Abstract
2,2,4-trimethyl, 1,3-pentanediol monoisobutyrate (TPM) is a widely used solvent found in water-based coatings. Ambient measurements of TPM are reported here for the first time. Although this compound has been previously measured in indoor air, this study illustrates successful detection and quantification of TPM in ambient air at three locations in Southern California: Pico Rivera, Azusa, and Riverside. TPM was detected in every sample collected, with concentrations ranging from 0.7 to 49.5 parts per trillion (ppt). Collections took place during summer 2009, fall 2009, winter 2009/2010, and spring 2010, for 5–7 days during each season. The highest mean concentrations were observed during the summer months for each city, when coating activities are typically at their highest.
2,2,4-Trimethyl, 1,3-pentanediol monoisobutyrate (TPM) is a widely used solvent found in water-based coatings. Ambient measurements of TPM are reported here for the first time. The highest mean concentrations were observed during the summer months for each city, when coating activities are typically at their highest. Unreacted TPM constitutes approximately 0.01% of the total nonmethane hydrocarbon (NMHC) concentrations in the Los Angeles air basin and given its slow reactivity rate in forming ozone, this would be an approximate upper limit for the fraction of ozone that it is responsible for forming.
Introduction
According to the U.S. Environmental Protection Agency (EPA), as of 2003–2005, 157 million people in the United States lived in areas in nonattainment for the National Ambient Air Quality Standards (NAAQS) for ozone (O3) under the 1997 standard (CitationEPA, 2007). O3 is formed in the atmosphere from the photochemical reaction of volatile organic compounds (VOCs) and nitrogen oxides (NOx). The Clean Air Act passed in 1970 was designed to address this problem, spawning an array of emission control technologies and rules such as the limits on the amount of VOCs that could be used in commercial paint. Many of the specific regulations related to the coatings industry were based on mass of VOC per volume of coating, without consideration of the reactivity of these compounds in the atmosphere. The overall mitigation efforts related to total VOC and NOx have greatly reduced air pollution levels, but the new PM2.5 (particulate matter with an aerodynamic diameter <2.5 μm) standard and the revision of the ozone standard in 1997 and again in 2008 have resulted in the designation of a larger number of nonattainment areas. The resulting regulatory responses will necessitate the development of new technologies and techniques for characterizing potential pollutants and assessing their environmental impacts. Further, these responses could result in revisions to the regulations concerning the coatings industry, particularly since the EPA is expected to lower the NAAQS standard for ozone again in the future.
Specifically, the Clean Air Act and its amendments have affected the coatings industries by requiring limits on the amount of VOCs allowed per gallon of paint. In some cases different federal and state VOC requirements have been promulgated, creating additional challenges, which have not been well characterized. Further, although the coatings industry has been working on perfecting lower-VOC products, the end users of coatings continue to demand high performance, especially in key characteristics such as coverage and durability. The impacts of currently available low-VOC coatings on all environmental media, however, are not well characterized in the literature. This study seeks to contribute to a better understanding of the issues.
The criteria air pollutants of major concern are ozone (O3) and PM2.5 due to their adverse effects on public health and because their NAAQS standards have been difficult to obtain. VOCs emitted from the application of coatings could contribute to the formation of ozone and particulate matter. The objective of this study was to quantify the concentrations of 2,2,4-trimethyl, 1,3-pentanediol monoisobutyrate (TPM) emitted into the atmosphere by coating activities that may undergo reactions to form O3.
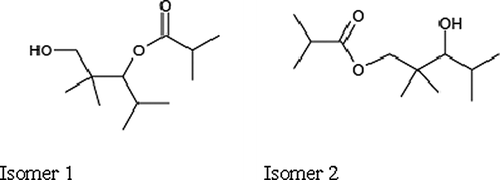
TPM is typically the most abundant VOC used in water-based architectural coatings. shows the structure of both isomers of this compound. TPM has no other commercial use other than in coatings. The percentage in coatings ranges from 0.3% to 2.5% by mass. TPM has a high boiling point (260 °C) with low vapor pressure (0.01 mm Hg at room temperature) and slowly evaporates from the coated surface as it dries. During the application process, the air concentrations of TPM are typically tens of parts per billion (ppb) to several hundred ppb, whereas in new houses concentrations are in the 10 ppb range (CitationBrown, 2003; CitationHodgson et al., 2000).
Figure 1. Isomers of TPM.
VOC control strategies aimed at reducing the photochemical formation of ozone have been primarily based on reducing the mass of VOC emitted regardless of the chemical nature of the VOC. However, it is well known that VOCs are oxidized in the atmosphere through very different mechanisms. The contribution of each VOC to the formation of ozone is different because each has a different oxidation mechanism in the atmosphere. The ozone formation potential for a representative urban airshed is often characterized by the maximum incremental reactivity (MIR) scale (CitationCarter, 1994). An incremental reactivity, IR, of an organic compound is the change in the peak ozone concentration, Δ[O3] in grams, divided by an incremental change in the initial concentration and emissions of the organic compound, Δ[VOC] in grams (eq 1).
MIRs are calculated for reference scenarios consisting of a specified meteorological situation, initial pollutant concentrations, and emission rates of NOx and VOC. If a series of simulations with differing NOx initial concentrations and emission rates are made, a scenario will be found in the VOC-limited region where the aggregate incremental reactivity reaches a maximum and this is defined as the MIR scenario (CitationCarter, 1994). Once the MIR scenario is known, incremental reactivities can be calculated through the simulation of cases with small variations in individual organic compounds.
The two isomers of TPM contain one hydroxy group (OH) and one ester group (C(O)O). The only atmospherically relevant reactions of saturated esters such as TPM are reactions with the hydroxyl radical (CitationAtkinson, 2000; CitationMellouki et al., 2003). Expected reaction products include acetone and other higher ketones (CitationCarter, 2010). CitationCarter and Malkina (2005) studied the environmental impact of TPM in a smog simulation chamber under NOx-limited conditions, and found that whereas the distribution of isomers in the liquid phase was 32% isomer 1 and 68% isomer 2, in the gas phase the distribution changed to 42% isomer 1 and 58% isomer 2. They also determined its maximum incremental reactivity (MIR) to be 0.88, and its rate constants for reaction with OH to be 1.30 × 10−11 cm3 molecule−1 sec−1 for isomer 1 and 1.68 × 10−11 cm3 molecule−1 sec−1 for isomer 2. If one assumes a concentration for hydroxyl radicals of 1 × 106 radicals cm−3, this gives a lifetime of 21.4 hr for isomer 1 and 16.5 hr for isomer 2 (CitationFinlayson-Pitts and Pitts, 1999).
Although concentrations have been measured for indoor applications to determine indoor concentrations and in flow-through chambers to measure the emission rate as a function of time (CitationHodgson, 1999; CitationBartekova et al., 2006), there have been few reported attempts to measure TPM in ambient air. CitationHodgson et al. (2003) reported concentrations in a call center building in the sub-ppb range and could not detect TPM in ambient air with an estimated detection limit of 80 ppt.
Experimental
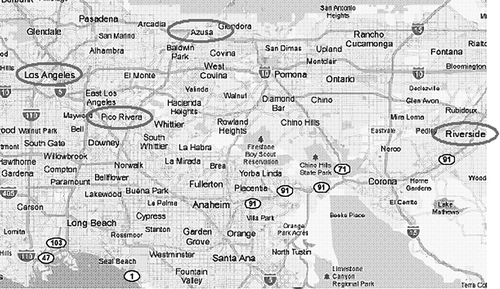
The prevailing on-shore wind in California's South Coast Air Basin (SoCAB) is from west to east in the daytime. Sampling took place at three monitoring locations: the first just downwind of the Los Angeles urban center (Pico Rivera, CA) operated by the South Coast Air Quality Management District (SCAQMD), approximately 13 km southeast of downtown Los Angeles (DTLA). The second location was near the middle of the SoCAB (Azusa, CA), another air monitoring station operated by the SCAQMD. This site is located approximately 32 km northeast of DTLA. The third location was on the eastern side of the SoCAB (Riverside, CA) at the College of Engineering Center for Environmental Research and Technology (CE‐CERT) facility at the University of California at Riverside (UCR), which is approximately 80 km east of DTLA (see ).
Figure 2. Map of Southern California with sampling locations circled, as well as the location of downtown Los Angeles.
The sampling strategy was to simultaneously collect sorbent tube samples at each site in triplicate over 6-hr periods. Samples were collected on Tenax-TA (Sigma Aldrich, St. Louis, MO) for gas chromatography–flame ionization detection (GC-FID) analysis. The sample collection tubes (made of either glass or stainless steel) were of 10 mm internal diameter (ID) with a length of 200 mm packed with 16 mL Tenax-TA. Samples were collected from 6 a.m. to noon, a period of relatively calm air. A second sample was collected from noon to 6 p.m., a period generally exhibiting strong on-shore flow. Samples were collected every other day to facilitate servicing of the samplers. For each sampling episode, 5–7 daily pairs were collected of triplicate sample sets. To evaluate seasonal variability, similar sampling episodes were conducted during all four seasons. A sampling rate of 1 L/min for this tube was employed for 6 hr. No indication of breakthrough of TPM was observed under these conditions during the previous phase of this project (unpublished data, University of California [UC] Riverside).
The first of the triplicate sample sets were analyzed for TPM (see below) at the Analytical Laboratory at CE-CERT at UC Riverside (conditions listed below). Half of the second of the triplicate sets were analyzed as replicates to determine measurement precision. The third of the triplicate sets was set aside for gas chromatography–mass spectrometry (GC-MS) analysis or for reanalysis, if needed. The five highest concentration samples from each sampling season were analyzed by high-resolution GC-MS. These analyses were conducted by the University of Nevada's Desert Research Institute's Organic Analytical Laboratory using the same chromatographic conditions as those used at UC Riverside (listed below).
For analysis at UC Riverside, tubes were thermally desorbed at 330 °C into a cooled microtrap at −50 °C using a Gerstel TDS 3 ThermoDesorption System (Gerstel, Linthicum, MD). The microtrap was then heated for injection into an Agilent model 6890N gas chromatograph with flame ionization detection (FID) (Agilent Technologies, Palo Alto, CA). The column used was a 50-m J&W Scientific DB-1701 column with a bore of 0.032 inches and a 1 μm film thickness (J&W Scientific, Folsom, CA). The GC oven was held at an initial temperature of 40 °C for 1 min, followed by a ramp up of 10 °C per minute up to 300 °C.
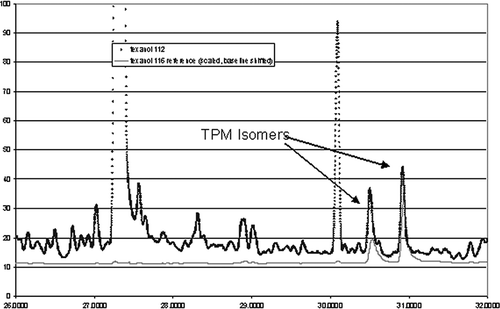
Although TPM may have significant concentrations in indoor air compared with other hydrocarbons, it is a very minor component of ambient air. Peaks were identified primarily by retention times. Peak identification was also aided by the relative ratio of the two TPM isomers, which is expected to be constant. Identification by retention time was confirmed by collecting collocated samples and then spiking one tube by sampling a calibration source of TPM ().
Figure 3. Typical ambient air chromatogram with reference TPM chromatogram superimposed.
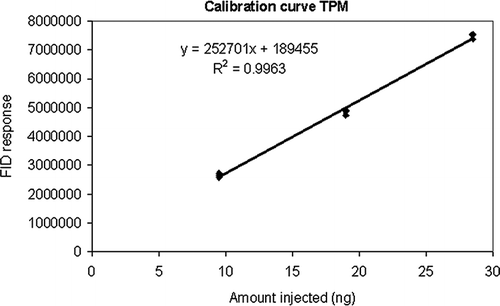
For calibration, a solution of TPM in methanol was prepared using TPM supplied by Eastman Chemical (Kingsport, TN) and then serially diluted to the concentration range observed in ambient air. Aliquots were then injected on the Tenax-TA cartridges for GC analysis. shows the calibration curve obtained. Calibration was verified by injecting a known amount of TPM into the CE-CERT 90-m3 environmental chamber (CitationCarter et al., 2005), and then analyzed with the same method as the ambient samples without informing the technician of the amount injected. The resulting analysis of the chamber sample was within 5% of the calculated amount injected.
Figure 4. Calibration curve for TPM analyzed by GC-FID.
Results
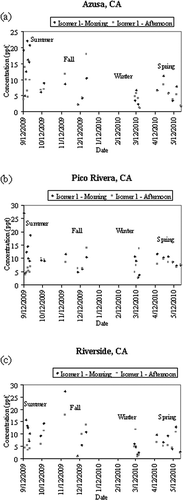
Ambient air samples were collected on Tenax-TA adsorbent tubes during each season: Summer 2009 (12–22 September), Fall 2009 (10 and 15 October, 18 November, and 8 and 15 December), Winter 2009/2010 (22 December and 10–17 March), and Spring 2010 (15 and 25 April and 3–23 May). Samples were collected twice per day: during morning hours (6 a.m. to noon) and afternoon hours (noon to 6 p.m.). TPM was detected in every ambient sample collected. For sample pairs, TPM values fell within ±20% for most samples, with a detection limit of 0.2 ng (0.07 ppt; based upon a sample volume of 360 L) per sample. Field blanks were also collected to test for diffusion of TPM onto the Tenax-TA, which was not observed. TPM was also detected by the Organic Analytical Laboratory at the Desert Research Institute using a thermal desorption/GC-MS method, confirming the presence of TPM in ambient air. shows the mean concentrations of both isomers of TPM for each season, each city, for mornings and afternoons. Higher concentrations of each isomer were observed during the morning hours of summer, which is when most exterior painting is typically done. The lowest values of TPM for Azusa, Pico Rivera, and Riverside were observed during winter mornings, when coating activity is lowest, and on summer afternoons, when conditions favor increased photooxidation. illustrates the time series of the observed TPM concentrations for isomer 1 for each city.
Table 1. Mean concentrations of TPM for each season in each city
Figure 5. Time-series data for each city for isomer 1. Morning hours in dark gray, afternoon hours in light gray. (a) Azusa, CA; (b) Pico Rivera, CA; (c) Riverside, CA.
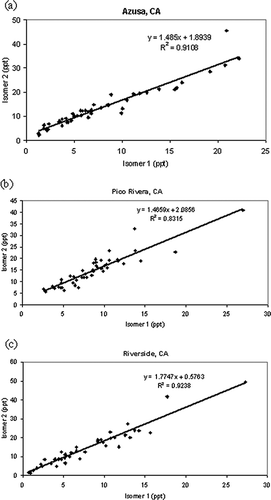
shows plots of isomer 2 versus isomer 1 for each city. The slope for Riverside, CA, is higher than for Pico Rivera or Azusa, indicating fresh emissions with respect to TPM, containing higher concentrations of the more reactive primary isomer. Total nonmethane hydrocarbons (NMHC) in the SoCAB are typically in the range of 360 ppb (CitationFujita et al., 2002). With a total mean concentration (isomer 1 + isomer 2) of about 30 ppt, unreacted TPM constitutes approximately 0.01% of the total NMHC. Typical concentrations of TPM are 1 order of magnitude lower than the BTEX (benzene, toluene, ethyl benzene, and xylenes) compounds (Fujita et al., 2002). m-Xylene has roughly the same atmospheric lifetime as TPM, but its MIR is 2.05, whereas the MIR for TPM is 0.88. Therefore, based on the MIR, TPM is not likely to be a large contributor to ozone formation. However, CitationCarter et al. (2005b) found TPM may have some small amount of PM formation, with its low volatility and ability to act as a radical inhibitor in a chamber environment.
Figure 6. Plots of isomer 2 versus isomer 1 for each city. (a) Azusa, CA; (b) Pico Rivera, CA; (c) Riverside, CA.
Box model simulations (http://www.cert.ucr.edu/~carter/SAPRC/SAPRCfiles.htm) using SAPRC07 (CitationCarter, 2010) were performed with and without TPM present in the VOC mixture to investigate the ability of TPM to act as a radical inhibitor in a real world scenario. Representative concentrations of typical VOC species found in the South Coast Air Basin were taken from Fujita et al. (2002). These simulations showed a difference in O3, NO, NO2, OH, and HO2 concentrations to be less than 1% with and without TPM present, indicating that the ability of TPM to act as a radical inhibitor is not significant at the TPM concentrations found in the ambient atmosphere.
Conclusions
An analytical system was developed based on adsorption on Tenax-TA followed by thermal volatilization that was capable of detecting TPM in ambient air. TPM was successfully detected and quantified in ambient air at three locations in the Southern California air basin. TPM was detected in all samples taken from ambient air, and is reported here for the first time. Ambient TPM concentrations (both isomers) ranged from 5 to 40 ppt. Unreacted TPM constitutes approximately 0.01% of the total NMHC concentrations in this air basin and given its slow reactivity rate in forming ozone, this would be an approximate upper limit for the fraction of ozone that it is responsible for forming. Airshed modeling could be used to better quantify this value.
Given the generally similar concentrations measured throughout the basin, it appears that the sources of TPM are fairly uniformly distributed. This would be expected in a highly developed area such as the Los Angeles metropolitan area. The change in the ratios of isomer 2 to isomer 1 between Pico Rivera and Riverside, since the two isomers have different reactivities (lifetime of approximately 16 versus 21 hr), indicates fresh emissions during transport between the two sites. Since the average concentrations are similar for all of the sites, the upwind sites would be expected to be a larger source of TPM than Riverside, since air parcels generally travel from west to east during daytime hours. This is also consistent with the higher intensity of development in the upwind areas.
Acknowledgments
This work was funded by the National Paint and Coatings Association. The authors would also like to acknowledge the generosity of the South Coast Air Quality Management District (SCAQMD) for permission for use of their facilities in Azusa and Pico Rivera, California.
References
- Atkinson , R. 2000 . Atmospheric chemistry of VOCs and NOx . Atmos. Environ. , 34 : 2063 – 2101 .
- Bartekova , A. , Lungu , C. , Shmulsky , R. , Huelman , P. and Park , J.Y. 2006 . Laboratory evaluation of volatile organic compounds emissions from coated and uncoated oriented strandboard . Forest Products J. , 56 : 85 – 90 .
- Brown, S.K. 2003. Indoor air pollution-lowering emissions of chemicals released from manufactured products. Presented at Hazmat 2003 Conference (Fire Protection Association Australia) Stamford Grande, N. Ryde, NSW Australia, April 29–30, 2003 http://www.carpetinstitute.com.au/downloads/indoor_quality_000.pdf (http://www.carpetinstitute.com.au/downloads/indoor_quality_000.pdf) (Accessed: June 2011 ).
- Carter , W.P.L. 1994 . Development of ozone reactivity scales for volatile organic compounds . J. Air Waste Manage. Assoc. , 44 : 881 – 899 .
- Carter , W.P.L. 2010 . Development of the SAPRC-07 chemical mechanism . Atmos. Environ. , 44 : 5324 – 5335 .
- Carter, W.P.L., D.R. Fitz, D.R. Cocker III, I.L. Malkina, K. Bumiller, C.G. Sauer, J.T. Pisano, C. Bufalino, and C. Song. 2005a. Development of a Next-Generation Environmental Chamber Facility for Chemical Mechanism and VOC Reactivity Research, Final Report. Cooperative Agreement CR 827331-01-0. Prepared by the Center for Environmental Research and Technology College of Engineering University of California Riverside for the U.S. Environmental Protection Agency, Research Triangle Park, NC. http://eprints.cert.ucr.edu/323/1/chamrpt.pdf (http://eprints.cert.ucr.edu/323/1/chamrpt.pdf) (Accessed: June 2011 ).
- Carter, W.P.L., and I.L. Malkina. 2005. Evaluation of Atmospheric Impacts of Selected Coatings VOC Emissions, Final Report. Contract No. 00–333. Prepared by the Center for Environmental Research and Technology College of Engineering University of California Riverside for the California Air Resources Board, Sacramento, CA http://www.cert.ucr.edu/~carter/coatings/coatrpt.pdf (http://www.cert.ucr.edu/~carter/coatings/coatrpt.pdf) (Accessed: June 2011 ).
- Carter, W.P.L., I.L. Malkina, D.R. Cocker, and C. Song. 2005b. Environmental Chamber Studies of VOC Species in Architectural Coatings and Mobile Source Emissions, Final Report. Contract No. 03468. Prepared by the Center for Environmental Research and Technology College of Engineering University of California Riverside for the South Coast Air Quality Management District, Diamond Bar, CA http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.81.305&rep=rep1&type=pdf (http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.81.305&rep=rep1&type=pdf) (Accessed: June 2011 ).
- Finlayson-Pitts , B.J. and Pitts , J.N. Jr . 1999 . Chemistry of the Upper and Lower Atmosphere , San Diego : Academic Press .
- Fujita, E.M., D.E. Campbell, W.R. Stockwell, R.E. Keislar, B. Zielinska, J.C. Sagebiel, W.S. Goliff, M. Keith, and J.L. Bowen. 2002. Weekend/Weekday Ozone Observations in the South Coast Air Basin: Volume II—Analysis Of Air Quality Data, Final Report. CRC Contract Number E-53. Prepared by the Division of Atmospheric Sciences, Desert Research Institute for the National Renewable Energy Laboratory, Golden, CA. http://http:ftp://preftp.arb.ca.gov/carbis/aqd/weekendeffect/final_wewd_5_1/nrelp3v2f.pdf (http://http:ftp://preftp.arb.ca.gov/carbis/aqd/weekendeffect/final_wewd_5_1/nrelp3v2f.pdf)
- Hodgson, A.T. 1999. Common Indoor Sources of Volatile Organic Compounds: Emission Rates and Techniques for Reducing Consumer Exposure. Final Report, No. 95–302. Prepared by the Lawrence Berkeley National Laboratory for the California Air Resources Board contract, Sacramento, CA http://eetd.lbl.gov/ie/pdf/LBNL-42402.pdf (http://eetd.lbl.gov/ie/pdf/LBNL-42402.pdf) (Accessed: June 2011 ).
- Hodgson , A.T. , Faulkner , D. , Sullivan , D.P. , Dibartolomeo , D.L. , Russell , M.L. and Fisk , W.J. 2003 . Effect of outside air ventilation rate on volatile organic compound concentrations in a call center . Atmos. Environ. , 37 : 5517 – 5527 .
- Hodgson , A.T. , Rudd , A.F. , Beal , D. and Chandra , S. 2000 . Volatile organic compound concentrations and emission rates in new manufactured and site-built houses . Indoor Air , 10 : 178 – 192 .
- Mellouki , A. , Le Bras , G. and Sidebottom , H. 2003 . Kinetics and mechanisms of the oxidation of oxygenated organic compounds in the gas phase . Chem. Rev. , 103 : 5077 – 5096 .
- U.S. Environmental Protection Agency. 2007. Report on Air Quality in Nonattainment Areas for 2003–2005 Covering Ozone, Particulate Matter, Carbon Monoxide, Sulfur Dioxide, Nitrogen Dioxide, and Lead, Technical Summary http://www.epa.gov/airtrends/pdfs/20070214_aq_na_2003-2005.pdf (http://www.epa.gov/airtrends/pdfs/20070214_aq_na_2003-2005.pdf) (Accessed: June 2011 ).