
Abstract
Direct decomposition of N2O by perovskite-structure catalysts including La2NiO4, LaSrNiO4, and La0.7Ce0.3SrNiO4 was investigated. The catalysts were prepared by the Pechini method and characterized by x-ray diffraction (XRD), BET, scanning electron microscopy (SEM), and O2-TPD. Experimental results indicate that the properties of La2NiO4 are significantly improved by partially substituting La with Sr and Ce. N2O decomposition efficiencies achieved with LaSrNiO4 and La0.7Ce0.3SrNiO4 are 44 and 36%, respectively, at 400ºC. As the temperature was increased to 600ºC, N2O decomposition efficiency achieved with LaSrNiO4 and La0.7Ce0.3SrNiO4 reached 100% at an inlet N2O concentration of 1,000 ppm, while the space velocity was fixed at 8,000 hr−1. In addition, effects of various parameters including oxygen, water vapor, and space velocity were also explored. The results indicate that N2O decomposition efficiencies achieved with LaSrNiO4 and La0.7Ce0.3SrNiO4 are not significantly affected as space velocity is increased from 8,000 to 20,000 hr−1, while La0.7Ce0.3SrNiO4 shows better tolerance for O2 and H2O(g). On the other hand, N2 yield with LaSrNiO4 as catalyst can be significantly improved by doping Ce. At a gas hour space velocity of 8,000 hr−1, and a temperature of 600ºC, high N2O decomposition efficiency and N2 yield were maintained throughout the durability test of 60 hr, indicating the long-term stability of La0.7Ce0.3SrNiO4 for N2O decomposition.
Implications:
Nitrous oxide (N2O) not only has a high global warming potential (GWP100 = 310), but also potentially destroys ozone in the stratosphere. Pervoskite-type catalysts including La2NiO4, LaSrNiO4, and La0.7Ce0.3SrNiO4 are applied for direct N2O decomposition. The results show that N2O decomposition can be enhanced as Sr and Ce are doped into La2NiO4. At 600ºC, N2O decomposition efficiencies achieved with LaSrNiO4 and La0.7Ce0.3SrNiO4 reach 100%, demonstrating high activity and good potential for direct N2O decomposition. Effects of O2 and H2O(g) contents on catalytic activities are also evaluated and discussed.
Introduction
Nitrous oxide (N2O) has been recognized as one of the major air pollutants with increasing atmospheric concentration year by year. It not only destroys the ozone layer in the stratosphere, but also is a strong greenhouse gas (Rodhe, Citation1990; Wójtowicz et al., Citation1993; Kramlich and Linak, Citation1994; Russo et al., Citation2007). Previous studies indicate that N2O can be converted into nitrogen oxide (NO) when it migrates into the stratosphere, furthermore, triggering ozone-destroying reactions (Jacob, Citation1999). In addition, N2O has a long lifetime in atmosphere (150 years) and a high global warming potential (GWP100 = 310), which aggravates the greenhouse effect (Russo et al., Citation2007). N2O emission may originate from natural and anthropogenic sources. Annual global N2O emission is approximately 4.7–7 million tons, with 30–40% coming from natural sources (Wójtowicz et al., Citation1993; Jacob, Citation1999). Among anthropogenic sources, adipic acid production, nitric acid manufacturing, and fossil fuel combustion are considered as significant. Therefore, developing effective control technology to reduce N2O emissions has become an emerging issue.
Generally speaking, methods for N2O removal include thermal decomposition, selective catalytic reduction with hydrocarbons (HC-SCR), adsorption, and direct catalytic decomposition into O2 and N2 (Kumar et al., Citation2012). Among these methods, direct catalytic decomposition of N2O into N2 and O2 is considered a relatively simple and economical option to reduce N2O emission. Previous studies indicate that catalysts available for direct N2O catalytic decomposition include noble metals (Hinshelwood and Prichard, Citation1925; Lintz and Riekert, Citation1984; Siva Sankar Reddy et al., Citation2007), metal oxide catalysts (Winter, Citation1974; Perez-Alonso et al., Citation2006; Granger et al., Citation2006), and zeolite-based catalysts (Pannov et al., Citation1990; Li and Armor, Citation1992; Sobolev et al., Citation1993). However, most zeolite-based catalysts are unpromising for practical use due to low hydrothermal stability and poisoning by sulfur-containing gases or water vapor (Li and Armor, Citation1992; Van den Brink et al., Citation2001). Noble metal catalysts are expensive although they show excellent performance; for example, the price of platinum (Pt) is about U$ 1,470 per ounce in 2014, while 1 ounce of rhodium (Rh) costs U$ 1,110 (NASDAQ, Citation2014). On the other hand, perovskite-type catalysts are interesting alternatives as they are low-cost materials and stable even at a temperature above 1,000ºC (Voorhoeve et al., Citation1972).
Perovskite-type catalysts are with a general formula of ABO3 or A2BO4, where the A site is either an alkaline metal or rare earth element and a relatively large ion—typically La is applied—while the B site is generally a 3d transition metal like Cu, Fe, Co, Ni, or Mn. The B site is mainly responsible for catalytic activity, because it is located next to the oxide anion and triggers desorption, while the A site can change the electronic state of the B site and influence catalysis indirectly. Perovskite-type catalysts with different physical properties can be synthesized by substituting different elements at the A and B sites and by varying the chemical compositions. The mechanisms of N2O decomposition with perovskite-type catalysts have been proposed in previous study as the reactions 1–3 (Kapteijn et al., Citation1996; Russo et al., Citation2007):
ABO3-type catalysts including LaMO3 (M = Cr, Co, Ni, Cr, Mn, and Fe) (Wang et al., Citation1995; Dacquin et al., Citation2009), and A2BO4-type catalysts including La2MO4 (M = Co, Ni, Cu) (Wang et al., Citation1995), M2NiO4 (M = La, Pr, Nd) (Ramanujachary et al., Citation1982), and M2CuO4 (M = La, Pr, Nd, Sm, Gd) (Ramanujachary et al., Citation1984) have been studied for N2O decomposition. Furthermore, modified perovskite-type catalysts were prepared by partial substitution at the A site and/or B site, including LaB1−x B′ xO3 (B = Mn, Fe and B′ = Cu, Ni) (Alini et al., Citation2007) and La1−xSrxM1−yMyO3 (M = Co, Ni, Cu) (Tofan et al., Citation2002a) for the improvement of catalyst activity. Overall, the highest N2O decomposition efficiencies achieved with perovskite-type catalysts could reach 100%, but they have to be operated at high temperatures (≥700°C) for high N2O decomposition activities. Additionally, oxygen and water vapor usually exist in industrial flue gases, so tolerances of oxygen and water vapor should be considered for good performance of catalyst.
Catalytic performance of perovskite-type catalysts can be enhanced by substituting different elements at the A and/or B sites. For example, strontium (Sr) can be applied to partially substitute La at the A site to improve the activity of the catalyst. Previous study indicates that nonstoichiometric perovskites (La1−xSrxM1−yMyO3−δ) show several advantages, such as fast electron transfer, high oxygen mobility, and easy oxygen desorption (Tabata and Misono, Citation1990; Kirchnerova and Klvana, Citation1994). As a low-valence cation (Sr2+) occupies the frame at the La3+ site, the oxide anion vacancies can be produced by charge compensation (Zhao et al., Citation1996). Recently, cerium (Ce) received much attention in preparing a catalyst because it has excellent performance for oxygen storing and scavenging. Catalytic activity can be enhanced when Ce is partially substituted for the A site due to its redox property. Kirchnerova et al. (Citation2002) found that the activity of LaCoO3 was significantly improved when LaCoO3 was doped with CeO2. Belessi et al. (Citation2001) indicate that the NO decomposition rate could be accelerated by increasing CeO2 content in La-Sr-Ce-Fe-O catalyst. Nitadori and Misono (Citation1985) applied Sr, Ce, and Th to substitute LaMO3 (M = Mn, Fe, Co) and indicated that Ce-substituted samples improved the property of oxygen mobility. This redox effect appears prominently on the A2BO4-type perovskite catalysts, because the lattice of the A2BO4-type catalyst has larger cavity space compared with the ABO3 type. A site of A2BO4 type can accommodate more relatively large ions of Ce, and previous study indicates that La1−xCexSrNiO4 lattice can store up to 10 times more cerium ions than can La1−xCexNiO3 (Imanaka and Masui, 1996). Another study also indicates that oxides with A2BO4 structure are more stable than that of ABO3 structure (Zhao et al., Citation1996).
In this study, the A2BO4-type perovskite catalyst (La2NiO4) is prepared and evaluated for effectiveness in direct N2O decomposition. Previous study has shown that nickel-based perovskites are excellent catalysts for a variety of oxygen-involving reactions (Kirchnerova and Klvana, Citation1994; Klvana et al., Citation1994). Furthermore, as the A site of La2NiO4 is partially substituted by doping Sr and Ce to prepare LaSrNiO4 and La0.7Ce0.3SrNiO4 catalysts, it is expected that properties and activities of the modified catalysts can be improved. In addition, the influences of space velocity, oxygen, and water vapor on N2O decomposition are also explored with a lab-scale experimental system.
Experimental
Catalyst preparation
The provskite-type catalysts tested are of A2BO4 structure, including La2NiO4, LaSrNiO4, and La0.7Ce0.3SrNiO4, which were prepared by the Pechini method (Pechini, Citation1967). The preparation method includes several steps; first, appropriate metal nitrates were dissolved in deionized water (DI water) as precursors for preparing the aqueous solution with appropriate stoichiometry, and this aqueous solution was continuously stirred at room temperature. Then citric acid and ethylene glycol were added into the mixed aqueous solution of metal nitrates. The mixed aqueous solution was stirred for 1 hr at 90ºC as citric acid was added into the mixed aqueous solution; subsequently, ethylene glycol was added and then stirred at 85ºC to evaporate the water. Citric acid was added into the mixed aqueous solution of metal nitrates with designated proportion so that the molar ratio of citric acid to the total metal cations was 4 to 1. Ethylene glycol was then added into the mixed aqueous solution to control the molar ratio of citric acid to the ethylene glycol as 1 to 1. Second, the mixture just described was dried at 110ºC for 24 hr, followed by heat treatment at 600ºC for 1 hr in air, and calcination in air at 950ºC for 6 hr. Finally, the samples were manually milled to powders by using a porcelain mortar and pestle; subsequently, the synthesized particles were then pulverized to the final 200 mesh size and used as such.
Characterization of catalysts
The specific surface areas of the catalysts were measured by N2 adsorption at –196ºC with a Micromeritics ASAP 2010 (ASAP2010, Micromeritics, USA), while measurements of the surface morphology and composition of catalyst were conducted using scanning electron microscopy (SEM) with associated energy-dispersive x-ray spectroscopy (EDS) (S80 JEOL, Japan). In addition, an x-ray diffractometer (D8AXRD, BRUKER, Germany) was utilized to identify the structure of catalyst, operating at 40 kV and 10 mA, by using Cu-Kα radiation. Diffraction patterns were obtained within a 2θ range of 5–80º at a scanning rate of 6º min−1. Additionally, O2-TPD was carried out with apparatus (AutoChem2920, Micromeritics, USA) equipped with a thermal conductivity detector (TCD). The samples (0.2 g) were first treated in O2 at 1,000ºC for 1 hr and cooled to room temperature in the same atmosphere, then swept with helium at a rate of 11.8 mL/min. Finally, the sample was heated at a rate of 20ºC/min in helium and the TPD profile was recorded.
Catalytic activity measurement
Experimental tests were carried out with a lab-scale experimental system, and catalytic activity of direct N2O decomposition was evaluated with a fixed-bed quartz reactor (ID 20 mm) containing 6 g catalyst. The feeding gas stream contained 1,000 ppm N2O with helium (He) as carrier gas. The total gas flow rate was fixed at ˜628 ccm (cm3/min), corresponding to a gas hourly space velocity (GHSV) of 8,000 hr−1. In addition, experimental tests were also conducted with the space velocity of 15,000 hr−1 and 20,000 hr−1, respectively, to evaluate the space velocity effect. In order to investigate the effects of O2 and H2O(g) contents on the activities of catalysts for direct N2O decomposition, 1–5% O2 and 5–10% H2O(g) were introduced into the feeding gas, respectively. Sources of N2O and O2 are from cylinders, and designated N2O and O2 concentrations can be obtained by adjusting the flow rate of individual gas stream. In addition, H2O is introduced into gas stream by a peristaltic pump. Experimental parameters are selected to simulate real conditions; that is, N2O concentration is in the range of 0–1,000 ppm, while O2 and H2O(g) concentrations are about 2–10% and 10% (Kapteijn et al., Citation1996), respectively. However, the space velocity applied in this study is smaller if compared with the field due to the limitation of the lab-scale experimental system.
Mass flow controllers (MFC) were utilized to adjust and control the gas flow rates, and all gases were mixed before reaching the reactor. Finally, operating temperature was varied from 300 to 700ºC to evaluate the temperature effect. The N2O concentration and products were analyzed before and after the reaction by using a Fourier-transform infrared spectrophotometer (Nicolet 6700, Thermo Scientific, USA) and NOx detector (Testo350, Testo, Germany), respectively, while analysis of N2 was conducted using gas chromatography (Technologies6890N, Agilent, USA) with a TCD and columns packed with 5A molecular sieve (capillary: 30.0 m × 530 μm × 25 μm nominal). All experimental tests were conducted three times to check the stability and repeatability, and the experimental data were taken when the catalytic reactions practically reached steady state. Decomposition efficiency of N2O and yields of N2 were calculated by eqs 4 and 5, respectively:
Results and Discussion
Characterization of the catalysts
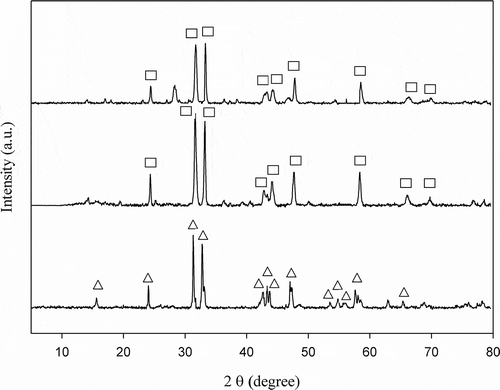
shows XRD patterns of three catalysts tested, indicating that all the catalysts are of perovskite-like structure (A2BO4-type). XRD patterns of La0.7Ce0.3SrNiO4, LaSrNiO4, and La2NiO4 are presented in , , nd 1c, respectively, while triangles and squares represent La2NiO4 and LaSrNiO4 crystalline phases. For c, there exist strong main diffraction peaks, which are assigned to those from La2NiO4. As Sr is partially substituted for the A site of La2NiO4, its main diffraction peaks are assigned to LaSrNiO4. For Ce-substituted catalyst, the diffraction peaks are mainly assigned to LaSrNiO4, but there exists one abrupt peak at 2θ= ∼28ο, generally, which is attributed to CeO2. However, 2θ = ∼28ο might imply the diffraction peak of La1−xCexSrNiO4. Previous study suggests that the peak of 2θ = ∼28οmay not be attributed to CeO2 when Ce is added into LaSrNiO4; that is, Ce may occupy the La-site without destroying the matrix structure (Zhu et al., Citation2005). In order to clarify whether this peak (2θ = ∼28ο) belong to CeO2, more information is needed for understanding the diffraction peaks of CeO2. According to Hernández-Enríquez et al. (Citation2012), the diffraction peaks of CeO2 are mainly distributed at 2θ = 28.5°, 33.0°, 47.6°, 56.4°, 59.1°, and 69.5°, respectively, which only belongs to the CeO2 phase. However, the Ce-substituted catalyst does not generate peaks of CeO2 at 2θ = 33°, 47.6°, 56.4°, 59.1°, and 69.5° in this study; hence, it is clear that 2θ = ∼28ο is ascribed to La1−xCexSrNiO4 but not to CeO2. These results demonstrate that Ce substitutes for lattice position, and this agrees with the literature (Zhu et al., Citation2005).
Figure 1. X-ray diffraction patterns of catalysts (a) La0.7Ce0.3SrNiO4, (b) LaSrNiO4, and (c) La2NiO4 (Δ:La2NiO4, □: LaSrNiO4) .
summarizes BET surface areas of the catalysts tested. The results indicate that the BET surface areas of La2NiO4, LaSrNiO4, and La0.7Ce0.3SrNiO4 are 3.7, 5.0, and 5.2 m2/g, respectively, indicating that all three catalysts have small surface area. Generally speaking, BET surface areas of perovskite-type catalysts are between 5 and 50 m2/g. displays the SEM patterns of La2NiO4, LaSrNiO4, and La0.7Ce0.3SrNiO4, respectively, and all three catalysts present as pellet with slight agglomeration, corresponding to the low BET surface area possibly due to high-temperature calcination. EDX results are shown in , indicating that the elemental composition of the catalysts deviates a little from the designated formula of catalysts.
Table 1. BET surface areas of the catalysts prepared
Figure 2. SEM patterns of catalysts (a) La2NiO4, (b) LaSrNiO4, and (c) La0.7Ce0.3SrNiO4.

Table 2. Elemental composition of the catalysts prepared
O2 Temperature-programmed desorption (O2-TPD)
O2 temperature-programmed desorption (TPD) is conducted for La2NiO4, LaSrNiO4, and La0.7Ce0.3SrNiO4 individually for better understanding the behavior of oxygen desorption, since it has important implications regarding the catalytic N2O decomposition. Typical perovskite-type catalysts have two kinds of oxygen desorption peaks for the O2-TPD test. The peak appearing at a relatively low temperature is denoted by α oxygen, which is relatively weak, while the peak appearing at higher temperatures is denoted as β oxygen. Generally, the temperatures from 40 to 500ºC are defined as the low temperature range, while those higher than 500ºC are defined as the high temperature range. However, peaks of α oxygen and β oxygen are variant with different perovskite-type catalysts, implying that their peaks may appear at lower or higher temperatures due to different properties of catalysts. The α oxygen is chemically desorbed from the surface, while β oxygen is desorbed from the oxygen of vacancy and lattice. Hence, the performance of the perovskite-type catalysts mainly depends on the intensity of β oxygen peak for N2O decomposition, namely, the β oxygen is released from the B site of perovskite-type catalysts.
The O2 desorption profiles of three selected catalysts are shown in . For La2NiO4, there exists one abrupt desorption peak of oxygen, which is β oxygen corresponding to T > 790ºC, while α and β oxygen of LaSrNiO4 are located in the regions of 220–450ºC and 520–925ºC, respectively. For Ce-improved catalyst, its desorption peak of β oxygen is between 510 and 805ºC. As presented in , it is clear that α oxygen does not appear as significant desorption peaks for La2NiO4 and La0.7Ce0.3SrNiO4; this result is different from previous studies (Zhao et al., Citation1996; Zhu et al., Citation2005). It is speculated that different preparing methods and conditions may change properties of catalysts. However, α oxygen is not mainly responsible for catalytic activity.
Figure 3. O2-TPD profiles for La2NiO4, LaSrNiO4, and La0.7Ce0.3SrNiO4, respectively.
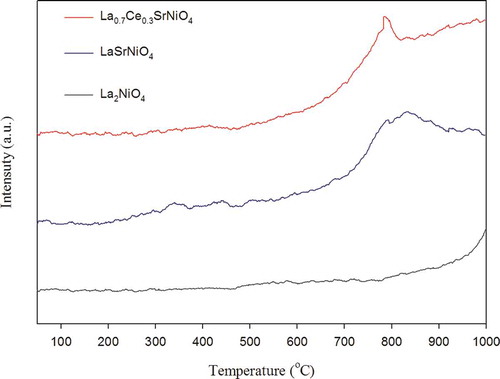
O2-TPD results indicate that adsorption of oxygen for La2NiO4 is strong and not easy to desorb at temperatures lower than 790ºC. If compared with the modified catalysts, thisd clearly shows that the β oxygen peak of La2NiO4 is shifted to lower temperature with partial substitution at the A site. Obviously, physicochemical properties of La2NiO4 are improved due to Sr substitution. In addition, we can see that the desorption area of β oxygen decreases when the A site of LaSrNiO4 is partially substituted with Ce, implying that the number of oxygen vacancies decreases due to Ce doping. Previous study suggests that existence of Ce in the frame of LaSrNiO4 may be of the Ce4+ form. On the contrary, the β oxygen desorption area does not decrease due to the same oxidative state of La3+ and Ce3+ (Zhu et al., Citation2005).
However, the highest β oxygen peak of LaSrNiO4 is shifted from 830 to 780ºC when Ce partially substitutes for the A site of LaSrNiO4; that is, oxygen desorption from the Ce-substituted catalyst is easier than that from LaSrNiO4. Therefore, the increasing order of the mobility of lattice oxygen in three perovskite-type catalysts tested is listed as La0.7Ce0.3SrNiO4 > LaSrNiO4 > La2NiO4. Possible enhancement of oxygen mobilization and desorption is discussed later for modified catalysts.
Activities of catalysts for N2O decomposition
The catalytic N2O decomposition activities of three perovskite-type catalysts prepared were first evaluated as a function of reaction temperature with He as the carrier gas. The reaction temperature was raised gradually from 300 to 700ºC. N2O decomposition efficiencies achieved with La2NiO4, LaSrNiO4, and La0.7Ce0.3SrNiO4, respectively, are presented in . The results indicate that the activities of three catalysts for N2O decomposition increase monotonously with increasing temperature. At initial temperature of 300ºC, N2O decomposition efficiencies achieved with La2NiO4, LaSrNiO4, and La0.7Ce0.3SrNiO4 are only 6.8%, 3.4%, and 6.6%, respectively. As temperature is increased to 400ºC, activity of La2NiO4 is significantly higher than that of LaSrNiO4 and La0.7Ce0.3SrNiO4, and their N2O decomposition efficiencies are 88.7%, 44.3%, and 36.5%, respectively. At 600ºC, the N2O decomposition efficiencies achieved with all three catalysts reached 100%, implying that the gap of N2O decomposition efficiencies becomes smaller with increasing temperature. In addition, the efficiency curves are frequently characterized by two parameters, that is, T50 and T90. T50 is defined as the temperature required to remove 50% of the pollutant, while T90 is the temperature needed to achieve 90% removal. As shown in , T50 for N2O decomposition with La2NiO4 is 350ºC, while T50 of LaSrNiO4 and La0.7Ce0.3SrNiO4 is 410ºC and 430ºC, respectively. The results indicate that La2NiO4 has the lowest T50. Likewise, T90 with La2NiO4 is also the lowest if compared with two other catalysts. The N2O decomposition efficiencies achieved with three perovskite-type catalysts rank in the order of La2NiO4 > LaSrNiO4 > La0.7Ce0.3SrNiO4 for the same operating condition.
Figure 4. Activities of La2NiO4, LaSrNiO4, and La0.7Ce0.3SrNiO4 for N2O decomposition at various temperatures ( = 0.1%, GHSV = 8,000 hr−1, He as carrier gas).
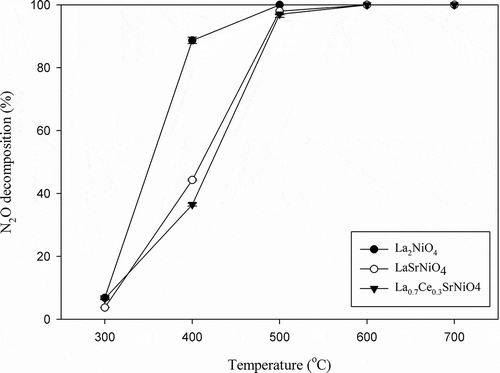
Table 3. Temperature for 50% and 90% N2O conversion
O2-TPD results show that desorption peak of β oxygen of La2NiO4 mainly appeared at temperatures higher than 790ºC. As previously mentioned, oxygen adsorption of La2NiO4 is strong and not easy to desorb at temperature lower than 790ºC. Inhibition of N2O decomposition by oxygen can be explained by the reversible dissociative adsorption of oxygen, that is, the back reaction of eq 2, to block active sites of catalyst, or via molecular adsorption of oxygen, as proposed in reaction 6 (Kapteijn et al., Citation1996):
On the other hand, activity of LaSrNiO4 is slightly higher than that of La0.7Ce0.3SrNiO4 at 400–500ºC for N2O decomposition. The areas of β oxygen of both catalysts indicate that the number of oxygen vacancies may decrease when Ce partially substitutes for the A site of LaSrNiO4, as shown in . However, N2O decomposition efficiencies achieved with three catalysts selected are close to 100% at 500ºC. Alini et al. (Citation2007) reported that N2O decomposition efficiencies achieved with LaMn0.7Cu0.3O3 and LaFe0.7Cu0.3O3 are less than 10% and 38%, respectively, at 500ºC with the GHSV of 22,100 hr−1. Dacquin et al. (Citation2009) applied LaCoO3 to decompose N2O at 600ºC with a space velocity of 10,000 hr−1, and indicate that N2O decomposition efficiency is 50%. Kumar et al. (Citation2012) studied the catalytic activity of Pr0.8Ba0.2O3 for N2O decomposition and reported that 92% of N2O decomposition efficiency could be achieved at 500ºC, with a GHSV of 7,500 hr−1.
Previous studies indicate that the perovskite-type catalysts have to be operated at a temperature higher than 700ºC for high N2O decomposition (Kapteijn et al., Citation1996), Overall, three catalysts selected (La2NiO4, LaSrNiO4, and La0.7Ce0.3SrNiO4) indeed show better performance for N2O decomposition at 500–600ºC compared with the catalysts tested in literature.
Effect of Sr and Ce on activity and properties of La2NiO4
The information on ion valence of catalyst is important for better understanding the N2O decomposition. As reported earlier, the La cation generally is trivalent (La3+) at the A site for perovskite-type catalysts, while Ni ion mainly presents as Ni2+ in La2NiO4. As La3+ at the A site is partially replaced by a lower valency cation (Sr2+), according to the principle of electroneutrality, the positive charge reduced could be balanced either via formation of higher oxidation state ion at the B site, that is, Ni2+ → Ni3+, or via formation of an oxygen vacancy (Vo). Thus, the mixed oxidation state of Ni and the presence of oxygen vacancy in LaSrNiO4 could be explained by reactions 7 and 8 (Zhao et al., Citation1996):
On the other hand, valence of Ni cannot be measured for La0.7Ce0.3SrNiO4, because valence of Ce is alterable (trivalent and tetravalent). As mentioned earlier, valence of Ni for La2NiO4 is mainly Ni2+, while it presents as Ni3+ in LaSrNiO4. From the formula of LaSrNiO4, La1−xCexSrNiO4, and La2NiO4 (according to the valence of Sr2+ and Ce4+), the amount of Ni3+ for La1−xCexSrNiO4 is between those of LaSrNiO4 and La2NiO4. Hence, it is clear that Ni3+ amount of LaSrNiO4 decreases as the high-valence cation (Ce4+) substitutes with the frame of La3+ of LaSrNiO4. Ladavos and Pornonis (Citation1991) plotted the reaction rate per unit area as a function of the content of Ni2+ to Ni3+, and indicate that the maximum activity appears in two cases, with one appearing at a low ratio of Ni2+ and Ni3+, implying that Ni exists in the Ni3+ state. In this case, the electronic configuration of the Ni ion (Ni3+, d7) should favor reaction. The other one is when the ratio of Ni2+ and Ni3+ is close to 1, and in such conditions, the catalyst possesses a higher percentage of “Ni2+-[ ]-Ni3+” pairs, which are active sites. This favors the occurrence of the surface reaction, resulting in higher activity (Ladavos and Pornonis, Citation1991).
In this study, Ce in the frame of LaSrNiO4 is of the Ce4+ form. Therefore, it decreases the content of Ni3+; furthermore, the ratio of Ni2+ and Ni3+ may be close to 1 by changing Ce4+. Generally, the active sites of perovskite-type catalysts modified are composed of oxygen vacancy for N2O decomposition, described as “Ni2+-[ ]-Ni3+”. However, for the La0.7Ce0.3SrNiO4 case, there may be a new complex active site designated as Ce3+-(O)-(Ni2+-[ ]-Ni3+) with the addition of Ce, of which the valence is alterable (Ce4+ ⇔ Ce3+). Hence, higher activity can be expected as the La site is replaced by the higher valency cation (Ce). Thus, the Ce in the La site would take part in the reaction and is one part of the active sites. In this study, the participation of Ce in N2O decomposition reaction may be described as (Zhu et al., Citation2005):
The results just described indicate that properties of La2NiO4 are significantly improved by doping with Sr and Ce; hence, the modified catalysts (LaSrNiO4 and La0.7Ce0.3SrNiO4) are mainly applied for the follow-up experimental tests.
Influence of space velocity on N2O decomposition
shows the effects of contact time on N2O decomposition achieved with LaSrNiO4 and La0.7Ce0.3SrNiO4, respectively. The results indicate that decomposition efficiency of N2O decreased slightly with increasing space velocity at 300–500ºC for both catalysts. Since the BET surface areas of LaSrNiO4 and La0.7Ce0.3SrNiO4 are only 5.0 and 5.2 m2/g, respectively, N2O decomposition reaction with a larger space velocity might be reduced due to diffusion limitations of reactant to the active site. However, N2O decomposition efficiencies achieved with both catalysts reached 100% at 600ºC, implying that activities of both catalysts are relatively high at T ≥ 600ºC. According to the reaction kinetics, the reaction rate increases with increasing temperature, because kinetic energy of pollutant molecule is enhanced as temperature is increased; furthermore, this can promote the chance of pollutant collision with catalyst.
Figure 5. Influences of gas residence time on activities of LaSrNiO4, and La0.7Ce0.3SrNiO4, respectively, at various temperatures ( = 0.1%, He as carrier gas).
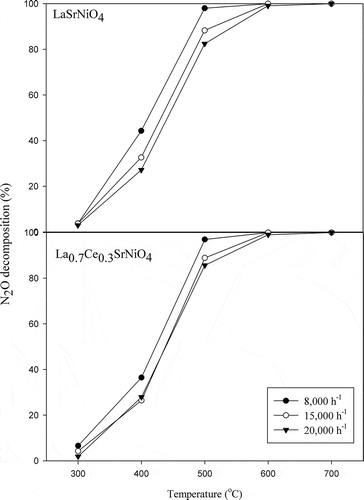
Overall, N2O decomposition efficiency achieved with LaSrNiO4 is affected slightly by decreasing residence time at temperatures less than 600ºC compared with La0.7Ce0.3SrNiO4.
Influence of O2 and water vapor on N2O decomposition activity
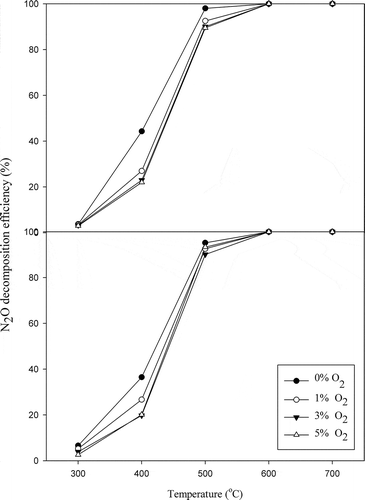
Generally, oxygen and water vapor exist in most flue gases, and their influences should be considered for field application. A catalyst is successfully evolved if it can effectively tolerate oxygen and water vapor. However, the suppression of N2O decomposition by oxygen remains one of the important obstacles to overcome in the effective N2O decomposition. Inhibition of catalytic activity by oxygen has been described in the previous section. However, as the oxygen removal from the catalyst surface is slow, oxygen inhibition may be significant (Hussain et al., Citation2014). The effects of oxygen content on the N2O decomposition over LaSrNiO4 and La0.7Ce0.3SrNiO4, respectively, are presented in . With 1–5% oxygen in the gas stream, the efficiencies of N2O decomposition decrease slightly for both catalysts if compared with the case without oxygen at low temperatures (300–500ºC). For LaSrNiO4, N2O decomposition efficiency decreases from 44% to 27% at 400ºC when 1% oxygen is introduced into the gas stream, while activity of La0.7Ce0.3SrNiO4 decreases from 37% to 27%. As oxygen content is further increased to 5%, N2O decomposition efficiencies achieved with LaSrNiO4 and La0.7Ce0.3SrNiO4 decrease to 22 and 20%, respectively. However, influence of O2 content gradually decreases with increasing temperature for both catalysts, and the N2O decomposition efficiencies achieved with both catalysts even reached 100% at 600ºC. The same trends also appear in the space velocity test, implying that both catalysts show excellent performance for N2O decomposition when operated at a temperature higher than 600ºC. On the other hand, activities of both catalysts for N2O decomposition would be substantially affected by oxygen at 400ºC.
Figure 6. Effects of oxygen contents on N2O decomposition at various temperatures ( = 0.1%,
= 1–5%, GHSV = 8,000 hr−1, He as carrier gas).
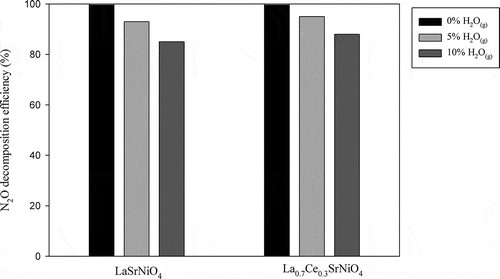
In order to investigate the effect of water vapor on N2O decomposition, 5 and 10% H2O(g) are introduced into system, respectively. shows the effect of 5 and 10% H2O(g) on N2O decomposition achieved with LaSrNiO4 and La0.7Ce0.3SrNiO4, respectively, at 600ºC. The results indicate that N2O decomposition efficiencies decrease from 100% to 94 and 95% as 5% H2O(g) is incorporated for LaSrNiO4 and La0.7Ce0.3SrNiO4, respectively. As H2O(g) is further increased to 10%, the N2O decomposition efficiencies achieved with LaSrNiO4 and La0.7Ce0.3SrNiO4 decrease to 86% and 88%, respectively. It is clear that activities of both catalysts suffer from H2O(g). Tofan et al. (Citation2002b) suggested that H2O(g) is easily adsorbed on all metal oxide catalysts, including perovskite-type catalysts used in this study, and H2O(g) adsorbed on catalyst surface causes a reduction of active sites for N2O decomposition. Another possibility is that the existence of H2O(g) may induce hydroxylation on catalyst surface and reduce active site density. However, the N2O decomposition efficiency achieved with LaSrNiO4 still maintains at 86%, while La0.7Ce0.3SrNiO4 maintains close to 90% at 10% H2O(g). Overall, both catalysts show good tolerance for H2O(g) when operated at 600ºC.
Figure 7. Effects of H2O(g) contents on N2O decomposition at 600ºC ( = 0.1%,
= 5–10%, GHSV = 8,000 hr−1, He as carrier gas).
Product analysis
summarizes N2, NO, and NO2 yields for three selected catalysts. The results indicate that NO and NO2 are not produced during N2O decomposition, and it demonstrates that N2O is purely decomposed into N2 and O2. N2 yield clearly depends on the temperature. Among the three catalysts, La2NiO4 shows the best N2O decomposition efficiency (88%) and the highest N2 yield (72%) at 400ºC. As temperature is increased to 700ºC, N2 yield achieved with La2NiO4 reached 90%. However, as shown in , the oxygen adsorbed by La2NiO4 is not easy to desorb; therefore, it may have a short lifetime, and its activity can also suffer from O2 seriously. On the other hand, N2 yield with LaSrNiO4 increases from 28% to 92% as temperature is increased from 400 to 700ºC, while N2 yield is increased from 30% to 100% as Ce is added into LaSrNiO4. Indeed, the activity of LaSrNiO4 at high temperatures can be improved by doping with Ce. Since La0.7Ce0.3SrNiO4 possesses better mobility of lattice oxygen, it promotes desorption of O2 and regeneration of the active sites.
Table 4. Products analysis and efficiencies of N2O decomposition achieved with three catalysts at various temperatures
Furthermore, with 5% oxygen in the gas stream, N2 yields are 28–99% at 400–700ºC for Ce improved catalyst, implying that it is not substantially affected by oxygen content when operated at 700ºC. In addition, N2 yields for both catalysts (LaSrNiO4 and La0.7Ce0.3SrNiO4) are only slightly reduced (82% to 80% and 93% to 89%, respectively) as 5% H2O(g) is introduced into the gas streams.
Durability test
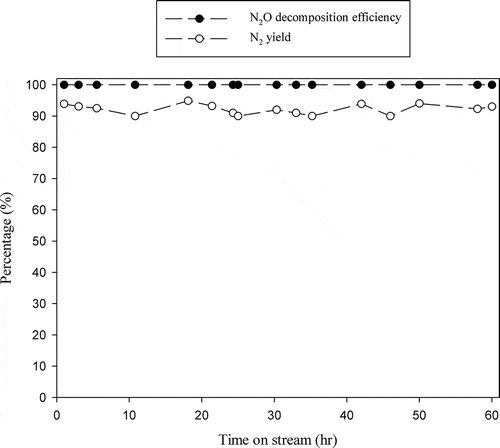
The durability of catalysts is important in determining their practical usefulness. indicates that N2O decomposition efficiency achieved with La0.7Ce0.3SrNiO4 maintains 100% at 600ºC, and no significant deactivation is observed for continuous operation for 60 hr, while N2 yield is between 90 and 94% for the durability test.
Figure 8. Durability test on the La0.7Ce0.3SrNiO4 catalyst for N2O decomposition ( = 0.1%, GHSV = 8,000 hr−1, reaction temperature = 600ºC, He as carrier gas).
Conclusion
Development of an effective N2O control technology is essential to reduce the environmental impact associated with N2O. In this study, we not only evaluate the effects of various parameters on N2O decomposition, but also explain oxygen mobility in bulk by doping various cations. Additionally, yields of N2 with different catalysts are also investigated.
The N2O decomposition efficiencies achieved with three perovskite-type catalysts rank in the order La2NiO4 > LaSrNiO4 > La0.7Ce0.3SrNiO4 for lower temperature operating conditions. However, La2NiO4 may have a shorter lifetime due to poisoning by oxygen. Indeed, physicochemical properties of La2NiO4 for N2O decomposition are significantly improved by partially substituting La with Sr. The mobilities of lattice oxygen with three perovskite-type catalysts are in the order La0.7Ce0.3SrNiO4> LaSrNiO4 >La2NiO4. Additionally, activity of LaSrNiO4 can be improved by doping Ce for N2 yield. Compared to previous studies using other compounds to dope La, reasonable N2O decomposition efficiencies (97–100%) are measured at lower temperatures (500-600ºC) for these three catalysts.
Moreover, contents of oxygen and water vapor are introduced into the reaction system to evaluate the effects, and La0.7Ce0.3SrNiO4 shows better tolerance for oxygen and water vapor, implying that the catalyst has a new highly active site Ce3+-(O)-(Ni2+-[ ]-Ni3+) due to the addition of Ce, which may facilitate oxygen mobilization and desorption at low temperature (˜300ºC). However, more studies are needed to further enhance the activity of these catalysts and to improve their tolerance to the complex composition for industrial application.
Additional information
Notes on contributors
Kuan Lun Pan
Kuan Lun Pan is a Ph.D. student and Moo Been Chang is a distinguished professor at the Graduate Institute of Environmental Engineering, National Central University, Chungli, Taiwan.
Sheng Jen Yu
Sheng Jen Yu and Shaw Yi Yan are researchers at the Industrial Technology Research Institute (ITRI), Hsinchu, Taiwan.
Shaw Yi Yan
Sheng Jen Yu and Shaw Yi Yan are researchers at the Industrial Technology Research Institute (ITRI), Hsinchu, Taiwan.
Moo Been Chang
Kuan Lun Pan is a Ph.D. student and Moo Been Chang is a distinguished professor at the Graduate Institute of Environmental Engineering, National Central University, Chungli, Taiwan.
References
- Alini, S., F. Basile, S. Blasioli, C. Rinaldi, and A. Vaccari. 2007. Development of new catalysts for N2O-decomposition from adipic acid plant. Appl. Catal. B: Environ. 70:323–329. doi:10.1016/j.apcatb.2005.12.031
- Belessi, V.C., A.K. Ladavos, and P.J. Pomonis. 2001. Methane combustion on La-Sr-Ce-FeO mixed oxides: bifunctional synergistic action of SrFeO3-x and CeOx phases. Appl. Catal. B Environ. 31:183–194. doi:10.1016/S0926-3373(00)00279-4
- Dacquin, J.P., C. Lancelot, C. Dujardin, P. Da Costa, G. Djega-Mariadassou, P. Beaunier, S. Kaliaguine, S. Vaudreuil, S. Royer, and P. Granger. 2009. Influence of preparation methods of LaCoO3 on the catalytic performances in the decomposition of N2O. Appl. Catal. B Environ. 91:596–604. doi:10.1016/j.apcatb.2009.06.032
- Granger, P., P. Esteves, S. Kieger, L. Navascues, and G. Leclercq. 2006. Effect of yttrium on the performances of zirconia based catalysts for the decomposition of N2O at high temperature. Appl. Catal. B Environ. 62:236–243. doi:10.1016/j.apcatb.2005.07.015
- Hernández-Enríquez, J.M., R. Silva-Rodrigo, R. García-Alamilla, L. A. García-Serrano, B.E. Handy, G. Cárdenas-Galindo, A. Cueto-Hernández. 2012. Synthesis and physico-chemical Characterization of CeO2/ZrO2-SO42- mixed oxides. J. Mex. Chem. Soc. 56:115–120.
- Hinshelwood, C.N., and C.R. Prichard. 1925. LI.—A comparison between the homogeneous thermal decomposition of nitrous oxide and its heterogeneous catalytic decomposition on the surface of platinum. J. Chem. Soc. Trans. 127:327–336. doi:10.1039/ct9252700327
- Hussain, M., D. Fino, and N. Russo. 2014. Development of modified KIT-6 and SBA-15-spherical supported Rh catalysts for N2O abatement: From powder to monolith supported catalysts. Chem. Eng. J. 238:198–323. doi:10.1016/j.cej.2013.06.032
- Imanaka, N., and T. Masui. 2012. Advances in direct NOx decomposition catalysts. Appl. Catal. A Gen. 431– 432:1–8. doi:10.1016/j.apcata.2012.02.047
- Jacob, D.J. 1999. Introduction to Atmosphere Chemistry. Princeton, NJ: Princeton University Press.
- Kapteijn, F., J. Rodriguez-Mirasol, and J.A. Moulijn. 1996. Heterogeneous catalytic decomposition of nitrous oxide. Appl. Catal. B: Environ. 9:25–64. doi:10.1016/0926-3373(96)90072-7
- Kirchnerova, J., M. Alifanti, and B. Delmon. 2002. Evidence of phase cooperation in the LaCoO3-CeO2-Co3O4 catalytic system in relation to activity in methane combustion. Appl. Catal. A Gen. 231:65–80. doi:10.1016/S0926-860X(01)00903-6
- Kirchnerova, J., and D. Klvana. 1994. Preparation and characterization of high surface perovskite electrocatalysts. Int. J. Hydrogen Energy. 19:501–506 doi:10.1016/0360-3199(94)90004-3
- Klvana, D., J. Vaillancourt, J. Kirchnerova, and J. Chaouki. 1994. Combustion of methane over La0.66Sr0.34Ni0.3Co0.7O3 and La0.4Sr0.6Fe0.4Co0.6O3 prepared by freeze-drying. Appl. Catal. A: Gen. 109:181–193. doi:10.1016/0926-860X(94)80117-7
- Kramlich, J.C., and W.P. Linak. 1994. Nitrous oxide behavior in the atmosphere, and in combustion and industrial systems. Prog. Energy Combust. Sci. 20:149–202. doi:10.1016/0360-1285(94)90009-4
- Kumar, S., A. Vinu, J. Subrt, S. Bakardjieva, S. Rayalu, Y. Teraoka, and N. Labhsetwar. 2012. Catalytic N2O decomposition on Pr0.8Ba0.2MnO3 type perovskite catalyst for industrial emission control. Catal. Today 198: 125–132. doi:10.1016/j.cattod.2012.06.015
- Ladavos, A.K., and P.J. Pomonis. 1991. Comparative study of the solid-state and catalytic properties of La2–xSrxNiO4λ perovskites (x = 0.00–1.50) prepared by the nitrate and citrate methods. J. Chem. Soc. Faraday Trans. 87: 3291–3297. doi:10.1039/ft9918703291
- Li, Y., and J.N. Armor. 1992. Catalytic decomposition of nitrous oxide on metal exchanged zeolites. Appl. Catal. B: Environ. 1:L21–L29. doi:10.1016/0926-3373(92)80019-V
- Lintz, H.-G., and L. Riekert. 1984. The decomposition of nitrous oxide on polycrystalline platinum. J. Catal. 88:244–245. doi:10.1016/0021-9517(84)90072-1
- NASDAQ (National Association of Securities Dealers Automated Quotations) System, USA. 2014. http://www.nasdaq.com ( accessed June 15, 2014).
- Nitadori, T. and M. Misono. 1985. Catalytic properties of La1-xA′xFeO3 (A′ = Sr, Ce) and La1 −xCexCoO3. J. Catal. 93:459–466. doi:10.1016/0021-9517(85)90193-9
- Pannov, G.I., V.I. Sobolev, and A.S. Kharitonov. 1990. The role of iron in N2O decomposition on ZSM-5 zeolite and reactivity of the surface oxygen formed. J. Mol. Catal., 61:85–97. doi:10.1016/0304-5102(90)85197-P
- Pechini, M.P., 1967. Method of preparing lead and alkaline earth titanates and niobates and coating method using the same to form a capacitor. U.S. Patent 3,330,697.
- Perez-Alonso, F.J., I. Melián-Cabrera, M. López Granados, F. Apteijn, and J.L.G. Fierro. 2006. Synergy of FexCe1−xO2 mixed oxides for N2O decomposition. J. Catal., 239:340–346.
- Ramanujachary, K.V., N. Kameswari, and C.S. Swamy. 1984. Studies on the catalytic decomposition of N2O on rare earth cuprates of the type Ln2CuO4. J. Catal. 86:121–128. doi:10.1016/0021-9517(84)90353-1
- Ramanujachary, K.V., K.M. Vijayakumar, and C.S. Swamy. 1982. Studies on the catalytic decomposition of N2O on rare earth nickelates. React. Kin. Catal. Lett. 20:233–238. doi:10.1007/BF02063615
- Rodhe, H. 1990. A Comparison of the contribution of various gases to the greenhouse effect. Science 248:1217–1219. doi:10.1126/science.248.4960.1217
- Russo, N., D. Fino, G. Saracco, and V. Specchia. 2007. N2O catalytic decomposition over various spinel-type oxides. Catal. Today 119:228–232. doi:10.1016/j.cattod.2006.08.012
- Russo, N., D. Mescia, D. Fino, G. Saracco, and V. Specchia. 2007. N2O decomposition over perovskite catalysts. Ind. Eng. Chem. Res. 46:4226–4231. doi:10.1021/ie0612008
- Siva Sankar Reddy, P., N. Pasha, M.G.V. Chalapathi Rao, N. Lingaiah, I. Suryanarayana, and P.S. Sai Prasad. 2007. Direct decomposition of nitrous oxide over Ru/Al2O3 catalysts prepared by deposition–precipitation method. Catal. Commun. 8:1406–1410. doi:10.1016/j.catcom.2006.11.039
- Sobolev, V.I., G.I. Panov, A.S. Kharitonov, V.N. Romannikov, A.M. Volodin, and K.G. Ione. 1993. Catalytic properties of ZSM-5 zeolites in N2O decomposition: The role of iron. J. Catal. 139:435–443. doi:10.1006/jcat.1993.1038
- Tabata, K., and M. Misono. 1990. Elimination of pollutant gases—Oxidation of CO, reduction and decomposition of NO. Catal. Today. 8:249–261. doi:10.1016/0920-5861(90)87021-T
- Tofan, C., D. Klvana, and J. Kirchnerova. 2002a. Decomposition of nitric oxide over perovskite oxide catalysts: Effect of CO2, H2O and CH4. Appl. Catal. B: Environ. 36:311–323. doi:10.1016/S0926-3373(01)00312-5
- Tofan, C., D. Klvana, and J. Kirchnerova. 2002b. Direct decomposition of nitric oxide over perovskite-type catalysts: Part I. Activity when no oxygen is added to the feed. Appl. Catal. A: Gen. 223:275–286. doi:10.1016/S0926-860X(01)00764-5
- Van den Brink, R.W., S. Booneveld, J.R. Pels, D.F. Bakker, and M.J.F.M. Verhaak. 2001. Catalytic removal of N2O in model flue gases of a nitric acid plant using a promoted Fe zeolite. Appl. Catal. B Environ. 32:73–81. doi:10.1016/S0926-3373(00)00294-0
- Voorhoeve, R.J.H., J.P. Remeika, P.E. Freeland, and B.T. Matthias. 1972. Rare-earth oxides of manganese and cobalt rival platinum for the treatment of carbon monoxide in auto exhaust. Mater. Res. Bull. 177:353–354. doi:10.1126/science.177.4046.353
- Wang, J., H. Yasuda, K. Inumaru, and M. Misono. 1995. Catalytic decomposition of dinitrogen oxide over perovskite-related mixed oxides. Bull. Chem. Sot. Jpn. 68:1226–1231. doi:10.1246/bcsj.68.1226
- Winter, E.R.S. 1974. The decomposition of N2O on oxide catalysts: III. The effect of O2. J. Catal. 34(3):431–439. doi:10.1016/0021-9517(74)90056-6
- Wójtowicz, M.A., J.R. Pels, and J.A. Moulijn. 1993. Combustion of coal as a source of N2O emission. Fuel Proc. Technol. 34:1–71. doi:10.1016/0378-3820(93)90061-8
- Zhao, Z., X. Yang, and Y. Wu. 1996. Comparative study of Nickel-based perovskite-like mixed oxide catalysts for direct decomposition of NO. Appl. Catal. B: Environ. 8: 28l–297. doi:10.1016/0926-3373(95)00067-4
- Zhu, J., D. Xiao, J. Li, X. Yang, and Y. Wu. 2005. Effect of Ce on NO direct decomposition in the absence/presence of O2 over La1−xCexSrNiO4 (0 ≤ x ≤ 0.3). J. Mol. Catal. A Chem. 234:99–105. doi:10.1016/j.molcata.2005.02.015