Tebbutt, and C. Carlsten. 2013. MicroRNA expression in response to controlled exposure to diesel exhaust: attenuation by the antioxidant N-acetylcysteine in a randomized crossover study. Environ. Health Perspect. 121 (6):670–75. doi:10.1289/ehp.1205963.
- Yamamoto-Taguchi, N., Y. Satou, P. Miyazato, K. Ohshima, M. Nakagawa, K. Katagiri, T. Kinashi, and M. Matsuoka. 2013. HTLV-1 bZIP factor induces inflammation through labile Foxp3 expression. PLoS Pathog. 9 (9):e1003630. doi:10.1371/journal.ppat.1003630.
- Yang, J., X. Zuo, W. Bai, P. Niu, L. Tian, and A. Gao. 2014. PTEN methylation involved in benzene-induced hematotoxicity. Exp. Mol. Pathol. 96 (3):300–06. doi:10.1016/j.yexmp.2014.03.008.
- Yano, M., S. Ohkoshi, Y. H. Aoki, H. Takahashi, S. Kurita, K. Yamazaki, K. Suzuki, S. Yamagiwa, A. Sanpei, S. Fujimaki, et al. 2013. Hepatitis B virus X induces cell proliferation in the hepatocarcinogenesis via up-regulation of cytoplasmic p21 expression. Liver Int. 33 (8):1218–29. doi:10.1111/liv.12176.
- Yauk, C., A. Polyzos, A. Rowan-Carroll, C. M. Somers, R. W. Godschalk, F. J. Van Schooten, M. L. Berndt, I. P. Pogribny, I. Koturbash, A. Williams, et al. 2008. Germ-line mutations, DNA damage, and global hypermethylation in mice exposed to particulate air pollution in an urban/industrial location. Proc. Natl. Acad. Sci. U.S.A. 105 (2):605–10. doi:10.1073/pnas.0705896105.
- Yoo, Y. G., T. Y. Na, H. W. Seo, J. K. Seong, C. K. Park, Y. K. Shin, and M. O. Lee. 2008. Hepatitis B virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene 27 (24):3405–13. doi:10.1038/sj.onc.1211000.
- Yoshioka, W., W. Higashiyama, and C. Tohyama. 2011. Involvement of microRNAs in dioxin-induced liver damage in the mouse. Toxicol. Sci. 122 (2):457–65. doi:10.1093/toxsci/kfr130.
- Yu, C. C., C. H. Tsai, H. I. Hsu, and Y. C. Chang. 2013. Elevation of S100A4 expression in buccal mucosal fibroblasts by arecoline: involvement in the pathogenesis of oral submucous fibrosis. PLoS ONE 8 (1):e55122. doi:10.1371/journal.pone.0055122.
- Yu, H., J. Lu, L. Zuo, Q. Yan, Z. Yu, X. Li, J. Huang, L. Zhao, H. Tang, Z. Luo, et al. 2012. Epstein-Barr virus downregulates microRNA 203 through the oncoprotein latent membrane protein 1: a contribution to increased tumor incidence in epithelial cells. J. Virol. 86 (6):3088–99. doi:10.1128/JVI.05901-11.
- Yu, R. A., L. F. He, and X. M. Chen. 2007. Effects of cadmium on hepatocellular DNA damage, proto-oncogene expression and apoptosis in rats. Biomed. Environ. Sci. 20 (2):146–53.
- Zekri Ael, R., A. A. Nassar, M. N. El-Din El-Rouby, H. I. Shousha, A. B. Barakat, E. D. El-Desouky, N. A. Zayed, O. S. Ahmed, A. S. El-Din Youssef, A. O. Kaseb, et al. 2014. Disease progression from chronic hepatitis C to cirrhosis and hepatocellular carcinoma is associated with increasing DNA promoter methylation. Asian Pac. J. Cancer Prev. 14 (11):6721–26. doi:10.7314/apjcp.2013.14.11.6721.
- Zhang, H., W. X. Hong, J. Ye, X. Yang, X. Ren, A. Huang, L. Yang, L. Zhou, H. Huang, D. Wu, et al. 2014. Analysis of trichloroethylene-induced global DNA hypomethylation in hepatic L-02 cells by liquid chromatography-electrospray ionization tandem mass spectrometry. Biochem. Biophys. Res. Commun. 446 (2):590–95. doi:10.1016/j.bbrc.2014.03.015.
- Zhang, H., X. Li, L. Ge, J. Yang, J. Sun, and Q. Niu. 2015. Methylation of CpG island of p14(ARK), p15(INK4b) and p16(INK4a) genes in coke oven workers. Hum Exp Toxicol 34 (2):191–97. doi:10.1177/0960327114533576.
- Zhang, X., X. Jia, L. Mei, M. Zheng, C. Yu, and M. Ye. 2016. Global DNA methylation and PTEN hypermethylation alterations in lung tissues from human silicosis. J Thorac Dis 8 (8):2185–95. doi:10.21037/jtd.2016.07.21.
- Zhang, Z. Z., X. Liu, D. Q. Wang, M. K. Teng, L. W. Niu, A. L. Huang, and Z. Liang. 2011. Hepatitis B virus and hepatocellular carcinoma at the miRNA level. World J. Gastroenterol. 17 (28):3353–58. doi:10.3748/wjg.v17.i28.3353.
- Zhao, Y., H. Liu, Y. Li, J. Wu, A. R. Greenlee, C. Yang, and Y. Jiang. 2011a. The role of miR-506 in transformed 16HBE cells induced by anti-benzo[a]pyrene-trans-7,8-dihydrodiol-9,10-epoxide. Toxicol. Lett. 205 (3):320–26. doi:10.1016/j.toxlet.2011.06.022.
- Zhao, Y., W. Zhong, X. Sun, Z. Song, D. L. Clemens, Y. J. Kang, C. J. McClain, and Z. Zhou. 2011b. Zinc deprivation mediates alcohol-induced hepatocyte IL-8 analog expression in rodents via an epigenetic mechanism. Am. J. Pathol. 179 (2):693–702. doi:10.1016/j.ajpath.2011.04.006.
- Zhou, F., S. Li, W. Jia, G. Lv, C. Song, C. Kang, and Q. Zhang. 2015. Effects of diesel exhaust particles on microRNA-21 in human bronchial epithelial cells and potential carcinogenic mechanisms. Mol Med Rep 12 (2):2329–35. doi:10.3892/mmr.2015.3655.
- Zhou, F., J. Liu, Q. M. Chen, X. L. Shan, L. L. Chen, H. Q. Quan, and N. Tang. 2012. Hepatitis C virus F protein-mediated inhibition of hepatoma cell proliferation. Zhonghua Gan Zang Bing Za Zhi 20 (5):368–71. doi:10.3760/cma.j.issn.1007-3418.2012.05.013.
- Zhou, Z. C., K. H. Norpoth, and E. Nelson. 1995. Genotoxicity of wood dust in a human embryonic lung cell line. Arch. Toxicol. 70 (1):57–60.
- Zhu, H., K. Li, J. Liang, J. Zhang, and Q. Wu. 2011. Changes in the levels of DNA methylation in testis and liver of SD rats neonatally exposed to 5-aza-2ʹ-deoxycytidine and cadmium. J Appl Toxicol 31 (5):484–95. doi:10.1002/jat.1673.