
ABSTRACT
Drug-Induced Liver Injury (DILI) constitutes hepatic damage attributed to drug exposure. DILI may be categorized as hepatocellular, cholestatic or mixed and might also involve immune responses. When DILI occurs in dose-dependent manner, it is referred to as intrinsic, while if the injury occurs spontaneously, it is termed as idiosyncratic. This review predominately focused on idiosyncratic liver injury. The established molecular mechanisms for DILI include (1) mitochondria dysfunction, (2) increased reactive oxygen species levels, (3) presence of elevated apoptosis and necrosis, (4) and bile duct injuries associated with immune mediated pathways. However, it should be emphasized that the underlying mechanisms responsible for DILI are still unknown. Prevention strategies are critical as incidences occur frequently, and treatment options are limited once the injury has developed. The aim of this review was to utilize retrospective cohort studies from across the globe to gain insight into epidemiological patterns. This review considers (1) what is currently known regarding the mechanisms underlying DILI, (2) discusses potential risk factors and (3) implications of the coronavirus pandemic on DILI presentation and research. Future perspectives are also considered and discussed and include potential new biomarkers, causality assessment and reporting methods.
Introduction
Drug-Induced Liver Injury (DILI), first described in the 1960s (Popper and Schaffner Citation1965) is a coverall term that describes the spectrum of pathological responses in the liver after exposure to potentially hepatotoxic chemical compounds (Katarey and Verma Citation2016; Ke et al. Citation2020). DILI is a significant concern in preclinical drug development and a leading cause of candidate drug attrition (Korver et al. Citation2021). It is believed that a frequent cause for acute liver failure (ALF) in developed countries is pharmacological intoxication followed by acute hepatitis B (Bechmann et al. Citation2014). DILI occurs when drug metabolism is altered and results in hepatic damage attributed to oxidative stress, inflammatory damage, apoptosis, necrosis or mitochondrial membrane breakdown and organ dysfunction (Garcia-Cortes et al. Citation2020).
Liver and its main functions
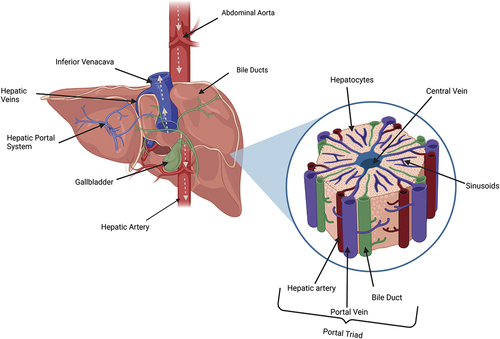
The liver depicted in is the largest internal organ within the body, making up approximately 2% of an adult’s bodyweight (Molina and DiMaio Citation2012). This multi-functional organ is involved in numerous physiological processes including macronutrient metabolism, filtration, detoxification of blood, bile formation, lipid and cholesterol homeostasis and processing xenobiotic compounds (Trefts, Gannon, and Wasserman Citation2017). The functional unit of the liver are hexagonal shaped lobules that communicate via the portal triad, comprising the portal vein, hepatic artery and bile ducts. Lobules contain three zones categorized by their perfusion status and which physiological processes occur within. The periportal zone (I) being the most proximal to the portal triad are highly perfused hepatocytes and perform beta-oxidation, bile and cholesterol formation, gluconeogenesis and amino acid catabolism (Trefts, Gannon, and Wasserman Citation2017). The pericentral zone (II) is a transitional area that carries out a mixture of zone I and III functions. The centrilobular zone (III) is adjacent to the central vein and experiences the lowest perfusion; therefore, this area is most susceptible to hypoxia and detoxification injury (Trefts, Gannon, and Wasserman Citation2017). The centrilobular zone is most involved in detoxification, ketogenesis, lipogenesis, glycolysis, glycogen synthesis and formation of glutamine (Ko et al. Citation2020). Drug metabolism is a biotransformation process that utilizes enzymes to convert insoluble compounds into more polar, soluble products to enable their elimination from the body (Almazroo, Miah, and Venkataramanan Citation2017) and primarily occurs in centrilobular hepatocytes. Some drug metabolites exhibit increased bioactivity compared to the parent compound prior to further metabolism. For example, diamorphine exerts minimal opioid agonist activity but is metabolized via deacetylation to produce active 6-monoacetyl morphine which is then further converted into morphine (Christrup Citation1997). However, metabolism of most drugs decreases their therapeutic effectiveness (World Federation of Societies of Anaesthesiologists Citation2023). The primary site of drug metabolism is within the smooth endoplasmic reticulum (ER) of the hepatocyte and usually occurs in phases. Phase I reactions involve oxidation, reduction or hydrolysis and introduction of reactive or polar groups to the compound – a process catalyzed by cytochrome P450 enzymes (Teschke and Danan Citation2021a). These modified compounds are then conjugated in Phase II reactions into hydrophilic compounds which are eliminated through efflux transporters. Phase II reactions are catalyzed by transferase enzymes (Zhao et al. Citation2021).
Figure 1. A schematic diagram of the liver (posterior view) showing the direction of blood flow indicated via arrows. Accompanied by a magnified representation of an individual lobule.
Intrinsic vs. idiosyncratic DILI
DILI may be classified as either predictable (intrinsic) or spontaneous (idiosyncratic) with the latter being associated with the most reported clinical presentations (Katarey and Verma Citation2016). Intrinsic reactions are less common, have a shorter latency period and are often described as being predictable, dose-dependent and reproducible in animal models (Yokoi and Oda Citation2021). It is understood that environmental and genetic sensitivity factors can influence the adverse effects of intrinsic toxicants. Some of the drugs often implicated with this are acetaminophen, aspirin and cocaine (Hamilton, Collins-Yoder, and Collins Citation2016). On the other hand, idiosyncratic reactions are not predictable, not dose-dependent, exhibit variable latency and are not reproducible experimentally in animal models (Fontana et al. Citation2023; Hosack, Damry, and Biswas Citation2023; Yokoi and Oda Citation2021).
The nature of drug-induced liver injury may further be classified according to the type of hepatic cells affected along with absence or presence of immunogenicity (Muscate, Woestemeier, and Gagliani Citation2021). DILI is reported to manifest as hepatocellular, cholestatic or mixed patterns and phenotypes present as any other hepatic disorder. Characteristics vary from being asymptomatic with only slightly elevated alanine aminotransferase (ALT) levels to jaundice, encephalopathy, and acute liver failure (Danan and Benichou Citation1993; Garcia-Cortes et al. Citation2020). Hepatocellular damage is associated with poor prognosis and may be diagnosed if liver function tests reveal ALT greater than 3-fold the upper limit of normal (ULN). These types of injuries are most common with idiosyncratic drug-induced liver injury (iDILI) (British Liver Trust Citation2023). Acute liver injury is characterized by significant hepatocyte necrosis while chronic cases tend to display hepatocytes fibrosis (David and Hamilton Citation2010; Fyfe, Zaldana, and Liu Citation2018). Cholestatic injury patterns present with elevated alkaline phosphate (ALP) levels as evidenced by more than 2-higher than ULN. These injuries present with abnormal biliary secretory function. Mixed (cholestatic hepatitis) injuries combine characteristics including hepatocyte damage, bile duct obstruction, degradation or problems with bile secretion (David and Hamilton Citation2010). Drug induced hepatic injury may be the result of direct toxicity from xenobiotics or their metabolites or indirectly through an aberrant immune system response (Gerussi et al. Citation2021). Direct damage occurs when compounds inhibit or block normal hepatocyte functions or when bile ducts are adversely affected. Sterile inflammation (triggered without an invading pathogen) might occur when damaged cells release proinflammatory mediators that engage leukocytes and initiate a cascade of immune response pathways (Gerussi et al. Citation2021). Stress arising from these immune processes and inflammation eventually leads to hepatocyte death. Immune-mediated responses might also occur in the form of dendritic cell activation in the liver and CD4+ T cell activity (Muscate, Woestemeier, and Gagliani Citation2021).
The current literature was thus reviewed to offer updated insights into the mechanisms underlying drug-induced liver injuries. Several risk factors involved in DILI were identified including genetic links to more severe outcomes, knowledge underlying disease prediction, disease determination, classification of incidences and how these epidemiological patterns differ globally. In addition, an extensive evaluation of how prescribed drugs might initiate DILI was included and clinical management processes for drugs are presented.
Methods
This review provides an extensive description of incidences of DILI, particularly (1) various molecular mechanisms associated with the use of commonly prescribed drugs, (2) biomarkers used in detection and (3) causality assessment methods that indicate prognosis. An extensive literature search was performed in PubMed and Cochrane Library. Keywords and phrases searched included “Drug Induced Liver Injury,” “DILI,” “DILI mechanisms,” “DILI incidence,” “Idiosyncratic DILI,” “DILI causality assessment,” “Drug Injury” and “Drug Discovery” in addition to various combinations of these phrases. The last review was conducted on 16th May 2023. The exclusion criteria applied to publications which focused on hepatic injury resulting primarily from alcohol and studies focusing on autoimmune hepatitis. Finally, due to the inevitable limitations and exclusionary nature of any literature search, investigations that did not include the search terms in the title or did not provide adequate information in the abstract might have been unintentionally omitted.
Patterns for global DILI
DILI accounts for approximately 15% of cases of acute liver failure in the United States and Europe (Reuben et al. Citation2010). It is not surprising that in countries with a higher usage of a particular compound there tends to be higher incidences of DILI attributed to those specific drugs. For example, investigations conducted in Africa demonstrated that the most common cause of DILI was due to anti-tuberculosis and anti-retroviral drugs utilization (Amadi and Orisakwe Citation2018; Riebensahm et al. Citation2019). It is no coincidence that this region exhibits high levels of human immunodeficiency virus (HIV) and tuberculosis (Riebensahm et al. Citation2019).
Approximately half of preclinical drugs with early evidence of hepatotoxicity are terminated prior to achieving market authorization (McGill and Jaeschke Citation2019). Continuous development and application of new drugs indicates the importance of accurately assessing hepatotoxic properties of novel compounds proposed for the expanding market. The drug discovery process is an intricate analysis of a potentially active compound to determine suitability for the treatment of diseases. Many different stages and checkpoints are required inferring that most novel drugs take over 10 years to gain market authorization (Hughes et al. Citation2011). Identifying and monitoring DILI is of upmost importance as new drugs come to market and an aging population is prescribed drugs more frequently. Molecular information on drugs has been compiled to assist in predicting toxicities and bioinformatic mining is commonplace throughout the drug discovery process, before commencing pre-clinical testing. Databases are also useful in recording incidences and case reports of DILI.
Drug induced liver injury (DILI)
Hepatic injuries are associated with one or more molecular pathways in response to xenobiotic compounds administration and vary between individuals depending upon host factors which makes predicting adverse drug reactions very difficult (McGill and Jaeschke Citation2018). Drug metabolism enzymes and transporters-particularly cytochrome P450 enzymes (CYP450) – convert drugs into metabolites through bioactivation (Russmann, Kullak-Ublick, and Grattagliano Citation2009). The metabolic products may be chemically reactive (CRM) and exert cellular stress through covalent bonding between host macromolecules such as proteins, DNA, enzymes, lipids, depletion of cellular anti-oxidants and mitochondrial dysfunction. This might lead to a loss of protein function, an increase in reactive oxygen species (ROS) levels further promoting oxidative stress damage (Villanueva-Paz et al. Citation2021), accumulation of toxic metabolites or trigger a cascade of immune responses ultimately leading to cell death (Hughes et al. Citation2021). Morphological changes in the hepatocytes might result in necrosis or apoptosis mediated by damage associated molecular patterns (DAMPs) initiated signaling pathways. Drug lipophilicity is positively associated with DILI risk as this might stimulate hepatocyte absorption, resulting in toxic levels of reactive metabolites (Teschke and Danan Citation2021a).
Metabolic dysfunction, cellular damage and oxidative stress
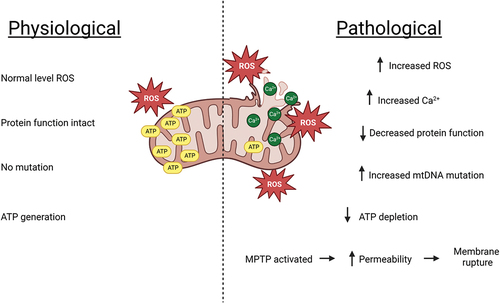
Dysfunction of the hepatic mitochondria might occur when reactive metabolites disrupt complexes involved in the mitochondrial electron transport chain, leading to an increased formation of ROS, which is a consequence of electrons prematurely reacting with molecular oxygen (Russmann, Kullak-Ublick, and Grattagliano Citation2009). Glutathione (GSH), an endogenous cellular anti-oxidant, neutralizes ROS via reduction (Zeke et al. Citation2016). GSH is a tripeptide (γ-L-glutamyl-L-cysteinyl-glycine) present in all mammalian tissues and especially highly concentrated in the liver (Lu Citation2009). Other intracellular processes utilizing GSH include cellular differentiation, proliferation and apoptosis, microtubule-related processes and immune function (Chen et al. Citation2022). GSH stores may be depleted over time or be insufficient in quantity to reduce free radicals to safe levels consequently resulting in cellular component damage. Another signaling pathway that might lead to an over-production of ROS levels is inflammation via release of pro-inflammatory mediators such as TNF-α and IFN-Y where ATP concentrations are ultimately decreased which occurs upon onset of hepatocyte damage, and this exacerbates mtROS production (Kozlov et al. Citation2017; Zhao et al. Citation2020). The concentrations of Bax (part of the Bcl-2 family of cell death regulating proteins) rise which promotes apoptosis. However, due to the depletion of ATP, the cell is unable to commit to cell death (usually ATP supplied through glycolysis or oxidative phosphorylation which is key in apoptotic cell death). The elevation in ROS levels activates mitochondrial permeability transition pores which further enhances ROS production and leads to mitochondrial swelling, apoptosis or necrosis (Yan et al. Citation2018). Enhanced generation of ROS levels activates c-Jun N-terminal kinase (JNK), a mitogen-activated protein kinase (MAPK) that is considered a critical control point between physiological and pathological cell states (Shi et al. Citation2019; Zeke et al. Citation2016). JNKs are activated via sequential phosphorylation of proteins in response to extracellular stimuli. JNK inhibitors were shown to attenuate hepatic injury by lowering oxidative stress and preventing apoptosis. On the other hand, phosphorylated JNK augments hepatic injury, as oxidative stress from inhibited anti-oxidant pathways or increased generation of ROS may enhance DILI severity (Shi et al. Citation2019). A visual generalized summary of mitochondrial damage is depicted in . Drugs associated with oxidative stress injury include flutamide, valproic acid, troglitazone and diclofenac (Chen et al. Citation2015).
Figure 2. Generalized hepatic pathological alterations associated with mitochondrial dysfunction resulting from an adverse drug reaction. Oxidative stress induces calcium over-load which initiates further subsequent accumulation of ROS. The oxygen species damages mitochondrial DNA, alter protein functions, limit mitochondrial respiration leading to lower levels of ATP and activate mitochondrial permeability transition pores. This increases permeability and eventual mitochondrial membrane rupture.
Endoplasmic reticulum stress
Endoplasmic reticulum stress is reported as a later event within metabolic dysfunction and is thought to occur due to (1) an over-accumulation of ROS, (2) an imbalance in intracellular Ca2+ levels and (3) activation of the unfolded protein response (Zhang et al. Citation2020). The unfolded protein response regulates through negative feedback by reducing protein synthesis, degrading mRNA, preventing translation and post-translational processes as well as transporting misfolded proteins to the cytoplasm for degradation (Zhang et al. Citation2020). Kozlov et al. (Citation2017) suggested that mitochondrial damage, JNK pathway activation and ER stress may be linked. ER stress is known to disrupt metabolism by inducing cell death.
Apoptotic and necrotic signaling pathways
Direct drug-mediated toxicity, intracellular organelle stress, or hepatocyte membrane damage might activate cell signaling pathways that results in apoptosis or necrosis (Krishna Citation2017). A balance between pro-apoptotic and anti-apoptotic Bcl-2 regulator proteins regulates the intrinsic pathway (Galluzzi et al. Citation2018). Pro-apoptotic BH3 interacting-domain death agonist (BID) is cleaved into truncated p15 BID during post-translation modification and then translocated to the mitochondria. In the mitochondrion BID interacts with Bax which subsequently initiates mitochondrial outer membrane permeabilization. This is considered the “point of no return” in apoptosis, whereby this enlarges mitochondrial membrane pores from approximately 5 kDa to over 100 kDa. This enables diffusion of proteins from the intermembrane space into the cytosol, one of which is cytochrome C (Bock and Tait Citation2020). Cytochrome C joins apoptotic protease activating factor 1 to form the apoptosome, a quaternary holoenzyme complex. The apoptosome activates caspase (CASP)-9 which subsequently activates CASP3 and CASP7, leading to cell death via nuclear fragmentation (karyorrhexis), protein breakdown (proteolysis) or chromatin condensation (pyknosis) (Iorga, Dara, and Kaplowitz Citation2017).
The extrinsic pathway is simpler and activated when tumor necrosis factor receptors, death receptors and Fas receptors are bound to their respective ligands (TNF-α, TRAIL, FASL). Subsequently, this activates FAS-associated death domain protein (FADD) which interacts with pro-caspase-8 and pro-caspase-10 that trigger production of CASP8 and CASP10. These caspases consequently activate CASP3 and CASP7 and converge the pathway into the aforementioned execution phase (Björnsson et al. Citation2015; Iorga, Dara, and Kaplowitz Citation2017).
The necrosis pathway is activated by mitochondrial permeability transition and results in cell swelling, a loss of plasma membrane integrity and eventually cell death (Shojaie, Iorga, and Dara Citation2020). Similar to apoptosis, necrosis may be elicited via cellular stress (particularly oxidative stress), ATP depletion and calpain, or death receptor activation (Fas and TNF-α), although the precise mechanism for necrosis via mitochondrial permeability transition has not yet been fully elucidated (Dara, Liu, and Kaplowitz Citation2016).
In recent years, ferroptosis has been identified as a plausible mechanism involved in certain types of DILI. Lorincz et al. (Citation2015) noted that challenge with ferrostatin-1 (specific ferroptosis inhibitor) in primary mouse hepatocytes treated with acetaminophen increased cell viability. It is of interest that (Lorincz et al. Citation2015) observed that ferrostatin-1 exerted no marked influence on CYP2E1 activity or altered intracellular GSH content. Therefore, it was concluded that protective effect on paracetamol-induced cell death is independent from interference of drug metabolism to N-acetyl-p-benzoquinone imine (NAPQI) (Lorincz et al. Citation2015).
Pyroptosis is an inflammatory cell death mechanism (cells under stress conditions, upregulate Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasomes) resulting in activation of caspase which was found to damage hepatic cells and augment organ inflammation (Luan and Ju Citation2018; Szabo and Petrasek Citation2015). In an interesting study, Xiang et al. (Citation2023) demonstrated that inflammation-related pyroptosis was implicated in thioacetamide and acetaminophen induced liver damage in mice. Xiang et al. (Citation2023) discovered that miR-29a-3p (believed to exhibit a protective role in liver cell damage) levels were decreased in DILI. Data suggested that miR-29a-3p was an activator of the PI3K/AKT signaling pathway which might inhibit pyroptosis to protect against DILI (Xiang et al. Citation2023).
Bile duct injury
Within the biliary system, cholangiocytes promote bile formation and regulation. Injuries of the bile duct present in cholestatic or mixed liver injuries and are characterized by the destruction of biliary epithelium which leads to biliary cirrhosis (Eiswerth et al. Citation2022). Obstructions in the ductal system produce interruptions in the flow of bile and accumulation of bile constituents within the blood (Visentin et al. Citation2018). These injuries most frequently affect the architecture of the interlobular ducts or the common hepatic ducts but might also affect the smaller canaliculi. Bile duct injuries are thought to be the result of T-cell mediated hypersensitivity but might also occur as biliary epithelium is directly exposed chemically reactive metabolites (CRMs) which are excreted following biotransformation (Grewal and Ahmad Citation2019). The canalicular bile salt export pump (BSEP) is a unidirectional efflux transporter located on the membrane of hepatocytes that enables drainage of bile salts into the canaliculus. Villanueva-Paz et al. (Citation2021) proposed that inhibition of BSEPs was a common mechanism underlying drug-induced cholestasis as it is understood that retention of bile acid contributes to hepatocellular stress. Frequently used drugs that are known to produce cholestatic injuries include carbamazepine, amoxicillin-clavulanate, sulfamethoxazole and floxuridine. A phenomenon rarely reported in literature known as vanishing bile duct syndrome might be present in severe cases of biliary destruction when histopathologic loss of intrahepatic ducts occurs (Eiswerth et al. Citation2022). There is a significant correlation between HLA predisposition and bile duct injury patterns (Nicoletti et al. Citation2017).
Immune-mediated injuries in DILI
Liver is fully enriched with diverse populations of immune cells of both innate and adaptive immune systems. The resident innate immune cells in liver include Kupffer cells (KC), dendritic cells (DC), natural killer cells, innate lymphoid cells, γδT cells, mucosal-associated invariant T cells, and adaptive immune cells such as T cells; regulatory T cells, cytotoxic T lymphocytes and helper T cells (Lee and Lawrence Citation2018). It is believed that immune system plays vital role in initiation and recovery from DILI (Cheng et al. Citation2021; Woolbright and Jaeschke Citation2018) ().
Table 1. Summary of mechanisms of hepatic immune tolerance in the healthy liver (Bottcher, Knolle, and Stabenow Citation2011; Doherty Citation2016; Heymann et al. Citation2015; Teigs and Lohse Citation2009).
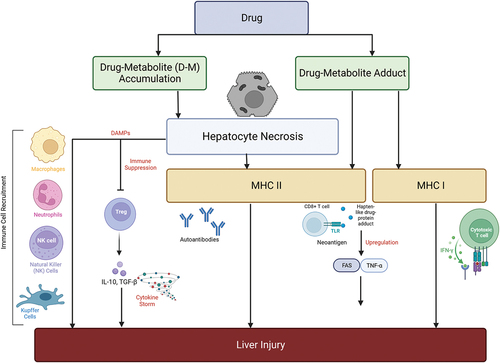
Immune-mediated injuries occur when a hypersensitivity reaction provokes an inflammatory response that involves both the innate and adaptive immune systems (Lee and Lawrence Citation2018). The liver is frequently exposed to foreign antigens due to its biological filtering role and therefore displays a predisposition toward immune tolerance to avoid excessive response. Iorga, Dara, and Kaplowitz (Citation2017) noted in DILI that this tolerance was hampered and inhibited. Drug metabolites may bind either covalently or non-covalently bind to CYP450 enzymes creating a hapten-like drug-protein adduct (Villanueva-Paz et al. Citation2021). These hapten-like drug-protein adducts might bind to endogenous proteins to form neoantigen structures that are presented to CD8+ cytotoxic T-Cells via MHC II complexes initiating upregulation of transcription factors Fas and TNF-α which ultimately lead to cell death after immune cell recruitment (Gerussi et al. Citation2021). Cellular stress might release damage associated molecular patterns (DAMPs) which bind to pattern-recognition receptors on the antigen presenting cells which subsequently stimulate the adaptive immune response.
Damage-associated molecular patterns are known to bind to toll-like receptors (TLRs) and purinergic receptors expressed by KCs which secrete an array of pro-inflammatory mediators such as IL6, IL18, TNF-α, IFN-Y and Fas ligands which further drive the proliferation of macrophages (Katarey and Verma Citation2016). Interestingly, TNF-α was found to be involved in hepatocyte proliferation in liver regeneration despite being a potent mediator in hepatotoxicity. In addition, the interaction of parental or metabolized drugs with T-cell receptors and MHC II and antigen interactions leads to activation of CD8+ cytotoxic cells and CD4+ helper cells which both play key role in idiosyncratic DILI. These drug-specific T-cells are triggered by liver dendritic cells which metabolize the drug to form drug-modified-peptide-HLA membrane molecules (Adair, Meng, and Naisbitt Citation2021; Gerussi et al. Citation2021) (). The molecular mechanisms that are involved in T-cell response in DILI are described by the pharmacological interaction p-i model, in which either i) the drug interacts directly with T cell immune receptors, whereas the drug forms a non-covalently bound self-peptide HLA complex without antigen processing ii) the drug is covalently bound to a self-protein, making the drug act as a hapten (the protein acts as a carrier), together forming a haptenized peptide that helps to stimulate antigen-specific T-cells, and iii) altered peptide model in which the activation or inhibition of particular T-cells might be influenced by changing the antigen’s peptide sequence. In contrast to the original peptide, these altered peptides may have altered MHC molecule binding affinities (Adair, Meng, and Naisbitt Citation2021; Gerussi et al. Citation2021; Pichler Citation2013).
Figure 3. Simplified diagrammatic representation of the causation and implications of MHC class I and II cells in immune-mediated DILI. The necrosis of hepatocytes might be directly induced by accumulation of toxic drug metabolites. Damage associated molecular patterns (DAMPs) recruit natural killer and Kupffer cells. Hapten-like drug-protein adducts bind with MHC II cells and this leads to upregulation of transcription factors such as FAS and TNF-α that initiate a further immune cascade increasing the severity of hepatocyte damage.
The altered peptide repertoire hypothesis suggests that drugs may induce mistargeting of endogenous peptides leading to HLA autoimmunity (Segovia-Zafra et al. Citation2021). Alternatively, chemically inert drugs may form non-covalent interactions with MHC II complexes, altering their binding pocket and encouraging an immune response (Kubes and Jenne Citation2018). However, it is postulated that multiple factors determine the immune-mediated response to a drug or its metabolite and this may account for the rarity in these types of injuries. Host factors such as age, gender, genetic polymorphisms, lifestyle, and immune status may also contribute to the adverse drug reaction (Villanueva-Paz et al. Citation2021). A summary and links between the types of injury are summarized in .
Figure 4. Molecular events involved in DILI during hepatocellular and cholestatic injuries. Hepatocyte injury is initiated by an increase in oxidative stress resulting from ROS imbalance, endoplasmic reticulum stress, ATP depletion, mitochondrial dysfunction, calcium overload, glutathione depletion, protein dysfunction and BSEP inhibition. BSEP inhibition may lead to bile duct injuries such as obstruction, accumulation of bile salts and interference in sinusoidal uptake. DAMPs are released upon hepatocyte damage and initiate further inflammation. Reactive metabolites might also activate the immune response via drug-protein adducts, haptens, TLR binding, mistargeting of peptides, MHC II binding release of pro-inflammatory cytokines in a positive loop with upregulation of the recruitment of additional immune cells.
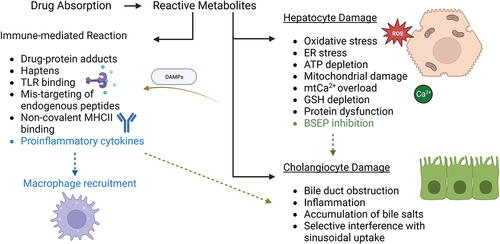
In iDILI, it is understood that activation of T lymphocytes is a critical process in hepatocellular damage. T cells are activated when antigen-presenting cells (APCs) present drug antigens or drug-modified self-antigens to these cells (Gerussi et al. Citation2021). Hepatocytes are then directly injured and undergo apoptosis due to CD8+ cytotoxic T cells activation. In parallel, pro-inflammatory cytokines are released by CD4+ helper T cells and other immune cells, including macrophages, which further promote inflammation and subsequent hepatic injury. T cell activation and antibody-mediated mechanisms are the predominant components of the immune response in cholestatic damage (Hosack, Damry, and Biswas Citation2023; Yu et al. Citation2017). Autoantibodies or drug-induced antibodies may combine with bile duct epithelial cells to generate immune complexes that activate complement and damage the bile duct. Antigens connected to bile duct epithelial cells may be recognized by CD4+ T lymphocytes, triggering an immunological response and production of cytokines that initiate bile duct damage. Further, bile salts accumulate due to medication or reactive metabolite blockage of the BSEP, which induces inflammation and immune-mediated bile duct damage (Grewal and Ahmad Citation2019). In both hepatocellular and cholestatic injuries, production of DAMPs from injured hepatocytes or bile duct cells further activates the immune system and stimulate inflammation (Allen, Jaeschke, and Copple Citation2011; Cai and Boyer Citation2021). Garcia-Cortes et al. (Citation2020) indicated that frequently used drugs thought to be associated with an immune response include halothane, sulindac, sulfamethoxazole, chlorpromazine, flucloxacillin and amoxicillin-clavulanate.
While acute DILI and immune-mediated DILI display distinct underlying causes, there are shared molecular mechanisms that contribute to hepatic injury in both presentations. Inflammation plays a key role in both types of injury. In direct injury toxic metabolites might overwhelm hepatic cells leading to an immune response, whereas immune-mediated DILI might occur without an accumulation of metabolites as immune mechanisms mistakenly identify drug-altered hepatic cells as foreign. Both types of injury involve release of cascade of pro-inflammatory cytokines (Tasnim et al. Citation2021). The inflammatory response is also enhanced by excess production of ROS in both processes. In direct DILI, MHC interactions and macrophage recruitment occur due to damaged hepatic cells releasing pro-inflammatory cytokines and chemokines and this elicits an immune response (Pu et al. Citation2023). ER stress is another shared mechanism in both presentations, but the specific triggers and pathways involved vary between the two conditions. In acute DILI, reactive metabolites may directly induce misfolded proteins resulting in unfolded protein response (UPR). In contrast, in immune-mediated DILI the elevated levels of pro-inflammatory cytokines might alter ER function. Similarly, sustained cytotoxic activity may lead to ER-induced stress (Björnsson and Björnsson Citation2022). It is noteworthy that while there are shared mechanisms, acute DILI and immune-mediated DILI are usually distinct in their clinical presentation and prognosis. Acute DILI is associated with recent drug exposure and may resolve quickly upon discontinuation of the offending agent, whereas immune-mediated DILI might be chronic requiring immunosuppressive treatment.
Global incidence of DILI
Studies into the incidence of DILI usually occur in the form of a retrospective database search or using information regarding patients with diagnosed liver damage from specific hospitals or areas (summarized in ). Findings are difficult to directly compare as various diagnostic parameters were employed, cohort demographics varied and epidemiological differences in prescriptions and drug uses contribute to varying contrasting results. In this review, in order to allow for the focus being placed on idiopathic DILI, paracetamol induced injury was excluded. An Icelandic study of patients with a DILI diagnosis from 2010 to 2011 using the Roussel Uclaf Causality Assessment Method (RUCAM) assessment method found amoxicillin-clavulanate to be the most implicated agent at 22% of DILI patients, followed by diclofenac (6%), azathioprine (4%), infliximab (4%) and nitrofurantoin (4%) (Björnsson et al. Citation2013). The highest risk of hepatotoxicity occurred with azathioprine (0.75%) and infliximab (0.68%), and the population-based incidence of idiopathic DILI was 19.1 per 100,000 inhabitants (Björnsson et al. Citation2013). Ninety-six individuals with DILI were included in this study using parameters of ALT > 3 × ULN and/or ALP > 2 × ULN with paracetamol-related injury excluded.
Table 2. Summary of global DILI incidence showing origin, most common offending drug and the population investigated.
A similar study originating in Sweden reviewed cases of DILI from 1995 to 2005 within a single hospital and measured injury based upon the aforementioned international criteria. A total of 1164 cases were analyzed leading to a DILI diagnosis in 77 cases (6.6%). De Valle et al. (Citation2006) found that DILI occurred more often in women and antibiotics were the most frequent agent (30%) followed by diclofenac (22%). Elsewhere, symptomatic drug-induced injuries were investigated in France between 1997 and 2000 by Sgro et al. (Citation2002) who reported that anti-microbials (19%) and non-steroidal anti-inflammatory drugs (NSAIDs) (16%) were associated with the highest incidence of DILI. Sgro et al. (Citation2002) also noted an annual incidence of 13.9% (±2.4%) per 100,000 inhabitants. The biological parameters used were ALT activity or conjugated bilirubin >2 × ULN or combined increase in ALT, ALP, and total bilirubin >2 × ULN (Sgro et al. Citation2002). Another study originating from the United Kingdom examined the General Practice Research Database (GPRD), a large database of anonymous medical records collected at GP Surgeries around the country from 1994 to 1999. This was one of the largest retrospective studies performed where a total of 1,636,792 individuals were followed for a total of 5,404,705 person-years with 128 confirmed cases of DILI studied (De Abajo et al. Citation2004). Data demonstrated a crude incidence rate of 2.4 per 100,000 person-years with the strongest drug associations consisting of anti-microbials (amoxicillin-clavulanic acid, flucloxacillin, macrolides, tetracyclines) and NSAIDs (De Abajo et al. Citation2004). In this investigation, DILI was classified by ALT > 2 × ULN, or a combined increase in AST, ALP, and total bilirubin – provided that one of these was >2 × ULN (De Abajo et al. Citation2004). Further, in a study conducted in Delaware (USA) via surveillance of DILI symptoms during 2014, an incidence rate of 2.7 per 100,000 adult residents was detected (Vega et al. Citation2017). Twenty patients met inclusion criteria of ALT/AST >5 × ULN, ALP > 2 × ULN on two consecutive occasions or any elevation in activities of ALT, AST or ALP with total bilirubin >2.5 mg/dl. Surprisingly, 43% of cases were attributed to herbal dietary supplements (HDS), while 36% were due to antibiotics (Vega et al. Citation2017).
While in western countries antibiotics and NSAIDs are the prominent cause in DILI, investigations originating from Asia tend to suggest higher incidences with exposure to herbal medicines and dietary supplements. Herbal supplement induced hepatic damage is often termed herb-induced liver injury (HILI). Suk et al. (Citation2012) focused on hospitals in South Korea from 2005 to 2007 and noted an incidence of 12 per 100,000 inhabitants with the frequent causative agents being herbal medications (27.5%). This study used the parameters of ALT > 3 × ULN or total bilirubin >2 × ULN to distinguish between cases of DILI and results were adjudicated by 7 review boards for confirmation of causality using the RUCAM. Herbal medications were also the most frequently implicated drug in studies in China resulting in DILI in as many as 53.63% of the cohort (Lai et al. Citation2012; Ou et al. Citation2015). One of these studies differed in biochemical parameters as the investigators focused on 129 patients with ALT > 10 × ULN, significantly higher levels than those reviewed within this paper (Ou et al. Citation2015).
In contrast, another study originating in China reported similar findings to western countries and noted antimicrobial incidence at 32% and glucocorticoids at 24% (Xu et al. Citation2012). In an Indian tertiary hospital studied 82 suspected DILI patients from 2014 to 2015 were detected and the most frequently implicated drugs were antitubercular therapy drugs (49%) and antiepileptic drugs (12%) (Rathi et al. Citation2017). Finally, Isa et al. (Citation2016) reported hepatotoxicity among hospitalized patients in Nigeria in 2013. These investigators demonstrated that hepatic injury attributed to anti-tuberculosis (TB) drugs occurred frequently (Isa et al. Citation2016). Similarly, a study carried out in 2009 at a hospital in South Africa showed the highest risk for DILI was antiretroviral therapy (ART) (35%) followed by anti-TB drugs (27%) (Schutz et al. Citation2012). This is indicative of higher rates of HIV and tuberculosis within these areas. Data seem to further suggest that immune-checkpoint inhibitors for HIV treatment are a significant enhanced risk for liver injury (Real et al. Citation2019).
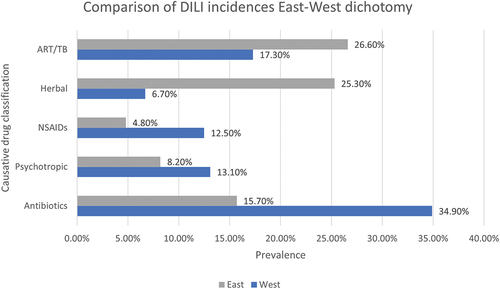
Meta-analysis conducted on the incidences of DILI around the world detected marked differences in the most frequently administered agents implicated in geographical locations. For greater ease in comparison, some studies focussed on “East” versus “West”- conceptual boundaries based upon culture and developmental status as opposed to geographical location alone. The prevalence of DILI induced by main hepatotoxic drug types (Anti acquired immunodeficiency syndrome and TB, herbal remedies and supplements, NSAIDs, psychotropic and Antibiotic drugs) (). ART/TB drugs and herbal compounds exhibit a significantly higher prevalence within the Eastern world, whereas antibiotics, NSAIDs and psychotropic drugs display a higher prevalence in the Western world (Low et al. Citation2020).
Figure 5. Causative drug classifications and their prevalence in “Eastern” locations compared to “Western” locations using meta-analysis data collected by (Low et al. Citation2020) using 28 studies focused on incidences of DILI initiated by anti-TB, anti-retroviral therapy, herbal drugs and supplements, NSAIDs, psychotropic and antibiotic drugs. In this instance, “East” includes data from Asia and Africa – frequently considered the Eastern world, while “West” includes data from Europe, United States of America, New Zealand and Australia – commonly referred to as the Western world.
Clinical assessment and classifications
Clinical intervention may often be delayed due to a lack of specific symptoms and lab testing. Clinical testing is useful for the detection of DILI but is not indicative of prognosis or etiology (McGill and Jaeschke Citation2019). Symptoms may mimic other hepatic disorders such as autoimmune hepatitis which emphasizes the need for DILI diagnosis by exclusion. A biomarker is a characteristic that is measured as an indicator of biological processes. Ideally, a functional biomarker needs exhibit high sensitivity and specificity (>90%) to achieve a high Positive Predictive Value and Negative Predictive Value (70–80%). This approach has limitations as predictive values are generally modest indicating low accuracy in predicting DILI. It has been widely reported that traditional liver enzyme biochemical markers have clinical limitations due to their restricted sensitivity, specificity, and accuracy (Garcia-Cortes et al. Citation2020).
Multiple drugs are frequently prescribed concurrently to treat several diseases and patients may take numerous drugs for their condition. This makes isolation of the offending agent increasingly difficult. Physicians may face further issues in discontinuing the offending agent as doing so may worsen the patient’s other co-morbidities. In 2011, the International Serious Adverse Events Consortium set biochemical thresholds at ALT or AST ≥ 5× ULN and ALP > 2 ULN which helps to provide a baseline reference for physicians (Jee, Sernoskie, and Uetrecht Citation2021). Elevations in activities of ALT, AST and ALP, and total bilirubin content are most frequently used for detection but do not reliably correlate with the degree of hepatic impairment, and do not show good prognostic value (McGill and Jaeschke Citation2018). Other biomarkers may be analyzed during diagnosis (acetaminophen-cysteine protein adducts), prediction (genetic markers), and prognosis (micro-RNA-122, keratin-18, glutamate dehydrogenase and mtDNA) (McGill and Jaeschke Citation2019). Recently, the FDA announced support for further clinical exploration of novel biomarkers microRNA-122 and glutamate dehydrogenase (Andrade et al. Citation2019).
With DILI symptoms being nonspecific, clinical assessment methods are frequently employed to diagnose cases (Björnsson Citation2021; European Association for the study of the liver 2019). RUCAM has been frequently utilized since its inception in 1993 (Danan and Benichou Citation1993). The method is a diagnostic tool based upon seven clinical observations that provide a framework for diagnosis and evaluation (Jee, Sernoskie, and Uetrecht Citation2021). An initial step in both methods is to determine the R ratio to classify the injury as either hepatocellular, cholestatic or mixed, depending upon serum enzyme activity elevations. The R ratio is calculated by: R = (ALT value/ALT ULN)/(ALP Value/ALP ULN). If the result is >5 it is considered hepatocellular, between 2 and 5 is mixed and >2 is considered cholestatic. However, it is worth mentioning that the ULN sometimes varies between different labs, which markedly alters the calculation. Further, there is no official recommendation for scenarios with preexisting liver impairment or abnormal baseline serum enzyme activity levels. It is worth noting that between 5% and 20% of a normal population fall into this category. Recently, Teschke and Danan (Citation2020) found that the RUCAM model was used to assess possible cases of idiosyncratic DILI in 81,856 published reports listed on PubMed since 1993. RUCAM provides (1) possible causative agent in the case of multiple drug administration, (2) unpredictability idiosyncratic liver events and (3) lack of diagnostic biomarkers inferring that clinical recognition is still troublesome (Hosack, Damry, and Biswas Citation2023). Taking this into account, it has been suggested that glutamate dehydrogenase might be a more specific marker of liver injury as compared to ALT (Clinton et al. Citation2021). In summary, RUCAM is a well established and trusted system of assigning points for clinical and biochemical features of liver injury which provides an overall assessment score indicating the likelihood of hepatic injury is due to the specific drug in question (Danan and Teschke Citation2016).
LiverTox (LiverTox Citation2022) is another multilayered and interactive website with evidence-based information on drug, dietary supplements, and herbal-induced liver injury. However, recently some issues have been raised regarding the appropriateness of this database as a means of predicting iDILI fundamentally due to concerns with factors such as quality of data, lack of evidence-based case studies and absence of standard definitions and categorization of liver injury itself (Teschke and Danan Citation2021b).
Risk factors of iDILI
There are numerous proposed risk factors for occurrence of iDILI or specific types of injury, which include but not limited to age, biological gender, genetics, preexisting disease state and drug characteristics.
Age and gender
A study that took place from 2001 to 2009, investigating age and gender as risk factors for DILI development (Lucena et al. Citation2009). In this study, patients aged 60+ accounted for 46% of cases with no cases recorded in the 20–29-year group (Lucena et al. Citation2009). These investigators also found that older age was associated with cholestatic injury type. However, it was concluded that neither old age nor female gender were pre-disposing factors to DILI overall, but rather a determinant for cholestatic injury with male dominance. Younger age was associated with cytolytic damage with a female over-representation (Lucena et al. Citation2009). In 2015, the Drug-Induced Liver Injury Network (DILIN) also stated that older age was not an overall risk factor for DILI but corroborates that older age is associated with cholestatic injury, especially resulting from amoxicillin-clavulanate, isoniazid and nitrofurantoin exposure (Chalasani et al. Citation2015). Fisher, Vuppalanchi, and Saxena (Citation2015) reported similar evidence and shared that valproate and aspirin were frequent agents associated with DILI in children, whereas amoxicillin-clavulanate was the most frequent causative agent in those over 50. Recent data from a World Health Organization safety report which examined 256 drugs known to be associated with DILI suggested that elderly patients (65+ years) were more likely to develop cholestatic injury (Ahmad and Odin Citation2017). Chalasani et al. (Citation2015) presented conflicting data regarding the female gender being an enhanced risk factor for DILI where a higher prevalence of females (59%) was noted. However, the Spanish DILI Registry recorded only 49% of cases occurring in females. It is believed that sex hormones are important in regulation of drug metabolism, transport and dimorphism in cellular stress pathways (Wang et al. Citation2021). In another study, Fisher, Vuppalanchi, and Saxena (Citation2015) suggested that females are not at an increased risk but are more likely to progress to acute liver failure, indicating that females are likely to exhibit worsened prognosis once DILI has occurred. Further, in an Icelandic investigation, Björnsson et al. (Citation2013) suggested that there was a 3-fold increased risk of DILI in older patients, although this may be attributed to higher prescription rates in the elderly.
Drug characteristics
Drug characteristics may also present as a risk factor for development of DILI, based upon their chemical structure, molecular weight, lipophilicity and known drug–drug interactions. Increased lipophilicity of a compound leads to increased permeability and uptake and increased levels of biotransformation and subsequent higher concentrations of toxic metabolites (Teschke and Danan Citation2021a). CRMs generated by CYP450 enzymes produce cellular stress and an association was identified between covalent binding and the risk of iDILI occurrence. This has led drug companies to start screening candidate drugs for their degree of covalent binding (Jee, Sernoskie, and Uetrecht Citation2021). There are also higher numbers of cases in which drugs undergo significant hepatic metabolism. This important consideration is emphasized in a 2021 review, where Teschke and Danan (Citation2021a) identified 61.1% of drugs implicated in iDILI being metabolized by CYP450 isoforms. Among these pharmaceuticals, almost half (49.6%) were shown to be metabolized by CYP3A4 and 5 and almost quarter (24.6%) were catabolized by CYP2C8 or 9 (Teschke and Danan Citation2021a). On the other hand, drugs with a carboxylic acid functional group may be associated with a lower risk of iDILI occurrence. This hypothesis considers that most of these drugs are metabolized to acyl glucuronides that freely undergo excretion (Teschke and Danan Citation2021a). Higher dosage has also been linked as a potential risk factor for DILI development. It is an interesting that almost all drugs with black box warnings have a daily dosage of >50 mg (Vuppalanchi et al. Citation2014). The Biopharmaceutics Drug Disposition Classification System (BDDCS) evaluates the severity of DILI warning drug labels. The system is a four-class approach based on water solubility and extent of metabolism. With the BDDCS, it is possible to predict the role of metabolic transporters and enzymes to calculate the new molecular entity prior to its administration to animals or humans (Bocci, Oprea, and Benet Citation2022).
Variations in the human leukocyte antigen in DILI
Host characteristics may also be considered as an elevated risk factor for DILI occurrence. Although preexisting liver disease is not necessarily an enhanced risk factor for DILI, these patients have been found to have a significantly higher risk of morbidity (Chalasani et al. Citation2015). Genetic risk factors most frequently present with variations of the HLA gene. HLA encodes for components of the MHC which are involved in immune processes that occur during DILI (Ahmad and Odin Citation2017) via surface proteins that bind to foreign molecules to elicit an immune response. The MHC consists of more than 200 genes on chromosome 6 and a total of over 1500 alleles of the HLA-B gene were identified (Perwitasari et al. Citation2018). A common HLA allele present in patients with DILI administered amoxicillin-clavulanate is HLA-DRB1 × 15 and HLA-DRB1 × 06. It has been demonstrated that flucloxacillin (an antibiotic widely used for staphylococcal infection) is a frequent cause of DILI and is associated with HLA-B × 57:01 (Ahmad and Odin Citation2017). This allele has been implicated in many DILI cases (OR 80.6); and therefore, it is prudent to carry out HLA genetic testing when treating patients with these types of injuries (Ahmad and Odin Citation2017). HLA genotyping is now widely accessible, affordable and might assist with diagnosis. Genetic susceptibility involving both candidate gene and genome-wide association approaches was previously examined (Daly and Day Citation2012). Evidence indicates that single nucleotide polymorphisms within the human leukocyte antigen (HLA) gene region may predict the development of DILI toxicity. In a 2016 genome-wide study, the most significant single nucleotide polymorphisms identified were rs114577328 in the major histocompatibility complex (MHC) region of chromosome 6 and rs72631567 on chromosome 2 (Nicoletti et al. Citation2016). These studies were international collaborative efforts involving many recruitment centers across the world and used retrospective data from a wide range of DILI investigations with varying clinical assessment criteria (Ahmad and Odin Citation2017; Daly and Day Citation2012; Nicoletti et al. Citation2016).
Non-HLA DILI associations
Polymorphisms within drug metabolizing enzymes are also associated with DILI (Perwitasari et al. Citation2018). The genetic polymorphisms most frequently associated with DILI are found in N-acetyl transferase 2, glutathione S-transferase M1, glutathione S-transferase T1, CYP2E1 and HLA genes (Bocci, Oprea, and Benet Citation2022). There is also a reported association between the microbiome of the gut and immune mediated DILI, particularly with the use of isoniazid when treating tuberculosis (Liu et al. Citation2023).
A rare complication occurs when Stevens-Johnson Syndrome (SJS) occurs concurrently with DILI. SJS is a rare, potentially fatal skin condition that can include toxic epidermal necrolysis. The organs implicated most commonly in SJS are the liver and skin with severe cutaneous adverse reactions (Ortega-Alonso et al. Citation2016). SJS is associated with immune-mediated DILI and patients with both SJS and DILI concurrently exhibit a higher risk of morbidity (Andrade et al. Citation2019). Individuals with SJS have a higher risk of liver injury due to exposure to xanthine oxidase inhibitors, dibenzoazepine and herbal medicines (Zhang et al. Citation2020).
Pre-existing disease
In 2023, metabolic dysfunction-associated steatotic liver disease (MASLD) (formally known as nonalcoholic fatty liver disease) displayed a high prevalence affecting 25% of the global population (McPherson et al. Citation2022), amongst whom nearly 30% progressed to metabolic dysfunction-associated steatohepatitis (MASH) (formally known as nonalcoholic steatohepatitis). It is now estimated that 2 out of every 3 adults in the UK exhibit early stages of MASLD. Mortality rates for liver disease have increased 400% since 1970, and in individuals aged younger than 65 years, mortality frequencies have risen almost 500% (British Liver Trust Citation2023). Recently, the Office for National Statistics (UK) reported that the average age of death attributed to liver disease in 2018 was 61 for males and 63 for females. In addition, approximately 75% of patients who will die from cirrhosis are currently unaware that these subjects are suffering from liver disease and the majority of individuals possess entirely normal liver serum chemistries until they present to hospital as an emergency. This important and additional complication is clinically significant as large numbers within the general population suffer from a wide spectrum of sub-clinical liver damage without any apparently visible disease manifestations which often remain clinically asymptomatic and undetected yet exhibit a high prevalence. Of note, in 2016, the USA’s economic burden of chronic liver diseases was $32.5 billion, with over 65% of spending on inpatient or emergency care, while in 2020, the clinical cost of liver disease in the UK alone was reported to be over £10 billion, accounting for 5% of the national health service budget (NHS Citation2023). At present, there are no approved therapies to manage MASLD. Importantly, 96% of adults with MASLD in the United States were unaware that these individuals were suffering from liver disease (Alqahtani et al. Citation2021).
Several investigators suggested that xenobiotic-induced adverse effects in the liver are aggravated in the diseased organ (Allard et al. Citation2021; Kermanizadeh et al. Citation2017, Citation2022; Lammert et al. Citation2019; Li, Gao, and Niu Citation2019; Vargas et al. Citation2017). The exposure to xenobiotics might aggravate preexisting liver conditions, related to the growing obesity pandemic, western diet and sedentary lifestyle. Metabolic liver diseases and exposure to xenobiotics might trigger similar cellular stress responses, with the capability to stimulate and accelerate further disease progression. Importantly, the form and extent of preexisting liver disease might also hamper the organ’s ability for regeneration. Indeed, hepatocyte proliferation is severely inhibited in chronic liver diseases, including MASLD, alcoholic liver disease, and chronic exposure to hepatotoxins (Fernandez-Checa et al. Citation2021; Michalopoulos Citation2013; Sancho-Bru et al. Citation2012).
COVID-19
The recent coronavirus pandemic raised a number of clinical concerns related to DILI. Liver serum enzyme abnormalities are common in hospitalized patients with COVID-19 (Gupta et al. Citation2020; Saini et al. Citation2020). The precise pathophysiological mechanism related to COVID is not yet fully understood, but current theories are centered on direct viral injury, treatment-related DILI and immune-mediated injury, where SARS-CoV-2 infect hepatocytes, directly via the angiotensin-converting enzyme 2 receptor, which the virus uses to infiltrate respiratory tissue cells. This direct infection results in liver injury, inflammation, cellular damage, treatment-related DILI and immune-mediated injury (Clinton et al. Citation2021; Marjot et al. Citation2021; McGrowder et al. Citation2021). Several investigators proposed that patients fighting an active COVID-19 infection may be relatively immunodeficient; however, when being treated with an array of drugs, the consequence is a greater risk of developing immune-mediated DILI (Bonaventura et al. Citation2020; Hui et al. Citation2020; Lu, Stratton, and Tang Citation2020; Naseralallah et al. Citation2022; Xie et al. Citation2020). In a recent investigation, Saha, Vij, and Rawat (Citation2022) using liver biopsies taken from COVID-19 patients showed microvesicular steatosis, patchy necrosis and infiltration of lymphocytes.
Liver injury in mild cases of COVID-19 is usually reversible. However, severe cases have required medical attention where liver functions are closely monitored and treated with hepatoprotective, anti-inflammatory agents such as glycyrrhizic acid, bicyclol and polyene phosphatidyl choline to diminish cytopathic effects of virus on the hepatocytes to reduce liver damage. Immunosuppressive drugs such as tocilizumab are used to treat cytokine storm syndrome and according to clinical guidelines (1) anti-viral drugs including remdesivir, favipiravir, lopinavir and ritonavir, (2) anti-microbial drugs such as azithromycin, (3) anti-malarial drugs such as hydroxychloroquine and chloroquine and (4) anti-pyretic drugs such as paracetamol, ibuprofen and acetaminophen are also frequently administrated in the treatment COVID-19 and related comorbidities (Li et al. Citation2022; McGrowder et al. Citation2021; Zhai et al. Citation2021; Zhao et al. Citation2023). The clinical efficacy and safety of these anti-viral drugs have shown potential for hepatotoxicity resulting in DILI after repeated dosing. Similar observations were found when anti-viral drugs were applied prophylactically, leading to subsequent liver damage. Inhibition of CYP3A4 enzyme activity and liver transporter proteins are suspected as being important factors leading to disruption of drug metabolism and subsequent manifestation of DILI. In severe cases, high dose use of tocilizumab, an IL-8 inhibitor is utilized to manage the cytokine storm in COVID-19 patients (Muhović et al. Citation2020; Vitiello et al. Citation2021).
Management and prognosis
iDILI is one of the more challenging forms of liver disease, both in terms of diagnosis and management. The prompt identification and discontinuation of the offending agent is crucial in early DILI management. Biochemical tests may aid diagnosis but are not reliably indicative of severity or prognosis (Rathi et al. Citation2017). Diagnosis is made further difficult as DILI follows diagnosis by exclusion. In addition, appropriate treatment may follow various pathways. Upon discontinuation, approximately 80% of DILI cases resolve spontaneously (Björnsson Citation2021). In these cases, active surveillance occurs while biochemical testing is repeated. In more severe injuries, a liver biopsy may be required to determine injury type and prognosis. The classification of injury needs to be determined as this dictates the treatment plan to follow. Unfortunately, a significant issue in treating DILI is that the therapeutic effect of the offending agent is lost upon the de-challenge. Physicians need to comprehensively assess the risk of primary disease progression when considering withdrawal. This is a particular issue in the treatment of TB where no alternative therapies are available. In this scenario, re-challenge strategies may be implemented with the aim of desensitizing patient reaction (Stine and Lewis Citation2016). DILI treatment has remained relatively unchanged for decades. Proactive measures may be taken for recent ingestion such as in the case of paracetamol overdose. Activated charcoal may provide therapeutic benefits, but its administration is restricted to within 3–4 hr since consumption (Hosack, Damry, and Biswas Citation2023). Hepatoprotective medications including N-acetylcysteine and L-carnitine may be used as an antidote, but these are only indicated for intrinsic DILI. Treatment is aimed at detoxification, scavenging free radicals, anti-oxidation, hepatocyte protection and immune regulation. A frequently utilized treatment for cholestatic injury is ursodeoxycholic acid which is a naturally occurring hydrophilic bile acid that decreases cholesterol saturation index of bile (Li et al. Citation2022; Robles-Diaz et al. Citation2021). Another frequently employed pharmacotherapy is bicyclol, an inflammatory cytokine inhibitor that suppresses activity of various necrosis factors and interleukins (Adam et al. Citation2018). Immune mediated DILI may benefit from a course of corticosteroids. Immune-checkpoint inhibitor (ICI) in hepatitis is rare, but cases are increasing as use of novel ICIs and combination with other chemotherapy drugs are being prescribed to oncology patients (Björnsson Citation2021).
The principal rule of managing suspected DILI is cessation of treatment with the drug in question. In addition, and unsurprisingly deliberate re-exposure to the drug suspected of initiating DILI is generally discouraged. However, for the overall well-being of the patient, it is vital that distinctions are made between instances where asymptomatic elevations in liver enzymes activities does not require immediate discontinuation of drug treatment (where enzyme activity increases return to norm despite continual drug treatment) or the cases in which the higher levels of liver enzyme activities might actually be indicative of severe liver damage.
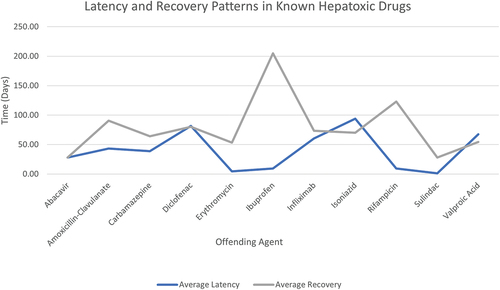
Unfortunately, some cases of DILI progress to acute liver failure (ALF) and require transplant or become fatal. Approximately 20% of DILI patients develop chronic liver disease (Wang et al. Citation2021) and large cohort studies reported DILI-related mortality to be 5–10% (Björnsson Citation2021). ALF patients may present with nausea and vomiting, hypoglycemia, coagulopathy and an elevated international normalized ratio (INR). In one study, iDILI showed a 3-week transplant-free survival rate of just 27% (Ostapowicz et al. Citation2002). Acute liver failure is defined as a permanent alteration in liver function with jaundice (Garcia-Cortes et al. Citation2020). Most cases of ALF in the Western world are produced by intrinsic DILI (paracetamol) (Chan and Benet Citation2017). Liver support devices such as hemodialysis and use of a Molecular Adsorbent Recirculating System (MARS) might replicate the detoxifying function of the liver and thus stabilize the patient’s health while awaiting transplant (Adam et al. Citation2018). Recent clinical practice guidelines have agreed that liver transplantation is still the primary resolution for severe ALF (Devarbhavi et al. Citation2021; Andrade et al. Citation2019). Studies indicate a 5-year post-transplant survival rate between 70% and 80% in DILI-ALF patients (Adam et al. Citation2018). illustrates the average latency from the start of drug administration to the onset of symptoms.
Figure 6. Average latency from drug start to onset of symptoms and mean recovery period to normal liver enzyme levels in patient case studies reported in LiverTox (Citation2012a, Citation2012b, Citation2012c, Citation2012d, Citation2012e, Citation2012f, Citation2012g, Citation2012h, Citation2012i). Incomplete data such as scenarios involving patient fatalities have been excluded.
Prevention
As treatment for DILI is limited, focus has shifted toward prevention. Strict screening criteria need to be met by a candidate drug during the discovery process before it progresses to clinical trials (Chan and Benet Citation2017). Assessing patients’ individual risk prior to commencing a known hepatotoxic drug is useful in preventing genetic associated adverse reactions. As one moves toward a future with personalized medicine, this might be considered for implementation. The restriction of the availability of known hepatotoxic drugs also prevents public misuse. In 1998, the United Kingdom imposed legislation to restrict pack sizes and sales of paracetamol to 16 in a regular store and 32 in a pharmacy. From 1998 to 2009 there was a 43% reduction in paracetamol-related death and a 61% fall in liver transplant following paracetamol overdose (Hawton et al. Citation2013). Ongoing monitoring of patients on longer duration or combinations of drugs might aid in preventing DILI-ALF as asymptomatic cases might be detected prior to lasting injury. Further, point of care testing of ALT similar to commonly used finger prick glucose monitoring has been proposed as a monitoring system for patients with a risk of hepatotoxicity (Pollock et al. Citation2013). Elsewhere, clinical monitoring has been advised for patients taking isoniazid. Avoiding the use of or substituting known hepatotoxic agents is a way that physicians might avoid development of DILI. This is currently implemented for patients with chronic liver disease as physicians are advised to avoid the use of valproic acid, methotrexate, ketoconazole and tolvaptan (Stine and Lewis Citation2016). Finally, the reporting of incidences of DILI is critical in identifying and emphasizing patterns as well as offering a wider view of DILI overall to aid in collating this information.
Conclusions and future perspectives
Drug-induced hepatotoxicity is a predominant response for agent withdrawal from global markets. Acute liver failure is an example of drug-induced hepatotoxicity which is known to occur frequently. Further, DILI is a significant source associated with drug recall and ALF. Although distinctions discussed above exist between the two classifications of DILI (intrinsic vs. idiosyncratic), hepatotoxicity continues to be a global concern. Further examination of the basic concepts of a dose response might also suggest that similarities exist between the two types of reactions. For a given drug that initiates idiosyncratic hepatotoxicity, the liver may not typically be a target for toxicity potentially due to the fact that the normal dose–response curve for hepatotoxicity is below the lethal dose in majority of the users. However, coinciding with an inflammatory episode in the organ, this might result in bringing hepatotoxic dose within a therapeutic range, and hence manifestation of a spontaneous toxic response.
As previously indicated, a growing body of literature demonstrated that xenobiotic-induced adverse effects in the liver are aggravated in a diseased organ. Therefore, it stands to reason that there is an increased health risk attributed toxic compounds exposure including drugs in patients with a metabolically compromised liver. If validated, this may have major implications for preclinical hepatotoxicity testing strategies in drug development and DILI where drug safety needs to be assessed not only in models of representative of healthy liver, but also in liver with altered metabolism. Assuming that clinically asymptomatic MASLD might render patients under medication to a higher risk for developing liver injury, then this cohort is clearly underrepresented in clinical safety studies. He, Morales, and Gutherie (Citation2020) reported that of a total of 305 investigated trials a third of the studies were at high risk of bias, most frequently because the clinical population used was not appropriate for the trial examined. It is not unreasonable to assume the same argument might be valid for heavy drinkers who might be more susceptible to developing acute DILI.
Evidence indicates that an important consideration from the scrutiny of the literature is that shortage of animal models exists that accurately capture iDILI, although a number of promising candidates do exist such as inflammagen model (Segovia-Zafra et al. Citation2021) or the Uetrecht-Pohl model (Gerussi et al. Citation2021). The prevailing view of iDILI is that drugs require transformation into metabolites by phase I enzymes with subsequent phase II conjugation. Biotransformation leads to chemically reactive or unstable metabolites with the potential to produce cellular dysfunction and liver injury. This emphasizes the necessity to be aware of the significant intra-species species differences in metabolism. Although the most common experimental animal models (particularly mice) express homologs of the major CYP450s in humans, not all those homologs possess the same substrate specificity (Turpeinen et al. Citation2007); hence, there is an overall poor human response predictivity. Therefore, it is critical that species differences in drug metabolism are fully considered.
The lack of specific biomarkers makes distinguishing DILI cases from other hepatic conditions difficult. This problem is worsened as the diagnosis of DILI relies upon the exclusion of alternative causes. Consequently, collaborative efforts to identify new biomarkers have been undertaken but thus far are only in the clinical exploration phase. Studies specifically on the incidences of DILI over time may show higher occurrences of DILI as chemotherapy drug usage increases. While international consortium thresholds helped somewhat, more specific assessment and reporting methods need to be implemented. Most cases of DILI are self-limiting and resolve upon withdrawal of the offending drug, but treatments for more serious reactions are limited. Large studies found DILI mortality rates to be approximately 5–10% and transplant is usually considered if symptoms progress to ALF. There is no standardized clinical guidance for the treatment of DILI as most treatments are nonspecific. Current causality assessment models may be ambiguous, lacking data to comprehensively evidence some of their components. Investigators demonstrated that while the female gender and increased age are not necessarily a risk factor for DILI overall, these demographics are more likely to lead to certain types of injury and greater risk of mortality. Drug characteristics such as lipophilicity and molecular structure were noted as risk factors in DILI development and these studies enabled enforcement of strict hepatoxicity criteria throughout the drug discovery process. As one moves toward a future of personalized medicine, genetic screening prior to commencement of usage of a potentially hepatotoxic drug may be necessary. Point of care testing for liver enzyme activity elevations may also aid in diagnosis and monitoring. There remains much left unknown regarding DILI and future research might certainly be of aid to physicians trying to manage this difficult diagnosis by exclusion. Due to the lack of specific diagnostic biomarkers, assessment of DILI presence relies on subjective expert consensus opinion. To this end, the use of the RUCAM scale as a validated liver-specific scale remains a highly valuable tool for causality assessment of DILI.
Over the last decade, significant advances in the field of nanomedicine offer real hope for the development of new prevention and treatment of liver damage including DILI. In this regard, the potential to deliver targeted therapeutics directly to site of injury is an attractive proposition. Further, with the continued and ever-growing body of investigations attempting to understand the link between gut microbes and host cells, new avenues may arise for the development of therapeutic applications in prevention and treatment of acute DILI.
The scrutiny of literature as well as the shortcomings in the available causality assessment tools discussed above suggest a real need for more well-designed mechanistic studies. As discussed, numerous investigations noted a link between DILI and polymorphisms of certain HLA genotypes as well as CYP450s; yet more investigations of better understanding of the mechanisms underlying DILI/iDILI are urgently needed.
Abbreviations
| ALF | = | Acute Liver Failure |
| ALP | = | Alkaline Phosphatase |
| ALT | = | Alanine Aminotransferase |
| APC | = | Antigen presenting cells |
| ART | = | Antiretroviral Therapy |
| AST | = | Aspartate Aminotransferase |
| ATP | = | Adenosine triphosphate |
| BDDCS | = | Biopharmaceutics Drug Disposition Classification System |
| BID | = | Pro-apoptotic BH3 interacting-domain death agonist |
| BSEP | = | Bile Salt Export Pump |
| CASP | = | Caspase |
| CMV | = | Cytomegalovirus |
| CRM | = | Chemically Reactive Metabolites |
| CYP | = | Cytochrome 450 |
| DAMPs | = | Damage Associated Molecular Patterns |
| DILI | = | Drug Induced Liver Injury |
| DILIN | = | Drug Induced Liver Injury Network |
| EBV | = | Epstein-Barr virus |
| ER | = | Endoplasmic reticulum |
| GM-CSF | = | Granulocyte-macrophage colony-stimulating factor |
| HIV | = | Human immunodeficiency virus |
| HLA | = | Human Leukocyte Antigen |
| ICI | = | Immune-Checkpoint Inhibitor |
| iDILI | = | Idiosyncratic Drug Induced Liver Injury |
| IFN | = | Interferon |
| IL | = | Interleukin |
| INR | = | International Normalized Ratio |
| MHC | = | Major Histocompatibility Complex |
| GPRD | = | General Practice Research Database |
| GSH | = | Glutathione |
| JNK | = | c-Jun N-terminal Kinase |
| MAPK | = | Mitogen-Activated Protein Kinase |
| MASLD | = | Metabolic dysfunction-Associated Steatotic Liver Disease |
| MASH | = | Metabolic dysfunction-Associated SteatoHepatitis |
| NSAID | = | Nonsteroidal Anti-Inflammatory Drug |
| ROS | = | Reactive Oxygen Species |
| RUCAM | = | Roussel Uclaf Causality Assessment Method |
| TNF | = | Tumor Necrosis Factor |
| TRAIL | = | Tumor Necrosis Factor-related Apoptosis-inducing Ligand |
| TB | = | Tuberculosis |
| ULN | = | Upper Limit of Normal |
| UPR | = | Unfolded Protein Response |
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Adair, K., X. Meng, and D. J. Naisbitt. 2021. Drug hapten‐specific T‐cell activation: Current status and unanswered questions. Proteomics 21 (17–18):e2000267. doi:10.1002/pmic.202000267.
- Adam, R., V. Karma, V. Cailliez, J. G. O Grady, D. Mirza, D. Cherqui, J. Klempnauer, M. Salizzoni, J. Pratschke, N. Jamieson, et al. 2018. Annual report of the European liver transplant registry (ELTR) - 50-year evolution of liver transplantation. Transpl. Int. 31:1293–317. doi:10.1111/tri.13358.
- Ahmad, J., and J. Odin. 2017. Epidemiology and genetic risk factors of drug hepatotoxicity. Clin. Liver Dis 21 (1):55–72. doi:10.1016/j.cld.2016.08.004.
- Allard, J., S. Bucher, J. Massart, P. J. Ferron, D. L. Guillou, R. Loyant, Y. Daniel, Y. Launay, N. Buron, K. Begriche, et al. 2021. Drug-induced hepatic steatosis in absence of severe mitochondrial dysfunction in HepaRG cells: Proof of multiple mechanism-based toxicity. Cell Biol. Toxicol. 37 (2):151–75. doi:10.1007/s10565-020-09537-1.
- Allen, K., H. Jaeschke, and B. L. Copple. 2011. Bile acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis. Am. J. Pathol. 178 (1):175–86. doi:10.1016/j.ajpath.2010.11.026.
- Almazroo, O., M. Miah, and R. Venkataramanan. 2017. Drug metabolism in the liver. Clin. Liver Dis 21 (1):1–20. doi:10.1016/j.cld.2016.08.001.
- Alqahtani, S. A., J. M. Pail, R. Biswas, T. Arshad, L. Henry, and Z. M. Younossi. 2021. Poor awareness of liver disease among adults with NAFLD in the United States. Hepatol. Commun. 5:1833–47. doi:10.1002/hep4.1765.
- Amadi, C., and O. Orisakwe. 2018. ‘Herb-induced liver injuries in developing nations: An update. Toxics 6 (2):24. doi:10.3390/toxics6020024.
- Andrade, R. J., G. P. Aithal, E. S. Björnsson, N. Kaplowitz, G. A. Kullak-Ublick, D. Larrey, T. H. Karlsen, and European Association of the Study of the Liver. 2019. EASL clinical practice guidelines: Drug-induced liver injury. J. Hepatol. 70 (6):1222–61. doi:10.1016/j.jhep.2019.02.014.
- Andrade, R., N. Chalasani, E. Björnsson, A. Suzuki, G. Kullak-Ublick, P. Watkins, H. Devarbhavi, M. Merz, M. Lucena, N. Kaplowitz, et al. 2019. Drug-induced liver injury. Nat. Rev. Dis. Prim. 5:58. doi:10.1038/s41572-019-0105-0.
- Bechmann, L., P. Manka, J. Best, F. Saner, A. Paul, A. Canbay, and G. Gerken. 2014. Medikamentös-toxische Schädigung als wichtigste Ätiologie des akuten Leberversagens. Dtsch. Med. Wochenschr. 139:878–82. doi:10.1055/s-0034-1369932.
- Björnsson, E. S. 2021. Clinical management of patients with drug-induced liver injury (DILI). Ueg. J 9:781–86. doi:10.1002/ueg2.12113.
- Björnsson, E., O. Bergmann, H. Björnsson, R. Kvaran, and S. Olafsson. 2013. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology 144 (7):1419. doi:10.1053/j.gastro.2013.02.006.
- Björnsson, K., and S. Björnsson. 2022. Drug-induced liver injury: Pathogenesis, epidemiology, clinical features, and practical management. Eur. J. Intern. Med. 97:26–31. doi:10.1016/j.ejim.2021.10.035.
- Björnsson, E., B. Gunnarsson, G. Gröndal, J. Jonasson, R. Einarsdottir, B. Ludviksson, B. Gudbjörnsson, and S. Olafsson. 2015. Risk of drug-induced liver injury from tumor necrosis factor antagonists. Clin. Gastroenterol. Hepatol. 13 (3):602–08. doi:10.1016/j.cgh.2014.07.062.
- Bocci, G., T. I. Oprea, and L. Z. Benet. 2022. State of the art and uses for the biopharmaceutics drug disposition classification system (BDDCS): New additions, revisions, and citation references. Aaps. J 24 (2):37. doi:10.1208/s12248-022-00687-0.
- Bock, F., and S. Tait. 2020. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21 (2):85–100. doi:10.1038/s41580-019-0173-8.
- Bonaventura, A., A. Vecchié, T. S. Wang, E. Lee, P. C. Cremer, B. Carey, P. Rajendram, K. M. Hudock, L. Korbee, B. W. Van Tassell, et al. 2020. Targeting GM-CSF in COVID-19 pneumonia: Rationale and strategies. Front. Immunol 11:1625. doi:10.3389/fimmu.2020.01625.
- Bottcher, J. P., P. A. Knolle, and D. Stabenow. 2011. Mechanisms balancing tolerance and immunity in the liver. Dig. Dis. 29:384–90. doi:10.1159/000329801.
- British Liver Trust. 2023. Accessed October 4 2023. https://britishlivertrust.org.uk/
- Cai, S. Y., and J. L. Boyer. 2021. The role of bile acids in cholestatic liver injury. Ann. Transl. Med. 9:737. doi:10.21037/atm-20-5110.
- Chalasani, N., H. Bonkovsky, R. Fontana, W. Lee, A. Stolz, J. Talwalkar, K. Reddy, P. Watkins, V. Navarro, H. Barnhart, et al. 2015. Features and outcomes of 899 patients with drug-induced liver injury: The DILIN prospective study. Gastroenterology 148:1340–52. doi:10.1053/j.gastro.2015.03.006.
- Chan, R., and L. Z. Benet. 2017. Evaluation of DILI predictive hypotheses in early drug development. Chem. Res. Toxicol. 30 (4):1017–29. doi:10.1021/acs.chemrestox.7b00025.
- Cheng, M. L., D. Nakib, C. T. Perciani, and S. A. MacParland. 2021. The immune niche of the liver. Clin. Sci. 135 (20):2445–66. doi:10.1042/CS20190654.
- Chen, M., A. Suzuki, J. Borlak, R. Andrade, and M. Lucena. 2015. Drug-induced liver injury: Interactions between drug properties and host factors. J. Hepatol. 63 (2):503–14. doi:10.1016/j.jhep.2015.04.016.
- Chen, Q., M. Yu, Z. Tian, Y. Cui, D. Deng, T. Rong, Z. Liu, M. Song, Z. Li, X. Ma, et al. 2022. Exogenous glutathione protects IPEC-J2 cells against oxidative stress through a mitochondrial mechanism. Molecules 27 (8):2416. doi:10.3390/molecules27082416.
- Christrup, L. 1997. Morphine metabolites. Acta. Anaesthesiol. Scand. 41 (1):116–22. doi:10.1111/j.1399-6576.1997.tb04625.x.
- Clinton, J., S. Kiparizoska, S. Aggarwal, S. Woo, W. Davis, and J. Lewis. 2021. Drug-induced liver injury: Highlights and controversies in the recent literature. Drug. Safety 44 (11):1125–49. doi:10.1007/s40264-021-01109-4.
- Daly, A. K., and C. P. Day. 2012. Genetic association studies in drug-induced liver injury. Drug Metab. Rev. 44 (1):116–26. doi:10.3109/03602532.2011.605790.
- Danan, G., and C. Benichou. 1993. Causality assessment of adverse reactions to drugs - I. A novel method based on the conclusions of international consensus meetings: Application to drug-induced liver injuries. J. Clin. Epidemiol 46:1323–30. doi:10.1016/0895-4356(93)90101-6.
- Danan, G., and R. Teschke. 2016. RUCAM in drug and herb induced liver injury: The update. Int. J. Mol. Sci. 17:14. doi:10.3390/ijms17010014.
- Dara, L., Z. Liu, and N. Kaplowitz. 2016. Questions and controversies: The role of necroptosis in liver disease. Cell. Death Discovery 2 (1):16089. doi:10.1038/cddiscovery.2016.89.
- David, S., and J. P. Hamilton. 2010. Drug-induced liver injury. US. Gastroenterol. Hepatol. Rev. 6:73–80.
- De Abajo, F., D. Montero, M. Madurga, and L. García-Rodríguez. 2004. Acute and clinically relevant drug-induced liver injury: A population-based case-control study. Br. J. Clin. Pharmacol 58:71–80. doi:10.1111/j.1365-2125.2004.02133.x.
- De Valle, M., V. Klinteberg, N. Alem, R. Olsson, and E. Björnsson. 2006. Drug-induced liver injury in a Swedish university hospital out-patient hepatology clinic. Aliment. Pharmacol. Ther. 24 (8):1187–95. doi:10.1111/j.1365-2036.2006.03117.x.
- Devarbhavi, H., G. Aithal, S. Treeprasertsuk, H. Takikawa, Y. Mao, S. M. Shasthry, S. Hamid, S. Tan, C. Philips, J. George, et al. 2021. Drug-induced liver injury: Asia pacific association of study of liver consensus guidelines. Hepatol. Int. 15:258–82. doi:10.1007/s12072-021-10144-3.
- Doherty, D. G. 2016. Immunity, tolerance and autoimmunity in the liver: A comprehensive review. J. Autoimmun. 66:60–75. doi:10.1016/j.jaut.2015.08.020.
- Eiswerth, M., M. Heckroth, A. Ismail, D. Gondim, and R. Kaufman. 2022. Infliximab-induced vanishing bile duct syndrome. Cureus 14. doi:10.7759/cureus.21940.
- Fernandez-Checa, J. C., P. Bagnaninchi, H. Ye, P. Sancho-Bru, J. M. Falcon-Perez, F. Royo, C. Garcia-Ruiz, O. Konu, J. Miranda, O. Lunov, et al. 2021. Advanced preclinical models for evaluation of drug-induced liver injury - consensus statement by the European drug-induced liver injury Network [PRO-EURO-DILI-NET]. J. Hepatol. 75:935–59. doi:10.1016/j.jhep.2021.06.021.
- Fisher, K., R. Vuppalanchi, and R. Saxena. 2015. Drug-induced liver injury. Arch. Pathol. Lab. Med. 139 (7):876–87. doi:10.5858/arpa.2014-0214-RA.
- Fontana, R. J., E. S. Bjornsson, R. Reddy, and P. J. Andrade. 2023. The evolving profile of idiosyncratic drug-induced liver injury. Clin. Gastroenterol. Hepatol. 8:2088–99. doi:10.1016/j.cgh.2022.12.040.
- Fyfe, B., F. Zaldana, and C. Liu. 2018. The pathology of acute liver failure. Clin. Liver Dis 22 (2):257–68. doi:10.1016/j.cld.2018.01.003.
- Galluzzi, L., I. Vitale, S. Aaronson, J. Abrams, D. Adam, P. Agostinis, E. Alnemri, L. Altucci, I. Amelio, D. Andrews, et al. 2018. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25:486–541. doi:10.1038/s41418-017-0012-4.
- Garcia-Cortes, M., M. Robles-Diaz, C. Stephens, A. Ortega-Alonso, I. Lucena, and R. Andrade. 2020. Drug induced liver injury: An update. Arch. Toxicol. 94 (10):3381–407. doi:10.1007/s00204-020-02885-1.
- Gerussi, A., A. Natalini, F. Antonangeli, C. Mancuso, E. Agostinetto, D. Barisani, F. Di Rosa, R. Andrade, and P. Invernizzi. 2021. Immune-mediated drug-induced liver injury: Immunogenetics and experimental models. Int. J. Mol. Sci. 22 (9):4557. doi:10.3390/ijms22094557.
- Grewal, P., and J. Ahmad. 2019. Bile duct injury due to drug induced liver injury. Curr. Hepatology. Rep 18:269–73. doi:10.1007/s11901-019-00474-0.
- Gupta, A., M. Madhavan, K. Sehgal, N. Nair, S. Mahajan, T. Sehrawat, B. Bikdeli, N. Ahluwalia, J. C. Ausiello, E. Wan, et al. 2020. Extrapulmonary manifestations of COVID-19. Nat. Med. 26:1017–32. doi:10.1038/s41591-020-0968-3.
- Hamilton, L. A., A. Collins-Yoder, and R. E. Collins. 2016. Drug-induced liver injury. Advan Crit. Care 27 (4):430–40. doi:10.4037/aacnacc2016953.
- Hawton, K., H. Bergen, S. Simkin, S. Dodd, P. Pocock, W. Bernal, D. Gunnell, and N. Kapur. 2013. Long term effect of reduced pack sizes of paracetamol on poisoning deaths and liver transplant activity in England and wales: Interrupted time series analyses. Br. Med. J 346 (feb07 1):f403. doi:10.1136/bmj.f403.
- He, J., D. R. Morales, and B. Gutherie. 2020. Exclusion rates in randomized controlled trials of treatments for physical conditions: A systematic review. Trials 21:228. doi:10.1186/s13063-020-4139-0.
- Heymann, F., J. Peusquens, I. Ludwig-Portugall, M. Kohlhepp, C. Ergen, P. Niemietz, C. Martin, N. van Rooijen, J. C. Ochando, G. J. Randolph, et al. 2015. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 62 (1):279–91. doi:10.1002/hep.27793.
- Hosack, T., D. Damry, and S. Biswas. 2023. Drug-induced liver injury: A comprehensive review. Therap. Adv. Gastroenterol. 16:17562848231163410. doi:10.1177/17562848231163410.
- Hughes, T., N. Flynn, N. Dang, and S. Swamidass. 2021. Modeling the bioactivation and subsequent reactivity of drugs. Chem. Res. Toxicol. 34 (2):584–600. doi:10.1021/acs.chemrestox.0c00417.
- Hughes, J., S. Rees, S. Kalindjian, and K. Philpott. 2011. Principles of early drug discovery. Br. J. Pharmacol. 162 (6):1239–49. doi:10.1111/j.1476-5381.2010.01127.x.
- Hui, D. S., E. I Azhar, T. A. Madani, F. Ntoumi, R. Kock, O. Dar, G. Ippolito, T. D. McHugh, Z. A. Memish, C. Drosten, et al. 2020. The continuing 2019-nCov epidemic threat of novel coronaviruses to global health - the latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 91:264–66. doi:10.1016/j.ijid.2020.01.009.
- Iorga, A., L. Dara, and N. Kaplowitz. 2017. Drug-induced liver injury: Cascade of events leading to cell death, apoptosis or necrosis. Int. J. Mol. Sci. 18 (5):1018. doi:10.3390/ijms18051018.
- Isa, S. E., A. O. Ebonyi, N. Y. Shehu, P. Idoko, J. A. Anejo-Okopi, G. Simji, R. U. Odesanya, I. O. Abah, and H. O. Jimoh. 2016. Antituberculosis drugs and hepatotoxicity among hospitalized patients in Jos, Nigeria. Int. J. Mycobacteriol 5 (1):21–26. doi:10.1016/j.ijmyco.2015.10.001.
- Jee, A., S. C. Sernoskie, and J. Uetrecht. 2021. Idiosyncratic drug-induced liver injury: Mechanistic and clinical challenges. Int. J. Mol. Sci. 22 (6):2954. doi:10.3390/ijms22062954.
- Katarey, D., and S. Verma. 2016. Drug-induced liver injury. Clin. Med (Northfield Il) 16:104–09. doi:10.7861/clinmedicine.16-6-s104.
- Ke, L., C. Lu, R. Shen, T. Lu, B. Ma, and Y. Hua. 2020. Knowledge mapping of drug-induced liver injury: A scientometric investigation (2010-2019). Front. Pharmacol 11:842. doi:10.3389/fphar.2020.00842.
- Kermanizadeh, A., N. R. Jacobsen, M. Roursgaard, S. Loft, and P. Møller. 2017. Hepatic hazard assessment of silver nanoparticle exposure in healthy and chronically alcohol fed mice. Toxicol. Sci. 158 (1):176–87. doi:10.1093/toxsci/kfx080.
- Kermanizadeh, A., J. Valli, K. Sanchez, S. Hutter, A. Pawlowska, G. Whyte, W. Moritz, and V. Stone. 2022. Particulate and drug induced toxicity assessed in novel quadruple cell human primary hepatic disease models of steatosis and pre-fibrotic NASH. Arch. Toxicol. 96:287–303. doi:10.1007/s00204-021-03181-2.
- Ko, S., J. Russell, L. Molina, and S. Monga. 2020. Liver progenitors and adult cell plasticity in hepatic injury and repair: Knowns and unknowns. Annu. Rev. Pathol 15 (1):23–50. doi:10.1146/annurev-pathmechdis-012419-032824.
- Korver, S., J. Bowen, K. Pearson, R. Gonzalez, N. French, K. Park, R. Jenkins, and C. Goldring. 2021. The application of cytokeratin-18 as a biomarker for drug-induced liver injury. Arch. Toxicol. 95 (11):3435–48. doi:10.1007/s00204-021-03121-0.
- Kozlov, A., J. Lancaster, A. Meszaros, and A. Weidinger. 2017. Mitochondria-mediated pathways of organ failure upon inflammation. Redox. Biol. 13:170–81. doi:10.1016/j.redox.2017.05.017.
- Krishna, M. 2017. Patterns of necrosis in liver disease. Clin. Liver Dis. 10 (2):53. doi:10.1002/cld.653.
- Kubes, P., and C. Jenne. 2018. Immune responses in the liver. Annu. Rev. Immunol. 36 (1):247–77. doi:10.1146/annurev-immunol-051116-052415.
- Lai, R. T., H. Wang, H. L. Gui, M. Z. Ye, W. J. Dai, X. G. Xiang, G. D. Zhao, W. J. Wang, and Q. Xie. 2012. Clinical and pathological features in 138 cases of drug-induced liver injury. Chin. J. Hepatol 20:185–89. doi:10.3760/cma.j.issn.1007-3418.2012.03.009.
- Lammert, C., T. Imler, E. Teal, and N. Chalasani. 2019. Patients with chronic liver disease suggestive of non-alcoholic fatty liver disease may be at higher risk for drug-induced liver injury. Clin. Gastroenterol. Hepatol. 17:2814–15. doi:10.1016/j.cgh.2018.12.013.
- Lee, F., and D. A. Lawrence. 2018. From infections to anthropogenic inflicted pathologies: Involvement of immune balance. J. Toxicol. Environ. Health - B. 21:24–46. doi:10.1080/10937404.2017.1412212.
- Li, X., P. Gao, and J. Niu. 2019. Metabolic comorbidities and risk of development and severity of drug-induced liver injury. Biomed. Res. Int. 18:8764093. doi:10.1155/2019/8764093.
- Li, M., Q. Luo, Y. Tao, X. Sun, and C. Liu. 2022. Pharmacotherapies for drug-induced liver injury: A current literature review. Front. Pharmacol 12:806249. doi:10.3389/fphar.2021.806249.
- Liu, N., J. Liu, B. Zheng, X. Zeng, Z. Ye, X. Huang, W. Liu, Y. Liu, Q. Fang, L. Chen, et al. 2023. Gut microbiota affects sensitivity to immune-mediated isoniazid-induced liver injury. Biomed. Pharmacother. 160:114400. doi:10.1016/j.biopha.2023.114400.
- LiverTox. 2022. Accessed November 10 2022. https://www.niddk.nih.gov/news/archive/2022/livertox-online-resource-information-drug-induced-liver-injury
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. 2012a. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. Abacavir. [Updated 2016 Jan 4]. https://www.ncbi.nlm.nih.gov/books/NBK548225/
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. 2012b. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. Amoxicillin-Clavulanate. [Updated 2020 Oct 20]. https://www.ncbi.nlm.nih.gov/books/NBK548517/
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. 2012c. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. Carbamazepine. [Updated 2017 Jan 15]. https://www.ncbi.nlm.nih.gov/books/NBK548097/
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. 2012d. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. Diclofenac. [Updated 2017b Dec 13]. https://www.ncbi.nlm.nih.gov/books/NBK547953/
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. 2012e. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. Erythromycin. [Updated 2017 (C) Aug 10]. https://www.ncbi.nlm.nih.gov/books/NBK547881/
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. 2012f. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. Ibuprofen. [Updated 2018 Apr 16]. https://www.ncbi.nlm.nih.gov/books/NBK547845/
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. 2012g. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. Sulindac. [Updated 2020 (B) Mar 20]. https://www.ncbi.nlm.nih.gov/books/NBK548315/
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. 2012h. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. Rifampin. [Updated 2018 Jun 10].
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury [Internet]. 2012i. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. Valproate. [Updated 2020 (C) Jul 31]. https://www.ncbi.nlm.nih.gov/books/NBK548284/
- Li, X., W. Wang, S. Yan, W. Zhao, H. Xiong, C. Bao, J. Chen, Y. Yue, Y. Su, and C. Zhang. 2022. Drug-induced liver injury in COVID-19 treatment: Incidence, mechanisms and clinical management. Front. Pharmacol 13:1019487. doi:10.3389/fphar.2022.1019487.
- Lorincz, T., K. Jemnitz, T. Kardon, J. Mandl, and A. Szarka. 2015. Ferroptosis is involved in acetaminophen induced cell death. Pathol. Oncol. Res. 21:1115–21. doi:10.1007/s12253-015-9946-3.
- Low, E., Q. Zheng, E. Chan, and S. Lim. 2020. Drug induced liver injury: East versus West - a systematic review and meta-analysis. Clin. Mol. Hepatol 26:142–54. doi:10.3350/cmh.2019.1003.
- Lu, S. 2009. Regulation of glutathione synthesis. Mol. Aspects Med. 30 (1–2):42–59. doi:10.1016/j.mam.2008.05.005.
- Luan, J., and D. Ju. 2018. Inflammasome: A double-edged sword in liver diseases. Front. Immunol. 9:2201. doi:10.3389/fimmu.2018.02201.
- Lucena, M. I., R. J. Andrade, N. Kaplowitz, M. García-Cortes, M. C. Fernández, M. Romero-Gomez, M. Bruguera, H. Hallal, M. Robles-Diaz, J. F. Rodriguez-González, et al. 2009. Phenotypic characterization of idiosyncratic drug-induced liver injury: The influence of age and sex. Hepatology 49 (6):2001–09. doi:10.1002/hep.22895.
- Lu, H., C. W. Stratton, and Y. W. Tang. 2020. Outbreak of pneumonia of unknown aetiology in Wuhan, China: The mystery and the miracle. J. Med. Virol. 92:401–02. doi:10.1002/jmv.25678.
- Marjot, T., G. J. Webb, A. S. Barritt, A. M. Moon, Z. Stamataki, V. W. Wong, and E. Barnes. 2021. COVID-19 and liver disease: Mechanistic and clinical perspectives. Nat. Rev. Gastro. Hepat. 18 (5):348–64. doi:10.1038/s41575-021-00426-4.
- McGill, M., and H. Jaeschke. 2018. Biomarkers of drug-induced liver injury: Progress and utility in research, medicine, and regulation. Exp. Rev. Molecular Diag. 18:797–807. doi:10.1080/14737159.2018.1508998.
- McGill, M., and H. Jaeschke. 2019. Biomarkers of drug-induced liver injury. Advan. Pharmacol. 85:221–39.
- McGrowder, D. A., F. Miller, M. Anderson Cross, L. Anderson-Jackson, S. Bryan, and L. Dilworth. 2021. Abnormal liver biochemistry tests and acute liver injury in COVID-19 patients: Current evidence and potential pathogenesis. Diseases 9 (3):50. doi:10.3390/diseases9030050.
- McPherson, S., M. J. Armstrong, J. F. Cobbold, L. Corless, Q. M. Anstee, R. J. Aspinall, S. T. Barclay, P. N. Brennan, T. M. Cacciottolo, R. D. Goldin, et al. 2022. Quality standards for the management of non-alcoholic fatty liver disease (NAFLD): Consensus recommendations from the British association for the study of the liver and British society of gastroenterology NAFLD special interest group. Lancet Gastroenterol. Hepatol. 7:755–69. doi:10.1016/S2468-1253(22)00061-9.
- Michalopoulos, G. K. 2013. Principles of liver regeneration and growth homeostasis. Compr. Physiol. 3:485–513.
- Molina, D. K., and V. J. M. DiMaio. 2012. Normal organ weights in men: Part II-the brain, lungs, liver, spleen, and kidneys. Am. J. Forensic Med. Pathol. 33 (4):368–72. doi:10.1097/PAF.0b013e31823d29ad.
- Muhović, D., J. Bojović, A. Bulatović, B. Vukčević, M. Ratković, R. Lazović, and B. Smolović. 2020. First case of drug‐induced liver injury associated with the use of tocilizumab in a patient with COVID‐19. Liver Int. 40 (8):1901–05. doi:10.1111/liv.14516.
- Muscate, F., A. Woestemeier, and N. Gagliani. 2021. Functional heterogeneity of CD4+ T cells in liver inflammation. Semin. Immunopathol 43 (4):549–61. doi:10.1007/s00281-021-00881-w.
- Naseralallah, L. M., B. A. Aboujabal, N. M. Geryo, A. Al Boinin, F. Al Hattab, R. Akbar, W. Umer, L. Abdul Jabbar, M. I. Danjuma, and E. Yunihastuti. 2022. The determination of causality of drug induced liver injury in patients with COVID-19 clinical syndrome. PLoS One 17 (9):e0268705. doi:10.1371/journal.pone.0268705.
- NHS. 2023. Accessed November 1 2023. https://www.nhs.uk/
- Nicoletti, P., G. P. Aithal, E. S. Bjornsson, R. J. Andrade, A. Sawle, M. Arrese, H. X. Barnhart, E. Bondon-Guitton, P. H. Hayashi, F. Bessone, et al. 2017. Association of liver injury from specific drugs, or groups of drugs, with polymorphisms in HLA and other genes in a genome-wide association study. Gastroenterology 152 (5):1078–89. doi:10.1053/j.gastro.2016.12.016.
- Nicoletti, P., A. Werk, A. Sawle, Y. Shen, T. Urban, S. Coulthard, E. Bjornsson, I. Cascorbi, A. Floratos, T. Stammschulte, et al. 2016. International drug-induced liver injury consortium. HLA-DRB1*16: 01-DQB1*05: 02 is a novel genetic risk factor for flupirtine-induced liver injury. Pharmacogenet. Genomics 26:218–24. doi:10.1097/FPC.0000000000000209.
- Ortega-Alonso, A., C. Stephens, M. Lucena, and R. Andrade. 2016. Case characterization, clinical features and risk factors in drug-induced liver injury. Int. J. Mol. Sci 17 (5):714. doi:10.3390/ijms17050714.
- Ostapowicz, G., R. J. Fontana, F. V. Schiødt, A. Larson, T. J. Davern, S. H. Han, T. M. McCashland, A. O. Shakil, J. E. Hay, L. Hynan, et al. 2002. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann. Intern. Med. 137:947–54. doi:10.7326/0003-4819-137-12-200212170-00007.
- Ou, P., Y. Chen, B. Li, M. Zhang, X. Liu, F. Li, Y. Li, C. Chen, Y. Mao, and J. Chen. 2015. Causes, clinical features and outcomes of drug-induced liver injury in hospitalized patients in a Chinese tertiary care hospital. SpringerPlus 4 (1):802. doi:10.1186/s40064-015-1600-8.
- Perwitasari, D. A., E. Darmawan, U. A. Mulyani, P. V. Vlies, J. C. Alffenaar, J. Atthobar, and B. Wilffert. 2018. Polymorphisms of NAT2, CYP2E1, GST, and HLA related to drug-induced liver injury in Indonesian tuberculosis patients. Int. J. Mycobacteriol 7:380–86. doi:10.4103/ijmy.ijmy_143_18.
- Pichler, W. J. 2013. Consequences of drug binding to immune receptors: Immune stimulation following pharmacological interaction with immune receptors (T-cell receptor for antigen or human leukocyte antigen) with altered peptide-human leukocyte antigen or peptide. Dermatol. Sin. 31 (4):181–90. doi:10.1016/j.dsi.2013.09.005.
- Pollock, N. R., S. McGray, D. J. Colby, F. Noubary, H. Nguyen, T. A. Nguyen, S. Khormaee, S. Jain, K. Hawkins, S. Kumar, et al. 2013. Field evaluation of a prototype paper-based point-of-care fingerstick transaminase test. PLoS One 8 (9):e75616. doi:10.1371/journal.pone.0075616.
- Popper, H., and F. Schaffner. 1965. The problem of chronicity in liver disease. Progr. Liver Dis. 2:519–38.
- Pu, S., Y. Pan, Q. Zhang, T. You, T. Yue, Y. Zhang, and M. Wang. 2023. Endoplasmic reticulum stress and mitochondrial stress in drug-induced liver injury. Molecules 28 (7):3160. doi:10.3390/molecules28073160.
- Rathi, C., N. Pipaliya, R. Patel, M. Ingle, A. Phadke, and P. Sawant. 2017. Drug induced liver injury at a tertiary hospital in India: Etiology, clinical features and predictors of mortality. Ann. Hepatol 16 (3):442–50. doi:10.5604/01.3001.0009.8600.
- Real, M., M. S. Barnhill, C. Higley, J. Rosenberg, and J. H. Lewis. 2019. Drug-induced liver injury: Highlights of the recent literature. Drug. Safety 42 (3):365–87. doi:10.1007/s40264-018-0743-2.
- Reuben, A., D. Koch, W. Lee, and Acute Liver Failure Study Group. 2010. Drug-induced acute liver failure: Results of a U.S. multicenter, prospective study. Hepatology 52:2065–76. doi:10.1002/hep.23937.
- Riebensahm, C., K. D, A. Sow, N. Semmo, and G. Wandeler. 2019. A closer look at the spectrum of drug-induced liver injury in sub-saharan Africa. Expert. Rev. Clin. Pharmacol 12:875–83. doi:10.1080/17512433.2019.1638251.
- Robles-Diaz, M., L. Nezic, V. Vujic-Aleksic, and E. Björnsson. 2021. Role of ursodeoxycholic acid in treating and preventing idiosyncratic drug-induced liver injury. A systematic review. Front. Pharmacol 12:744488. doi:10.3389/fphar.2021.744488.
- Russmann, S., G. Kullak-Ublick, and I. Grattagliano. 2009. Current concepts of mechanisms in drug-induced hepatotoxicity. Curr. Med. Chem. 16 (23):3041–53. doi:10.2174/092986709788803097.
- Saha, L., S. Vij, and K. Rawat. 2022. Liver injury induced by COVID-19 treatment - what do we know? World. J. Gastroenterol. 28:6314–27. doi:10.3748/wjg.v28.i45.6314.
- Saini, R. K., N. Saini, S. Ram, S. L. Soni, V. Suri, P. Malhotra, J. Kaur, I. Verma, S. Sharma, and D. Zohmangaihi. 2020. COVID-19 associated variations in liver function parameters: A retrospective study. Postgrad. Med. J. 98:91–97. doi:10.1136/postgradmedj-2020-138930.
- Sancho-Bru, P., J. Altamirano, D. Rodrigo-Torres, M. Coll, C. Millan, J. J. Lozano, R. Miquel, V. Arroyo, J. Caballeria, P. Gines, et al. 2012. Liver progenitor cell markers correlate with liver damage and predict short-term mortality in patients with alcoholic hepatitis. Hepatology 55 (6):1931–41. doi:10.1002/hep.25614.
- Schutz, C., Z. Ismail, C. J. Proxenos, S. Marais, R. Burton, C. Kenyon, G. Maartens, R. J. Wilkinson, and G. Meintjes. 2012. Burden of antituberculosis and antiretroviral drug-induced liver injury at a secondary hospital in South Africa. S. Afr. Med. J. 102:506–11. doi:10.7196/SAMJ.5650.
- Segovia-Zafra, A., D. Zeo-Sánchez DE, C. López-Gómez, Z. Pérez-Valdés, E. García-Fuentes, R. J. Andrade, M. I. Lucena, and M. Villanueva-Paz. 2021. Preclinical models of idiosyncratic drug-induced liver injury (iDILI): Moving towards prediction. Acta. Pharm. Sin B. 11:3685–726. doi:10.1016/j.apsb.2021.11.013.
- Sgro, C., F. Clinard, K. Ouazir, H. Chanay, C. Allard, C. Guilleminet, C. Lenoir, A. Lemoine, and P. Hillon. 2002. Incidence of drug-induced hepatic injuries: A French population-based study. Hepatology 36 (2):451–55. doi:10.1053/jhep.2002.34857.
- Shi, C., B. Hao, Y. Yang, I. Muhammad, Y. Zhang, Y. Chang, Y. Li, C. Li, R. Li, and F. Liu. 2019. JNK signalling pathway mediates acetaminophen-induced hepatotoxicity accompanied by changes of glutathione S-transferase A1 content and expression. Front. Pharmacol 10:1092. doi:10.3389/fphar.2019.01092.
- Shojaie, L., A. Iorga, and L. Dara. 2020. Cell death in liver diseases: A review. Int. J. Mol. Sci. 21 (24):9682. doi:10.3390/ijms21249682.
- Stine, J. G., and J. H. Lewis. 2016. Current and future directions in the treatment and prevention of drug-induced liver injury: A systematic review. Expert. Rev. Gastroenterol. Hepatol. 10 (4):517–36. doi:10.1586/17474124.2016.1127756.
- Suk, K. T., D. J. Kim, C. H. Kim, S. H. Park, J. H. Yoon, Y. S. Kim, G. H. Baik, J. B. Kim, Y. O. Kweon, B. I. Kim, et al. 2012. A prospective nationwide study of drug-induced liver injury in Korea. Am. J. Gastroenterol. 107:1380–87. doi:10.1038/ajg.2012.138.
- Szabo, G., and J. Petrasek. 2015. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 12 (7):387–400. doi:10.1038/nrgastro.2015.94.
- Tasnim, F., X. Huang, C. Lee, F. Ginhoux, and H. Yu. 2021. Recent advances in models of immune-mediated drug-induced liver injury. Front. Toxicol. 3:605392. doi:10.3389/ftox.2021.605392.
- Teigs, G., and A. W. Lohse. 2009. Immune tolerance: What is unique about the liver. J. Autoimmun. 34:1–6. doi:10.1016/j.jaut.2009.08.008.
- Teschke, R., and G. Danan. 2020. Worldwide use of RUCAM for causality assessment in 81,856 idiosyncratic DILI and 14,029 HILI cases published 1993-mid 2020: A comprehensive analysis. Medicines 7:62. doi:10.3390/medicines7100062.
- Teschke, R., and G. Danan. 2021a. Idiosyncratic drug induced liver injury, cytochrome P450, metabolic risk factors and lipophilicity: Highlights and controversies. Int. J. Mol. Sci. 22 (7):3441. doi:10.3390/ijms22073441.
- Teschke, R., and G. Danan. 2021b. The LiverTox paradox-gaps between promised data and reality check. Diagnostics 11 (10):1754. doi:10.3390/diagnostics11101754.
- Trefts, E., M. Gannon, and D. Wasserman. 2017. The liver. Curr. Biol. 27:1147–51. doi:10.1016/j.cub.2017.09.019.
- Turpeinen, M., C. Ghiciuc, M. Opritoui, L. Tursas, O. Pelkonen, and M. Pasanen. 2007. Predictive value of animal models for human cytochrome P450 (CYP)-mediated metabolism: A comparative study in vitro. Xenobiotica 37 (12):1367–77. doi:10.1080/00498250701658312.
- Vargas, J. I., J. P. Arab, F. Bessone, M. I. Lucena, R. J. Andrade, and M. Arrese. 2017. Hepatotoxicity in patients with metabolic syndrome: Causes and consequences. Curr. Hepatology. Rep. 16:286–92. doi:10.1007/s11901-017-0380-8.
- Vega, M., M. Verma, D. Beswick, S. Bey, J. Hossack, N. Merriman, A. Shah, V. Navarro, and Drug Induced Liver Injury Network (DILIN). 2017. The incidence of drug - and herbal and dietary supplement-induced liver injury: Preliminary findings from gastroenterologist-based surveillance in the population of the state of Delaware. Drug. Safety 40 (9):783–87. doi:10.1007/s40264-017-0547-9.
- Villanueva-Paz, M., L. Morán, N. López-Alcántara, C. Freixo, R. Andrade, M. Lucena, and F. Cubero. 2021. Oxidative stress in drug-induced liver injury (DILI): From mechanisms to biomarkers for use in clinical practice. Antioxidants 10 (3):390. doi:10.3390/antiox10030390.
- Visentin, M., D. Lenggenhager, Z. Gai, and G. A. Kullak-Ublick. 2018. Drug-induced bile duct injury. BBA- Molecular. Basis Dis. 1864:1498–506. doi:10.1016/j.bbadis.2017.08.033.
- Vitiello, A., R. La Porta, V. D’Aiuto, and F. Ferrara. 2021. The risks of liver injury in COVID-19 patients and pharmacological management to reduce or prevent the damage induced. Egypt Liver. J 11:11. doi:10.1186/s43066-021-00082-y.
- Vuppalanchi, R., R. Gotur, K. R. Reddy, R. J. Fontana, M. Ghabril, A. S. Kosinski, J. Gu, J. Serrano, and N. Chalasani. 2014. Relationship between characteristics of medications and drug-induced liver disease phenotype and outcome. Clin. Gastroenterol. Hepatol: Off Clin Prac J. Amer Gastroenterol. Ass. 12 (9):1550–55. doi:10.1016/j.cgh.2013.12.016.
- Wang, Q., A. Huang, J. B. Wang, and Z. Zou. 2021. Chronic drug-induced liver injury: Updates and future challenges. Front. Pharmacol 12:627133. doi:10.3389/fphar.2021.627133.
- Woolbright, B. L., and H. Jaeschke. 2018. Mechanisms of inflammatory liver injury and drug-induced hepatotoxicity. Curr. Pharmacol. Rep. 4:346–57. doi:10.1007/s40495-018-0147-0.
- World Federation of Societies of Anaesthesiologists. 2023. The role of the liver in drug metabolism anaesthesia. Accessed March 2 2023. https://resources.wfsahq.org/wp-content/uploads/179_english-1.pdf
- Xiang, D. D., J. T. Liu, Z. B. Zhong, Y. Xiong, H. Y. Kong, H. J. Yu, T. Peng, and J. Q. Hunag. 2023. MicroRNA-29a-3p prevents drug-induced acute liver failure through inflammation-related pyroptosis inhibition. Cur. Med. Sci. 43:456–68. doi:10.1007/s11596-023-2734-5.
- Xie, H., J. Zhao, N. Lian, S. Lin, Q. Xie, and H. Zhuo. 2020. Clinical characteristics of non-ICU hospitalized patients with coronavirus disease 2019 and liver injury: A retrospective study. Liver Int. 40 (6):1321–26. doi:10.1111/liv.14449.
- Xu, H. M., Y. Chen, J. Xu, and Q. Zhou. 2012. Drug-induced liver injury in hospitalized patients with notably elevated alanine aminotransferase. World J. Gastroenterol. 18:5972–78. doi:10.3748/wjg.v18.i41.5972.
- Yan, M., Y. Huo, S. Yin, and H. Hu. 2018. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox. Biol. 17:27–283. doi:10.1016/j.redox.2018.04.019.
- Yokoi, T., and S. Oda. 2021. Models of idiosyncratic drug-induced liver injury. Annu. Rev. Pharmacol. Toxicol. 61 (1):247–68. doi:10.1146/annurev-pharmtox-030220-015007.
- Yu, Y. C., Y. M. Mao, C. W. Chen, J. J. Chen, J. Chen, W. M. Cong, Y. Ding, Z. P. Duan, Q. C. Fu, X. Y. Guo, et al. 2017. Drug-induced liver injury (DILI) study group, Chinese Society of Hepatology (CSH), Chinese medical association. CSH guidelines for the diagnosis and treatment of drug-induced liver injury. Hepatol. Int. 11:221–41. doi:10.1007/s12072-017-9793-2.
- Zeke, A., M. Misheva, A. Reményi, and M. Bogoyevitch. 2016. JNK signaling: Regulation and functions based on complex protein-protein partnerships. Microbiol. Mol. Biol. R. 80 (3):793–835. doi:10.1128/MMBR.00043-14.
- Zhai, G., M. Li, Y. Wang, and J. Wu. 2021. Drug-induced liver disturbance during the treatment of COVID-19. Front. Pharmacol. 12:719308. doi:10.3389/fphar.2021.719308.
- Zhang, Z., S. Li, Z. Zhang, K. Yu, X. Duan, L. Long, S. Zhang, M. Jiang, and O. Liu. 2020. Clinical features, risk factors, and prognostic markers of drug-induced liver injury in patients with Stevens-Johnson syndrome/toxic epidermal necrolysis. Indian J. Dermatol. 65 (4):274–78. doi:10.4103/ijd.IJD_217_19.
- Zhao, S., J. Jiang, Y. Jing, W. Liu, X. Yang, X. Hou, L. Gao, and L. Wei. 2020. The concentration of tumour necrosis factor-α determines its protective or damaging effect on liver injury by regulating Yap activity. Cell. Death Dis. 11:1–13. doi:10.1038/s41419-020-2264-z.
- Zhao, M., M. Jingsong, M. Li, Y. Zhang, B. Jiang, X. Zhao, C. Huai, L. Shen, N. Zhang, L. He, et al. 2021. Cytochrome P450 enzymes and drug metabolism in humans. Int. J. Mol. Sci 22:1280. doi:10.3390/ijms222312808.
- Zhao, S. W., Y. M. Li, Y. L. Li, and C. Su. 2023. Liver injury in COVID-19: Clinical features, potential mechanisms, risk factors and clinical treatments. World. J. Gastroenterol 29:241–56. doi:10.3748/wjg.v29.i2.241.