Abstract
In this study, removing sulfur dioxide (SO2), nitrogen oxides (NOx), and mercury (Hg) from simulated flue gas was investigated in two laboratory-sized bubbling reactors that simulated an oxidizing reactor (where the NO and Hg0 oxidation reactions are expected to occur) and a wet limestone scrubber, respectively. A sodium chlorite solution was used as the oxidizing agent. The sodium chlorite solution was an effective additive that enhanced the NOx, Hg, and SO2 capture from the flue gas. Furthermore, it was discovered that the location of the sodium chlorite application (before, in, or after the wet scrubber) greatly influences which pollutants are removed and the amount removed. This effect is related to the chemical conditions (pH, absence/presence of particular gases) that are present at different positions throughout the flue gas cleaning system profile. The research results indicated that there is a potential to achieve nearly zero SO2, NOx, and Hg emissions (complete SO2, NO, and Hg removals and ∼90% of NOx absorption from initial values of 1500 ppmv of SO2, 200 ppmv of NOx, and 206 μg/m3 of Hg0) from the flue gas when sodium chlorite was applied before the wet limestone scrubber. However, applying the oxidizer after the wet limestone scrubber was the most effective configuration for Hg and NOx control for extremely low chlorite concentrations (below 0.002 M) and therefore appears to be the best configuration for Hg control or as an additional step in NOx recleaning (after other NOx control facilities). The multipollutant scrubber, into which the chlorite was injected simultaneously with the calcium carbonate slurry, appeared to be the least expensive solution (when consider only capital cost), but exhibited the lowest NOx absorption at ∼50%. The bench-scale test results presented can be used to develop performance predictions for a full- or pilot-scale multipollutant flue gas cleaning system equipped with wet flue gas desulfurization scrubber.
The control of NOx, Hg, and SO2 emissions from coal-fired combustors is an important, worldwide concern affecting environmental pollution. In both the European Union and the United States, NOx and SO2 emissions are subject to newly tightened emission limits. This paper investigated the NOx, Hg, and SO2 removals in a flue gas cleaning system equipped with a classical wet limestone scrubber, enhanced by a solution of sodium chlorite. This paper demonstrated that the location at which the sodium chlorite is applied in a flue gas cleaning system affects the control of these pollutants and discusses how possible solutions could be practically implemented.
Introduction
Coal usage is on the rise and will likely remain the world's main source of power into the mid-21st century (CitationU.S. Energy Information Administration, 2010). Society, in general, does not approve of coal combustion as a source of energy because of the large amount of pollutants, such as sulfur dioxide (SO2), nitrogen oxides (NOx), particulate matter (PM), mercury (Hg), and carbon dioxide (CO2), that are emitted and, subsequently, are responsible for decreasing quality of life and destroying ecosystems. However, coal is uniquely capable of satisfying the growing electricity demands. Renewable resources, such as solar and wind power, are not currently able to totally replace coal because they cannot provide a constant, steady supply of electricity that must always be available to ensure that the electricity transition grid functions properly. Moreover, producing electricity from coal is less expensive than using most other fuels. Today, coal continues to be the fuel of choice to generate electricity because it is abundant and affordable (CitationAmerican Coalition for Clean Coal Electricity, 2011). Therefore, it is important to use this source of energy in a way that protects human health and ecosystems. Coal combustion could be environmentally friendly and fulfill the current and future stringent environmental standards, but it requires the development of new and more economically affordable clean coal technologies as well as efficient air pollution control technologies.
Presently, there are many different control technologies in various stages of development (CitationChmielewski et al., 2010; CitationChu et al., 2001; CitationEllison, 2003; CitationHutson et al., 2008; CitationJin et al., 2006; CitationLee et al., 2005; CitationLiu et al., 2010; CitationSuchecki et al., 2005) that will allow these goals to be achieved. The most common and effective technology for SO2 removal is flue gas desulfurization, in which limestone slurry is used as a scrubbing solution. It is estimated that approximately one-third of the United States' coal-fired capacity is using some types of flue gas desulfurization technology (FGD), where nearly 90% of them are wet FGD scrubbers, with nearly 70% of those being limestone-base systems (CitationSrivastava et al., 2006). FGD technologies have been shown to remove more than 95% of the SO2 emissions (CitationSrivastava and Jozewicz, 2001). NOx emissions can be decreased through combustion and postcombustion treatments. Combustion treatments are routinely used in boilers, in which the goal is to decrease the production of NOx while the fuel is being combusted (CitationDabrowski and Piecuch, 2011). Postcombustion technologies reduce NOx from the flue gas after fuel is combusted. There are many different postcombustion technologies, such as selective catalytic reduction (SCR), selective noncatalytic reduction (SNCR), wet scrubbing, electron beam irradiation, and absorption, among which SCR and SNCR are the most effective and commercially available technologies for NOx removal. NOx removal efficiencies using SCR are high, between 80% and 90% (CitationSmith, 1992), but SCR is relatively expensive. In addition, some of the excess ammonia used in SCR may combine with sulfur trioxide (SO3) to form ammonium bisulfate (NH4HSO4), which condenses in the heat exchanger. Moreover, catalysts are affected by dust and are poisoned by sulfur and chlorine compounds (CitationChirona and Altshuler, 1998).
The aforementioned problems and the newly tightened NOx and SO2 emission limits in the United States and the European Union (EU) (CitationThe European Parliament and the Council of the European Union, 2001, Citation2010; CitationU.S. Environmental Protection Agency [EPA], 2011a) encourage scientists to develop alternative techniques to control NOx emissions. In 2011, the EPA proposed a new the Cross-State Air Pollution Rule (CSAPR) to replace the Clean Air Interstate Rule (CAIR) (CitationEPA, 2011a). According to the CSAPR, SO2 emissions should be reduced in 28 states by 73% from the 2005 levels by 2014, and NOx emissions should be reduced by 54% in the same period of time. Similar action is being taken in Europe (CitationThe European Parliament and the Council of the European Union, 2001, Citation2010). In the European Union, emissions of coal-fired power plants have to be reduced according to the Large Combustion Plants (LCP) Directive (2001/80/EC), which specifies emission limits (in mg/Nm3 for SO2, NOx, and dust) for plant that use combustible fuels to generate electricity and/or heat, and have a maximum thermal input greater than 50 MW. In 2 years' time, the new Industrial Emission Directive (IED) (2010/75/EU) (CitationThe European Parliament and the Council of the European Union, 2010) will replace LCP. The new directive sets stricter than LCP emission limits for NOx, SO2, and dust from the largest plants. The IED requires, for example, to limit NOx and SO2 to 200 mg/Nm3 from new combustion power plants with thermal effect >100 MWth, and 150 mg/Nm3 for >300 MWth (CitationThe European Parliament and the Council of the European Union, 2010). Thus, because of these more stringent emission requirements, a significant increase in the use of wet-FGD and NOx control technologies is expected in the new decade. Moreover, many countries work together to reduce mercury emission from various sources (CitationSloss, 2008). The U.S. Environmental Protection Agency (EPA) issued Mercury and Air Toxics Standards (MATS) for power plants to limit mercury, acid gases, and other toxic pollution from power plants, keeping 91% of the mercury in coal from being released to the air (CitationEPA, 2011b). Other countries have also indicated an intention to do it (CitationSloss, 2008). Mercury also comes from coal-fired electric utility boilers (CitationPawlowski, 2011; CitationSloss, 2008), and needs to be controlled because of its well-known, high toxicity.
Researchers have focused on the concept of multipollutant control in wet scrubber systems, as it is theoretically possible to remove NOx in wet scrubbers (CitationChu et al., 2001; CitationEllison, 2003; CitationHutson et al., 2008; CitationJin et al., 2006; CitationJozewicz et al., 2004; CitationLee et al., 2005; CitationSada et al., 1978). Because NO has an extremely low solubility, removing NOx effectively by wet scrubbing demands the oxidation of NO to NO2 or higher nitrogen oxides, which are highly water-soluble and could be easily and efficiently removed in the wet scrubbing system. There have been many bench-scale experiments performed worldwide on NOx and NOx/SO2 removal using different types of oxidant additives, where sodium chlorite (NaClO2) was one of the most effective types (CitationChu et al., 2001; CitationHutson et al., 2008; CitationJin et al., 2006; CitationLee et al., 2005; CitationSada et al., 1978). It is worth emphasizing that the oxidation and absorption processes of pollutants, such as SO2, NOx, and Hg, depend on many parameters that all contribute to the final removal effects (CitationHutson et al., 2008). After a series of experiments, we found that understanding the chemical process parameters and their proper applications is the key to optimize the SO2, NOx, and Hg removal efficiencies. The chemical conditions and the flue gas composition are different at different locations, before, in, or after the wet scrubber.
This paper demonstrates how the location of the sodium chlorite injection affects the removal of SO2, NOx, and Hg. The presented research may be useful in pilot- or full-scale tests, where the placement of lances and/or additional reactors must be considered.
Experimental Methods and Procedures
As shown in , the bench-scale experimental setup, in which an oxidizer was used before the wet limestone scrubber, is marked as “Configuration 1.” The system consisted of four parts: the flue gas simulation system, the flue gas cleaning system, the chemistry blending system, and the analysis system. The flue gas simulation system generated a simulated flue gas from a controlled mixture of the cylinder gases. Mass-flow controllers and valves were used to control the flow of the component gases, where a standard flue gas comprised of approximately 81 vol % N2, 11 vol % CO2, 8 vol % O2, 1500 ppmv of SO2, 200 ppmv of NO, and 206 μg/m3 Hg0 was simulated. These concentrations simulate the pollutant concentrations of a real flue gas from a coal-fired power plant. Elemental mercury vapor was supplied using a VICI Metronics Dynacalibrator permeation oven (Valco Instruments Co. Inc., Poulsbo, WA) held at 100 °C, where N2 was used as the carrier gas. The mercury concentration was 10 times greater than is typical for a coal combustion flue gas to accommodate the sensitivity of the mercury analyzer. The experimental process parameters are listed in
Table 1. Experimental process parameters
Figure 1. Schematic diagram for the removal of SO2, NOx, and Hg from the simulated flue gas—Configuration 1.
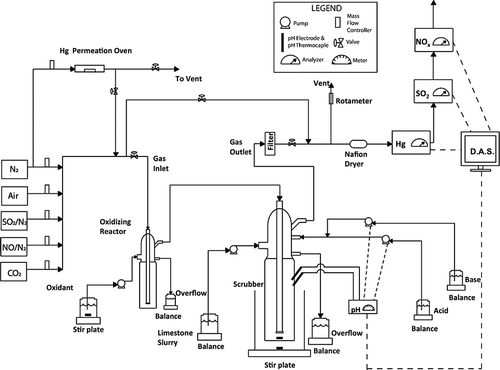
The total flow rate of the simulated flue gas was 2 L/min (standard temperature and pressure [STP] conditions). The simulated gas was heated to 90 °C by an electrical heating tape before entering into the oxidation/absorption impingers. This temperature represents a temperature of flue gas, which might be seen before wet-FGD scrubber in a coal-fired power plant. As shown in , the flue gas cleaning system consisted of two impingers in Configuration 1. The simulated flue gas continuously flowed through the first small oxidizing reactor (20 mL to overflow level), which was a flow-through, gas-liquid impinger. After the oxidizing reactor, the flue gas flowed through the larger (600 mL to overflow level) flow-through, gas-liquid impinger, which simulated a wet limestone scrubber. The oxidizing reactor was at ambient temperature (22 °C), and the wet scrubber temperature was maintained at 55 °C by immersing the scrubber to the overflow from a temperature-controlled water bath. The temperature of wet scrubber was maintaining at 55 °C, because this temperature represents a temperature existing in the real limestone wet-FGD scrubbers.
The chlorite solution was pumped into the oxidizing impinger, which overflowed to maintain a constant chlorite level and a volume of ∼20 mL. Calcium carbonate slurry was pumped into the limestone scrubber to maintain a constant slurry level and a volume of ∼600 mL. The addition rate of the oxidant solution was monitored via weight loss in a beaker. The alkali solution addition rate was monitored via the weight gain from the overflow of the scrubber. The alkali scrubber solution was constantly mixed to ensure a homogeneous 0.5-wt% solid slurry. Different concentrations of sodium chlorite were tested, ranging from 0 to 0.008 M.
The exhaust gas after the impingers passed through mini-impingers containing quartz wool to remove any water mist before entering the various gas analyzers. The scrubbed gas then passed through heat-traced lines to a Naftion dryer, which removed any remaining water vapor. The flue gas then flowed through a series of analyzers, where any excess gas was vented to an exhaust hood. The NOx species were measured using a continuous chemiluminescence NOx analyzer (Teledyne; Advanced Pollution Instrumentation [API], Model 200AH; San Diego, CA, USA). The Teledyne API Model 200AH analyzer is designed to measure the concentration of nitric oxide [NO], total oxides of nitrogen [NOx] and, by calculation, nitrogen dioxide [NO2]. The [NO] component is calculated by subtracting [NOx] − [NO] = [NO2] using the built-in computer.
The SO2 concentration was measured using a continuous fluorescence analyzer (Teledyne; Advanced Pollution Instrumentation, Model 100AH). The Hg0 vapor was measured using a continuous cold vapor atomic absorption (CVAA) analyzer (BUCK Model 400A; Buck Scientific, East Norwalk, CT), which used atomic absorption to quantify the elemental mercury by producing an output signal between 0 and 250 mV that is proportional to the concentration. Both water and SO2 interfere with the mercury measurement by absorbing light at the wavelength being measured. Water was eliminated from the sample with an in-line Permapure Naftion dryer (Perma Pure LLC, Toms River, NJ). SO2 was an integral part of the flue gas being tested, and it produced a CVAA signal proportional to the SO2 concentration. The SO2 concentration was determined using the same sample stream as the analyzed mercury concentration (i.e., the SO2 analyzer was installed in series, downstream of the CVAA analyzer). The CVAA response to the SO2 was determined daily, and the mercury concentrations were subsequently corrected for the SO2 interference. To prevent long-term instrument drift, the sample cell from the CVAA analyzer was removed, soaked in a 10% nitric acid solution for at least 4 hr, rinsed, and dried between each experiment. All the analyzer results were automatically logged to a data acquisition system every 10 sec.
Before the experiments, the simulated oxidizing reactor and wet scrubber were filled to their overflow levels, whereas the simulated flue gas bypassed the system (see ). The flue gas was then introduced to the impingers and scrubbed for 60 min. Fresh chlorite solution and calcium carbonate slurry were continuously added at a constant rate to the impingers during the experiment. The effluents from the oxidizing reactor and limestone scrubber were collected every 10 min. The samples were automatically weighed and placed in ice to quench or slow any oxidation reactions. The samples were then immediately analyzed for chloride (Cl−), chlorite (ClO2 −), chlorate (ClO3 −), sulfate (SO4 2−), nitrate (NO3 −), and nitrite (NO2 −) by ion chromatography (IC) using the EPA Reference Method 300.0 (EPA, 1993). The total Hg content of the samples was also analyzed according to Ontario Hydro Method D6784-02, as described for the potassium chloride fraction. Mercury analysis of each digest was performed by the CVAA according to the EPA SW846 Method 7470A, “Mercury in Liquid Waste (manual cold vapor technique),” where a Perkin-Elmer FIMS 100 flow injection mercury system (PerkinElmer, Waltham, MA) was used for the analysis. The pH in the wet scrubber was measured continuously during the experiment and was recorded by a data acquisition system every 10 sec. An independent sample from the oxidizing reactor was collected to measure the pH during the experiment. The tests labeled “without chlorite solution” were conducted using deionized water.
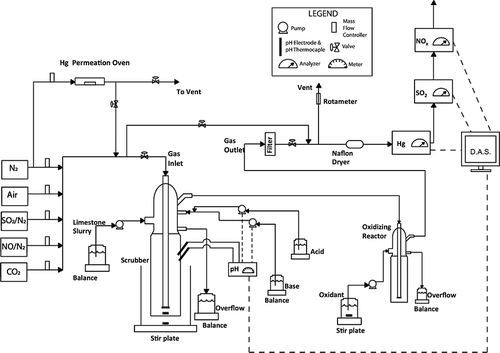
shows the bench-scale experimental setup, where an oxidizer was injected after the wet limestone scrubber, marked as “Configuration 2.” All the experimental parameters presented in and experimental procedures were identical to those described for Configuration 1, except for the placement of the oxidizing reactor, which was after the wet scrubber.
Figure 2. Schematic diagram for the removal of SO2, NOx, and Hg from the simulated flue gas—Configuration 2.
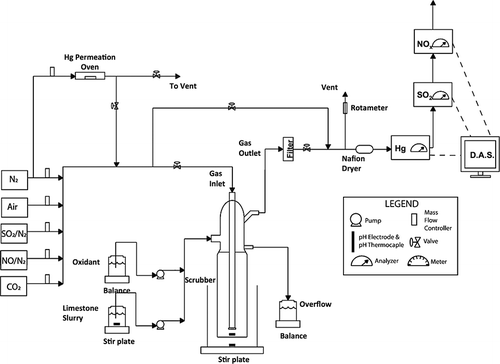
shows the bench-scale experimental setup, where an oxidant was injected simultaneously with a calcium carbonate slurry into the wet scrubber, marked as “multipollutant scrubber.” All the experimental parameters presented in and procedures were identical to those described for “Configuration 1” except that an oxidant was blended with a calcium carbonate slurry before entering the wet scrubber and injected simultaneously at a constant rate.
Figure 3. Schematic diagram for the removal of SO2, NOx, and Hg from the simulated flue gas—Configuration 3 (multipollutant scrubber).
Experimental Results
Nitric oxide (NO) oxidation
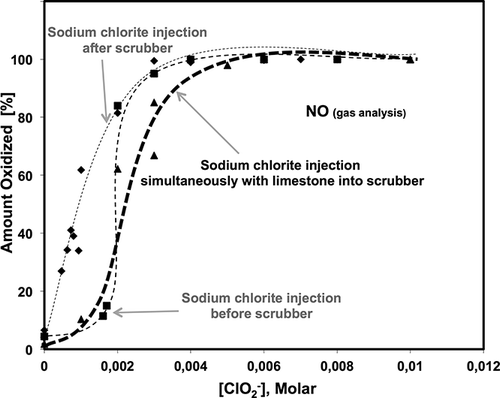
Typically, 90–95% of NOx in flue gas is present as NO. The results for the NO oxidation (to NO2 mainly) in Configurations 1, 2, and 3 are shown in In Configuration 1, the oxidation of NO increased only slightly when the chlorite concentrations were below 0.002 M, then sharply increased to above 80% when the chlorite concentration was at 0.003 M, further increasing the chlorite concentration did not affect the results. In Configuration 2, the amount of NO oxidized to NO2 increased rapidly as the chlorite (ClO2 −) concentration increased. The NO oxidation initially increased as the chlorite concentration increased up to a concentration of 0.003M sodium chlorite. The NO oxidation was essentially complete at 0.003 M of NaClO2, and further increasing the chlorite concentration did not significantly affect the results. In Configuration 3, where when sodium chlorite was injected simultaneously with calcium carbonate slurry, the process of NO oxidation was less effective at low chlorite concentrations (less than ∼0.004 M) compared with the other two configurations. However, at higher sodium chlorite concentrations (greater than 0.006 M), the NO oxidation quickly increased to 100% ().
Figure 4. Amount of NO oxidized for Configurations 1, 2, and 3 at different chlorite concentrations.
Nitrogen oxides (NOx) absorption
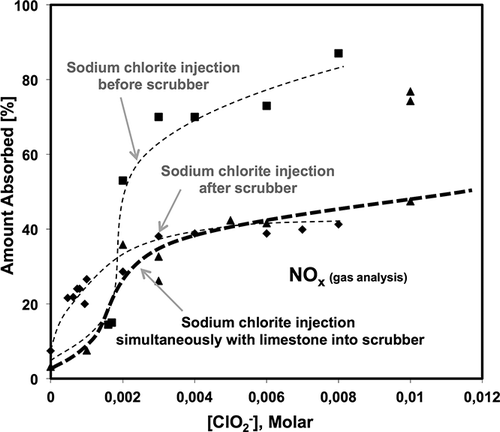
shows the absorption of the NOx species from the flue gas for Configurations 1, 2, and 3. For Configuration 1, when sodium chlorite was injected before the wet scrubber, the NOx absorption increased slowly as the chlorite concentration increased, and achieved only a ∼15% NOx absorption when the NaClO2 concentration was ∼0.0016 M; the NOx absorption then increased sharply when the chlorite concentration reached 0.002 M, and finally reached a NOx absorption of ∼90% at 0.008 M of NaClO2. Nearly all of the NOx was removed from the flue gas when a sufficient amount of sodium chlorite was injected before the wet scrubber. For Configuration 2, when the sodium chlorite was injected after the wet scrubber, the NOx absorption increased as the chlorite concentration increased, from ∼7.0% (no chlorite) to ∼40% (0.003 M of NaClO2); and further increasing the chlorite concentration did not affect the results. The NOx absorption trend in for both Configurations 1 and 2 corresponded well with the NO oxidation trend in For Configuration 3, when sodium chlorite was injected simultaneously with calcium carbonate slurry, the NOx absorption was similar to Configuration 1 at a low chlorite concentration (less than 0.002 M). However, the NOx removal increased slowly when the chlorite concentration was greater than 0.002 M in the multipollutant scrubber, whereas the NOx absorption for Configuration 1 increased sharply for the same chlorite concentration. At higher chlorite concentrations (greater than 0.003 M), the NOx absorption in the multipollutant scrubber followed a similar trend seen in Configuration 2. Certainly, the highest NOx absorption (∼90%) was observed for Configuration 1, in which the oxidizing reactor was injected before the wet scrubber.
Figure 5. Amount of NOx absorbed for Configurations 1, 2, and 3 at different chlorite concentrations.
Mercury removal
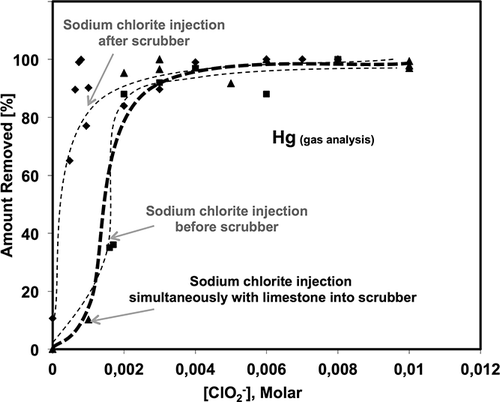
shows the amount of Hg removed from the flue gas for Configurations 1, 2, and 3. As can be seen from , the Hg removal increased rapidly as the chlorite concentration increased when sodium chlorite was injected after the wet scrubber. In this case, for an extremely low chlorite concentration of ∼0.0005 M, the Hg removal was ∼65% and reached over 90% at ∼0.0007 M of NaClO2 and further increasing the chlorite concentration resulted in complete Hg capture. As shown in , different Hg removal trends were observed when sodium chlorite was injected before the wet scrubber. The Hg removal at 0.0016 M of NaClO2 was only ∼35% when it was injected before the scrubber and ∼90% when it was injected after the wet scrubber. However, in Configuration 1, the Hg capture increased sharply when the chlorite concentration was slightly higher than 0.002 M, achieving ∼90% Hg capture, and continued to increase at higher chlorite concentrations.
Figure 6. Amount of Hg removed for Configurations 1, 2, and 3 at different chlorite concentrations.
Increasing the temperature in the oxidizing reactor for Configuration 2
To enhance the NOx absorption in Configuration 2, when the oxidizer was injected after the wet scrubber, the temperature in the oxidizing reactor was increased to 35 and 55 °C. Generally, increasing the temperature can accelerate the reaction rate but can also decrease the gas solubility in the liquid. shows how increasing the temperature affects the pollutant removal efficiencies. As shown in , increasing the temperature did not significantly affect the NO/NOx results (). The NO removal was complete at both temperatures.
Figure 7. Effect of the temperature in the oxidizing reactor.
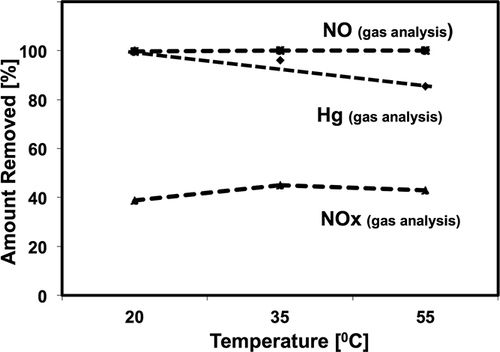
The NOx absorption was approximately 45%, essentially the same for all the tested temperature ranges. However, increasing the temperature to 55 °C resulted in decrease in Hg removal (to ∼85%). When the temperature in the oxidation scrubber was 35 °C, the Hg removal efficiency was ∼96%. Thus, the results showed that increasing the temperature of the oxidizing reactor above 35 °C results in less Hg removal. It should also be noted that SO2 capture was complete at all tested temperatures.
Discussion of the Results
The location at which the oxidant is injected is important, but the final removal efficiency depends on the conditions of the system, which can vary greatly between different flue gas cleaning systems. The presence or lack of certain flue gas components, such as SO2 or NOx, can change the chemical conditions, which are extremely important in the pollutant removal process. For example, the pH of the solution could be different. The pH in the oxidizing reactor applied before the wet scrubber was ∼2.0 in the presented research, whereas in the oxidizing reactor located after the wet scrubber was ∼6.0. The multipollutant scrubber slurry pH varies between 6.0 and 6.7, depending on an initial sodium chlorite concentration used. As was presented earlier (CitationHutson et al., 2008; CitationKrzyzynska and Hutson, 2012), parameters, such as the pH, flue gas pollutant concentrations, and the chlorite concentration, can greatly influence the multipollutant process. Furthermore, the effect of the aforementioned parameters is different when the oxidant is injected before, after, or inside a traditional limestone scrubber.
The authors of this paper tried to further improve the pollutant removal efficiencies by determining the optimum position of the oxidizer application in a flue gas cleaning system profile. In this study, the oxidizer and the sodium chlorite were injected at three different locations: before, after, and in the wet scrubber at otherwise identical experimental conditions.
NO oxidation
The highest amount of NO oxidized for the low chlorite concentrations (less than ∼0.003 M) was achieved when the sodium chlorite was injected after the wet scrubber. A low chlorite concentration positively influenced the overall economy of the process. The lowest chlorite consumption was related to a lack of SO2 gas species, where the oxidizer was not consumed, resulting in higher amount of NO oxidized. The pH of the solution in the oxidizing reactor was in that case ∼6.0. However, most of the NO was oxidized for all the configurations when the amount of available chlorite was relatively high (greater than 0.005 M). The results also showed that the highest chlorite concentration for NO oxidation is necessary if the oxidizer is injected before the wet scrubber. The higher oxidizer consumption is because the chlorite may be consumed by not only the NO and Hg but also by the SO2 species to a certain degree. However, most of the NO was oxidized when the chlorite concentration was ∼0.003 M. A higher chlorite concentration, the low pH (pH ∼2.0 in the oxidizing reactor), which is a favorable environment for NO oxidation, and the presence of chlorine dioxide (ClO2(gas)) helped achieve complete NO oxidation relatively fast. It is worth mentioning that at such a low pH (i.e., 2.0), ClO2(gas) is present, which is an additional strong oxidizer that reacts quickly with the other flue gas components (reactions 1 and 3) (CitationJin et al., 2006; CitationKrzyzynska and Hutson, 2012). The presence of ClO2(gas) enhances the NO oxidation according to reaction 1:
Because ClO2(gas) is consumed quickly by the flue gas components, it is not detected at the outlet of a multipollutant scrubber (CitationKrzyzynska and Hutson, 2012). The presence of chlorine dioxide in the system when the chlorite was injected before the wet scrubber can be explained by reaction 2, which can occur when the pH of the solution is low (CitationKrzyzynska and Hutson, 2012; CitationWhite et al., 1942). A low-pH solution (∼2.0) will cause NaClO2 decomposition according to reaction 2:
Generally, NO reacts with NaClO2 via many pathways, as described in the literature (CitationHutson et al., 2008; CitationLee et al., 2005; CitationSada et al., 1978;), but can also react with ClO2(gas), which enhanced the amount of NO oxidized when sodium chlorite was injected before the wet scrubber.
NOx absorption
The highest NOx absorption (∼90%) was achieved when chlorite was injected before the wet scrubber with a concentration of ∼0.008 M. Such a significant NOx absorption could be caused by a possible reaction between ClO2(gas) and NO2 according to reaction 3. Also, some additional NOx absorption was possible in the wet scrubber. The ion analysis showed that an insignificant amount of NOx (1–2% of the total N) was absorbed in the wet scrubber.
The NOx absorptions in the other configurations were ∼50% for the high chlorite concentrations. Lower NOx absorption could be caused by less favorable chemical conditions (no ClO2(gas) and higher pH) existing in the multipollutant scrubber and in the oxidizing reactor placed after the wet scrubber. The ion analysis showed that all oxidation of NO and absorption of NOx were taking place in the oxidizing reactor when sodium chlorite was injected after the wet scrubber. It was quite predictable, because typical wet limestone scrubbers are not designed for NOx removal.
Hg removal
The results showed that Hg was effectively removed in all the tested cases, though applying the oxidizer after the wet scrubber appeared to be the best at low chlorite concentrations (∼0.003 M). Mercury removal rapidly increased as the chlorite concentration was lowered when the sodium chlorite was injected after the wet scrubber. This rapid Hg removal increase is related to the lack of SO2, which would consume some of the oxidizer. Thus, more oxidant for the NO and Hg0 oxidations was available. Moreover, the pH in the oxidizing reactor remained sufficient for the NO and Hg oxidation/removal (∼6.0) (CitationKrzyzynska and Hutson, 2012).
A low pH is favorable for NO and Hg0. However, an extremely low pH (i.e., 2.0) appeared more favorable for NO than for Hg0 (CitationKrzyzynska and Hutson, 2012), which may be due to the lower amount of available sodium chlorite. Sodium chlorite would decompose into ClO2(gas), which reacts more quickly with NO than for Hg. Thus, reaction 4 will occur slower than reactions 1, 2, and 3.
At higher chlorite concentrations in the reactor, the mercury removal increased sharply. Moreover, the ion analysis indicated that all the mercury was oxidized and removed in the oxidizing reactor when the sodium chlorite was injected before the wet limestone scrubber. In all the tested samples, no mercury was detected in the wet scrubber. All mercury oxidation/removal processes occurred in the oxidizing reactor in both configurations.
SO2 removal
SO2 capture was complete for all the configurations and at all the chlorite concentrations. The graphs are not presented because there was no difference between any of the configurations. SO2 reacts with the calcium carbonate slurry (CaCO3), which occurs in the traditional wet scrubber systems, and may react with the sodium chlorite and chlorine dioxide in Configuration 1 according to reactions 5 and 6 to a certain degree. The ion analysis indicated that most of the SO2 was removed in the oxidizing reactor positioned before the wet scrubber when the sodium chlorite concentration was relatively high (greater than 0.004 M). There was evidence that SO2 reacted with the sodium chlorite and/or chlorine dioxide (gas) at a low pH, which were present in the reactor, according to reactions 5 and 6.
When the sodium chlorite was injected after the wet scrubber, the SO2 removal followed the traditional mechanism common in real scrubbers, because the ion analysis indicated that all of the SO2 was removed in the wet scrubber for that case. The pH in the wet scrubber was between 6.0 and 7.0, depending on applied configuration.
As described in previous section, the lowest NO/NOx and Hg pollutant removal efficiencies among all the tested configurations were when the sodium chlorite was injected simultaneously with the calcium carbonate slurry at a low chlorite concentration (∼0.004 M). Higher chlorite concentrations improved the results significantly. For the same case, the lowest pollutants removal efficiencies at the low chlorite concentrations can be explained by the additional reactions that occurred in the scrubber, which consumed the oxidizer. In the multipollutant scrubber, sodium chlorite can be consumed by NO and Hg0 but can also be consumed by a reaction with SO2. Furthermore, the sodium chlorite may additionally react, to a certain degree, with calcium sulfite (CaSO3), which results in the creation of calcium sulfate (CaSO4), as presented in reaction 7.
Moreover, there was no ClO2(gas) present (because of the higher pH of the slurry liquor), which was discovered to enhance the pollutant removal efficiency in case when the sodium chlorite was injected before the wet scrubber. However, when there was a higher sodium chlorite concentration (greater than 0.005 M) in the multipollutant scrubber, the NO and mercury removal efficiency quickly increased to 100%. The highest NOx absorption (∼90%) was clearly observed for Configuration 1, in which the sodium chlorite was injected before the wet scrubber.
Comparison of the three configurations
This study demonstrated that the location at which the chlorite is injected with respect to the wet scrubber affects how much of the pollutants are removed. Average pollutant removal efficiencies for all configurations and three different chlorite concentrations are presented in The best configuration (in respect to pollutants removal) for each of three chlorite concentrations is bolded, what might help in ranking of the three options presented. However, it is difficult to report the best solution because every tested configuration exhibited pros and cons. The least expensive solution, in a practical application (when consider only capital cost) would be when the chlorite is simultaneously injected with the limestone into the multipollutant scrubber because no other reactors or space is required. However, the operating cost might be higher than in other configurations because of higher oxidant usage. Moreover, it is difficult to achieve a NOx absorption greater than ∼50–60%. A multipollutant scrubber might be the least expensive choice initially, but the removed products would be contaminated with mercury. Mercury could potentially be recovered from the spent slurry, but the configuration would be more complicated and more expensive.
Table 2. Average pollutant removal efficiencies for all configurations and three different chlorite concentration (%)
The highest NOx absorption (∼90%) was achieved when the chlorite was injected before the wet scrubber. It was possible to achieve complete NO oxidation and nearly complete Hg removal at a relatively low chlorite concentration (∼0.003 M). Also, it would be possible to achieve the cleanest flue gas from that configuration, and the gypsum from the wet scrubber would contain no mercury. Additionally, the experiments showed that Configuration 1 resulted in the best NOx absorption, but the cost of its potential installation at a large scale would likely incur the highest cost. The high cost is due to the cost of an additional oxidizing reactor, which must be made from anti-corrosive steel because of the extremely low pH that would be present. In previous experiments (CitationKrzyzynska and Hutson, 2012), ClO2(gas) was not detected in the outlet flue gas, but ClO2(gas) existed in the oxidizing reactor and reacted with the flue gas components; therefore, ClO2(gas) detectors should be installed to fulfill safety requirements. An additional oxidizing reactor would also require additional space.
Configuration 2, in which the sodium chlorite was injected after the limestone scrubber, removed the most mercury. Mercury was removed much more quickly than in the other configurations. The mercury could be completely removed (with ∼40% NO oxidation and ∼25% NOx absorption) at low chlorite concentrations (∼0.0007 M). For the same chlorite concentration, the multipollutant scrubber was only able to remove less than 10% of the Hg (with ∼8% NO oxidation and approximately 5% NOx absorption). Also, similar pollutant removal efficiencies at low chlorite concentrations were observed for Configuration 1. These results indicate that applying the oxidizer after the wet scrubber could control mercury perfectly or could be used as an additional step in removing the mercury and NOx from the gases after other NOx control facilities. Lower concentrations of chlorite were necessary to achieve similar Hg and NO removals compared with the other configurations. A lower oxidant consumption would allow a smaller oxidizing reactor design, which could be constructed from lower quality material, compared with that of Configuration 1 because of the difference in pH, which would subsequently result in lowering the cost. However, despite these theoretical considerations regarding construction on a large scale, more detailed, pilot-scale tests on real flue gas is necessary for practical application considerations.
Conclusions
Sodium chlorite is an effective additive to enhance NOx, Hg, and SO2 capture. However, it has been discovered that the position at which the sodium chlorite is injected (before, in, or after the wet scrubber) greatly influences pollutant control. The effect of sodium chlorite is related to the different chemical conditions (i.e., pH, absence/presence of some particular gases) that will occur in different locations of the flue gas cleaning system profile. The location at which the sodium chlorite is injected affects the amount of the pollutants removed. The results showed that for low chlorite concentrations (less than ∼0.003 M), the highest amount of NO oxidized was achieved when sodium chlorite was injected after the wet scrubber, which subsequently influences, in a positive way, the overall economy of the process. The highest NOx absorption (∼90% and complete NO and Hg removal also occurred) was achieved when the sodium chlorite was injected before the wet scrubber and the chlorite concentration was ∼0.008 M. This configuration resulted in the lowest Hg, NOx, and SO2 emissions. However, the configuration would be the most expensive solution among the others because an additional anti-corrosive oxidizing reactor with a higher chlorite concentration and ClO2(gas) detectors would be required.
Mercury was removed with a high efficiency in all the tested cases; however, applying the sodium chlorite after the wet scrubber seemed to be the best at low chlorite concentrations (∼0.003 M). Mercury was removed completely much more quickly than in the other configurations. This result suggests that injection of sodium chlorite after the wet scrubber could perfectly control the mercury or could be used as an additional cleaning step to remove the mercury and NOx from the gases after other NOx control facilities. Theoretically, it is thought that the multipollutant scrubber would be the least expensive solution in a practical application, when consider only capital cost, because no other reactors or space would be required. However, the operating cost might be higher than in other configurations because of higher sodium chlorite usage. Moreover, results showed that it was difficult to achieve a NOx removal efficiency greater than ∼50–60% in that case.
Acknowledgments
This work was supported by funding from the U.S. Environmental Protection Agency (Office of Air and Radiation [OAR] and Office of Research and Development [ORD]) and by Polish Ministry of Science and Higher Education. Mention of trade names of commercial products and companies does not constitute endorsement or recommendation for use.
Related Research Data
References
- American Coalition for Clean Coal Electricity (ACCCE). 2011. Coal keeps electricity prices affordable http://www.cleancoalusa.org/ (http://www.cleancoalusa.org/) (Accessed: 6 December 2011 ).
- American Society for Testing and Materials International . 2002 . ASTM Standard D6784 – 02, Standard Test Method for Elemental, Oxidized, Particle-Bound and Total Mercury in Flue Gas Generated from Coal-Fired Stationary Sources (Ontario Hydro Method) , West Conshohocken , PA : ASTM International . doi: 10.1520/D6784-02
- Chirona , R.J. and Altshuler , B. 1998 . Chemical aspects of NOx scrubbing . Pollut. Eng. , 31 : 32 – 37 .
- Chu , H. , Chien , T.W. and Li , S.Y. 2001 . Simultaneous absorption of SO2 and NO from flue gas with KMnO4/NaOH solutions . Sci. Total Environ. , 257 : 127 – 135 .
- Chmielewski , A.G. , Ostapczuk , A. and Licki , J. 2010 . Electron beam technology for multipollutant emissions control from heavy fuel oil-fired boiler . J. Air Waste Manage. Assoc , 62 ( 8 ) : 932 – 938 .
- Dabrowski , J. and Piecuch , T. 2011 . Mathematical description of combustion process of selected groups of waste . Annu. Set Environ. Prot. (Rocznik Ochrony Srodowiska) , 13 : 253 – 268 .
- Ellison , W. Chemical process techniques for simultaneous NOx removal in existing FGD installations . Proceedings of DOE/NETL 2003 Conference on Selective Catalytic Reduction and Non-Catalytic Reduction for NOx Control . October 29–30 2003 , Pittsburgh , PA .
- Hutson , N.D. , Krzyzynska , R. and Srivastava , R.K. 2008 . Simultaneous removal of SO2, NOx and Hg from coal flue gas using a NaClO2-enhanced wet scrubber . Ind. Eng. Chem. Res. , 47 : 5825 – 5831 .
- Jin , D.S. , Deshwal , B.R. , Park , Y.S. and Lee , H.K. 2006 . Simultaneous removal of SO2 and NO by wet scrubbing using aqueous chlorine dioxide solution . J. Hazard. Mater. , 135 : 412 – 417 .
- Jozewicz , W. , Srivastava , R.K. , Ellison , W. and Ferrell , R.J. Investigation of multipollutant scrubbing by wet flue gas desulfurization systems . Proceedings of 29th International Technical Conference on Coal Utilization & Fuel Systems . April 18–22 2004 , Clearwater , FL . Washington , DC : U.S. Department of Energy .
- Krzyzynska , R. and Hutson , N.D. 2012 . Effect of solution pH on SO2, NOx, and Hg removals from simulated coal combustion flue gas in an oxidant-enhanced wet scrubber . J. Air Waste Manage. Assoc. , 62 : 212 – 220 . doi: 10.1080/10473289.2011.642951
- Lee , H.K. , Deshwal , B.R. and Yoo , K.S. 2005 . Simultaneous removal of SO2 and NO by sodium chlorite solution in wetted-wall column . Korean J. Chem. Eng. , 22 : 208 – 213 .
- Liu , Y. , Bisson , T.M. , Yang , H. and Xu , Z. 2010 . Recent developments in novel sorbents for flue gas clean up . Fuel Proc. Techn , 91 ( 10 ) : 1175 – 1197 .
- Pawlowski , L. 2011 . How heavy metals affect sustainable development . Annu. Set. Environ. Prot. (Rocznik Ochrony Srodowiska). , 13 : 51 – 64 .
- Sada , E. , Kumazawa , H. , Yamanaka , Y. , Kudo , I. and Kondo , T. 1978 . Absorption of sulfur dioxide and nitric oxide in aqueous mixed solutions of sodium chlorite and sodium hydroxide . J. Chem. Eng. Jpn. , 11 : 276 – 282 .
- Sloss , L.L. 2008 . “ Economics of Mercury Control ” . In Report of Clean Coal Center and International Energy Agency, CCC/134 , London : IEA Clean Coal Centre .
- Smith , D.J. 1992 . NOx emission control demands a range of solutions . Power Eng. , 96 : 44 – 48 .
- Srivastava , R.K. and Jozewicz , W.S. 2001 . Flue gas desulfurization: The state of the art . J. Air Waste Manage. Assoc. , 51 : 1676 – 1688 .
- Srivastava , S.K. , Hutson , N.D. , Martin , G.B. , Princiotta , F. and Staudt , J. 2006 . Control of mercury emissions from coal-fired electric utility boilers . Environ. Sci. Technol. , 40 : 1385 – 1393 .
- Suchecki , T.T. , Mathews , B. and Kumazawa , H. 2005 . Kinetic study of ambient-temperature reduction of FeIIIedta by Na2S2O4 . Ind. Eng. Chem. Res , 44 ( 12 ) : 4249 – 4253 .
- The European Parliament and the Council of the European Union . 2001 . Directive 2001/80/EC of the European Parliament and of the Council of 23 October 2001 on the limitation of emission of certain pollutants into air from large combustion plants . Official Journal of the European Communities L 309, 27.11.2001 , 44 : 1 – 21 .
- The European Parliament and the Council of the European Union . 2010 . Directive 2010/75/EU of the European Parliament and of the Council of 24 November 2010 on industrial emissions (integrated pollution prevention and control) . Official Journal of the European Communities L 334, 17.12.2010 , 53 : 17 – 119 .
- U.S. Environmental Protection Agency 1993. EPA Method 300.0. Determination of Inorganic Anions by Ion Chromatography. Methods for the Determination of Inorganic Substances in Environmental Samples. EPA/600/R-93/100.
- U.S. Environmental Protection Agency. 1994. EPA Method 7470A. Mercury in Liquid Waste (Manual Cold-Vapor Technique). SW-846 Online: Test Methods for Evaluating Solid Waste, Physical/Chemical Methods.
- U.S. Energy Information Administration. 2010. Independent Statistics And Analysis. International Energy Outlook 2010—Highlights. Report DOE/EIA-03 83(2010). Washington, DC: U.S. Department of Energy http://www.eia.gov/ (http://www.eia.gov/) (Accessed: 6 December 2011 ).
- U.S. Environmental Protection Agency. 2011a. Cross-State Air Pollution Rule (CSAPR) http://epa.gov/airtransport/index.html (http://epa.gov/airtransport/index.html) (Accessed: 1 September 2012 ).
- U.S. Environmental Protection Agency. 2011b. Mercury and Air Toxics Standards (MATS) for power plants http://www.epa.gov/mats/ (http://www.epa.gov/mats/) (Accessed: 1 December 2012 ).
- White , J.F. , Taylor , M.C. and Vincent , G.P. 1942 . Chemistry of chlorites . Ind. Eng. Chem. , 34 : 782 – 792 .